

Abstract
Cerebrospinal fluid (CSF) biomarkers based on the core pathological proteins associated with Alzheimer’s disease (AD), i.e., amyloid-β (Aβ) and tau protein, are widely regarded as useful diagnostic biomarkers. However, a lack of biomarkers for monitoring the treatment response and indexing clinical severity has proven to be problematic in drug trials targeting Aβ. Therefore, new biomarkers are needed to track non-Aβ and non-tau pathology. Many proteins involved in the pathophysiological progression of AD have shown promise as new biomarkers. Neurodegeneration- and synapse-related biomarkers in CSF (e.g., neurofilament light polypeptide [NFL], neurogranin, and visinin-like protein 1) and blood (e.g., NFL) aid prediction of AD progress, as well as early diagnosis. Neuroinflammation, lipid dysmetabolism, and impaired protein clearance are considered important components of AD pathophysiology. Inflammation-related proteins in the CSF, such as progranulin, intercellular adhesion molecule 1, and chitinase-3-like protein 1 (YKL-40), are useful for the early detection of AD and can represent clinical severity. Several lipid metabolism-associated biomarkers and protein clearance-linked markers have also been suggested as candidate AD biomarkers. Combinations of subsets of new biomarkers enhance their utility in terms of broadly characterizing AD-associated pathological changes, thereby facilitating precise selection of susceptible patients and comprehensive monitoring of the treatment response. This approach could facilitate the development of effective treatments for AD.
Subject terms: Alzheimer's disease, Predictive markers, Alzheimer's disease, Predictive markers
Alzheimer’s disease: New ways to track disease progression
Finding new biomarkers for Alzheimer’s disease (AD) may help in tracking disease progression and identifying optimal patient-specific treatments. Although useful markers are available for diagnosis of AD, they are unreliable for tracking disease progression. Looking for better ways to track disease progression, Sun Ah Park at the Ajou University School of Medicine, Suwon, South Korea, and coworkers have reviewed alternative AD markers. They report that several markers for axonal degeneration, synaptic loss, brain inflammation and lipid metabolism show promise for tracking AD. Some of these markers can be obtained from blood samples, which are minimally invasive to collect. Use of combinations of markers is especially promising for estimating a patient’s disease stage. These results will contribute to developing tailored treatments for this common cause of dementia.
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder that eventually results in dementia. The initial pathologic definition of AD constitutes accumulations of amyloid-β (Aβ) and pathologically modified tau proteins to form senile plaque and neurofibrillary tangles, respectively, which are regarded as core pathologic features in AD1. The measurements of Aβ1-42 (Aβ42), total tau (tTau), and phosphorylated tau at Thr181 (pTau181) in cerebrospinal fluid (CSF), as well as the visualization of fibrillar Aβ protein loads in the brain using a radioactive ligand, have proven useful in the early diagnosis of AD, which leads to their inclusion in diagnostic guidelines2,3 and biological definitions of AD4. Current clinical trials use measurements of Aβ protein and tau proteins in CSF and/or blood to guide participant recruitment and outcome measures5. This practice permits the enrollment of patients with AD pathology, even at the preclinical stage, and allows monitoring of treatment effects on Aβ- and tau-pathology. However, early saturation of Aβ accumulation in the brain, indicated by plateaus in CSF Aβ42 levels and amyloid PET uptake after clinical symptom onset, limits the usefulness of Aβ biomarkers for monitoring disease progression and drug response6–8. Tau protein levels are more likely than Aβ to reflect the clinical status; however, their clinical correlations are also lost with the advancement of neurodegeneration, revealing stabilization or a reduction in protein levels8,9. The shortage of Aβ and tau biomarkers is a serious problem during successive failures of Aβ-targeting drug trials. Treatment-responsive improvements in Aβ biomarkers (e.g., reduced Aβ uptake on amyloid PET and increased levels of CSF Aβ)10–13 and tau biomarkers12 were not accompanied by clinical benefits13–16. This finding clearly showed that changes in Aβ and tau biomarkers are not reliable in terms of predicting disease progression and monitoring clinical status. Therefore, new biomarkers are needed to resolve this shortage. Ideally, new biomarkers should represent Aβ- and tau-independent AD pathology, thereby enabling monitoring of clinical and biological benefits during Aβ- and tau-targeting therapies, in which changes in Aβ- and tau-biomarkers are inevitable.
The heterogeneity of AD is a considerable obstacle for the development of efficient disease-modifying treatments and the establishment of ideal disease-tracking biomarkers. Large neuropathological studies have demonstrated that pure AD pathology is infrequent in elderly patients with cognitive decline17. The precise pathophysiology, which determines the probability of developing clinical symptoms of dementia, is diverse in terms of the presence of Aβ and tau pathology. The actual contribution of AD pathology to cognitive loss has been estimated to vary from 22 to 100%17. This suggests that Aβ and tau pathologies alone cannot sufficiently represent the clinical severity of AD. Considerations of other biomarkers that directly signify pathologic substrates indicative of cognitive dysfunction are necessary.
Overview of biological targets for non-Aβ and -tau biomarkers
Proteins related to various aspects of AD pathophysiological progression have been suggested as new fluid biomarkers in AD (Fig. 1). In addition to senile plaque and neurofibrillary tangles, dystrophic axons and dendrites surrounded by activated glial cells are abundant in the AD brain, which directly represent neurodegeneration and synapse loss18. Synapse loss is closely correlated with cognitive dysfunction18. Thus, neurodegeneration-related biomarkers may most closely indicate cognitive status. However, these biomarkers have received minimal attention in the field of AD biomarker development because they represent nonspecific and event-ending pathologies that are commonly observed in many neurodegenerative disorders. Nonetheless, accumulating evidence supports their clinical usefulness in diagnosis and clinical staging19–23.
Fig. 1. Overview of the pathophysiological process in Alzheimer’s disease.

BBB, blood–brain barrier; CSF, cerebrospinal fluid.
Although astrogliosis and neuroinflammation are prevalent features in the AD brain18, the roles of microglia and astrocytes in AD pathophysiology have received less attention than neurons. However, many AD-related risk genes identified in genome-wide association studies (e.g., ABCA7, CD33, CR1, EPHA1, MS4A, and TREM2) are reportedly expressed in microglia and involved in neuroinflammation24. Accumulating experimental data support the active role of neuroinflammation in AD pathogenesis; moreover, it is currently regarded as a target for the development of AD treatments25. Various markers signifying the activation of inflammatory brain cells and the release of neuroinflammation-modulating factors have been suggested as potential biomarkers.
The APOE ε4 allele is the strongest and most prevalent risk gene for AD24. The primary biological role of the apolipoprotein E (ApoE) protein is to transport lipids and regulate cholesterol metabolism26. Lipid homeostasis is important in the physiological functions of the brain, including cellular membrane function, synaptic integrity, neuronal regeneration, and neuronal plasticity27. The disturbed lipid metabolism in AD is evidenced by many lipid droplets within glial cells28, as well as altered lipid content and distribution29, which are expected to contribute to AD pathogenesis. Proteins involved in lipid metabolism have been suggested as biomarkers for the diagnosis and monitoring of disease progression in AD.
