

Abstract
Cancer-promoting inflammation is an important event in cancer development. Canine urothelial carcinoma (cUC) overexpresses prostaglandin E2 (PGE2) and has a unique sensitivity to cyclooxygenase 2 (COX2)-inhibiting therapy. In addition, majority of cUC harbour BRAFV595E mutation. However, mechanisms underlying aberrant PGE2 production in BRAFV595E cUC patients remain unclear. Drug screening revealed that inhibition of RAF/MEK/ERK pathway, p38 and JNK pathway reduced PGE2 production in cUC cells. By pharmacological inhibition of the multiple components in the pathway, activation of the ERK MAPK pathway was shown to mediate overexpression of COX2 and production of PGE2 in BRAFV595E cUC cells. In silico gain-of-function analysis of the BRAF mutation also implicated involvement of mutation in the process. The positive association between ERK activation and COX2 expression was further validated in the clinical patients. Moreover, it was also suggested that p38 and JNK regulates PGE2 production independently of ERK pathway, possibly through COX2-dependent and COX1-/COX2- independent manner, respectively. In conclusion, this study demonstrated that activation of ERK induces production of PGE2 in BRAFV595E cUC cells, which is also independently regulated by p38 and JNK. With its unique vulnerability to COX-targeted therapy, BRAFV595E cUC may serve as a valuable model to study the tumour-promoting inflammation.
Subject terms: Bladder cancer, Bladder cancer
Introduction
Inflammation occurring in cancer tissues, referred to as cancer-promoting inflammation, promotes cancer progression by providing various factors, such as growth factors, pro-angiogenic factors, enzymes required for cancer invasion and metastasis, and immune-suppressive factors1,2. Being recognised as one of the hallmarks of cancer, cancer-promoting inflammation is a promising target for cancer therapy1. Among various processes causing cancer-promoting inflammation, the pathway of cyclooxygenase 2 (COX2) and its metabolite prostaglandin E2 (PGE2) has been widely accepted to be important in human cancers2. COXs are rate-limiting enzymes required for the biosynthesis of PGs in the arachidonic acid cascade, and PGE2 is the most abundant COX metabolite. Under physiological conditions, COX2 and PGE2 are induced during inflammatory processes and act as pro-inflammatory factors3. COX2 overexpression and subsequent PGE2 overproduction are observed in various human cancers4–7 and play a crucial role in the development of the tumour-promoting inflammatory microenvironment2,8.
Canine urothelial carcinoma (cUC) is the most common malignancy affecting the lower urinary tract of dogs. cUC is a unique tumour often well managed using COX inhibitors or non-steroidal anti-inflammatory drugs (NSAIDs)9. In addition, COX2 is overexpressed in cUC10–12. We previously reported that cUC cell lines overexpress PGE2 in vitro compared to other canine tumour cell lines with different tissues of origin13. Further, we suggested that aberrant PGE2 production is important for the development of tumour microenvironment and not for cell proliferation or survival13. However, the pathway that induces upregulation of COX2/PGE2 axis in cUC cells was not elucidated.
Another characteristic of cUC is that a single nucleotide mutation in the BRAF gene, V595E, is detected in 70%–80% of canine patients14,15. BRAF is an isoform of RAF serine/threonine kinase, which belongs to the RAF/MEK/ERK mitogen-activated protein kinase (MAPK) pathway. This pathway is one of the most important signalling pathways that transmit extracellular signals to cell nuclei, thereby regulating cell proliferation, differentiation, survival and various other cellular functions. The human counterpart of this mutation, which is recognised as BRAFV600E, is frequently observed in a variety of human malignancies such as malignant melanoma, colorectal cancer and papillary thyroid cancer16–18. The BRAFV600E mutation reportedly induces oncogenic cellular proliferation via constitutive activation of the ERK MAPK pathway16,19. Therefore, several molecular targeting drugs against BRAFV600E have been established and have improved the prognosis of patients with cancer20,21. Although canine BRAFV595E is also suggested to contribute to constitutive activation of the ERK MAPK signalling cascade, its importance in cUC progression remains unclear.
In this study, we screened molecular targeting agents to determine the pathways involved in PGE2 production in a BRAF mutant cUC cell line. We investigated the contribution of the ERK MAPK pathway in the regulation of the COX2/PGE2 axis including various cUC cell line, most of which harboured BRAFV595E mutation. Next, we investigated the relationship between BRAF genotype, ERK phosphorylation and COX2 expression in cUC tissues. Eventually, involvement of the other two MAPK pathways has been also evaluated. Our findings indicate a novel association between the activation of the ERK MAPK pathway in BRAF mutant cUC cells and dysregulation of the COX2/PGE2 axis.
