

Summary
The mechanisms that enable immune evasion at metastatic sites are poorly understood. We show that the Polycomb Repressor Complex 1 (PRC1) drives colonization of the bones and visceral organs in Double-Negative Prostate Cancer (DNPC). In vivo genetic screening identifies CCL2 as the top prometastatic gene induced by PRC1. CCL2 governs self-renewal and induces the recruitment of M2-like tumor-associated macrophages and regulatory T cells, thus coordinating metastasis initiation with immunosuppression and neoangiogenesis. A catalytic inhibitor of PRC1 cooperates with immune checkpoint therapy to reverse these processes and suppress metastasis in genetically-engineered mouse transplantation models of DNPC. These results reveal that PRC1 coordinates stemness with immune evasion and neoangiogenesis and point to the potential clinical utility of targeting PRC1 in DNPC.
Keywords: PRC1, DNPC, metastasis, epigenetic regulation, tumor microenvironment, double checkpoint immunotherapy
Graphical Abstract
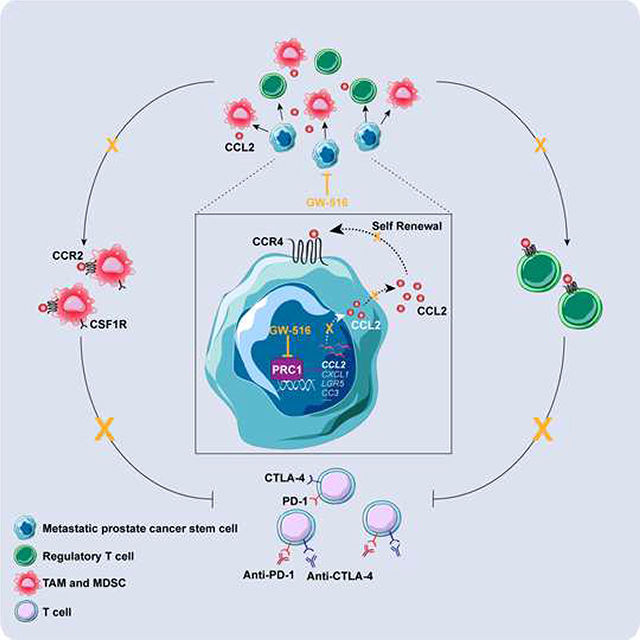
Introduction
Cancer cells exploit several mechanisms to evade destruction by the immune system and resist to therapy. However, it is unclear if and to what extent these mechanisms operate also during metastatic colonization of distant organs. Separate lines of inquiry have documented a role for stemness, encompassing both self-renewal and aberrant differentiation, and immune evasion in metastatic colonization (Giancotti, 2013; Gonzalez et al., 2018). However, it is not known if a common regulatory mechanism orchestrates both functions.
In prostate cancer, resistance to hormone deprivation therapy is intimately linked to the development of metastasis. Potent androgen receptor (AR) signaling inhibitors, such as enzalutamide and abiraterone, can induce durable responses in a fraction of metastatic castration-resistant prostate cancer (M-CRPC) patients. However, the remainder exhibits a transient and often partial response or are completely insensitive to these agents (Attard et al., 2016). Inactivation of TP53 and exposure to abiraterone or simultaneous inactivation of TP53 and RB1 can reprogram prostate adenocarcinomas to AR-negative neuroendocrine prostate cancer (NEPC) in mice (Mu et al., 2017; Zou et al., 2017). Moreover, experiments with LNCaP-AR cells have specifically implicated the Polycomb Repressor Complex 2 (PRC2, comprising EED, EZH2 and SUZ12) in transdifferentiation to NEPC and resistance to enzalutamide (Ku et al., 2017). However, as the use of abiraterone and enzalutamide has become widespread, the incidence of AR pathway-negative M-CRPC devoid of neuroendocrine traits (Double-Negative Prostate Cancer, DNPC) has risen substantially, highlighting the need to understand the origin and therapeutic vulnerabilities of these cancers (Bluemn et al., 2017).
Canonical and non-canonical variants of PRC1 consist of several subunits, each encoded by multiple paralogs, and share the ability to promote monoubiquitination of histone H2A through their common catalytic subunit RNF2 (Wang et al., 2004). PRC1 complexes are defined by a core heterodimeric subcomplex, RING-PCGF, which induces monoubiquitination of histone H2A. Canonical PRC1 (cPRC1) comprises CBX, HPH and RING-PCGF and is targeted to chromatin through CBX, which recognizes the H3K27me3 mark created by PRC2. Non-canonical variants of PRC1 (ncPRC1) are targeted to chromatin through an interaction mediated by specific constituent subunits (Figure 1A). Often acting in tandem to silence target genes, PRC1 and PRC2 promote de-differentiation and stemness during development and in cancer (Schuettengruber et al., 2017). Mouse genetic studies have specifically implicated the cPRC1 component BMI1 in prostate development and susceptibility to malignant transformation (Lukacs et al., 2010). Here, we investigate the role of PRC1 in prostate cancer metastasis.
Figure 1. Activation of PRC1 in DNPC.

(A) cPRC1 and ncPRC1 complexes.
(B and C) Genetic aberrations of PRC1 and PRC2 components in primary and metastatic prostate cancers in the Grasso dataset (n=94) (Grasso et al., 2012) (B) and the SU2C dataset (n=118) (Robinson et al., 2015) (C).
(D) Similarity matrix of the expression of AR and its target genes (n=11), RNF2-regulated genes (n=20), and NE signature genes (n=10) in the SU2C dataset.
(E) Scatter plot of the expression of AR and NE signatures in M-CRPC samples (Kumar et al., 2016), prostate cancer cell lines, and PtenPC-/- and PtenPC-/-Smad4PC-/- tumors (Wang et al., 2015).
(F) Immunoblotting of the indicated cell lines.
See also Figure S1.
Results
Activation of PRC1 in DNPC
To examine the potential role of PRC1 in prostate cancer metastasis, we examined patient datasets including non-castrate primary and castrate metastatic samples. We found that the genes encoding several canonical and non-canonical components are selectively amplified or overexpressed in a large fraction of metastases but not in localized tumors in the Grasso et al. (2012) dataset (Figure 1B and S1A). Analysis of the SU2C/PCF (Robinson et al., 2015) and UCSF (Quigley et al. 2018) datasets, which include only castration-resistant metastatic samples, confirmed these findings. Of note, PRC1 components were altered more frequently than PRC2 components, including EZH2, in both datasets (Figure 1C and S1B). Consistently, analysis of a tissue microarray demonstrated that the levels of the core PRC1 components RNF2 and BMI1 are elevated in invasive and metastatic prostate cancers as compared to organ-confined primary tumors with no positive locoregional lymph nodes (Figure S1C).
To gauge the level of PRC1 activity across M-CRPC subtypes, we classified the SU2C/PCF dataset using previously defined AR_score and NE_score genesets (Bluemn et al., 2017). The results indicated that this dataset consists of 64% ARPC (AR-pathway positive prostate cancer), 12% NEPC, and 23% DNPC. These frequencies are similar to those observed in the nearly contemporaneous FHCRC necropsy series (Kumar et al., 2016). Similarity matrix clustering of the dataset revealed that the expression of a previously defined set of RNF2 target genes (Rai et al., 2015) negatively correlates with the expression of AR_score and the NE_score genes, indicating that the activation of PRC1 is largely confined to DNPC (Figure 1D).
Further analysis revealed that PRC1 activity correlates with Epithelial-to-Mesenchymal Transition (EMT) and stemness signatures in M-CRPC (Figure S1D). To study this connection, we examined a panel of prostate cancer cell lines, which we had molecularly classified as ARPC, NEPC, and DNPC based on GSEA (Figure 1E). Immunoblotting and qRT-PCR revealed that the PC3 and PC3M cells, which possess DNPC traits and metastatic potential, exhibit elevated levels of cPRC1 and ncPRC1 components as compared to other subtypes of prostate cancer cells (Figure 1F and S1E). Further analysis indicated that the AR- DU145, PC3 and PC3M cells do not express E-cadherin but express vimentin, suggesting that they had acquired mesenchymal traits (Figure 1F). In addition, they exhibited elevated levels of CD44 and of the β4 integrin (ITGB4) (Figure 1F, S1F and S1G). Of note, ITGB4 marks normal and neoplastic prostate stem cells and is upregulated in breast cancer cells that have acquired stemness features by entering into a hybrid Epithelial/Mesenchymal state (Yoshioka et al., 2013, Bierie et al., 2017). Consistently, we found that the PC3 and PC3M cells form a larger number of tumor spheres and invade through Matrigel in vitro more efficiently as compared to DU145 and LNCaP cells (Figure S1H). These results suggest that PRC1 components are elevated in prostate cancer cells that exhibit mesenchymal and stem cell traits and are endowed with metastatic capacity.
PRC1 is required for tumor initiation and metastasi
To investigate the role of PRC1 in prostate cancer metastasis, we inactivated the E3 ligase RNF2 or the activating subunit BMI1 in PC3 cells. Depletion of RNF2 de-stabilized BMI1, as anticipated, but it did not reduce the expression of mesenchymal or stem cell markers (Figure 2A). Intracardiac injection (i.c.) experiments indicated that the PC3 cells colonize efficiently the bone, producing osteolytic lesions similar to those occurring in patients with AR-independent M-CRPC (Beltran et al., 2014). Depletion of RNF2 suppressed metastasis in this model (Figure 2B and 2C) as well as in mice inoculated i.c. with PC3M cells (Figure S2A).
Figure 2. PRC1 is required for tumor initiation and metastasis.

(A) Control or RNF2- and BMI1-silenced PC3 cells were subjected to immunoblotting.
(B) Representative images (top) and quantification of luciferase counts (bottom) of male nude mice injected intracardially (i.c.) with 5×105 luciferase labeled control and RNF2-silenced PC3 cells at week 4; Error bars, mean±SEM.
(C) H&E (top) and IHC (middle) staining of bone sections and X ray imaging of hindlegs (bottom) of mice in (B). Scale bar=50 μm. Dash oval indicates osteolytic lesion.
(D and E) Representative images (D) and quantification of luciferase counts (E) of male nude mice at 3 weeks after injected i.c. with 1×105 PtenPC-/-Smad4PC-/- cells expressing the indicated constructs. Bars, mean±SEM.
(F and G) Quantification (F) and representative images (G) of control and RNF2-silenced PC3 cells subjected to sphere assay at day 7. Scale bar=200 μm. Error bars, mean±SD of triplicate experiments, **** p<0.0001 two-tailed Student t test.
(H) Control and BMI1-silenced PC3 cells were subjected to sphere assay and the results quantified at day 7. Bars, mean±SD.
(I) Control and RNF2-silenced PC3 cells were transfected with empty, WT Rnf2-, or Ring domain-truncated Rnf2-expressing vector and subjected to sphere assay. Bars, mean±SD, n.s.: not significant.
(J and K) Quantification (J) and representative images (K) of control and RNF2-silenced PC3 cells subjected to 3D growth for 14 days. Error bars, mean±SD. Scale bar=200 μm.
(L and M) Quantification of Ki67 and CC3 positive cells (L) and representative images (M) of PC3 cells from 3D growth. Error bars, mean±SD. Scale bar=50 μm.
(N) Tumor volumes of control and RNF2-silenced PC3 cells injected subcutaneously in NOG mice as indicated. Bars, mean±SD, *** p < 0.001, **** p < 0.0001.
See also Figure S2.