Late and sporadic onset occurs in more than 90% of patients with AD. While synthesis of Aβ is the primary problem in AD with genetic mutation, impaired abnormal protein clearance is the main pathogenesis in sporadic AD30. Inside neurons and other brain cells, abnormal protein burdens are diminished by the autophagy–lysosomal system, ubiquitin–proteasome system and chaperone-mediated autophagy to maintain intracellular homeostasis31. In the extracellular space, protein clearance is mediated via protease, phagocytosis by astrocytes and microglia and exportation through the glymphatic system and blood–brain barrier (BBB) into CSF and systemic circulation31. Therefore, checking protein degradation machinery-related proteins might provide information regarding disease status caused by abnormal protein accumulation.
The frequent coexistence of vascular pathology and other neurodegenerative disorders, such as TDP-43 (e.g., frontotemporal dementia) and α-synuclein proteinopathy (e.g., Parkinson’s disease, Lewy body dementia, and multiple system atrophy), makes the pathophysiology and clinical manifestations of AD more variable17. The identification of combined brain pathology is necessary to correctly estimate AD status and predict disease progression because enhanced neurodegeneration can result in augmented cognitive dysfunction when other brain disorders are added. Several fluid biomarkers specific to core pathologic proteins of other neurodegenerative disorders, such as TDP-43 and α-synuclein protein levels, have emerged as potential biomarkers in frontotemporal dementia and Lewy body dementia/Parkinson’s disease dementia, respectively32,33. This review does not extend to non-AD-specific biomarkers because these have not yet yielded consistent results32,34. Instead, the differential diagnostic values of new biomarkers are discussed in the context of AD vs. non-AD pathologies.
To develop novel biomarkers, two types of approaches have been conducted in large studies: targeted and nontargeted. The targeted approach uses hypothesis-driven methodology to verify candidate proteins that are preselected following basic experiments and bioinformatics analyses. In contrast, the nontargeted approach is purely data-driven. If candidate biomarkers are suggested by an explorative study, a subsequent validation study is needed using targeted measurement to confirm the validities of the biomarkers. This review is focused on new biomarkers (e.g., non-Aβ and non-tau biomarkers) for which the significance is supported by two or more independent studies involving different cohorts. Based on the most relevant biological pathways, candidate biomarkers are grouped and described for easy understanding (Fig. 2).
Fig. 2.
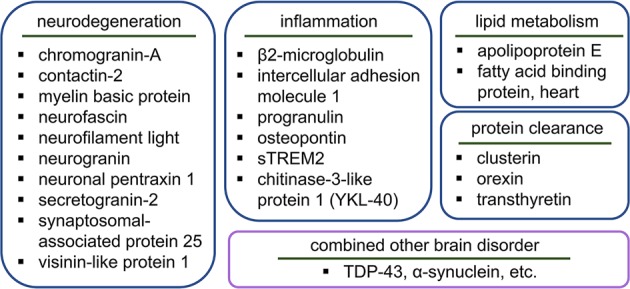
Overview of candidate non-Aβ and non-tau fluid biomarkers.
Neurodegeneration-related biomarkers: synaptic loss and axonal degeneration
Proteins that exhibit changing expression in CSF and blood during the development and progression of AD are valuable as disease-tracking biomarkers. Because neurodegeneration is inevitable in AD and increases with AD progression, many synapse- and axon-related protein levels in CSF or blood have been closely investigated in many studies. Several proteins have been identified as promising AD biomarkers (Table 1 and Supplementary Table 1).
Table 1.
Utility of new fluid biomarkers in Alzheimer’s disease.
| Early diagnosis | Specific diagnosis | Prediction | Correlation | |
|---|---|---|---|---|
| Neurodegeneration-related | ||||
| Chromogranin-A (CSF) |
↑MCI vs. CON ↓/→AD vs. CON |
NA | NA | ? Brain atrophy |
| Contactin-2 (CSF) |
↑MCI vs. CON ↑/↓AD vs. CON |
NA | NA | ? Cognition |
| Myelin basic protein (CSF) | ↑AD vs. CON | NA | NA | NA |
| Neurofascin (CSF) |
↑MCI vs. CON ↓AD vs CON |
NA | NA | NA |
| Neurofilament light (CSF) |
↑Preclinical AD vs. CON ↑MCI vs. CON ↑AD vs. CON |
Not specific |
Cognitive decline Brain atrophy |
Cognition Brain atrophy & hypometabolism |
| Neurofilament light (blood) |
↑preclinical AD vs. CON ↑preclinical MC vs. NC ↑MCI vs. CON ↑AD vs. CON |
Not specific |
Cognitive decline Brain atrophy |
Cognition Brain atrophy |
| Neurogranin (CSF) |
↑preclinical AD vs. CON ↑MCI vs. CON ↑AD vs. CON |
Specific to AD |
Cognitive decline Brain atrophy |
Cognition Brain atrophy |
|
Neurogranin (NDE in blood) |
↓/→AD vs. CON | ? Not specific | ? Cognitive decline in MCI | NA |
| Neuronal pentraxin 1 (CSF) |
↑MCI vs. CON ↓AD vs. CON |
NA | NA | NA |
| Secretogranin-2 (CSF) |
↑MCI vs. CON ↓AD vs. CON |
NA | ? Cognitive decline in MCI | NA |
| SNAP-25 (CSF) |
↑MCI-Aβ (+) vs. CON ↑AD vs. CON |
NA | NA | NA |
| VILIP-1 (CSF) |
↑MCI vs. CON in most ↑AD vs. CON in most |
Possible | Cognitive decline | Brain atrophy |
| Inflammation-related | ||||
| β2-microglobulin (CSF) | ↑MCI vs. CON | NA | ? Cognitive decline in MCI | NA |
| ICAM1 (CSF) |
↑ preclinical AD vs. CON ↑MCI vs. CON ↑AD vs. CON |
NA | ? Cognitive decline & its rapidity | Cognition |
| Progranulin (CSF) |
↑MC vs. NC ↑AD vs. preclinical AD |
Not specific | NA |
Cognition Brain atrophy Brain hypometabolism |
| Osteopontin (CSF) |
↑MCI vs. CON ↑AD vs. CON |
Controversial | Cognitive decline | ? Acuteness of cognitive dysfunction |
| sTREM2 (CSF) |
↑/→ preclinical AD vs. CON ↑MCI vs. CON ↑peak at MCI > AD > CON ↑AD vs. CON |
Not specific | NA |
Age No association with cognitive function |
| YKL-40 (CSF) |
↑preclinical AD vs. CON ↑MCI vs. CON ↑AD vs. CON ↑AD vs. MCI |
Not specific | Maybe cognitive decline |
Maybe cognition Gray matter atrophy Advancement of disease stage |
| YKL-40 (blood) | ↑AD vs. CON | Not specific | NA |
? Cognition Age |
| Lipid metabolism-related | ||||
| Apolipoprotein E (CSF) |
↓/↑AD vs. CON ↓AD vs. MCI |
Controversial | Cognitive decline in APOEε4 noncarriers | Brain atrophy in APOEε4 noncarriers |
| FABP3 (CSF) |
↑MCI vs. CON ↑AD vs. CON |
Controversial | Cognitive decline in MCI |
Cognition Brain atrophy |
| Protein clearance-related | ||||
| Clusterin (CSF) | ↑AD vs. CON | ? Not specific | NA | ? Cognition |
| Clusterin (blood) | → AD vs. CON | NA | NA | ? Cognition & brain atrophy |
| Orexin (CSF) |
↑MCI vs. CON ↑/→AD vs. CON |
? Possible | NA | NA |
| Transthyretin (CSF) | ↑/→AD vs. CON | Controversial | NA | NA |
| Transthyretin (blood) | ↓AD vs. CON | NA | NA | ? Rapidity & severity of cognitive decline |
AD Alzheimer’s disease, CON control, CSF cerebrospinal fluid, FABP-3 fatty acid binding protein, heart, ICAM1 intercellular adhesion molecule 1, MC mutation carrier, MCI mild cognitive impairment, NA not applicable due to lack of evidence, NC noncarrier, NDE neuron-derived exosome, SNAP-25 synaptosomal-associated protein 25, sTREM2 soluble triggering receptor expressed on myeloid cells 2, VILIP-1 visinin-like protein 1, YKL-40 chitinase-3-like protein 1.