Results
In vitro drug screening for disruption of PGE2 production in BRAF mutant cUC cells
We previously reported that cUC cell lines overexpress PGE213. To elucidate the mechanisms underlying aberrant PGE2 production in cUC cells, we screened 331 inhibitor compounds using SCADS inhibitor kit 1–4 obtained from Molecular Profiling Committee, Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Animal Model Support (AdAMS)” from The Ministry of Education, Culture, Sports, Science and Technology, Japan (KAKENHI 16H06276; see Supplementary Table S1). A BRAF mutant cUC cell line, Sora, was treated with each inhibitor compound at 10 µM for 12 h. A concentration of 10 µM was used during the screening process according to manufacturer instructions in consideration of the IC50 value of each reagent to inhibit its target molecule(s). The amount of PGE2 in the medium was quantified after the treatment, and percent change in PGE2 production with respect to that in vehicle control (DMSO) was calculated (Fig. 1A and see Supplementary Table S1). Eighty compounds showed ≥ 50% reduction in PGE2 production in the cUC cells. After categorisation of all the compounds into their specific targeting biological pathways, enrichment of each category for the PGE2-suppressing compounds was analysed. Statistical analysis revealed that compounds targeting the arachidonic acid cascade (FDR = 0.086), ERK MAPK pathway (FDR = 0.067) and p38 and JNK MAPK pathways (FDR = 0.067) were enriched in these 80 compounds (Table 1 and Fig. 1A,B). In addition, the compounds against the enriched pathways did not show strong cytotoxic effects on BRAF mutant cUC cells (Fig. 1B and Supplementary Fig. S1). Since the arachidonic acid cascade falls directly upstream of PGE2 production, it was considered that the inhibitory effect observed in the screening does not explain mechanisms for the induction of COX2/PGE2 in cUC cells. As the cell line harboured BRAF mutation, we initially focused on a role of the ERK MAPK pathway in the regulation of COX2/PGE2. Later on, the investigation was extended to p38 and JNK MAPK pathways.
Figure 1.

PGE2 production in cUC cells (Sora) during drug screening. (A) Y axis represents log 10 values of changes in PGE2 production for each inhibitor (n = 331). The drugs which belong to selected pathways are coloured as indicated. (B) Visualisation and comparison of the results from drug screening for PGE2 production and cell growth52. Drugs which showed ≥50% reduction in PGE2 production and cell density were included in statistical analysis. Categories which have more than 3 inhibitors are presented in this graph. Node size represents proportion of the drugs which showed reduction more than the threshold in each category. Colour represents statistical significance.
Table 1.
Percent change in PGE2 by the treatment of the inhibitors targeting Arachidonic acid cascade, ERK MAPK pathway, and p38 and JNK MAPK pathway.
| Pathway | Target | Compound | Percent change (%) |
|---|---|---|---|
| Arachidonic acid cascade (FDR = 0.086) | COX-1 | Sulindac sulphide | −89.1 |
| COX-1 | Valeryl salicylate | −55.4 | |
| COX-2 | NS-398 | −90.2 | |
| COX | Sodium salicylate | −8.7 | |
| PLA2 | cPLA2inhibitor | −78.9 | |
| PLA2 | OBAA | −29.9 | |
| lipoxygenase | Nordihydroguaiaretic acid (NDGA) | −28.7 | |
| 12, 15-lipoxygenase | ETYA | −73.4 | |
| 12-lipoxygenase | Baicalein | −68.0 | |
| MAPK (ERK) pathway (FDR = 0.067) | MEK | PD 98059 | −76.3 |
| MEK | U0126 | −69.1 | |
| MAPK | ERK inhibitor II | −50.0 | |
| MEK | MEK inhibitor I | −78.2 | |
| Raf | RAF1 kinase inhibitor I | −72.5 | |
| Raf | ZM 336372 | −48.2 | |
| Tpl2 | Tpl2 kinase inhibitor | 26.2 | |
| BRAF | Vemurafenib | −69.1 | |
| MAPK (p38 and JNK) pathway (FDR = 0.067) | JNK | SP600125 | −53.9 |
| p38 (MAPK) | PD169316 | −82.0 | |
| p38 (MAPK) | SB 203580 | −73.0 | |
| JNK | JNK inhibitor VIII | −12.4 | |
| p38 | SB202190 | −68.3 | |
| p38 | SB239063 | −57.6 |
Effects of ERK MAPK inhibition on the expression of the COX2/PGE2 axis
The inhibitors, aim at various components of the ERK MAPK pathway, were used for the experiments; dabrafenib (Dab) (BRAF inhibitor), LY3009120 (LY) (pan-RAF inhibitor), PD0325901 (PD) (MEK inhibitor) and SCH772984 (SCH) (ERK inhibitor). All compounds were used at 1 µM for 0, 6, 12 and 24 h. After 6 h of treatment, it was noted that COX2 expression and PGE2 production were decreased by ERK MAPK inhibitors accompanying dephosphorylation of ERK (Fig. 2A). Next, cells were exposed to each compound at varying concentrations for 12 h. It was confirmed that ERK MAPK inhibitors suppressed COX2 expression and PGE2 production in cUC cells in a dose-dependent manner (Fig. 2B). In the both experiments, RAF inhibitors (Dab and LY) induced dephosphorylation of MEK and ERK while MEK and ERK inhibitors (PD and SCH) caused overphosphorylation of MEK, which could be attributed to the release from the negative feedback loop of the pathway22. PD is a non-ATP-competitive inhibitor which inhibits phosphorylation of ERK by MEK.