To confirm and extend these results, we developed a genetically engineered transplantation model of DNPC metastasis. Transcriptomic analysis indicated that the tumors arising in Ptenpc-/- mice cluster between ARPC and DNPC samples, whereas the invasive and potentially metastatic tumors from Pten pc-/-Smad4pc-/- mice (Ding et al., 2011) overlap with DNPC (Figure 1E). In agreement with our findings and their DNPC nature, Ptenpc-/-Smad4pc-/- tumors exhibited higher expression of mesenchymal and stem cell transcripts as compared to Ptenpc-/- tumors (Figure S2B). Moreover, late stage tumors from Ptenpc-/-Smad4pc-/- mice consisted of large areas of AR- and SYP- DNPC and smaller areas of AR+ adenocarcinoma, consistent with progression from ARPC to DNPC in this model (Figure S2C). Transcriptional analysis and immunoblotting further indicated that tumor cells isolated from these mice exhibit DNPC features (Figure 1E, S1F and S2D). Importantly, whereas Pten pc-/-Smad4pc-/- cells rapidly produced bone and liver metastases in syngeneic FVB/NJ mice upon i.c. injection, those depleted of RNF2 exhibited reduced capacity to metastasize to these organs (Figure 2D, 2E and S2E). Of note, analysis of the SU2C (Robinson et al., 2015) and UCSF (Quigley et al. 2018) datasets indicated that DNPC metastases are prevalent in bone and liver but not in other distant organs (Figure S2F), indicating that the Ptenpc-/-Smad4pc-/- transplantation model mimics the organotropism of human DNPC. These findings indicate that inactivation of PRC1 inhibits metastasis in a genetically engineered transplantation model of DNPC.
To further corroborate the role of PRC1 in metastasis, we tested the RM1 cells, which are derived from v-HRas v-gag-Myc transgenic prostatic tumors and exhibit activation of signaling pathways and transcriptional programs prevalent in DNPC (Power et al., 2009; Thompson et al., 1989) (Figure 1E, S1F and S2D). Depletion of RNF2 inhibited their capacity to colonize multiple organs upon i.c. injection (Figure S2G–S2I) and the bone upon intra-femoral artery injection, confirming that inactivation of Rnf2 can suppress bone colonization even when the tumor cells are directly targeted to the bone (Figure S2J and S2K). These findings confirm that PRC1 is required for metastatic initiation and outgrowth in the bone and visceral organs across transplantation models.
Given the connection between stemness and metastasis initiation, we evaluated if PRC1 promotes metastasis by regulating stemness capacity. Indicative of a role for PRC1 in self-renewal, depletion of RNF2 or BMI1 suppressed the ability of PC3, Ptenpc-/-Smad4pc-/-, and RM1 cells to form tumor spheres (Figure 2F–2H and S2L). To more accurately determine if PRC1 affects self-renewal in vitro, we stained control and RNF2-silenced cells with PKH-26 and subjected them to serial sphere assay. Consistent with the notion that the slowly cycling, label-retaining cells possess the highest self-renewal capacity (Cicalese et al., 2009), the PKHHIGH tumor cells formed a higher number of tumor spheres at each passage as compared to the PKHPOS and PKHNEG tumor cells. Silencing of RNF2 inhibited sphere formation at each passage (Figure S2M and S2N). Rescue experiments with a WT or a Ring domain deleted-RNF2 demonstrated a requirement for RNF2 catalytic activity for sphere formation (Figure 2I). Since silencing of neither RNF2 nor BMI1 reduced CD44 or ITGB4 expression (Figures 2A, S2E, and S2O), we infer that PRC1 promotes self-renewal but is not required for the specification of cancer stem cells or the expression of these markers.
Control experiments indicated that RNF2 depletion does not affect the proliferation of Ptenpc-/-Smad4pc-/-, RM1 or PC3 cells in standard culture (Figure S2P and S2Q). This latter result does not contradict the observation that inactivation of RNF2 inhibits LNCaP cell proliferation by stabilizing p53 (Su et al., 2013) because the PC3 and RM1 cells are TP53 mutant and the Pten pc-/-Smad4pc-/- cells do not exhibit detectable p53 (Figure S2E and S2R–S2T). These results suggest that PRC1 promotes metastasis in the context of loss of TP53, which has been linked to metastasis in genomic studies of human prostate cancer (Turajlic and Swanton, 2016).
To further investigate the role of PRC1 in prostate cancer stemness, we examined PC3 cells placed in 3D Matrigel. Whereas control PC3 cells formed invasive outgrowths in 14 days, the RNF2-depleted cells formed abortive structures containing a large fraction of apoptotic cells (Figure 2J–2M). Finally, tumor initiation experiments showed that RNF2 depletion inhibited tumor outgrowth when limiting numbers of tumor cells were inoculated in mice, and this effect was also linked to increased apoptosis (Figure 2N; p<0.01). Based on these results, we conclude that PRC1 sustains multiple stem cell traits in DNPC cells.
PRC1 promotes the expression of CCL2 and other pro-metastatic genes
To examine the mechanism through which PRC1 regulates the acquisition of stemness and metastatic traits, we first conducted RNA sequencing studies and found that depletion of RNF2 modifies the expression of about 500 genes by >1.0 log2 fold in PC3 cells, with 49% reduced and 51 % increased (Table S1). We then performed ChIP-seq analysis for RNF2 (cPRC1 and ncPRC1), BMI1 and PHC2 (cPRC1), and KDM2B (ncPRC1.1) and integrated the results with the known occupancy data for the repressive histone mark H3K27me3 and the activation mark H3K4me3 (GSE57498) in PC3 cells (Taberlay et al., 2014). Hierarchical clustering of RNF2-induced and suppressed genes based on H3K27me3 and H3K4me3 promoter occupancy yielded two subsets in each class (Figure 3A). Amongst the top 100 induced genes, 42% were found in cluster 1 and 58% in cluster 2, and amongst the top 100 repressed genes, 33% in cluster 3 and 67% in cluster 4 (Figure S3A). Cluster 1 and 3 genes were constitutively expressed at higher levels as compared to cluster 2 and 4 (Figure 3B). In spite of their divergent direction of regulation by RNF2, the promoters of cluster 1 and 3 genes were characterized by a higher level of the H3K4me3 as compared to those of 2 and 4. However, cluster 1 promoters, which were induced by RNF2, exhibited a lower level of H3K27me3 and of KDM2B as compared to cluster 3, consistent with a repressive role for KDM2B (Figure 3A–3D). In contrast, cluster 2 and 4 promoters were characterized by lower levels of RNF2 occupancy and both H3K27me3 and H3K4me3 as compared to 1 and 3 (Figure 3A, S3B and S3C).
Figure 3. PRC1 promotes the expression of CCL2 and other pro-metastatic genes.

(A) Hierarchical clustering of H3K4me3 and H3K27me3 read densities (Taberlay et al., 2014) across the promoters of RNF2-induced or repressed genes and the heatmap of the read densities of indicated histone modifications and PRC1 subunits.
(B) Global analysis of gene expression in each cluster in control and RNF2-silenced PC3 cells. A standard boxplot was applied to display the z-score of the value of “fragments per kilobase of transcript per million mapped reads” (FPKM) based on a 5-number summary (“minimum”, first quartile (Q1), median, third quartile (Q3), and “maximum”). The median value is depicted as a line splitting the box in half. The middle “box” represents the middle 50% of scores for the group. The upper and lower whickers represent scores outside the middle 50%. Outlier as points extending beyond the whiskers.
(C) Boxplot of the average read densities for H3K4me3, H3K27me3, RNF2 and KDM2B among the core promoters of genes in clusters 1 and 3. Standard boxplot was applied to display the distribution of the values of average reads densities based on a 5-number summary as described in (B).
(D) UCSC Genome Browser view of the H3K4me3, H3K27me3, H2AUb, RNF2, BMI1, PHC2 and KDM2B peaks for the indicated representative genes in clusters 1 and 3.
(E) GO enrichment and KEGG pathway analysis of genes differentially expressed upon RNF2 knockdown. Heatmap represents the top 29 genes from the ES pathway.
(F) ssGSEA score correlations of RNF2-activated geneset and EMT, AR, Stemness, NEPC geneset in the SU2C dataset (n=118).
(G) Representative bioluminescent images of male nude mice injected i.c. with 5×105 RNF2-silenced PC3 cells infected with the custom library sacrificed after 4 weeks.
(H) Heatmap of the library gene expression fold change in bone metastases from (G).
(I) Relative expression levels of CCL2 in control and RNF2- or BMI1-silenced PC3 and RM1 cells. Bars, mean±SD.
(J and K) Representative images (J) and quantification of luciferase counts (K) of male nude mice at 4 weeks after injected i.c. with 5×105 PC3 cells expressing the indicated constructs. Bars, mean±SEM.
(L) Survival analysis of mice from (J).
(M and N) Relative occupancy of RNF2, H2AK119Ub, H3K27me3, H3K9Ac, H3K27Ac and IgG control on the CCL2 (M) or ATF3 (N) promoter from ChIP-qPCR in control or RNF2-silenced PC3 cells; bars, mean±SD. * p < 0.05, ** p < 0.01, *** p < 0.005.
See also Figure S3.
Pathway analysis revealed that clusters 1 and 2 (induced by PRC1) are dominated by genes involved in cell adhesion and migration and genes belonging to the Extracellular Space (ES), which includes cytokines, components of the extracellular matrix, and their regulators. In contrast, cluster 3 and 4 (repressed by PRC1) comprised genes involved in metabolic pathways and genes belonging to the ES pathways, respectively (Figure S3D). Consistently, pathway analysis of the global gene expression program regulated by RNF2 indicated that a large majority of genes induced by PRC1 belong to the ES category (Figures 3E and S3E).
To validate the importance of the RNF2-dependent gene expression in prostate cancer, we examined patient datasets using a signature comprising upregulated or downregulated genes from the ES pathway (Table S2). Increased expression of the upregulated geneset correlated with poor disease-free survival in the TCGA and Taylor datasets (Cancer Genome Atlas Research, 2015; Taylor et al., 2010) (Figure S3F and S3G), but increased expression of the repressed geneset did not (TCGA p=0.217; Taylor p=0.25). GSEA indicated that the expression of RNF2-activated genes correlated positively with EMT and stemness signatures and negatively with the AR signature in the SU2C dataset (Figure 3F). This result suggests that the capacity of PRC1 to positively control gene expression is associated with the acquisition of mesenchymal and stem-like traits and progression to metastasis in DNPC.
To identify PRC1 target genes involved in metastasis, we conducted a focused genetic screen in vivo by injecting RNF2-silenced PC3 cells transduced with a pool of vectors encoding the ORFs of top 5 RNF2-activated genes and multiple shRNAs targeting the top 10 RNF2-repressed genes. Four out of 10 mice developed bone metastases in 4 weeks (Figure 3G). Tumor cells were isolated and subjected to qRT-PCR to identify the genes more consistently up- or down-regulated. Expression levels of the CC chemokine CCL2 were upregulated by about 5-fold from all 4 metastatic samples (Figure 3H). Other mediators that have been implicated in tumorigenesis and metastasis, including CXCL1, LGR5, LCN2 and C3 (Acharyya et al., 2012; Boire et al., 2017; de Lau et al., 2014; Jung et al., 2016), were not as highly or reproducibly up-regulated in these lesions as CCL2. Moreover, none of the repressed genes in the custom library scored positive in the screen. These findings suggest that CCL2 rescues metastatic capacity after silencing of RNF2, identifying this cytokine as the top pro-metastatic mediator of PRC1.
CCL2 and the second top ranked target, CXCL1, mediate recruitment of inflammatory monocytes and their conversion into myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs), which suppress immunity and promote angiogenesis and metastasis (Noy and Pollard 2014; Quail and Joyce, 2013). Moreover, both cytokines have been linked to bone colonization in prostate cancer (Loberg et al. 2007; Lu et al. 2009). qRT-PCR analysis of a panel of prostate cancer cells revealed that CCL2 mRNA levels were 50 folds higher in the PC3 and PC3M cells as compared to the ARPC LNCaP cells (Figure S3H). These elevations in CCL2 expression correlated positively with those in RNF2 expression but were larger, as anticipated from an inducer-target relationship. Silencing of RNF2 or BMI1 suppressed the expression of CCL2 in both PC3 and RM1 cells, consistent with the potential identification of CCL2 as a PRC1 target gene (Figure 3I). Silencing of PCGF1, PHC2 and KDM2B exerted a similar effect, suggesting a participation of the ncPRC1 complex KDM2B-PRC1 in the regulation of CCL2 (Figure S3I). Additional experiments indicated that depletion of RNF2, RING1A, PHC2 or KDM2B also suppresses the expression of CXCL1. As anticipated, ATF3, one of the downregulated genes, responded in opposite fashion (Figure S3J). Further analysis of the relative roles of cPRC1 and ncPRC1.1 in prostate cancer metastasis revealed that depletion of BMI1 suppresses bone colonization by PC3 cells, whereas inactivation of KDM2B almost completely blocks this process (Figure 3J and 3K). Survival analysis confirmed that silencing of KDM2B exhibits a more profound inhibitory effect on metastasis (Figure 3L). We speculate that the greater effect of KDM2B inactivation may at least in part result from its ability to attenuate cell growth (Figure S3K). These results suggest that both cPRC1 and ncPRC1.1 promote prostate cancer metastasis.