↑, increased protein level; ↓, decreased protein level; →, no change in protein level; ?, not sure due to insufficient number of studies.
Neurofilament light polypeptide
Neurofilament light polypeptide (NFL) is the most abundant component of large myelinated axons, which is released into CSF and systemic circulation when neurodegeneration occurs35. NFL has been extensively examined in terms of its clinical utility, and many studies have demonstrated its high degree of usefulness in clinical applications (Supplementary Table 1). It has also been documented that CSF NFL levels are well correlated with plasma NFL levels, although the levels in plasma are 50-fold lower than those in CSF19,36,37. Both CSF and plasma NFL levels are increased in relation to AD progression, revealing a high degree of correlation with cognitive functions and a good predictive value for future cognitive decline20–22,36. However, in terms of the differentiation of AD from other neurodegenerative disorders, NFL levels are less likely to be beneficial. High plasma NFL levels were frequently observed in progressive supranuclear palsy38, frontotemporal dementia39, multiple system atrophy, and corticobasal degeneration40. The capability of plasma NFL levels to predict cognitive decline and cortical regional atrophy was noticeable in both progressive supranuclear palsy and in AD38. Therefore, NFL levels are currently regarded as representative of neurodegeneration itself, independent of Aβ and tau pathology, which can be useful for proper disease tracking in both AD and non-AD dementia. Increases in NFL levels are evident well before the clinical onset of cognitive impairments: a distinct increase in serum NFL in AD-causative mutation carriers was observed 16.2 years earlier than clinical symptoms began, according to the findings of the Dominant Inherited Alzheimer’s Disease Network study19. In a study of normal elderly individuals without cognitive impairment, increased CSF NFL levels were observed in those who developed cognitive decline during follow-up9,22. The measurement of NFL in body fluids, especially easily accessible blood, is therefore expected to be applicable for preventative screening of preclinical stages of AD.
Neurogranin
Neurogranin is a postsynaptic protein that is abundant in dendritic spines and plays a role in synaptic activity and plasticity. In contrast to NFL, which represents axonal degeneration, neurogranin signifies synaptic degeneration. In cross-sectional comparisons, many reports have demonstrated that neurogranin increases in CSF from patients with AD and patients with mild cognitive impairment (MCI) relative to that from healthy controls41–44 (Supplementary Table 1). Furthermore, increased CSF neurogranin is specific to AD among the various neurodegenerative disorders23,43,44. The value of CSF neurogranin for the prediction of future cognitive decline was also identified, although the direction differed among studies. Many studies have suggested that increased baseline neurogranin levels are indicative of future cognitive deterioration in patients with MCI20,41,42, while a few studies have suggested that low baseline neurogranin levels are indicative of future cognitive deterioration22. This discrepancy may be caused by dynamic changes in CSF neurogranin levels, depending on disease stage9, which has also been demonstrated for other synapse-related proteins (i.e., an early increase above and subsequent gradual reduction below the levels of controls, corresponding to disease progression)45. The early transient increase in CSF level is presumably due to the active degradation of synapses and compensatory enlargement of the remaining synapses18. Measurements of neurogranin levels in peripheral blood were performed, including neuron-derived exosomes; reduced levels of neurogranin in neuron-derived exosomes were reported in patients with AD compared with controls, but further validation is needed46.
Visinin-like protein 1
Visinin-like protein 1 (VILIP-1) is a neuronal calcium sensor protein that is exclusively expressed at high levels in neurons47. Its release into CSF and systemic circulation is regarded as a marker of neuronal injury. Increased levels of VILIP-1 in CSF have often been found in patients with AD compared with healthy controls and patients with other neurodegenerative disorders, such as Lewy body dementia, frontotemporal dementia, and progressive supranuclear palsy; this finding suggests that increased VILIP-1 levels may constitute a specific marker for AD (Supplementary Table 1). However, no differences in VILIP-1 levels between patients with AD and controls48 or between patients with AD and those with vascular dementia or frontotemporal dementia have been reported49. Considering that longitudinal reduction in VILIP-1 levels occurs with disease progression after the initial increase in AD9, the increased levels of VILIP-1 in CSF could be unclear at certain stages of advanced clinical disease, which could weaken the validity of VILIP-1 as a biomarker. Therefore, stage-dependent interpretation of VILIP-1 levels is needed. At early stages of AD, such as preclinical and MCI stages, high CSF VILIP-1 levels predict future cognitive decline48,50 and brain atrophy51.
Other candidate neurodegeneration-related biomarkers
Chromogranin-A and secretogranin-1 (also known as chromogranin-B) are well-known soluble components of large dense-core vesicles that play critical roles in the formation of secretory vesicles. Granin proteins are involved in various biological functions, including vasodilation, antiapoptosis, mast cell migration, microglial activation, neurotransmitter release, and synaptic function52. Altered CSF levels of granins are reportedly correlated with brain regional atrophy in patients with AD45,53,54. Chromogranin-A and secretogranin-1 exhibit characteristically dynamic changes in CSF expression according to disease stage in a manner similar to that of neurogranin: increased levels during early stages (i.e., MCI) and reduced levels during advanced dementia. Contactin-2 organizes the Ranvier nodes of axons and cell adhesion and was identified as a potential CSF biomarker in AD55. Myelin basic protein has a role in the formation and maintenance of the myelin sheath and was identified as a candidate CSF biomarker in AD and subcortical vascular disease56. Proteins involved in neurite outgrowth and synaptic stabilization, such as neurofascin and neuronal pentraxin 1, were suggested as CSF AD biomarkers45,54. Synaptosomal-associated protein 25, which has a role in neurotransmitter release, was also suggested as a candidate CSF biomarker9. These synapse-related proteins have the potential to be valuable disease-tracking biomarkers, as well as diagnostic biomarkers, considering that synapse loss is the factor most representative of clinical severity18.
Neuroinflammation-related biomarkers
Modulators of inflammation and markers of activated inflammatory cells have been suggested as new biomarkers in AD (Table 1 and Supplementary Table 2).
β2-Microglobulin and intercellular adhesion molecule 1
β2-Microglobulin is involved in the innate immune system through antigen presentation to the immune system57. Intercellular adhesion molecule 1 (ICAM1) is a cell-surface glycoprotein in endothelial cells and immune cells, which provides ligands that facilitate adhesion of leukocytes to endothelial cells; this allows leukocyte trafficking into the brain58. Both β2-microglobulin and ICAM1 in CSF are reportedly increased in patients with AD at the early, preclinical, and MCI stages45,59. Moreover, ICAM1 levels in CSF have been correlated with the severity of cognitive decline60.