Figure 2.

Effect of ERK MAPK inhibition on COX2 expression and PGE2 production. Protein levels in whole cell lysates were detected by Western blotting with Actin as loading control. Amount of PGE2 in culture medium was measured by enzyme-linked immunosorbent assay and normalised to cell number. Bar graph represents % control of PGE2 production. (A) cUC cells (Sora) were treated with vehicle (dimethyl sulfoxide; Cont) and inhibitors of BRAF (Dabrafenib; Dab), pan-RAF (LY3009120; LY), MEK (PD0325901; PD) and ERK (SCH772984; SCH) at 1 μM for indicated time. (B) cUC cells (Sora) were treated with vehicle (Cont), Dabrafenib (Dab), LY3009120 (LY), PD0325901 (PD) and SCH772984 (SCH) for 12 h at indicated dose. (C) cUC cell lines (TCCUB, Love, Nene, NMTCC, LTCC, MCTCC and OMTCC) were treated with vehicle (Cont), Dabrafenib (Dab) at 10 μM, LY3009120 (LY) at 1 μM, PD0325901 (PD) at 10 μM and SCH772984 (SCH) at 10 μM for 12 h. Data are presented as mean ± SD of three experiments. *Indicates p < 0.05, **p < 0.01, ‡p < 0.001 compared to vehicle control (Dunnett’s test).
To generalise the finding that inhibition of ERK MAPK pathway leads to suppression of COX2/PGE2 in cUC cells, another six BRAF mutant cUC cell lines (TCCUB, Love, Nene, NMTCC, LTCC and MCTCC) and one BRAF wild type cUC cell line (OMTCC) were further included in the study. The characteristics of the cell lines at basal state has been evaluated in advance (Supplementary Fig. S2A and B). Four of eight cUC cell lines (Sora, TCCUB, Nene, NMTCC) showed increased PGE2 production as we have published previously13. Two (Love, LTCC) showed moderate and the other two (MCTCC, OMTCC) did low production of PGE2. Phosphorylation of ERK was observed in all the cell line including BRAFWT OMTCC to some extent. Activation of p38 and JNK pathways and expression of COX1 were also observed in several cell lines.
These additional seven cell lines were treated with the ERK MAPK inhibitors with the varying doses for 12 h. In five cell lines (TCCUB, Love, Nene, NMTCC, LTCC), which showed comparatively higher PGE2 production at basal state (see Supplementary Fig. S2B), decrease in COX2 expression and PGE2 production was observed when the various components of ERK MAPK pathway was inhibited (Fig. 2C). It is noted that this effect was not observed in the low PGE2-producing cell lines (OMTCC and MCTCC) although the drugs successfully inhibited phosphorylation of ERK (Fig. 2C). One reason for this observation may be that these two cell lines do not express COX2 at basal state (Fig. 2C and Supplementary Fig. S2A). Collectively, it can be said that inhibition of ERK MAPK leads to decrease in COX2 expression in the BRAF mutant cUC cell lines.