To validate if CCL2 is a direct target of PRC1, we performed ChIP-qPCR. We found that the chromatin surrounding the CCL2 promoter is decorated by activating modifications, including H3K9ac and H3K27ac, in control PC3 and PC3M cells. RNF2 depletion removed these modifications, consistent with a role for PRC1 in CCL2 induction. In contrast, the repressive marks H2AK119ub and H3K27me3 were very low on the CCL2 promoter in control cells and did not change upon RNF2 knockdown (Figure 3M and S3L). As anticipated, silencing of RNF2 caused a decrease of the H2AK119ub mark and an increase of the H3K9ac and H3K27ac marks on the promoter of the PRC1-repressed gene ATF3 (Figure 3N). These results indicate that PRC1 directly promotes the expression of CCL2 in prostate cancer cells.
Targeting PRC1-CCL2 signaling impairs bone metastasis
Having verified that RNF2 inactivation decreases CCL2 and CD68+ TAMs in subcutaneous PC3 tumors (Figure S4A), we analyzed bone metastases and found that RNF2 depletion not only suppresses the recruitment of TAMs but also decreases microvessel density and increases NK cells (Figure 4A). These findings indicate that PRC1 promotes the recruitment of TAMs to the tumor stroma, creating an immunosuppressive and proangiogenic microenvironment for metastatic outgrowth.
Figure 4. Targeting PRC1-CCL2 signaling impairs bone metastasis.

(A) Bone tissues from mice injected with control and RNF2-silenced PC3 cells were subjected to IHC staining. CD68+ TAMs, CD31+ endothelial cells, and NKp46+ NK cells were imaged (left, bar=50 μm) and quantified (right, bars, mean±SD, * p < 0.05, ** p < 0.01).
(B) Representative images (top) and quantification of luciferase counts (bottom) of male nude mice injected i.c. with 2.5×105 control and CCR4-silenced PC3 cells at week 5; bars, mean±SEM.
(C) Representative images (top) and quantification of luciferase counts (bottom) at week 5 of male nude mice injected i.c. with 2.5×105 PC3 cells and treated with vehicle (captisol), RS504393 (10 mg/kg, twice/week) or BLZ945 (200 mg/kg, QD) 7 days after injection. Bars, mean±SEM.
(D) Bone tissues from mice injected i.c. with control and RNF2-silenced RM1 cells were subjected to IHC staining. TAMs, endothelial cells, Foxp3+ Tregs, and B220+ B cells were imaged (left, bar=50 μm) and quantified (right, bars, mean±SD).
(E and F) ssGSEA of RNF2 activity (E) or CCL2 mRNA levels (F) in ARPC, DNPC and NEPC in the SU2C dataset. Bars, mean±SD, one-way ANOVA and Tukey's multiple comparisons test, * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001;
(G) Correlation of RNF2 activity with intratumoral abundancy of immune cell subsets. ** p < 0.01; * p < 0.05 (two-tailed).
See also Figure S4.
Since PC3 cells express the CCL2 receptor CCR4, we asked if CCL2 could promote self-renewal by an autocrine mechanism. Depletion of CCL2 or CCR4 inhibited tumor sphere formation by a similar degree (Figure S4B and S4C) although not as effectively as silencing of RNF2 (Figure 2F), suggesting that PRC1 promotes self-renewal at least in part by inducing CCL2. To examine the relative roles of the autocrine and paracrine effect of CCL2, we inactivated CCR4 in PC3 cells or targeted the CCL2/CCR2 axis in monocytes/macrophages. Silencing of CCR4 compromised bone metastasis, providing evidence that the increased self-renewal capacity conferred by CCL2 signaling is necessary for successful bone colonization (Figure 4B). To block the CCL2/CCR2 axis and inhibit macrophage recruitment, we used the selective CCR2 antagonist RS504393 or the CSF-R1 inhibitor BLZ945, respectively. Both compounds effectively inhibited the outgrowth of macroscopic bone lesions (Figure 4C). These results indicate that CCL2 promotes bone colonization by inducing autocrine self-renewal and by recruiting pro-tumorigenic macrophages.
To examine the consequences of PRC1 inactivation in immune competent mice, we stained bone sections from C57BL/6 mice inoculated with RM1 cells. Silencing of RNF2 not only reduced TAMs infiltration and suppressed neoangiogenesis but also inhibited recruitment of regulatory T cells (Tregs) and B cells to bone metastases (Figure 4D). Whereas it is well established that Tregs mediate immunosuppression in cancer (Plitas and Rudensky, 2016), B cells have also been implicated in prostate cancer progression (Ammirante et al., 2013). Depletion of RNF2 also induced an increase of NK cells and CD4+ T cells but not of CD8+ T cells, suggesting that this manipulation can reverse the immunosuppressive microenvironment but is insufficient to drive infiltration of effector T cells (Figure S4D).
To further study the connection between PRC1 activity and DNPC, we built a prostate cancer specific RNF2 activity score consisting of genes robustly downregulated following RNF2 depletion and categorized the SU2C dataset into ARPC, DNPC, and NEPC (Figure S4E and S4F). Single sample GSEA showed that the RNF2 activity geneset is enriched in DNPC but not in NEPC as compared to ARPC (Figure 4E). Moreover, although CCL2 is not a component of the RNF2 score defined above, we found that its expression is significantly higher in DNPC but not in NEPC (Figure 4F). Finally, to analyze the immune cell subsets present in DNPC, we used Cibersort, a deconvolution method that infers the abundance of immune cell subsets from bulk-tissue transcriptome data (Newman et al., 2015). The RNF2 score positively correlated with infiltration by various classes of immunocytes, including dendritic cells and M2 macrophages (Figure 4G). Together, these data support the conclusion that PRC1 and CCL2 drive development of an immunosuppressive tumor microenvironment in DNPC metastases.
Development of a Catalytic Inhibitor of PRC1
Since PRC1 promotes the expression of multiple prometastatic genes in addition to CCL2 (Table S2), inhibition of PRC1 should exert a higher therapeutic efficacy as compared to inhibition of the CCL2-CCR4 axis. Prior studies have identified the small molecule PRT4165 as an inhibitor of the E3 ligase of PRC1 (Figure 5A) (Alchanati et al., 2009). However, this compound inhibited PRC1 activity, as assessed by H2AUb and growth of oncosphere only at 25 μM (Figure 5B, 5C, S5A, and S5B). To identify a more potent inhibitor, we screened a private library of small molecule compounds and identified GW-516 as the most potent (Figure 5A; Ouerfelli at al., unpublished data). Titration experiments revealed that GW-516 inhibits H2AUb and sphere formation in PC3 cells ≥7.5 fold more efficiently as compared to PRT4165 (Figure 5B and 5C). GW-516 inhibited oncosphere to a similar extent in RM1 cells (Figure S5C and S5D). As anticipated from the selective role of PRC1 in self-renewal, GW-516 did not inhibit 2D cell growth when used at concentrations up to 1 μM (Figure S5E). Importantly, GW-516 inhibited RNF2-mediated H2AUb in a dose-dependent fashion in a cell-free system (Figure 5D). We also compared GW-516 to PTC209, which has been proposed to function by targeting BMI1 translation (Yong et al., 2016). Of note, GW-516 inhibited PRC1 activity more effectively as compared to PTC209 (Figure 5E). Moreover, the inhibitory effect of GW-516 persisted for at least 48 hours similarly to that of PTC209 (Figure 5F). These results identify GW-516 as a RNF2 inhibitor with an apparent IC50 in cells and on target of ~ 0.47 μM.
Figure 5. Development of a Catalytic Inhibitor of PRC1.

(A) Chemical structures of PRT4165 and GW-516.
(B) Acid-extracted histones from PC3 cells treated as indicated for 24 hours were subjected to immunoblotting followed by densitometry. H2AUb was normalized to control and the IC50 determined.
(C) PC3 cells were subjected to sphere assay in the presence of PRT4165 or GW-516. The inhibition of sphere formation was normalized to control and the IC50 determined.
(D) Histone H2A was incubated with RNF2 in the presence of E1 and E2. Reactions were carried out in the presence of PRT4165 or GW-516.
(E and F) Cells were treated with GW-516 or PTC209 at the indicated concentrations for 6 hours (E), or treated with GW-516 or PTC209 for the indicated times (F).
(G and H) Expression of RNF2 target genes upon RNF2 depletion or GW-516 treatment in PC3 (G) and RM1 cells (H). Bars, mean±SD.
(I and J) Representative images (I) and quantification of luciferase counts (J) of mice injected i.c. with PC3 cells and treated with vehicle (DMSO) or GW-516 as indicated. Mice were dosed twice per week. Bars, mean±SEM.
(K) Representative images (left, bar=50 μm) and percentages of staining intensity (right, Bars, mean±SD) of IHC staining for CCL2 or H2AUb of bone tissues from (I).
See also Figure S5.
To estimate the selectivity of GW-516, we compared the gene expression changes induced by GW-516 or PRT4165 treatment with those observed after silencing of RNF2. Pathway analysis indicated that the two molecules modified the expression of genes associated with specific cancer-relevant pathways in a similar fashion. GW-516 also induced changes in pathways that appeared to be not affected by RNF2 depletion and vice versa, possibly reflecting off-target effects of the molecule or differences between genetic and pharmacological modulation (Figure S5F). Further attesting to the potency of GW-516, qRT-PCR of key PRC1 targets confirmed the ability of GW-516 to either downregulate or upregulate them as effectively as silencing of RNF2 (Figure 5G and 5H).
To examine the preclinical activity of GW-516 as a single agent in the metastatic setting, we injected PC3 cells in mice and delivered GW-516 at 25 mg/kg starting from either day 7, when micrometastases can be detected histologically, or from day 21, when bioluminescent macrometastases are evident in the bones. Administration of GW-516 from day 7 prevented formation of bone metastases, whereas treatment starting from day 21 resulted in a suppression of their expansion (Figure 5I and 5J). Analysis of bone sections showed that GW-516 substantially decreases nuclear H2AUb levels and secretion of CCL2 in the tumor microenvironment, confirming target inhibition in vivo (Figure 5K). GW-516 also inhibited the outgrowth of bone, brain and liver metastases when administered to C57BL/6 mice injected with RM1 cells (Figure S5G and S5H). FACS analysis on peripheral blood leukocytes showed a significant decrease of macrophages and increase of T cells and NK cells in treated mice, suggesting that targeting PRC1 with GW-516 can reverse immunosuppression systemically (Figure S5I).
Pharmacological inhibition of PRC1 reverses immune suppression and cooperates with immunotherapy to suppress metastasis
To examine the hypothesis that targeting PRC1 improves the efficacy of double checkpoint immunotherapy (DCIT), we used the syngeneic Ptenpc-/-Smad4pc-/- and RM1 mouse models. FVB mice were inoculated i.c. with Ptenpc-/-Smad4 pc-/- cells and dosed with GW-516 or DCIT (anti-CTLA-4 + anti-PD-1), singly or in combination. GW-516 was used at 10 mg/kg to minimize potential toxicity and better reveal cooperation with DCIT. The combination treatment completely suppressed metastasis, whereas GW-516 or DCIT alone only inhibited this process (Figure 6A and 6B). Survival analysis confirmed the superiority of the combination treatment (Figure 6C). Detailed analysis of multi-organ site metastasis showed that bone and liver metastases almost disappear from the combination treatment group (Figure 6D–6F). FACS analysis of peripheral blood and bone marrow indicated that GW-516 reduces the numbers of MDSCs and TAMs, DCIT increases the number of T cells, and the combination exerts additive effects, indicating that the two treatment modalities have complementary systemic effects (Figure 6G). Similar effects were observed in RM1-injected C57BL/6 mice (Figure S6A–6G).