Progranulin
Progranulin (encoded by the GRN gene) is a growth factor that is expressed in neurons and microglia and is released from these cells. Progranulin is involved in neuroinflammatory modulation, specifically toward reducing microgliosis and astrogliosis61; moreover, it enhances neuronal outgrowth and neuronal survival62. Its expression is highly increased during microglial activation and neuronal maturation. Because GRN gene mutations are pathogenic with respect to the development of frontotemporal dementia spectrum disorders, the relation of progranulin with AD has received less attention. However, clinical manifestations of GRN mutations can also extend to AD63. The possibility of using progranulin as an AD biomarker was recently investigated in a large population of patients with familial and late-onset sporadic AD (Dominant Inherited Alzheimer’s Disease Network and Alzheimer’s Disease Neuroimaging Initiative cohorts)64. The CSF levels of progranulin were reportedly increased 10 years before the clinical onset of disease in AD mutation carriers. In patients with sporadic AD, increased CSF levels of progranulin were evident when neurodegeneration developed64. Increased levels of progranulin in CSF were also detected in suspected non-AD pathophysiology (SNAP) cases (i.e., normal Aβ biomarkers despite abnormalities in tau or neurodegeneration biomarkers; A−/TN+4)64. However, correlations of CSF progranulin levels with cognitive functions and CSF tau protein levels were present only in patients with AD, not in patients with SNAP64.
Soluble triggering receptor expressed on myeloid cells 2 (sTREM2)
sTREM2 is an ectodomain of triggering receptor expressed on myeloid cells 2 (TREM2) that is released following proteolytic cleavage by α-secretases, disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and ADAM1765. TREM2 is expressed on the surface of microglia and is involved in innate immunity through modulation of microglial activity66. Several loss-of-function genetic variants of TREM2 have been shown to increase the risk of AD, including a variant at the His157 site, which affects the rate of release of sTREM2 into the extracellular space65,67. The role of sTREM2 in the progression of AD pathogenesis is under active investigation; it is potentially involved in modulating the survival and activity of microglia68,69. The released sTREM2 can be measured in both CSF and blood. Most studies have shown that increased levels of CSF sTREM are indicative of AD; however, a few studies showed no change in these levels in patients with AD (Supplementary Table 2). Longitudinal measurement in AD mutation carriers revealed altered CSF levels 5 years before expected clinical onset, when signs of brain amyloidosis and neurodegeneration were already obvious70. CSF sTREM2 levels were highest at the clinical stage of MCI, compared with other stages of AD71,72; increases in these levels were most pronounced immediately before the onset of dementia symptoms, when widespread neurodegeneration and synaptic loss were ongoing. Alterations in CSF sTREM2 levels were also identified in patients with other brain disorders, as well as in patients with SNAP71. Thus, there may not be a specific relationship between sTREM2 and AD. Extensive blood measurements of sTREM2 have rarely been performed; in the few studies involving these measurements, differences based on AD diagnosis were not evident, and whether there was a correlation with CSF sTREM2 levels was unclear73. A recent longitudinal follow-up study in Japan demonstrated that high serum sTREM2 levels were associated with future overall development of dementia, rather than AD or vascular dementia74. Additional studies with more refined measurement tools are needed to elucidate the precise value of blood sTREM2 levels as a biomarker for AD.
Chitinase-3-like protein 1 (YKL-40)
YKL-40 is a carbohydrate-binding protein that is secreted by activated macrophages and microglia. It is thought to be involved in the modulation of inflammation, migration of astrocytes, and remodeling of tissue. Increased expression of YKL-40 has mainly been identified in reactive astrocytes in various neurological disorders, including AD, which suggests that YKL-40 is important in the astrocyte response to disease-related environmental conditions75. Elevated YKL-40 levels in CSF have often been identified and suggested to represent AD-related increased inflammation and astrocytosis in the earlier stages of AD, such as MCI or subclinical disease (Supplementary Table 2). However, there may not be a specific relationship between YKL-40 levels and Aβ pathology because no differences in YKL-40 levels have been detected between patients with AD and patients with other neurodegenerative dementias, such as Lewy body dementia, vascular dementia, and frontotemporal dementia. The progression of clinical symptoms and brain cortical atrophy are more closely associated with increases in YKL-40 levels22,76,77. Therefore, increases in YKL-40 are presumably linked to a common pathway that results in neurodegeneration itself, rather than a specific disease process.
Other candidate inflammation-related biomarkers
In addition to the above candidates, other inflammation-related proteins have been suggested as possible biomarkers. Osteopontin is a glycophosphoprotein with roles in cell-matrix interactions and innate immunity. It is involved in inflammatory processes as a proinflammatory cytokine and modulates the activity of immune cells such as macrophages and microglia78. Increased CSF osteopontin levels have been reported in patients with AD and MCI compared with healthy controls (Supplementary Table 2). Larger increases in CSF osteopontin may represent disease progression and acute-phase disease79. However, reports have been contradictory in terms of specificity for AD. Levels of various complement proteins59, including fms-related tyrosine kinase60, fractalkine72, interleukin-1080, interleukin-1560, lysozyme C45, macrophage migration inhibitory factor53 and monocyte chemoattractant protein 172, are significantly altered in patients with AD. These findings should be further explored using refined measurement tools and large sample sizes of patients with AD at various stages to confirm their usefulness as AD biomarkers.
Lipid metabolism-related biomarkers
Lipid transport is essential for neuronal survival, synaptic activity and immune responses of glial cells in the brain81. Proteins linked to lipid metabolism have been suggested as candidate AD biomarkers (Table 1 and Supplementary Table 3). ApoE is involved in lipid homeostasis through regulation of the production, conversion, and clearance of lipoprotein, as well as in lipid transport via lipidation and subsequent binding to cell-surface receptors (e.g., LDL receptor family members)82. The presence of ApoE4 isoforms is known to increase AD risk due to the altered physiological function of the ApoE protein in the brain82. Measurements of ApoE protein levels in CSF have been performed in relation to the diagnosis of AD; notably, contradictory results have been demonstrated (Supplementary Table 3). ApoE protein levels have been suggested for use in the differential diagnosis of AD from Lewy body dementia and other disorders; however, further validation is needed due to the lack of supporting evidence.
Heart fatty acid-binding protein (FABP3) is another lipid-binding protein that plays a role in lipid transport. FABP3 is released from myocytes during the early stages of myocardial infarction; thus, blood levels of FABP3 constitute a useful biomarker for early diagnosis of heart attack83. In patients with AD, FABP3 levels in CSF have been reported to increase as early as the MCI stage (Supplementary Table 3). Higher baseline levels of FABP3 in patients with MCI could predict conversion to AD during follow-up84. FABP3 levels exhibit a weak ability to discriminate AD from other brain disorders involving dementia. However, consideration of pTau181 CSF levels in combination with FABP3 levels has been shown to increase the accuracy of differentiating AD from Lewy body dementia85.
Based on the results of a nonbiased proteomic study and subsequent quantitative selective reaction monitoring that included small numbers of CSF samples from patients with AD, Parkinson’s disease, Lewy body dementia, and nonneurodegenerative conditions, levels of other lipid metabolism-linked proteins (e.g., beta-2-glycoprotein 1 [also known as apolipoprotein H], ectonucleotide pyrophosphatase/phosphodiesterase family member 2 [also known as autotaxin], prosaposin, and vitamin D-binding protein) were reportedly increased in the CSF of patients with AD and have been suggested as possible biomarkers in AD86.
Biomarkers related to the clearance of neurotoxic proteins
Proteins related to the degradation and removal of abnormal proteins, Aβ and pathologic tau proteins, have been suggested as candidate biomarkers (Table 1 and Supplementary Table 4). Clusterin is a secretory glycoprotein, is mainly produced by astrocytes in the brain87, and serves as a molecular chaperone; it binds to partially unfolded proteins, thereby preventing their aggregation88. Clusterin also has a neuronal differentiation-promoting effect and neuroprotective properties87. Missense and small deletion polymorphisms in the clusterin gene increase the risk of AD24; these findings suggest that it plays a role in AD pathology. In CSF, levels of clusterin were reported to increase in patients with AD and Lewy body dementia and were correlated with cognitive decline.
Orexin is a neuropeptide that regulates circadian rhythm and related physiological homeostasis89. Its reduction in CSF is a well-established biomarker for narcolepsy. In relation to AD, altered levels of orexin in CSF have generally been reported to increase, with a few exceptions (Supplementary Table 4). Furthermore, infusion of orexin from patients with AD led to increased Aβ production and amyloid deposition in human AβPP transgenic mice carrying the Swedish mutation, which suggested that it has a pathophysiological role in AD90.
Transthyretin is an Aβ-binding molecule that has been reported to inhibit Aβ aggregation, thereby reducing Aβ-induced cellular toxicity91. It has been reported to increase or remain stable in CSF, whereas it has been reported to decrease in blood and in patients with AD who exhibited rapid and severe cognitive decline (Supplementary Table 4). In addition, cystatin C, GM2 ganglioside activator, LAMP-1, ubiquitin, ubiquitin carboxyl-terminal esterase L1, carboxypeptidase E, carnosine dipeptidase 1, and ectonucleotide pyrophosphatase/phosphodiesterase were found to be higher in the CSF of patients with AD than in that of controls86. Matrix metalloproteinase proteins are potentially altered in the CSF of patients with AD56.
The combinations of new biomarkers
The simultaneous consideration of new biomarkers is beneficial in concurrent assessment of various aspects of AD pathophysiology, thereby correctly estimating disease status92. There have been multiple recent investigations to establish reliable and useful combinations of CSF biomarkers (Table 2). The co-consideration of neurodegeneration and inflammation markers, neurogranin and YKL-40, improved the accuracy of differential diagnosis of AD from non-AD dementia, achieving an area under the receiver operating characteristic curve of 85%93. The co-consideration of neurogranin and NFL levels (neurogranin represents synaptic damage while NFL reflects axonal damage) demonstrated improved diagnostic accuracy of AD relative to that of each marker alone20; both proteins showed significant predictive associations with cognitive decline and brain atrophy. However, NFL levels were significantly associated with cognitive decline and brain atrophy in all patients, regardless of amyloid pathology, while neurogranin levels were significantly associated with cognitive decline and brain atrophy only in patients with amyloid pathology20. When both biomarkers were compared directly, NFL levels were superior to neurogranin as a prognostic biomarker in MCI patients with positive Aβ biomarker results94 and in normal elderly individuals (mean age, 59.3 ± 6.3 years)95.
Table 2.
Sets of new biomarker combinations.
| Combination | Findings |
|---|---|
| CSF neurogranin, YKL-4093 |
▪ Both increased in AD, but not correlated with each other: represent different pathology in AD ▪ Higher differential diagnostic value of neurogranin than YKL-40, AD vs. non-AD (85% of AUC) |
|
CSF chromogranin-A, FABP-3, matrix metalloproteinase-2, pancreatic polypeptide levels + regional brain volume on MRI + CSF Aβ42, pTau181, tTau levels53 |
▪ Improved accuracy in the prediction of MCI conversion to AD on 12 m FU (95% accuracy) when combined |
|
CSF neurogranin, NFL + CSF tTau levels20 |
▪ Improved diagnostic accuracy in AD vs. CON when combined (neurogranin, NFL, tTau), showing highest AUC (85.5%) ▪ tTau and neurogranin: strongly associated with cognitive decline and brain atrophy in case of Aβ (+) on 2 yr FU ▪ NFL: associated with cognitive decline and brain atrophy independent of Aβ pathology, in Aβ (+)/(−) on 2 yr FU |
| CSF FABP-3, IL-10, NFL80 |
▪ Different pattern over longitudinal change: (1) increased FABP-3: more sensitive to milder AD stages, (2) increased IL-10: associated with rate of longitudinal cognitive decline at MCI stage, (3) increased NFL: most strongly associated with the dementia stage of AD ▪ These are complementary to each other in AD clinical staging |
| CSF neurogranin, NFL94 | ▪ NFL: highest accuracy in prediction of MCI conversion to AD compared to neurogranin, Aβ42, pTau181, and tTau levels, on >1 yr FU |
| CSF neurogranin, SNAP-25, VILIP-1, YKL-409,98 |
▪ Different pattern in longitudinal change, on 1-7 yr FU-LP ▪ Complementary to each other in AD clinical staging ▪ Combination of baseline Aβ42, neurogranin, SNAP-25, VILIP-1 and YKL-40; combination of baseline pTau, neurogranin and SNAP-25: good correlation with baseline cognition in MC ▪ Combination of baseline Aβ42, tTau, neurogranin, SNAP-25 and VILIP-1: prediction of EYO |
|
CSF neurogranin, NFL + CSF tTau levels95 |
▪ NFL: stronger correlation with cognitive decline at FU than neurogranin and tTau levels |
| CSF clusterin, fractalkine, MCP-1, sTREM2, YKL-4071 |
▪ All increased in subjects with neurodegeneration ▪ Different starting time of level change: (1) sTREM2 from subclinical stage, (2) MCP-1 from MCI stage, (3) YKL-40 and clusterin from dementia stage |
|
CSF neurogranin, NFL, YKL-40 + CSF tTau22 |
▪ Different prediction accuracy of cognitive decline depending on clinical stage (2.3 yr FU at mean): (1) in CON-Aβ (+) group: high baseline NFL levels predicts cognitive decline. (2) in MCI-Aβ (+) group: high baseline NFL and tTau and decreased neurogranin levels can predict cognitive decline. (3) in AD-Aβ (+) group: increased baseline NFL and neurogranin levels can predict cognitive decline. (4) in MCI-Aβ (−) group: increased baseline NFL and tTau levels can predict cognitive decline. |
AD Alzheimer’s disease, AUC area under the curve, CON control, CSF cerebrospinal fluid, EYO expected year of onset of AD in mutation carrier, FABP-3 fatty acid binding protein, heart, FU follow-up, IL-10 interleukin-10, LP lumbar puncture, m month, mc mutation carrier, MCI mild cognitive impairment, MCP-1 monocyte chemoattractant protein 1, MRI magnetic resonance imaging, NFL neurofilament light polypeptide, SNAP-25 synaptosomal-associated protein 25, sTREM2 soluble triggering receptor expressed on myeloid cells 2, tTau total tau protein, pTau phosphorylated tau protein, VILIP-1 visinin-like protein 1, YKL-40 chitinase-3-like protein 1, yr year.
Aβ (+)/(−), positive (+) or negative (−) Aβ biomarker, either on CSF or amyloid positron emission tomography (PET).
Combinations of synaptic degeneration markers (e.g., neurogranin, synaptosomal-associated protein 25, and VILIP-1) with the inflammation marker YKL-40 revealed a remarkable and differential longitudinal change across the clinical spectrum of AD patients9. This finding suggests that specific biomarkers become more useful at particular disease stages9,72,80. Therefore, synaptic, axonal, and inflammation-related biomarkers are complementary to each other in terms of staging AD and predicting clinical progression, in addition to representing different aspects of AD-related brain pathology.