Figure 6. Pharmacological inhibition of PRC1 cooperates with immunotherapy to suppress metastasis.

(A-C) Representative images (A), cumulative luciferase counts (B) and survival analysis (C) of FVB mice injected i.c. with 1×105 PtenPC-/-Smad4PC-/- cells and treated with vehicle (control IgG), PD-1 + CTLA-4 antibodies (PD-1: 200 μg/mouse; CTLA-4: 250 μg/mouse), GW-516 (10 mg/kg) or combination starting at day 7 post-injection. Mice were dosed twice per week. Bars, mean±SEM. * p < 0.05; ** p < 0.01; **** p < 0.0001.
(D-F) Quantification of luciferase counts in the bone (D), liver (E), and brain (F) of end point mice from (A); bars, mean±SEM.
(G) FACS analysis of immune cell subsets in the blood and bone marrow of end point mice from (A). Bars, mean±SD. * p < 0.05, ** p < 0.01.
See also Figure S6.
On-treatment staining of bone lesions revealed that GW-516, alone or in combination with DCIT, reduces the numbers of TAMs and Tregs in the bone lesions generated by Ptenpc-/-Smad4pc-/- and RM1 cells (Figure 7A and S7A). Double staining for CD68 and Arg1/iNOS further showed that GW-516 treatment decreases the percentage of M2-like TAMs and increases the number of M1-like macrophages present at bone metastatic sites in both models (Figure 7A and S7A). In contrast, DCIT, alone or in combination with GW-516, increased the recruitment of CD4+ and CD8+ T cells, whereas the combination treatment inhibited the recruitment of potentially pro-tumorigenic B cells (Figure 7A, 7B, S7A and S7B). Moreover, although we did not observe a significant reduction of tumor cell proliferation in any of the treatment groups, each treatment induced apoptosis with the combination exerting the largest effect (Figure 7C and S7C). Overall, the combination treatment resulted in a more profound reduction of TAMs and Tregs and inhibition of neoangiogenesis and a larger increase in CD4+ and CD8+ T cells in both models, highlighting the complementary effects of the two treatments.
Figure 7. Pharmacological inhibition of PRC1 reverses immune suppression and cooperates with immunotherapy to suppress metastasis.

(A-C) Representative staining images (left, bar=50 μm) and quantifications of positive cells (right, bars, mean±SD, **** p < 0.0001) for immune-suppressive cells (A), T cells (B), and endothelial cells and proliferating/apoptotic tumor cells (C) in bone tissues from mice inoculated i.c. with PtenPC-/-Smad4PC-/- cells collected after 1 week of treatment and subjected to IHC or IF staining.
See also Figure S7.
Discussion
The mechanisms that enable metastatic prostate cancer cells to overcome secondary immunoediting and avert elimination at metastatic sites are poorly understood. In this study, we show that PRC1 not only controls self-renewal and metastasis initiation but also governs the recruitment of MDSCs, TAMs and Tregs, thus creating a profoundly immunosuppressive and pro-angiogenic microenvironment in DNPC. Pharmacological inhibition of PRC1 reverses these processes and cooperates with DCIT to suppress multi-organ metastasis. These results reveal a link between epigenetic regulation of stemness and molding of an immunosuppressive microenvironment and identify PRC1 as a therapeutic target in M-CRPC.
We found recurring cases of amplification and overexpression of genes encoding multiple PRC1 components in M-CRPC but not in primary tumors. These alterations potentially function in concert with upstream stimuli, such as those impinging on IKK α (Ammirante et al., 2013), to selectively elevate PRC1’s activity in DNPC. Moreover, PRC1 activity correlated with EMT and stemness traits in DNPC patient samples and metastatic cell lines, suggesting that PRC1 may sustain the oncogenicity of prostate cancer cells that have acquired mesenchymal and stem-like transcriptional traits in support of metastatic capacity. Consistently, inactivation of PRC1 inhibited tumor sphere formation, invasive outgrowths in 3D Matrigel, and metastatic colonization of the bone and visceral organs.
PRC1 performs complex roles in gene regulation. In addition to cPRC1, biochemical and functional analysis has defined several ncPRC1 complexes, including the cancer-relevant KDM2B-PRC1 complex (ncPRC1.1) (Banito et al., 2018; Van den Boom et al., 2016). By combining genome-wide occupancy analysis with expression profiling, we found that cPRC1 associates more robustly with the promoter of RNF2-activated genes, whereas KDM2B binds more extensively to the promoters of RNF2-repressed genes. This suggests that at least in prostate cancer cells, cPRC1 promotes gene expression at a genome-wide level, whereas ncPRC1.1 is predominantly involved in gene repression. This said, qRT-PCR analysis revealed that the induction of the major pro-metastatic targets of PRC1, CCL2 and CXCL1, requires not only cPRC1 but also ncPRC1.1 and, in consonance with these results, depletion of either BMI1 or KDM2B suppressed prostate cancer metastasis. Given the multitude of PRC1 targets and the existence of additional ncPRC1, we cannot exclude their participation in the prometastatic program governed by PRC1.
Through a focused genetic screen and subsequent mechanistic studies, we identified CCL2 as the major target of PRC1 and showed that this cytokine functions in an autocrine fashion to promote self-renewal and in a paracrine fashion to recruit TAMs at metastatic sites. Extensive evidence implicates TAMs, which descend from myeloid progenitors in the bone marrow and circulate as inflammatory monocytes, in paracrine interactions that support cancer stem cells and their ability to colonize target organs (Quail and Joyce, 2013). In particular, M2-type TAMs, which are prevalent in advanced tumors, impair the maturation of dendritic cells and the activity of effector T cells, promote cancer proliferation by secreting EGF, and induce matrix remodeling and angiogenesis through production of matrix metalloproteases (Kessenbrock et al., 2010; Mantovani et al. 2017; O'Sullivan et al., 1993). Consistently, we found that inhibition of PRC1 reverses immunosuppression at bone metastatic sites and suppresses angiogenesis in models of DNPC. In addition to switching macrophage polarization from M2 to M1, inhibition of PRC1 blocked recruitment of Tregs, which have also been shown to participate in immune suppression (Plitas and Rudensky, 2016). These findings illustrate the ability of PRC1 to mold an immunosuppressive microenvironment at metastatic sites and to potentially overcome the barrier imposed by secondary immunoediting.
Antibodies blocking CCL2 have not proven to be effective in prostate cancer due to rebound production of CCL2 upon cessation of therapy (Pienta et al., 2013). Moreover, newly produced CCL2 releases inflammatory monocytes from the bone marrow and promotes angiogenesis and metastasis in mouse models of breast cancer, suggesting that anti-CCL2 monotherapy may paradoxically have harmful consequences (Bonapace et al., 2014). In contrast, GW-516 suppressed the recruitment of total TAMs and increased the number of M1-like antigen presentation-competent macrophages present at metastatic sites, removing a block to immune response and curbing neo-angiogenesis. In addition, GW-516 suppressed the recruitment of immunoinhibitory Tregs, presumably as a result of reduced CCL2 and CCL5 (Chang et al., 2016; Tan et al., 2009), and reduced infiltration by B cells. Of note, both types of immunocytes have been implicated in prostate cancer progression (Ammirante et al., 2013; Flammiger et al., 2013). More important, although DCIT was modestly effective when used alone, it provoked a substantial recruitment of CD4+ and CD8+ T cells and induced tumor cell apoptosis and metastasis regression in combination with GW-516. These results indicate that targeting PRC1 catalytic activity reverses immunosuppression at metastatic sites.
Developmental studies have revealed that a variety of immune cells, including macrophages and Tregs, regulate the self-renewal and activation of adult stem cells to coordinate tissue homeostasis and wound repair (Naik et al., 2018). Although the mechanisms that regulate the interaction of normal prostate stem cells with the immune system are not yet known, we speculate that metastatic stem cells hijack the normal function of PRC1 to induce immunosuppression during prostate cancer metastasis. More broadly, our results indicate that a master epigenetic regulator, PRC1, coordinates metastasis initiation and outgrowth with suppression of both the innate and adaptive immune system and induction of neoangiogenesis. We envision that targeting PRC1 may sensitize M-CRPC and other immunologically ‘cold’ cancer types to immunotherapy.
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Filippo G. Giancotti (FGGiancotti@MDAnderson.org).
Experimental Model and Subject Details
Cell Lines and Reagents
LNCaP, 22rv1, VCaP, DU145, PC3, Myc-Cap, TRAMP-C1 and PC12 cells were obtained from ATCC, 293FT packaging cells were from Invitrogen and cultured according to manufacturers' instructions. PC3M cells were a gift from Dr. Raymond Bergan (formerly of Northwestern University, now OHSU Knight Cancer Institute) and cultured in RPMI-1640 supplemented with 10% Fetal Bovine Serum, 2 mM L-Glutamine (Glu), 100 IU/ml Penicillin/Streptomycin. PtenPC-/-Smad4PC-/- cells from Guocan Wang’s lab at MDACC were cultured in RPMI supplemented with 10% Fetal Bovine Serum, 2 mM L-Glutamine (Glu), 100 IU/ml Penicillin/Streptomycin. RM1 cells from Timothy Thompson’s laboratory at MDACC were cultured in DMEM supplemented with 10% Fetal Bovine Serum, 2 mM L-Glutamine (Glu), 100 IU/ml Penicillin/Streptomycin. The RNF2 inhibitor PRT4165 (5047), CCR2 antagonist RS504393 (2517), and BMI1 inhibitor PTC209 were from Tocris. The CSF-1R inhibitor BLZ945 (S7725) was from Selleckchem.
Mice
For all the animal studies in the present study, the study protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Memorial Sloan Kettering Cancer Center and UT MD Anderson Cancer Center. Male BALB/c nude mice, male NOD SCID gamma mice, male C57BL/6J mice, and male FVB/NJ mice (aged 4–6 weeks) were obtained from The Jackson Laboratory. For localized tumor growth assay, cells were resuspended in 100 μl PBS with Matrigel in 1:1 ratio and subcutaneously injected into both rear flanks. The volume of the s.c. xenograft was calculated as V = L × W2/2, where L and W stand for tumor length and width, respectively. For experimental metastasis assays, cells were resuspended in 100 μl PBS and intracardially injected into the left ventricle with a 26G tuberculin syringe. For bone colonization, RM1 cells were resuspended 100 μl PBS and injected into the intra-femoral artery. Metastatic burden was detected through non-invasive bioluminescence imaging of experimental animals using an IVIS Spectrum. To investigate the effect of drug treatment, compounds or antibodies were delivered twice every week or every three days through i.p. injection except BLZ945, which was delivered orally. Bioluminescence signal was measured using the ROI tool in Living Image software (Xenogen).
Establishment of prostate cancer cells from Ptenpc-/-Smad4pc-/- mice
The PtenPC-/-Smad4PC-/- prostate cancer cells were isolated from a 33-weeks old Ptenloxp/loxpSmad4loxp/loxpPB-Cre4+ (PtenPC-/-Smad4PC-/-) FVB mouse. Tumor tissue was dissected from the prostate of the mouse, minced, and digested with 0.5% type I collagenase as described previously (Ding et al., 2011). After filtering through a 40-μm mesh, the trapped fragments were plated in tissue culture dishes coated with type I collagen. Cells were cultured and expanded in DMEM plus 10% fetal bovine serum. Luciferase labeled PtenPC-/-Smad4PC-/- cells were generated by infecting the cells with a lenti-virus vector expressing luciferase and mCherry followed by FACS selection.
Human Pathology
Paraffin-embedded tissue microarray sections with multiple cores of prostate tumors were obtained from US Biomax. Inc. The levels of expression of RNF2 and BMI1 were determined by immunohistochemical staining. RNF2 and BMI1 immunoreactivity was evaluated and scored. The expression score was determined by combining staining intensity and the percentage of immunoreactive cells.