Perspective on the utility of new biomarkers
Proper disease-tracking and clinical trial design
The staging of AD has mainly relied on clinical data, cognitive impairment, and activities of daily living, which can divide AD into the following stages: preclinical AD, MCI due to AD (or prodromal AD), and AD dementia2,3. Recently, the A/T/N system has provided more precise AD staging based on AD-related pathological processes, i.e., the deposition of Aβ (A) and abnormal tau (T) proteins and neurodegeneration (N)4. For clinical application of the A/T/N system, sufficient evidence supporting the utility of CSF and neuroimaging biomarkers is needed, including CSF Aβ42, the CSF Aβ42/Aβ40 ratio and amyloid PET for A; CSF pTau and Tau PET for T; and structural MRI, fluorodeoxyglucose PET and CSF tTau levels for N4. Since its introduction, new biomarkers to support the A/T/N system have been required4. For N, which relies on CSF tTau levels (which can be affected by tau pathology), the identification of new biomarkers of neurodegeneration, such as CSF NFL and neurogranin levels, may now be imminent. In many studies, CSF NFL better reflected clinical severity and predicted future cognitive decline more accurately than Aβ and tau proteins22,94,95. Therefore, CSF NFL could improve our ability to track disease progression.
Characterizing AD-related pathophysiology through biomarkers linked to diverse aspects of AD pathophysiology (e.g., neurodegeneration, synaptic dysfunction, neuroinflammation, lipid dysmetabolism and disturbed protein clearance) would be helpful for predicting the progression of individual facets of the pathology and for understanding their relative contributions to clinical deterioration96. A comprehensive understanding of disease status will aid in the selection of patients who are most likely to have a favorable response to specific disease-modifying therapies. Since other brain disorders commonly cooccur with AD7, considering both disease-specific and more general biomarkers of pathology may increase the likelihood of realizing efficient disease-modifying therapeutics. This approach could lead to more efficient treatment regimens and allow monitoring of the response to treatment on an individual patient basis, which is important given the diversity of AD pathology. Such precision medicine can maximize the therapeutic effect (Fig. 3).
Fig. 3. Perspectives on future clinical utility of new biomarkers.

AD, Alzheimer’s disease; MCI, mild cognitive impairment.
Readily accessible blood biomarkers
CSF is continuous with the interstitial fluid of the brain and directly reflects chemical changes in the brain. Most fluid biomarkers that have proven valuable for the diagnosis or monitoring of AD have been measured in CSF. Lumbar puncture, which is necessary to obtain CSF, is generally safe and well tolerated; however, it is time consuming and sometimes results in postpuncture headache and other side effects97. There are increasing efforts to develop easily assessable fluid biomarkers (e.g., from peripheral sources). Specifically, blood levels of NFL have shown strong evidence of usefulness, as described above, which are well correlated with CSF levels of NFL. With advances in ultrasensitive measurement tools, additional useful biomarkers from peripheral sources are likely to be identified. These promising peripheral fluid biomarkers will aid in increasing the efficiency of clinical trials with fewer costs and difficulties and provide mass screening of early AD for prevention.
Concluding remarks
The value of new biomarkers demonstrates that the consideration of diverse AD pathophysiology (e.g., other than Aβ- and tau-centered aspects, such as neurodegeneration, synaptic dysfunction, neuroinflammation, lipid dysmetabolism, and disturbed protein clearance) would help to develop useful disease-tracking AD biomarkers. Neurodegeneration-related biomarkers represent axonal injury, synaptic dysfunction, or synaptic loss. Early diagnostic and prognostic values of these markers are well established. However, in terms of the differentiation of AD from other brain disorders, synaptic proteins (e.g., neurogranin and VILIP-1) are useful, whereas axonal injury markers (e.g., NFL) are not. However, NFL is excellent for predicting disease progression and tracking disease severity. Therefore, co-consideration of NFL with AD-specific biomarkers may be complementary and increase clinical utility. Neuroinflammation is regarded as a main component of AD pathophysiology. CSF levels of ICAM1, progranulin, sTREM2, and YKL-40 are useful in early AD diagnosis. However, their specificities for AD are controversial, and disease-tracking characteristics are unclear for some biomarkers. In particular, dynamic changes in the levels of sTREM2 based on clinical stage make its value unclear. Stage-dependent changes in biomarkers are also prevalent among synaptic markers during longitudinal assessment, which complicates the clinical applications of these biomarkers; however, these changes provide clues that can be used for disease staging and tracking following appropriate interpretation. Lipid metabolism-related biomarkers (e.g., ApoE and FABP3) and several protein clearance-related markers may have advantages, but more convincing data are needed for some of them. Considering that these mechanisms are intimately related to AD pathogenesis and are candidate targets of disease-modifying AD therapeutics, some of these biomarkers are expected to be useful in the future after more evidence is obtained.
The ideal combination of new biomarkers for enhanced clinical application has not yet been determined; however, the simultaneous incorporation of neurodegeneration- and neuroinflammation-related biomarkers is likely to be optimal. For NFL, neurogranin, VILIP-1, YKL-40, and FABP3 levels in CSF and for NFL levels in blood, there is substantial evidence supporting their value as diagnostic and prognostic biomarkers in patients with AD. In the near future, some of these new biomarkers may be incorporated into diagnostic criteria and research frameworks for AD, in combination with the current Aβ and tau biomarkers, to improve the staging and prediction of disease progression in patients with AD. Consideration of new biomarkers that represent different pathological changes would aid in the precise application of disease-modifying therapies for the most susceptible individuals, as well as comprehensive monitoring of the treatment response. These would jointly contribute to the development of efficient disease-modifying treatments, which cleverly target AD pathophysiology in the appropriate patients at the correct stage of disease.
Supplementary information
Supplementary table 1, table 2, table 3, table 4
Acknowledgements
This work was supported by the Basic Science Research Program (NRF-2018R1A2B6009439 and 2019R1A5A2026045) and the Original Technology Research Program for Brain Science (NRF-2018M3C7A1056293) of the National Research Foundation of Korea (NRF) funded by the Korean government, MSIT.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Sources of support for the work: This work was supported by the Basic Science Research Program (NRF-2018R1A2B6009439 and 2019R1A5A2026045) and the Original Technology Research Program for Brain Science (NRF-2018M3C7A1056293) of the National Research Foundation of Korea (NRF) funded by the Korean government, MSIT.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s12276-020-0418-9.
References
- 1.Hyman BT, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKhann GM, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dubois B, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–629. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- 4.Jack CR, Jr., et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mattsson N, et al. Revolutionizing Alzheimer’s disease and clinical trials through biomarkers. Alzheimers Dement. 2015;1:412–419. doi: 10.1016/j.dadm.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bateman RJ, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Bastard N, et al. Longitudinal stability of cerebrospinal fluid biomarker levels: fulfilled requirement for pharmacodynamic markers in Alzheimer’s disease. J. Alzheimers Dis. 2013;33:807–822. doi: 10.3233/JAD-2012-110029. [DOI] [PubMed] [Google Scholar]
- 8.Toledo JB, Xie SX, Trojanowski JQ, Shaw LM. Longitudinal change in CSF Tau and Abeta biomarkers for up to 48 months in ADNI. Acta Neuropathol. 2013;126:659–670. doi: 10.1007/s00401-013-1151-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sutphen CL, et al. Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer’s disease. Alzheimers Dement. 2018;14:869–879. doi: 10.1016/j.jalz.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rinne JO, et al. 11C-PiB P. E. T. assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 11.Farlow M, et al. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 2012;8:261–271. doi: 10.1016/j.jalz.2011.09.224. [DOI] [PubMed] [Google Scholar]
- 12.Blennow K, et al. Effect of immunotherapy with bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer disease. Arch. Neurol. 2012;69:1002–1010. doi: 10.1001/archneurol.2012.90. [DOI] [PubMed] [Google Scholar]
- 13.Egan MF, et al. Randomized trial of verubecestat for prodromal Alzheimer’s disease. N. Engl. J. Med. 2019;380:1408–1420. doi: 10.1056/NEJMoa1812840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doody RS, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014;370:311–321. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- 15.Salloway S, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014;370:322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honig LS, et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N. Engl. J. Med. 2018;378:321–330. doi: 10.1056/NEJMoa1705971. [DOI] [PubMed] [Google Scholar]
- 17.Boyle PA, et al. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann. Neurol. 2018;83:74–83. doi: 10.1002/ana.25123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Preische O, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med. 2019;25:277–283. doi: 10.1038/s41591-018-0304-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mattsson N, et al. Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer’s disease. EMBO Mol. Med. 2016;8:1184–1196. doi: 10.15252/emmm.201606540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mattsson N, Cullen NC, Andreasson U, Zetterberg H, Blennow K. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019;76:791–799. doi: 10.1001/jamaneurol.2019.0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bos I, et al. Cerebrospinal fluid biomarkers of neurodegeneration, synaptic integrity, and astroglial activation across the clinical Alzheimer’s disease spectrum. Alzheimers Dement. 2019;15:644–654. doi: 10.1016/j.jalz.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Portelius E, et al. Cerebrospinal fluid neurogranin concentration in neurodegeneration: relation to clinical phenotypes and neuropathology. Acta Neuropathol. 2018;136:363–376. doi: 10.1007/s00401-018-1851-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuyvers E, Sleegers K. Genetic variations underlying Alzheimer’s disease: evidence from genome-wide association studies and beyond. Lancet Neurol. 2016;15:857–868. doi: 10.1016/S1474-4422(16)00127-7. [DOI] [PubMed] [Google Scholar]
- 25.Ardura-Fabregat A, et al. Targeting neuroinflammation to treat Alzheimer’s disease. CNS Drugs. 2017;31:1057–1082. doi: 10.1007/s40263-017-0483-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 27.Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol. Life Sci. 2003;60:1158–1171. doi: 10.1007/s00018-003-3018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige erkankung der hirnrinde”. Clin. Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 29.Wong MW, et al. Dysregulation of lipids in Alzheimer’s disease and their role as potential biomarkers. Alzheimers Dement. 2017;13:810–827. doi: 10.1016/j.jalz.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 30.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 31.Boland B, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018;17:660–688. doi: 10.1038/nrd.2018.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feneberg E, Gray E, Ansorge O, Talbot K, Turner MR. Towards a TDP-43-based biomarker for ALS and FTLD. Mol. Neurobiol. 2018;55:7789–7801. doi: 10.1007/s12035-018-0947-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldman JG, et al. Cerebrospinal fluid, plasma, and saliva in the BioFIND study: relationships among biomarkers and Parkinson’s disease features. Mov. Disord. 2018;33:282–288. doi: 10.1002/mds.27232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parnetti L, et al. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 2019;18:573–586. doi: 10.1016/S1474-4422(19)30024-9. [DOI] [PubMed] [Google Scholar]
- 35.Zetterberg H, et al. Association of cerebrospinal fluid neurofilament light concentration with alzheimer disease progression. JAMA Neurol. 2016;73:60–67. doi: 10.1001/jamaneurol.2015.3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mattsson N, Andreasson U, Zetterberg H, Blennow K. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2017;74:557–566. doi: 10.1001/jamaneurol.2016.6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pereira JB, Westman E, Hansson O. Association between cerebrospinal fluid and plasma neurodegeneration biomarkers with brain atrophy in Alzheimer’s disease. Neurobiol. Aging. 2017;58:14–29. doi: 10.1016/j.neurobiolaging.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 38.Rojas JC, et al. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann. Clin. Transl. Neurol. 2016;3:216–225. doi: 10.1002/acn3.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rohrer JD, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016;87:1329–1336. doi: 10.1212/WNL.0000000000003154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hansson O, et al. Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology. 2017;88:930–937. doi: 10.1212/WNL.0000000000003680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kester MI, et al. Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol. 2015;72:1275–1280. doi: 10.1001/jamaneurol.2015.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kvartsberg H, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement. 2015;11:1180–1190. doi: 10.1016/j.jalz.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 43.Janelidze S, et al. Cerebrospinal fluid neurogranin and YKL-40 as biomarkers of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2016;3:12–20. doi: 10.1002/acn3.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wellington H, et al. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology. 2016;86:829–835. doi: 10.1212/WNL.0000000000002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duits FH, et al. Synaptic proteins in CSF as potential novel biomarkers for prognosis in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2018;10:5. doi: 10.1186/s13195-017-0335-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goetzl EJ, et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016;30:4141–4148. doi: 10.1096/fj.201600816R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Braunewell KH. The visinin-like proteins VILIP-1 and VILIP-3 in Alzheimer’s disease-old wine in new bottles. Front. Mol. Neurosci. 2012;5:20. doi: 10.3389/fnmol.2012.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kester MI, et al. Cerebrospinal fluid VILIP-1 and YKL-40, candidate biomarkers to diagnose, predict and monitor Alzheimer’s disease in a memory clinic cohort. Alzheimers Res. Ther. 2015;7:59. doi: 10.1186/s13195-015-0142-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Babic Leko M, Borovecki F, Dejanovic N, Hof PR, Simic G. Predictive value of cerebrospinal fluid visinin-like protein-1 levels for Alzheimer’s disease early detection and differential diagnosis in patients with mild cognitive impairment. J. Alzheimers Dis. 2016;50:765–778. doi: 10.3233/JAD-150705. [DOI] [PubMed] [Google Scholar]
- 50.Tarawneh R, et al. Visinin-like protein-1: diagnostic and prognostic biomarker in Alzheimer disease. Ann. Neurol. 2011;70:274–285. doi: 10.1002/ana.22448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tarawneh R, et al. Cerebrospinal fluid markers of neurodegeneration and rates of brain atrophy in early Alzheimer disease. JAMA Neurol. 2015;72:656–665. doi: 10.1001/jamaneurol.2015.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bartolomucci A, Pasinetti GM, Salton SR. Granins as disease-biomarkers: translational potential for psychiatric and neurological disorders. Neuroscience. 2010;170:289–297. doi: 10.1016/j.neuroscience.2010.06.057. [DOI] [PubMed] [Google Scholar]