Method Details
MTT assay
Control and RNF2-silenced PC3 cells were plated at 1 × 103 per well in 96 wells plates for 24 hours. After 24 hours, cells were incubated in 0.5 mg/ml MTT (3-{4,5-dimethylthiazol-2-yl}-2,5-diphenyl tetrazolium bromide; Invitrogen) for 2h at 37°C. MTT crystals were dissolved in DMSO and absorbance was measured in a plate reader at 540 nm.
Tumor Sphere Assay
Single cells suspensions of LNCaP, DU145, PC3, PC3M or RM1 cells (1,000 cells/ml) were plated on ultra-low attachment plates and cultured in serum-free PrEGM (Lonza) supplemented with 1:50 B27, 20 ng/ml basic fibroblast growth factor (bFGF) and 40 ng/ml epidermal growth factor (EGF) for 10 days. Tumor spheres were visualized under phase contrast microscope, photographed, and counted. For serial passage, tumor spheres were collected using 70-μm cell strainers and dissociated with Accutase for 30 min at 37°C to obtain single-cell suspensions.
Cell Invasion Assay
Cell invasion was assayed using Matrigel-coated BioCoat Cell Culture Inserts from Corning. After Matrigel was rehydrated at room temperature, 2 × 104 cells suspended in 0.5 ml medium without FBS were plated into each insert. 0.5 ml medium with 10% FBS were added into the bottom of each well. Non-invading cells were removed after 16 hours culture. The cells on the lower surface of the membrane were stained with crystal violet.
Matrigel 3D Culture
Dissociated cells were incubated in PrEGM medium (Lonza) supplemented with 1:50 B27, 20 ng/ml basic fibroblast growth factor (bFGF) and 40 ng/ml epidermal growth factor (EGF). Matrigel bed was made in 6 well plate by putting 4 separate drops of matrigel per well (50 μl Matrigel per drop). Plates were placed in 37 °C CO2 incubator for 30 min to allow the Matrigel to solidify. For each sample, 100 μl of cell suspension was mixed with 100 μl cold Matrigel, and pipetted on top of the Matrigel bed (50 μl each). The plates were then incubated at 37 °C for another 30 min. Warm PrEGM (2.5 ml) was then added to each well. The cells were cultured and monitored for 10–14 days with 50% medium change every 3 days. For immunostaining experiments, the cells were cultured in 8 well chamber slide. Cells were fixed with 4% paraformaldehyde for 20 min and proceed to standard immunostaining protocol.
Biolominescent and X-ray Imaging
For bioluminescent imaging, mice were anesthetized and injected with 1.5 mg of D-luciferin retro-orbitally at the indicated times. Animals were imaged in an IVIS 100 chamber within 5 min after D-luciferin injection, and data were recorded using Living Image software (Xenogen). To measure bone colonization after intracardiac injection, photon flux was calculated by using the ROI tool in Living Image software. Bone metastases were further confirmed by X-Ray imaging. Mice were anesthetized with Ketamine (100 mg/kg) and xylazine (10 mg/kg), placed on digital X ray Film (Scan X) and exposed at 25kV for 15s using a Faxitron instrument (Model MX-20; Faxitron Corp. Buffalo, IL).
Immunostaining
Immunohistochemistry on paraffin-embedded sections and immunofluorescent staining were performed using a Discovery XT processor (Ventana Medical Systems). The tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems), antigen retrieval was performed with CC1 buffer (Ventana Medical Systems). Sections were blocked for 30 minutes with Background Buster solution (Innovex), followed by avidin-biotin blocking for 8 minutes (Ventana Medical Systems) (except for slides stained with CD4 and NKp46 antibodies). Sections were incubated with anti-RNF2 (Proteintech, cat#160311-AP, 2 μg/ml); anti-BMI1 (Cell Signaling, cat#6964, 1 μg/ml); anti-AR (Abcam, ab133273 1 μg/ml); Vimentin (Cell Signaling, cat# 5741, 0.5 μg/ml); anti-Synaptophysin (Abcam, ab32127, 1 μg/ml); anti-Phospho AKT (Cell Signaling, cat# 4060, 1 μg/ml); anti-Ki67 (Abcam, cat# ab16667, 2.5 μg/ml); anti-Cleaved Caspase 3 (Cell Signaling, cat# 9661, 0.1 μg/ml); anti-CD11b (Abcam, cat# ab133357, 1 μg/ml); anti-CCL2 (Invitrogen, MA5–17040, 1:200); anti-CD68 (Boster, cat# PA1518, 5 μg/ml); anti-CD8 (Cell Signaling, cat# 98941, 2.4 μg/ml); anti-CD31 (Dianova, cat# DIA-310, 1 μg/ml); anti-B220 (BD Biosciences, cat# 550286, 0.3 μg/ml); anti-FoxP3 (eBioscience, cat# 14–577382, 5 μg/ml); anti-CD4 (R&D Systems, cat# AF554, 2 μg/ml); anti-NKp46 (R&D Systems, cat#AF2225, 2 μg/ml) for 5 hours, followed by 60 minutes incubation with biotinylated horse anti-rabbit (Vector Labs, cat# PK6101) at 1:200 dilution (for Ki67, Cleaved Caspase 3, CD11b, CD8, AR, ITGB4, Vimentin, and phosphor-AKT), HRP-conjugated goat anti-rabbit (PI-1000) at 1:250 dilution (for synaptophysin), biotinylated goat anti-rat IgG (Vector labs, cat# PK-4004) at 1:200 dilution (for CD31, B220 and FoxP3), or biotinylated horse anti-goat IgG (Vector labs, cat#BA-950) at 1:200 dilution (for CD4 and NKp46). The detection was performed with DAB detection kit (Ventana Medical Systems) according to manufacturer instruction. Slides were counterstained with hematoxylin and coverslipped with Permount (Fisher Scientific).
For immunofluorescent staining, the tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems) and antigen retrieval was performed with CC1 buffer (Ventana Medical Systems). Sections were blocked for 30 minutes with Background Buster solution (Innovex), followed by avidin-biotin blocking for 8 minutes (Ventana Medical Systems).
For iNOS/CD68 or Arg1/CD68 staining, slides were first incubated with anti-iNOS (Abcam, cat#ab15323, 5 μg/ml) or anti-Arg1 (Cell Signaling cat#93668) for 5 hours, followed by 60 minutes incubation with biotinylated goat anti-rabbit IgG (Vector, cat # PK6101) at 1:200 dilution. The detection was performed with Streptavidin-HRP D (part of DABMap kit, Ventana Medical Systems), followed by incubation with Tyramide Alexa Fluor 488 (Invitrogen, cat# T20922) prepared according to manufacturer instruction with predetermined dilutions. Next, sections were incubated with anti-CD68 (Boster, cat#PA1518, 5 μg/ml) for 5 hours, followed by 60 minutes incubation with biotinylated goat anti-rabbit IgG (Vector, cat # PK6101) at 1:200 dilution. The detection was performed with Streptavidin-HRP D (part of DABMap kit, Ventana Medical Systems), followed by incubation with Tyramide Alexa CF 594 (Biotium, cat# 92174) prepared according to manufacturer instruction with predetermined dilutions. After staining, the slides were counterstained with DAPI (Sigma Aldrich, cat# D9542, 5 μg/ml) for 10 min and coverslipped with Mowiol.
Oncoprint and Hierarchical Clustering
Prostate cancer patient sample gene expression and amplification data were acquired from the Oncomine database and the cBioportal database. Additionally, the UCSF metastatic prostate cancer patient dataset was kindly provided by the authors (Quigley et al., Cell, 2018, referred as Feng’s dataset in manuscript). Z-score 2.0 was used as cut-off value to determine mRNA up/downregulation in a given sample. For the UCSF dataset, copy number alteration was called using following log2 ratio bounds, as used in the original paper:
- chr1-chr22 Gain / shallow loss / deep loss: 3 / 1.65 / 0.6
- chrX, chrY Gain / loss: 1.4, 0.6
Oncoprint (http://www.cbioportal.org/public-portal/oncoprinter.jsp) was generated using sorted data of mRNA up/downregulation and gene amplification/deletion information, ordered by aberration rate (%) and classified by tumor site (primary vs. metastatic). Morpheus was used for clustering and heatmap generation (https://software.broadinstitute.org/morpheus).
ARPC, NEPC and DNPC Classification and AR/NE Score
We followed the AR/NE/DN prostate cancer subtype classification proposed by Dr. Nelson’s group (Bluemn et al., 2017). Briefly, Androgen receptor (AR) and downstream target gene KLK3, neuroendocrine prostate cancer (NEPC) representative markers SYP and CHGA were used as determination markers. mRNA expression z-score (calculated from RPKM) was acquired from cBioportal.org. ARPC was defined by those whose AR and/or KLK3 mRNA z-score > 0. NEPC was defined by those whose SYP and/or CHGA mRNA z-score >0. If there is overlap with ARPC and NEPC, AR score and NE score were compared and determined by the larger score. DNPC was defined by those were not ARPC nor NEPC. AR score and NE score were calculated by using the mRNA z-score of 10 AR activity genes (KLK3, KLK2, TMPRSS2, FKBP5, NKX3–1, PLPP1, PMEPA1, PART1, ALDH1A3, STEAP4) and 10 NE signature genes (SYP, CHGA, CHGB, ENO2, CHRNB2, SCG3, SCN3A, PCSK1, ELAVL4, NKX2–1).
RNA-seq Analysis
Data were analyzed in Partek. Total RNA was isolated from PC3 cells. Libraries were prepared by using the standard methodology from Illumina and run on a HiSeq2500 system. Raw reads were quality-checked and subsequently mapped to the human genome (hg19) using Tophat2 (2.2.4) with default settings (Langmead and Salzberg, 2012). Differential gene expression was analyzed using the DESeq2 (1.8.1) package in R using default settings (Love et al., 2014). Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) was performed on a pre-ranked gene list that was generated based on the gene expression changes between the RNF2 knockdown and control cells. The hallmark gene sets and GO gene sets from the Molecular Signatures Database (MSigDB v5.1) (Subramanian et al., 2005) were evaluated by GSEA with 1,000 permutations, and those significantly (FDR < 0.1) enriched pathways and GO were reported using ggplot2 R package. Heatmap analysis was performed to show the gene expression patterns between the RNF2 knockdown and control repeats, using heatmap3 R package with ward2 as distance function. Gene expressions in the heatmap were transformed in logarithm scale and normalized accordingly.
ChIP-seq Analysis and Data Visualization
Cell nuclei from approximately 20 million formaldehyde crosslinked PC3 cells (1%; 10 minutes at room temperature) were isolated and chromatin was fragmented using sonicator (bioruptor). Lysate were cleared and protein-DNA complexes were isolated using target antibodies and protein-G coated magnetic beads. Chromatin IP was conducted following the standard protocol from ActiveMotif ChIP-IT High Sensitivity® (HS) Kit. Libraries were prepared according to standard Illumina protocol. Samples were sequenced at the Integrated Genomics Operation Core at MSKCC. ChIP-Seq analysis and data visualization ChIP-seq reads were trimmed by trimmomatic (v0.33; http://www.usadellab.org/cms/?page=trimmomatic) (Bolger et al., 2014) prior to alignment, as recommended by the ChIP kit manufacturer. The trimmed reads were then aligned to the hg19 reference genome using bowtie2 (v2.3.4.2, http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) (Langmead and Salzberg, 2012). Only uniquely aligned reads were kept for downstream analysis, with duplicate reads removed by the samtools software v1.9 (Li et al., 2009). The read density matrix (+/- 5kb from the transcription start sites (TSS) of the corresponding genes) from the HOMER software (v4.10, http://homer.ucsd.edu/homer/) (Heinz et al., 2010) was imported to the R package pheatmap for drawing heatmaps, with signal of input subtracted. Hierarchical clustering of H3K4me3 read densities and H3K27me3 read densities across the promoter regions of RNF2 active genes or the promoter regions of RNF2 repressed genes. To visualize ChIP-seq signal at individual genomic regions, we used the UCSC Genome Browser (https://genome.ucsc.edu/) (Kent et al., 2002). Identification of significantly over-represented functional categories was done using function of “Investigate Gene Sets” from GSEA (http://software.broadinstitute.org/gsea/msigdb/annotate.jsp) (Mootha et al., 2003).
Immune Cell Subset Deconvolution Analysis
Intratumoral immune cell subsets from the SU2C M-CRPC dataset were analyzed by using CIBERSORT bulk transcriptome deconvolution technique (Newman et al., 2015). CIBERSORT is a computational framework for accurately quantifying the relative levels of distinct cell types within a complex gene expression admixture. We used the LM22 signature genes file consisting of 547 genes that accurately distinguish 22 mature human hematopoietic populations and activation states, including seven T cell types, naïve and memory B cells, plasma cells, NK cells, and myeloid subsets. We used those p < 0.05 (n=86) from the total deconvolution data output (n=118).
Gene Set Enrichment Analysis
We used v3.0 of java GSEA program (Subramanian et al., 2005).Transcriptomic data of established human prostate cancer cell lines were from the Cancer Cell Line Encyclopedia, PtenPC-/-Smad4PC-/- tumor data were from GSE25140 (Wang et al., 2015), otherwise were from this study.
Single Sample GSEA Projections and Visualizations
We carried out ssGSEA (Barbie et al., 2009) using the GenePattern module ssGSEA Projection (v9) (www.genepattern.org). We used Prism (v7) for data visualization and related statistical analysis. Genesets used for the analysis are from the Molecular Signature Database, including their hallmark genesets (Liberzon et al., 2015). The geneset used for prostate cancer specific RNF2 activity is: LCN2, SEMA6A, IGF2, ANGPT1, PI3, MCAM, VNN2, ZNF91, KDR, ZNF681, ANGPTL4, DOK3, TRIM31, EMP2, DBC1, C3, SYNGR4, CHI3L2, RAB36, WISP1, TLR4, SLCO2B1, HR, LHX1, DIO3.
Customized Library Screen
shRNA and cDNA pools were generated based on RNA-seq data from RNF2-silenced PC3 cells. shRNAs were cloned into LENG (pMSCV) vector. The number of shRNAs targeting each gene is between 3 to 6. cDNAs were cloned into pCW-neo vector. 48 hours after virus infection, PC3 cells were resuspended in 100 μl 1× PBS and intracardially injected into the left ventricle. Mice were sacrificed 4 weeks after injection. Tumor cells isolated from bone lesions were subjected to qRT-PCR gene expression analysis.
Chromatin Immunoprecipitation
Cell nuclei from approximately 20 million formaldehyde crosslinked PC3 cells (1%; 10 minutes at room temperature) were isolated and chromatin was fragmented using sonicator (bioruptor). Lysate were cleared and protein-DNA complexes were isolated using the indicated antibodies and protein-G coated magnetic beads. Chromatin IP was conducted following the standard protocol from ActiveMotif ChIP-IT High Sensitivity® (HS) Kit. Eluted DNA were then subjected to qPCR (SYBR green system) with primers targeting human CCL2 or ATF promoter regions.
Candidate Library Compound Screening
A private library of compounds was provided by the Organic Synthesis Core Facility of MSKCC. The testing concentration of candidate compounds on PC3 cells was 1 μM. RNF2 target gene expression change was used as a readout for the first round of screening. Cell viability, tumor sphere formation assay and histone modification changes were then used to further confirm the activity of the compounds. GW-516 was obtained through rounds of SAR.
FACS Analysis
Control and RNF2-silenced PC3 cells were detached with Accutase and washed in blocking solution (HBSS supplemented with 10% FBS). Cell suspensions were incubated with the indicated antibodies for 45 minutes at 4 °C and analyzed by FACS. At the end point of the in vivo experiment, blood and bone marrow cells were collected from each mouse and treated with Red Blood Cell lysis buffer for 5 minutes. Cells were then washed once with RPMI supplemented 2% FBS, stained with indicated antibodies for 45 minutes and analyzed by FACS.
GW-516 Synthesis
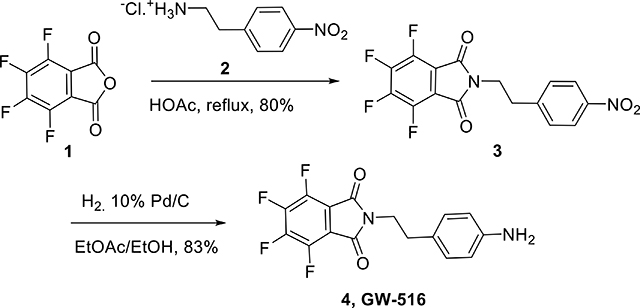
Tetrafluoro phthalic anhydride 1 (1.13 g, 6.09 mmol, 1.3 equiv.) was added to a solution of 4-nitrophenetlyl amine hydrochloride 2 (1 g, 4.68 mmol) in acetic acid (40 ml), and the resulting mixture was refluxed overnight. After cooling down to room temperature, acetic acid was distilled out under vacuum. The residue was then dissolved in EtOAc (100 ml), washed with water, dried over Na2SO4 and concentrated under vacuum. The mixture was purified by flash silica gel chromatography eluting with EtOAc/Hexane gradient, 40–50%, to afford the title compound 3 (1.39 g, yield 80%) as a yellow powder. 1H NMR (DMSO-d6, 600 MHz): δ 8.17 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H), 3.97 (t, J = 7.3 Hz, 2H), 3.1 (t, J = 7.5 Hz, 2H); 19F NMR (proton decoupled, DMSO-d6, 600 MHz): δ -139.00 (m), -144.62 (m).
Catalytic hydrogenation: The nitro substrate 3 (330 mg, 0.897 mmol) was dissolved in 10 ml of a mixture ethanol: EtOAc (1:1, v/v). The resulting solution was degassed with argon before it was quickly treated with 10% (by weight) Pd/C (90 mg, 20 wt. % loading) and the reaction was purged with hydrogen. Then a hydrogen balloon was applied to the reaction mixture through a three-way adapter under vigorous stirring. Reaction evolution was monitored by TLC. The mixture was then degassed with argon, filtered through a thick pad of celite and washed with methanol. The filtrate was transferred to a round bottomed flask and solvents were removed under vacuo. The residue was purified by flash chromatography eluting with 50–70% EtOAc in hexanes to afford amine 4 (250 mg, 83%) as a yellow powder. 1H NMR (DMSO-d6, 600 MHz): δ 6.84 (d, J = 8.3 Hz, 2H), 6.46 (d, J = 8.3 Hz, 2H), 4.90 (brs, 2H), 3.67 (t, J = 7.2 Hz, 2H), 2.7 (t, J = 7.5 Hz, 2H); 13C NMR (125 MHz): δ 168.03, 152.12, 134.62, 130.10, 119.64, 44.81, 38.20; 19F NMR (proton decoupled, DMSO-d6, 600 MHz): δ - 135.62 (q, J = 9.6, 21.4 Hz), -142.48 (q, J = 9.4, 21.6 Hz); EIMS: m/z 339.1 [M+H]+, calcd for C16H11F4N2O2: 339.08.
Analysis of protein and mRNA expression
For immunoblotting, cells were washed with PBS and lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS) supplemented with protease inhibitors (Calbiochem) and phosphatase inhibitors (PhosSTOP, Roche Life Science). Protein concentrations were measured by using the DC Protein Assay. Total RNA was extracted using the RNeasy Mini kit coupled with RNase-free DNase set (Qiagen) and reverse transcribed with SuperScript III First-Strand Synthesis SuperMix (Invitrogen). cDNA corresponding to approximately 10 ng of starting RNA was used for one reaction. qPCR was performed with Taqman Gene Expression Assay (Applied Biosystems). All quantifications were normalized to endogenous GAPDH.
Acid Extraction of Histones
PC3 cells were exposed to drugs for the indicated hours, then harvested using 0.53 mm EDTA in PBS, and washed once with cold PBS. Nuclear extracts were prepared and histones were extracted using 0.4N sulfuric acid. H2A or H2AUb was measured using the indicated antibodies.
In vitro Ubiquitination Assay
RNF2-PRC1 complex was immunoprecipitated from one 15 cm plate of PC3 cells. After extensive washing, the complex was pre-incubated with drugs at 4 °C for 30 minutes. UBC2D3 and E1 were from Boston Biochem. Reactions were performed in 30 μl of ubiquitilation buffer (50 mM Tris, pH 7.5, 2.5 mM MgCl2, 0.5 mM DTT) containing ubiquitin-activating enzyme 100 ng E1, 200 ng UBC2D3, 10 μg ubiquitin, 0.2 mM ATP, 1 μg H2A, and the indicated RNF2-PRC1 complex. After incubation at 37 °C for 60 min, the reactions were stopped by the addition of Laemmli sample buffer, and proteins were resolved by SDS-PAGE and immunoblotted with anti-H2A antibodies.
Quantification and Statistical Analysis
Statistical analyses used R and GraphPad Prism 7 software, with a minimum of three biologically independent samples for significance. For animal experiments with subcutaneous injections, each subcutaneous tumor was an independent sample. For intracardiac injection and survival analysis, each mouse was counted as a biologically independent sample. Results are reported as mean ± SD or mean ± SEM. Comparisons between two groups were performed using an unpaired two-sided Student’s t test (p < 0.05 was considered significant). Comparison of multiple conditions was done with Oneway or two-way ANOVA test. For correlation analysis, the Pearson coefficient was used. All experiments were reproduced at least three times, unless otherwise indicated.
Data Availability
The RNA sequencing data has been deposited in the GEO under ID code GSE103074. The ChIP sequencing data has been deposited in the GEO under ID code GSE131830. The RNA sequencing data for PtenPC-/- and Pten PC-/-Smad4PC-/- tumors was from Wang et al., 2015, with the GEO ID code GSE25140. The ChIP sequencing data for nucleosome occupancy in PC3 cells was from Taberlay et al., 2014, with the GEO ID code GSE57498. The Grasso prostate cancer dataset was from Grasso et al., 2012, with the GEO ID code GSE35988. The SU2C prostate cancer dataset was from Robinson et al., 2015, with the accession number dbGap: phs000915.v1.p1. The UCSF prostate cancer dataset was from Quigley et al., 2018, with the accession number dbGap: phs001648.v1.p1. The TCGA prostate cancer dataset was from The Cancer Genome Atlas Research, 2015, with the accession from http://www.cbioportal.org/study.do?cancer_study_id=prad_tcga_pub. The Taylor prostate cancer dataset was from Taylor et al., 2010, with the GEO ID code GSE21032. The FHCRC prostate cancer dataset was from Kumar et al., 2016, with the GEO ID code GSE77930.