- 53.Khan W, et al. A subset of cerebrospinal fluid proteins from a multi-analyte panel associated with brain atrophy, disease classification and prediction in Alzheimer’s disease. PLoS ONE. 2015;10:e0134368. doi: 10.1371/journal.pone.0134368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brinkmalm, G. et al. A parallel reaction monitoring mass spectrometric method for analysis of potential CSF biomarkers for Alzheimer’s disease. Proteomics Clin. Appl. 10.1002/prca.201700131 (2018). [DOI] [PubMed]
- 55.Chatterjee M, et al. Contactin-2, a synaptic and axonal protein, is reduced in cerebrospinal fluid and brain tissue in Alzheimer’s disease. Alzheimers Res. Ther. 2018;10:52. doi: 10.1186/s13195-018-0383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bjerke M, et al. Cerebrospinal fluid matrix metalloproteinases and tissue inhibitor of metalloproteinases in combination with subcortical and cortical biomarkers in vascular dementia and Alzheimer’s disease. J. Alzheimers Dis. 2011;27:665–676. doi: 10.3233/JAD-2011-110566. [DOI] [PubMed] [Google Scholar]
- 57.Zijlstra M, et al. Beta 2-microglobulin deficient mice lack CD4-8+ cytolytic T cells. Nature. 1990;344:742–746. doi: 10.1038/344742a0. [DOI] [PubMed] [Google Scholar]
- 58.Dietrich JB. The adhesion molecule ICAM-1 and its regulation in relation with the blood-brain barrier. J. Neuroimmunol. 2002;128:58–68. doi: 10.1016/s0165-5728(02)00114-5. [DOI] [PubMed] [Google Scholar]
- 59.Simonsen AH, et al. Novel panel of cerebrospinal fluid biomarkers for the prediction of progression to Alzheimer dementia in patients with mild cognitive impairment. Arch. Neurol. 2007;64:366–370. doi: 10.1001/archneur.64.3.366. [DOI] [PubMed] [Google Scholar]
- 60.Janelidze S, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91:e867–e877. doi: 10.1212/WNL.0000000000006082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahmed Z, et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 2010;177:311–324. doi: 10.2353/ajpath.2010.090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van Damme P, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rademakers R, et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C–>T (Arg493X) mutation: an international initiative. Lancet Neurol. 2007;6:857–868. doi: 10.1016/S1474-4422(07)70221-1. [DOI] [PubMed] [Google Scholar]
- 64.Suáarez-Calvet M, et al. CSF progranulin increases in the course of Alzheimer’s disease and is associated with sTREM2, neurodegeneration and cognitive decline. EMBO Mol. Med. 2018;10:e9712. doi: 10.15252/emmm.201809712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thornton P, et al. TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol. Med. 2017;9:1366–1378. doi: 10.15252/emmm.201707673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neumann H, Takahashi K. Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J. Neuroimmunol. 2007;184:92–99. doi: 10.1016/j.jneuroim.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 67.Guerreiro R, et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhong L, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017;214:597–607. doi: 10.1084/jem.20160844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhong L, et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 2019;10:1365. doi: 10.1038/s41467-019-09118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suáarez-Calvet M, et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med. 2016;8:369ra178. doi: 10.1126/scitranslmed.aag1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Suáarez-Calvet M, et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016;8:466–476. doi: 10.15252/emmm.201506123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nordengen K, et al. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflammation. 2019;16:46. doi: 10.1186/s12974-019-1399-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Piccio L, et al. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016;131:925–933. doi: 10.1007/s00401-016-1533-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ohara T, et al. Serum soluble triggering receptor expressed on myeloid cells 2 as a biomarker for incident dementia: the Hisayama study. Ann. Neurol. 2019;85:47–58. doi: 10.1002/ana.25385. [DOI] [PubMed] [Google Scholar]
- 75.Bonneh-Barkay D, Wang G, Starkey A, Hamilton RL, Wiley CA. In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J. Neuroinflammation. 2010;7:34. doi: 10.1186/1742-2094-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gispert JD, et al. CSF YKL-40 and pTau181 are related to different cerebral morphometric patterns in early AD. Neurobiol. Aging. 2016;38:47–55. doi: 10.1016/j.neurobiolaging.2015.10.022. [DOI] [PubMed] [Google Scholar]
- 77.Gispert JD, et al. The APOE epsilon4 genotype modulates CSF YKL-40 levels and their structural brain correlates in the continuum of Alzheimer’s disease but not those of sTREM2. Alzheimers Dement. 2017;6:50–59. doi: 10.1016/j.dadm.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lund SA, Giachelli CM, Scatena M. The role of osteopontin in inflammatory processes. J. Cell Commun. Signal. 2009;3:311–322. doi: 10.1007/s12079-009-0068-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun Y, et al. Elevated osteopontin levels in mild cognitive impairment and Alzheimer’s disease. Mediators Inflamm. 2013 doi: 10.1155/2013/615745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gangishetti U, et al. Non-beta-amyloid/tau cerebrospinal fluid markers inform staging and progression in Alzheimer’s disease. Alzheimers Res. Ther. 2018;10:98. doi: 10.1186/s13195-018-0426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 2019;15:501–518. doi: 10.1038/s41582-019-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012;2:a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McMahon CG, et al. Diagnostic accuracy of heart-type fatty acid-binding protein for the early diagnosis of acute myocardial infarction. Am. J. Emerg. Med. 2012;30:267–274. doi: 10.1016/j.ajem.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 84.Olsson B, et al. Cerebrospinal fluid levels of heart fatty acid binding protein are elevated prodromally in Alzheimer’s disease and vascular dementia. J. Alzheimers Dis. 2013;34:673–679. doi: 10.3233/JAD-121384. [DOI] [PubMed] [Google Scholar]
- 85.Chiasserini D, et al. Differential role of CSF fatty acid binding protein 3, alpha-synuclein, and Alzheimer’s disease core biomarkers in Lewy body disorders and Alzheimer’s dementia. Alzheimers Res. Ther. 2017;9:52. doi: 10.1186/s13195-017-0276-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Heywood WE, et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol. Neurodegener. 2015;10:64. doi: 10.1186/s13024-015-0059-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cordero-Llana O, et al. Clusterin secreted by astrocytes enhances neuronal differentiation from human neural precursor cells. Cell Death. Differ. 2011;18:907–913. doi: 10.1038/cdd.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kumita JR, et al. The extracellular chaperone clusterin potently inhibits human lysozyme amyloid formation by interacting with prefibrillar species. J. Mol. Biol. 2007;369:157–167. doi: 10.1016/j.jmb.2007.02.095. [DOI] [PubMed] [Google Scholar]
- 89.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 90.Kang JE, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Costa R, Goncalves A, Saraiva MJ, Cardoso I. Transthyretin binding to A-Beta peptide-impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008;582:936–942. doi: 10.1016/j.febslet.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 92.Dhiman K, Blennow K, Zetterberg H, Martins RN, Gupta VB. Cerebrospinal fluid biomarkers for understanding multiple aspects of Alzheimer’s disease pathogenesis. Cell Mol. Life Sci. 2019;76:1833–1863. doi: 10.1007/s00018-019-03040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hellwig K, et al. Neurogranin and YKL-40: independent markers of synaptic degeneration and neuroinflammation in Alzheimer’s disease. Alzheimers Res. Ther. 2015;7:74. doi: 10.1186/s13195-015-0161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kern S, et al. Association of cerebrospinal fluid neurofilament light protein with risk of mild cognitive impairment among individuals without cognitive impairment. JAMA Neurol. 2019;76:187–193. doi: 10.1001/jamaneurol.2018.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Merluzzi AP, et al. Differential effects of neurodegeneration biomarkers on subclinical cognitive decline. Alzheimers Dement. 2019;5:129–138. doi: 10.1016/j.trci.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Molinuevo JL, et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018;136:821–853. doi: 10.1007/s00401-018-1932-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moulder KL, et al. Factors influencing successful lumbar puncture in Alzheimer research. Alzheimer Dis. Assoc. Disord. 2017;31:287–294. doi: 10.1097/WAD.0000000000000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schindler SE, et al. Emerging cerebrospinal fluid biomarkers in autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2019;15:655–665. doi: 10.1016/j.jalz.2018.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1, table 2, table 3, table 4