Supplementary Material
Table S1, related to Figure 3. List of genes with changed expression upon RNF2 depletion.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD44 (FACS) | BD | 555478 |
| ITGB4 (FACS) | MSKCC Antibody Facility | |
| ITGB4 (western blot) | Santa Cruz | Sc-9090 |
| RNF2 (western blot) | Proteintech | 16031-1-AP |
| RNF2 (CHIP) | MBL | D139-3 |
| BMI1 (western blot) | Cell Signaling | 6964 |
| BMI1 (CHIP) | Active Motif | 39993 |
| AR (western blot) | Cell Signaling | 5153 |
| AR (western blot) | Santa Cruz | Sc-816 |
| AR (IHC) | Abcam | ab133273 |
| Synaptophysin | Abcam | ab32127 |
| p-AKT | Cell Signaling | 4060 |
| E-cadherin | Cell Signaling | 3195 |
| Vimentin | Cell Signaling | 5741 |
| CD44 (western blot) | Cell Signaling | 3570 |
| GR | Cell Signaling | 3660 |
| PCGF1 | Santa Cruz | Sc-515371 |
| PHC2 | Active Motif | 39661 |
| KDM2B | Millipore | 09-864 |
| RING1 | Cell Signaling | 13069 |
| EZH2 | Cell Signaling | 4905 |
| SUZ12 | Cell Signaling | 3737 |
| RhoGDI | Santa Cruz | Sc-360 |
| P53 | Cell Signaling | 9282 |
| P53(S15) | Cell Signaling | 9284 |
| CC3 | Cell Signaling | 9661 |
| Ki67 | Abcam | Ab16667 |
| CCL2 | Invitrogen | MA5-17040 |
| H2AK119Ub | Millipore | 05-678 |
| H3K27Me3 | Millipore | 07-449 |
| H3K9Ac | Millipore | 07-352 |
| H3K27Ac | Cell Signaling | 07-360 |
| H2A | Abcam | Ab18255 |
| mCherry | Abcam | Ab167453 |
| CD45 | BioLegend | 103125 |
| CD3ε | BioLegend | 100327 |
| F4/80 | BioLegend | 123113 |
| NK1.1 | BioLegend | 108715 |
| CD11b | BioLegend | 101239 |
| Ly6G | BioLegend | 127607 |
| Ly6C | BioLegend | 128035 |
| Gr-1 | BioLegend | 108443 |
| Anti-mouse CTLA-4 | Bio X Cell | BE0131 |
| Anti-mouse PD-1 | Bio X Cell | BE0146 |
| IgG2a Isotype control | Bio X Cell | BE0089 |
| Hamster IgG Isotype control | Bio X Cell | BE0087 |
| CD68 | Boster | PA1518 |
| B220 | BD | 550286 |
| CD11b | Abcam | 133357 |
| CD4 | R&D | AF554 |
| CD8 | Cell Signaling | 98941 |
| Foxp3 | eBioscience | 14-5773-82 |
| CD31 | Dianova | DIA-310 |
| NKp46 | R&D | AF2225 |
| iNOS | Abcam | Ab15323 |
| Arg1 | Cell Signaling | 93668 |
| Biological Samples | ||
| Paraffin-embedded tissue microarray | BIOMAX.US | PR8011a PR484 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | ThermoFisher Scientific | 11965-092 |
| RPMI 1640 | ThermoFisher Scientific | 61870-036 |
| Ham’s F-12K | ThermoFisher Scientific | 21127022 |
| PrEGM BulletKit | Lonza | CC-3166 |
| L-glutamine | Corning | 25005CI |
| B27 supplement | ThermoFisher Scientific | 17504044 |
| penicillin G-streptomycin | Corning | 30004CI |
| Accutase | Innovative Cell Technologies | AT104 |
| Trypsin-EDTA (0.05%) | ThermoFisher Scientific | 25300054 |
| Recombinant human EGF | R&D systems | 236-EG-200 |
| Recombinant human FGF | ThermoFisher Scientific | PHG0261 |
| UBE1 | BostonBiochem | E-305 |
| UBE2D3 | BostonBiochem | E2-627 |
| Ubiquitin | BostonBiochem | U-110 |
| Histone H2A | BioLabs | M2502S |
| PRT4165 | Tocris | 5047 |
| PTC209 | Tocris | 5191 |
| RS504393 | Tocris | 2517 |
| BLZ945 | Selleckchem | S7725 |
| Captisol | Captisol® | RC-0C7-020 |
| GW-516 | This paper | |
| Critical Commercial Assays | ||
| CHIP-IT High Sensitivity Kit | ActiveMotif | 53040 |
| MTT | ThermoFisher Scientific | M6494 |
| CellTiter-Glo | Promega | G7573 |
| BioCoat Matrigel Invasion Chamber | Corning | 354480 |
| Deposited Data | ||
| Raw and analyzed RNAseq data | This paper | GEO: GSE103074 |
| Raw and analyzed CHIPseq data | This paper | GEO: GSE131830 |
| Raw RNAseq data of the Ptenpc−/−Smad4pc−/− tumors | Wang et al., 2015 | GEO: GSE25140 |
| Raw CHIPseq data of nucleosome occupancy in PC3 cells | Taberlay at al., 2014 | GEO: GSE57498 |
| Grasso et al. prostate cancer dataset | Grasso et al., 2012 | GSE35988 |
| SU2C prostate cancer dataset | Robinson et al., 2015 | dbGap: phs000915.v1.p1 |
| UCSF prostate cancer dataset | Quigley et al. 2018 | dbGAP: phs001648.v1.p1 |
| TCGA prostate cancer dataset | The Cancer Genome Atlas Research Network | http://www.cbioportal.org/study.do?cancer_study_id=prad_tcga_pub |
| Taylor prostate cancer dataset | Taylor et al., 2010 | GEO: GSE21032 |
| FHCRC prostate cancer dataset | Kumar et al., 2016 | GEO: GSE77930 |
| Experimental Models: Cell Lines | ||
| LNCaP | ATCC | CRL-1740 |
| 22RV1 | ATCC | CRL-2505 |
| VCaP | ATCC | CRL-2876 |
| DU145 | ATCC | HTB-81 |
| PC3 | ATCC | CRL-1435 |
| Myc-Cap | ATCC | CRL-3255 |
| TRAMP-C1 | ATCC | CRL-2730 |
| PC12 | ATCC | CRL-1721 |
| RM1 | From Timothy Thompson | Thompson et al., 1989 |
| Ptenpc−/−Smad4pc−/− | From Guocan Wang | Wang et al., 2015 |
| Experimental Models: Organisms/Strains | ||
| BALB/c Nude mice | The Jackson Laboratory | 000711 |
| Nod SCID gamma mice | The Jackson Laboratory | 005557 |
| C57BL/6J mice | The Jackson Laboratory | 000664 |
| FVB/NJ mice | The Jackson Laboratory | 001800 |
| Oligonucleotides | ||
| Human RNF2 short hairpin | Sigma | TRCN0000033696 |
| Human RNF2 short hairpin | Sigma | TRCN0000033697 |
| Human BMI1 short hairpin | Sigma | TRCN0000020155 |
| Human BMI1 short hairpin | Sigma | TRCN0000020156 |
| Human KDM2B short hairpin | Sigma | TRCN0000234588 |
| Human KDM2B short hairpin | Sigma | TRCN0000118437 |
| Human CCL2 short hairpin | Sigma | TRCN0000381382 |
| Human CCL2 short hairpin | Sigma | TRCN0000338480 |
| Human CCR4 short hairpin | Sigma | TRCN0000356811 |
| Human CCR4 short hairpin | Sigma | TRCN0000356812 |
| Mouse RNF2 short hairpin | Sigma | TRCN0000226018 |
| Mouse RNF2 short hairpin | Sigma | TRCN0000040579 |
| Mouse Bmi1 short hairpin | Sigma | TRCN0000012563 |
| Mouse Bmi1 short hairpin | Sigma | TRCN0000012565 |
| Mouse CCL2 short hairpin | Sigma | TRCN0000301702 |
| Mouse CCL2 short hairpin | Sigma | TRCN00000301701 |
| Human RNF2 smart pool | Dharmacon | L-006556-00-0005 |
| Human RING1 smart pool | Dharmacon | L-006554-00-0005 |
| Human PCGF1 smart pool | Dharmacon | L-007094-00-0005 |
| Human PHC2 smart pool | Dharmacon | L-021410-00-0005 |
| Human KDM2B smart pool | Dharmacon | L-014930-00-0005 |
| RNF2 Taqman probe | ThermoFisher Scientific | Hs00200541_m1 |
| BMI1 Taqman probe | ThermoFisher Scientific | Hs00180411_m1 |
| AR Taqman probe | ThermoFisher Scientific | Hs00171172_m1 |
| P53 Taqman probe | ThermoFisher Scientific | Hs01034249_m1 |
| PHC1 Taqman probe | ThermoFisher Scientific | Hs01863307_s1 |
| PHC2 Taqman probe | ThermoFisher Scientific | Hs00189460_m1 |
| PHC3 Taqman probe | ThermoFisher Scientific | Hs01118132_m1 |
| PCGF1 Taqman probe | ThermoFisher Scientific | Hs01016642_g1 |
| PCGF2 Taqman probe | ThermoFisher Scientific | Hs00810639_m1 |
| PCGF3 Taqman probe | ThermoFisher Scientific | Hs00196998_m1 |
| PCGF5 Taqman probe | ThermoFisher Scientific | Hs00737074_m1 |
| PCGF6 Taqman probe | ThermoFisher Scientific | Hs00827882_m1 |
| KDM2B Taqman probe | ThermoFisher Scientific | Hs00404800_m1 |
| RING1 Taqman probe | ThermoFisher Scientific | Hs00968517_m1 |
| L3MBTL1 Taqman probe | ThermoFisher Scientific | Hs00210032_m1 |
| RYBP Taqman probe | ThermoFisher Scientific | Hs00393028_m1 |
| YAF2 Taqman probe | ThermoFisher Scientific | Hs00994514_m1 |
| BCOR Taqman probe | ThermoFisher Scientific | Hs00372378_m1 |
| CCL2 Taqman probe | ThermoFisher Scientific | Hs00234140_m1 |
| CYR61 Taqman probe | ThermoFisher Scientific | Hs00998500_g1 |
| LIF Taqman probe | ThermoFisher Scientific | Hs01055668_m1 |
| IL7R Taqman probe | ThermoFisher Scientific | Hs00233682_m1 |
| ATF3 Taqman probe | ThermoFisher Scientific | Hs00231069_m1 |
| LXN Taqman probe | ThermoFisher Scientific | Hs00220138_m1 |
| PLAU Taqman probe | ThermoFisher Scientific | Hs01547054_m1 |
| GDF15 Taqman probe | ThermoFisher Scientific | Hs00171132_m1 |
| FGFBP1 Taqman probe | ThermoFisher Scientific | Hs01921428_s1 |
| RELN Taqman probe | ThermoFisher Scientific | Hs01022646_m1 |
| NTS Taqman probe | ThermoFisher Scientific | Hs00175048_m1 |
| C3 Taqman probe | ThermoFisher Scientific | Hs00163811_m1 |
| LGR5 Taqman probe | ThermoFisher Scientific | Hs00969422_m1 |
| LCN2 Taqman probe | ThermoFisher Scientific | Hs01008571_m1 |
| CXCL1 Taqman probe | ThermoFisher Scientific | Hs00236937_m1 |
| GAPDH Taqman probe | ThermoFisher Scientific | Hs02786624_g1 |
| RNF2 Taqman probe | ThermoFisher Scientific | Mm00803321_m1 |
| BMI1 Taqman probe | ThermoFisher Scientific | Mm03053308_g1 |
| CCL2 Taqman probe | ThermoFisher Scientific | Mm00441242_m1 |
| CXCL1 Taqman probe | ThermoFisher Scientific | Mm04207460_m1 |
| ATF3 Taqman probe | ThermoFisher Scientific | Mm00476033_m1 |
| NTS Taqman probe | ThermoFisher Scientific | Mm00481140_m1 |
| LGR5 Taqman probe | ThermoFisher Scientific | Mm00438890_m1 |
| GAPDH Taqman probe | ThermoFisher Scientific | Mm99999915_g1 |
| ATF3_up for CHIP | This paper | AGGCAGGTGCGAAAGTCCCAG |
| ATF3_down for CHIP | This paper | CAATGACCACAGGCGGAACCC |
| CCL2_up for CHIP | This paper | AGAGGTAATGAGTATCAGGGAAACT |
| CCL2_down for CHIP | This paper | GCTAGATAATGCCTACAAGTGTC |
| Recombinant DNA | ||
| pRK-zrnf2 | This paper | N/A |
| pRK-zmutrnf2 | This paper | N/A |
| Softerware and Algorithms | ||
| GraphPad Prism 7 | ||
| Living Image software | Xenogen | IVIS Spectrum |
| Morpheus | Broad Institute | https://software.broadinstitute.org/morpheus |
| Samtools software v1.9 | Li et al., 2009 | |
| HOMER software v4.10 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| UCSC Genome Brower | Kent et al., 2002 | http://genome.ucsc.edu/ |
| GSEA | Mootha et al., 2003 | http://software.broadinstitute.org/gsea/msigdb/annotate.jsp |
Significance.
Chronic inflammation and immunosuppression constitute a significant barrier to the development of effective immunotherapies for metastatic castration-resistant prostate cancer. Here we show that PRC1 not only controls self-renewal and metastasis initiation but also governs the recruitment of M2-like tumor-associated macrophages and regulatory T cells, thus creating a profoundly immune suppressive and pro-angiogenic microenvironment in metastatic sites. Pharmacological inhibition of PRC1 reverses these processes and cooperates with immune checkpoint blockade to suppress multi-organ site metastasis. These results reveal a link between epigenetic regulation of cancer stem cells and molding of the tumor microenvironment and identify PRC1 as a therapeutic target in metastatic prostate cancer.
Acknowledgements
This work was supported by NIH grants R35 CA197566 (Outstanding Investigator Award to F.G.G.), P01 CA094060 (Project 4 to F.G.G.), P30 CA016672 (MDACC Cancer Center Support Grant), P50 CA92629 SPORE in Prostate Cancer (H.I.S.), and P30 CA008748 (MSKCC Cancer Center Support Grant), by CPRIT Recruitment of Established Investigators Award RR160031 (to F.G.G.), by the generous philanthropic contributions to The University of Texas MD Anderson Moon Shots Program, and by fellowships from the Department of Defense Prostate Cancer Research Program (W81XWH-15–1-0275) (to W.S.) and the Metastasis and Tumor Ecosystems Center of MSKCC (to W.S.). We thank members of the Giancotti’s lab for discussions and L. Tian for help with the submission of RNAseq data.
Declaration of Interests
F.G. receives research funding from Janssen. N.R. is on the scientific advisory board (SAB) and receives research funding from Chugai; on the SAB and owns equity in Beigene and Fortress; on the SAB of Daiichi-Sankyo, Astra-Zeneca-MedImmune, and F-Prime; and is a past SAB member of Millenium-Takeda, Kadmon, Kura, and Araxes. N.R. is a consultant to Novartis, Boehringer Ingelheim, Tarveda, and Foresight and consulted in the last three years with Eli Lilly, Merrimack, Kura Oncology, Araxes, and Kadman. N.R. owns equity in ZaiLab, Kura Oncology, Araxes, and Kadman. H.S. received personal fees from Astellas, Clovis Oncology, Merck, OncLive Insights, Physicians Education Resource, Sanofi Aventis, WCG Oncology, and Asterias Biotherapeutics; nonfinancial support as an uncompensated consultant from Ferring Pharmaceuticals, Janssen Research & Development LLC, and Sanofi Aventis; and grants from Illumina Inc, Innocrin Pharma, and Janssen. H.S. has also acted as an uncompensated consultant for Epic Sciences. F.G., O.O., W.S., G.Y., and H.S. are filing a provisional patent for the GW-516 compound.
References
- Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, et al. (2012). A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alchanati I, Teicher C, Cohen G, Shemesh V, Barr HM, Nakache P, Ben-Avraham D, Idelevich A, Angel I, Livnah N, et al. (2009). The E3 ubiquitin-ligase Bmi1/Ring1A controls the proteasomal degradation of Top2alpha cleavage complex - a potentially new drug target. PloS one 4, e8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammirante M, Kuraishy AI, Shalapour S, Strasner A, Ramirez-Sanchez C, Zhang W, Shabaik A, and Karin M (2013). An IKKalpha-E2F1-BMI1 cascade activated by infiltrating B cells controls prostate regeneration and tumor recurrence. Genes & development 27, 1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attard G, Parker C, Eeles RA, Schroder F, Tomlins SA, Tannock I, Drake CG, and de Bono JS (2016). Prostate cancer. Lancet 387, 70–82. [DOI] [PubMed] [Google Scholar]
- Banito A, Li X, Laporte AN, Roe JS, Sanchez-Vega F, Huang CH, Dancsok AR, Hatzi K, Chen CC, Tschaharganeh DF, et al. (2018). The SS18-SSX Oncoprotein Hijacks KDM2B-PRC1.1 to Drive Synovial Sarcoma. Cancer Cell 34, 346–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, Huang J, True L, Gleave ME, Soule H, et al. (2014). Aggressive variants of castration-resistant prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research 20, 2846–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierie B, Pierce SE, Kroeger C, Stover DG, Pattabiraman DR, Thiru P, Liu Donaher J, Reinhardt F, Chaffer CL, Keckesova Z, and Weinberg RA (2017). Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proceedings of the National Academy of Sciences of the United States of America 114, E2337–E2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, Bianchi-Frias D, Dumpit RF, Kaipainen A, Corella AN, et al. (2017). Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 32, 474–489 e476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boire A, Zou Y, Shieh J, Macalinao DG, Pentsova E, and Massague J (2017). Complement Component 3 Adapts the Cerebrospinal Fluid for Leptomeningeal Metastasis. Cell 168, 1101–1113 e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, and Bentires-Alj M (2014). Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 515, 130–133. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2015). The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, Kanojia D, Pituch KC, Qiao J, Pytel P, et al. (2016). CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer research 76, 5671–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, Brisken C, Minucci S, Di Fiore PP, and Pelicci PG (2009). The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 138, 1083–1095. [DOI] [PubMed] [Google Scholar]
- de Lau W, Peng WC, Gros P, and Clevers H (2014). The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes & development 28, 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, Perry SR, Labrot ES, Wu X, Lis R, et al. (2011). SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 470, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flammiger A, Weisbach L, Huland H, Tennstedt P, Simon R, Minner S, Bokemeyer C, Sauter G, Schlomm T, and Trepel M (2013). High tissue density of FOXP3+ T cells is associated with clinical outcome in prostate cancer. Eur J Cancer 49, 1273–1279. [DOI] [PubMed] [Google Scholar]
- Giancotti FG (2013). Mechanisms governing metastatic dormancy and reactivation. Cell 155, 750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez H, Hagerling C, and Werb Z (2018). Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes & development 32, 1267–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. (2012). The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Oren B, Mora J, Mertens C, Dziumbla S, Popp R, Weigert A, Grossmann N, Fleming I, and Brune B (2016). Lipocalin 2 from macrophages stimulated by tumor cell-derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci Signal 9, ra64. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, and Werb Z (2010). Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbe DP, Gomez EC, Wang J, et al. (2017). Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, et al. (2016). Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nature medicine 22, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loberg RD, Ying C, Craig M, Yan L, Snyder LA, and Pienta KJ (2007). CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia 9, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Chen Q, Corey E, Xie W, Fan J, Mizokami A, and Zhang J (2009). Activation of MCP-1/CCR2 axis promotes prostate cancer growth in bone. Clin Exp Metastasis 26, 161–169. [DOI] [PubMed] [Google Scholar]
- Lukacs RU, Memarzadeh S, Wu H, and Witte ON (2010). Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell stem cell 7, 682–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Marchesi F, Malesci A, Laghi L, and Allavena P (2017). Tumour-associated macrophages as treatment targets in oncology. Nature reviews Clinical oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z, et al. (2017). SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Larsen SB, Cowley CJ, and Fuchs E (2018). Two to Tango: Dialog between Immunity and Stem Cells in Health and Disease. Cell 175, 908–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nature methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy R, and Pollard JW (2014). Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan C, Lewis CE, Harris AL, and McGee JO (1993). Secretion of epidermal growth factor by macrophages associated with breast carcinoma. Lancet 342, 148–149. [DOI] [PubMed] [Google Scholar]
- Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, et al. (2013). Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs 31, 760–768. [DOI] [PubMed] [Google Scholar]
- Plitas G, and Rudensky AY (2016). Regulatory T Cells: Differentiation and Function. Cancer Immunol Res 4, 721–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power CA, Pwint H, Chan J, Cho J, Yu Y, Walsh W, and Russell PJ (2009). A novel model of bone-metastatic prostate cancer in immunocompetent mice. Prostate 69, 1613–1623. [DOI] [PubMed] [Google Scholar]
- Quail DF, and Joyce JA (2013). Microenvironmental regulation of tumor progression and metastasis. Nature medicine 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, Foye A, Kothari V, Perry MD, Bailey AM, et al. (2018). Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 175, 889. [DOI] [PubMed] [Google Scholar]
- Rai K, Akdemir KC, Kwong LN, Fiziev P, Wu CJ, Keung EZ, Sharma S, Samant NS, Williams M, Axelrad JB, et al. (2015). Dual Roles of RNF2 in Melanoma Progression. Cancer discovery 5, 1314–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. (2015). Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Bourbon HM, Di Croce L, and Cavalli G (2017). Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 171, 34–57. [DOI] [PubMed] [Google Scholar]
- Su WJ, Fang JS, Cheng F, Liu C, Zhou F, and Zhang J (2013). RNF2/Ring1b negatively regulates p53 expression in selective cancer cell types to promote tumor development. Proceedings of the National Academy of Sciences of the United States of America 110, 1720–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlay PC, Statham AL, Kelly TK, Clark SJ, and Jones PA (2014). Reconfiguration of nucleosome-depleted regions at distal regulatory elements accompanies DNA methylation of enhancers and insulators in cancer. Genome Res 24, 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, Eberlein TJ, Hsieh CS, and Linehan DC (2009). Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol 182, 1746–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson TC, Southgate J, Kitchener G, and Land H (1989). Multistage carcinogenesis induced by ras and myc oncogenes in a reconstituted organ. Cell 56, 917–930. [DOI] [PubMed] [Google Scholar]
- Turajlic S, and Swanton C (2016). Metastasis as an evolutionary process. Science 352, 169–175. [DOI] [PubMed] [Google Scholar]
- van den Boom V, Maat H, Geugien M, Rodriguez Lopez A, Sotoca AM, Jaques J, Brouwers-Vos AZ, Fusetti F, Groen RW, Yuan H, et al. (2016). Non-canonical PRC1.1 Targets Active Genes Independent of H3K27me3 and Is Essential for Leukemogenesis. Cell reports 14, 332–346. [DOI] [PubMed] [Google Scholar]
- Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, Fang Z, Zhao K, Konaparthi R, Hua S, et al. (2015). Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer discovery 6, 80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, and Zhang Y (2004). Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–878. [DOI] [PubMed] [Google Scholar]
- Yong KJ, Basseres DS, Welner RS, Zhang WC, Yang H, Yan B, Alberich-Jorda M, Zhang J, de Figueiredo-Pontes LL, Battelli C, et al. (2016). Targeted BMI1 inhibition impairs tumor growth in lung adenocarcinomas with low CEBPalpha expression. Science translational medicine 8, 350ra104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka T, Otero J, Chen Y, Kim YM, Koutcher JA, Satagopan J, Reuter V, Carver B, de Stanchina E, Enomoto K, et al. (2013). beta4 Integrin signaling induces expansion of prostate tumor progenitors. The Journal of clinical investigation 123, 682–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, Sun Y, Le Magnen C, Chester D, Mostaghel EA, Califano A, et al. (2017). Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer discovery 7, 736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Figure 3. List of genes with changed expression upon RNF2 depletion.
Data Availability Statement
The RNA sequencing data has been deposited in the GEO under ID code GSE103074. The ChIP sequencing data has been deposited in the GEO under ID code GSE131830. The RNA sequencing data for PtenPC-/- and Pten PC-/-Smad4PC-/- tumors was from Wang et al., 2015, with the GEO ID code GSE25140. The ChIP sequencing data for nucleosome occupancy in PC3 cells was from Taberlay et al., 2014, with the GEO ID code GSE57498. The Grasso prostate cancer dataset was from Grasso et al., 2012, with the GEO ID code GSE35988. The SU2C prostate cancer dataset was from Robinson et al., 2015, with the accession number dbGap: phs000915.v1.p1. The UCSF prostate cancer dataset was from Quigley et al., 2018, with the accession number dbGap: phs001648.v1.p1. The TCGA prostate cancer dataset was from The Cancer Genome Atlas Research, 2015, with the accession from http://www.cbioportal.org/study.do?cancer_study_id=prad_tcga_pub. The Taylor prostate cancer dataset was from Taylor et al., 2010, with the GEO ID code GSE21032. The FHCRC prostate cancer dataset was from Kumar et al., 2016, with the GEO ID code GSE77930.