We further explored the association between BRAFV595E gain-of-function mutation and expression of COX2. Public database (PubMed and GREIN) search was conducted and data from the four literature were accumulated, in which transfection of BRAFV600E into human normal cells was performed and changes in the transcriptome were evaluated23–27. The information of the retrieved data was summarised in Supplementary Table S2. By comparing the fold changes of normalised COX2 (PTGS2) expression, it was revealed that forced expression of BRAFV600E in human normal cells frequently induced strong transcription of PTGS2 in more than one type of cell (Supplementary Fig. S3A). Conversely, there was no clear trend regarding COX1 (PTGS1) expression (Supplementary Fig. S3B). This result suggests a hypothesis that BRAFV595E mutation is a direct cause of COX2 overexpression and subsequent PGE2 overproduction in cUC cells, which needs to be further examined by future transfection and loss of function studies using canine cell lines.
Association between BRAF mutation, ERK phosphorylation and COX2 expression in cUC patients
To confirm our findings in patients, we performed immunohistochemical analysis of COX2 and pERK expression, and genotyping of the BRAF gene using the clinical tumour tissues from cUC patients. The levels of COX2 and pERK expression were analysed using a semi-quantitative method described in previous reports (Fig. 3 and Supplementary Fig. S4)10,28–32. Digital PCR-based BRAF genotyping identified BRAFV595E mutation in 33 cUC tissues, while 10 tumours were BRAF wild-type. cUC tissues with BRAF mutation tended to have higher COX2 expression, although the difference was not statistically significant (p = 0.0569; Fig. 3A,B and Supplementary Fig. S4A). Moreover, phosphorylation of ERK was significantly stronger in BRAF mutant cUC tissue and strong phosphorylation of ERK was significantly associated with COX2 expression (Fig. 3C–E and Supplementary Fig. S4B).
Figure 3.

Levels of COX2 and pERK expression in cUC tissue. (A) Representative image of the COX2 expression in normal urothelium (top left) and blood vessel (top right) as negative and positive control, respectively, and cUC tissues (middle and bottom) at 100-fold magnification. Immunohistochemical score (IHS) was determined by a semi-quantitative method as described in Materials and Methods. IHSs of presented pictures were 12 (middle left), 9 (middle right), 6 (bottom left), 2 (bottom right), respectively. Scale bar: 200 μm. (B) A summarising box plot for COX2 expression in BRAF wild-type (WT; blue) and mutant cUC tissue (Mutant; red). Y axis represents IHS. p = 0.0569 (Wilcoxon rank sum test). (C) Representative image of the pERK expression in normal urothelium (top left) and blood vessel (top right) as negative and positive control, respectively, and cUC tissues (middle and bottom) at 200-fold magnification. IHSs of presented pictures were 12 (middle left), 9 (middle right), 4 (bottom left), 0 (bottom right), respectively. Scale bar: 100 μm. (D) A summarising box plot for COX2 expression in BRAF wild-type (WT; blue) and mutant cUC tissue (Mutant; red). Y axis represents IHS. *Indicates p < 0.05 (Wilcoxon rank sum test). (E) A summarising box plot for COX2 expression in cUC tissues which showed weak pERK expression with IHS < 6 (pERK_low; grey) and strong pERK expression with IHS ≥ 6 (pERK_high; orange). Y axis represents IHS for COX2. *Indicates p < 0.05 (Wilcoxon rank sum test).
Involvement of p38 and JNK MAPK pathways in the regulation of COX2/PGE2
The results of the drug screening and the characterisation of the cUC cell lines also indicated that involvement of the other MAPK pathways in regulation of COX2/PGE2 in BRAFV595E cUC cells. Therefore, additional in vitro experiments were performed using a p38 inhibitor (SB239063, SB) and a JNK inhibitor (SP600125, SP).
First, the BRAFV595E Sora cell line was treated with the drugs at the varying concentrations for 12 h (Fig. 4A). As observed in the screening, inhibition of both pathways led to a reduction in PGE2 production in a dose-dependent manner. The two drugs showed the effects without changing the phosphorylation level of ERK, which indicated independency of the p38 and JNK pathways from the ERK pathway regarding the cellular control of PGE2. The involvement of p38 and JNK pathways were further generalised by the time-course experiment and demonstrations in the other cUC cell lines (Supplementary Fig. S5A and B). Inhibition of p38 and JNK was able to decrease PGE2 production in COX2-expressing BRAFV595E cell lines without affecting ERK phosphorylation. Interestingly, throughout the experiments, inhibition of JNK increased COX2 at the protein level despite the decrease in PGE2 while p38 inhibition resulted in reduced COX2 expression. Inhibition of p38 and JNK pathways, as well as ERK pathway, did not change COX1 expression (Supplementary Fig. S6). These results suggested that the JNK pathway regulates PGE2 production in a COX1/COX2-independent manner while the ERK and p38 pathways were considered to exert the effects in a COX2-dependent manner.
Figure 4.
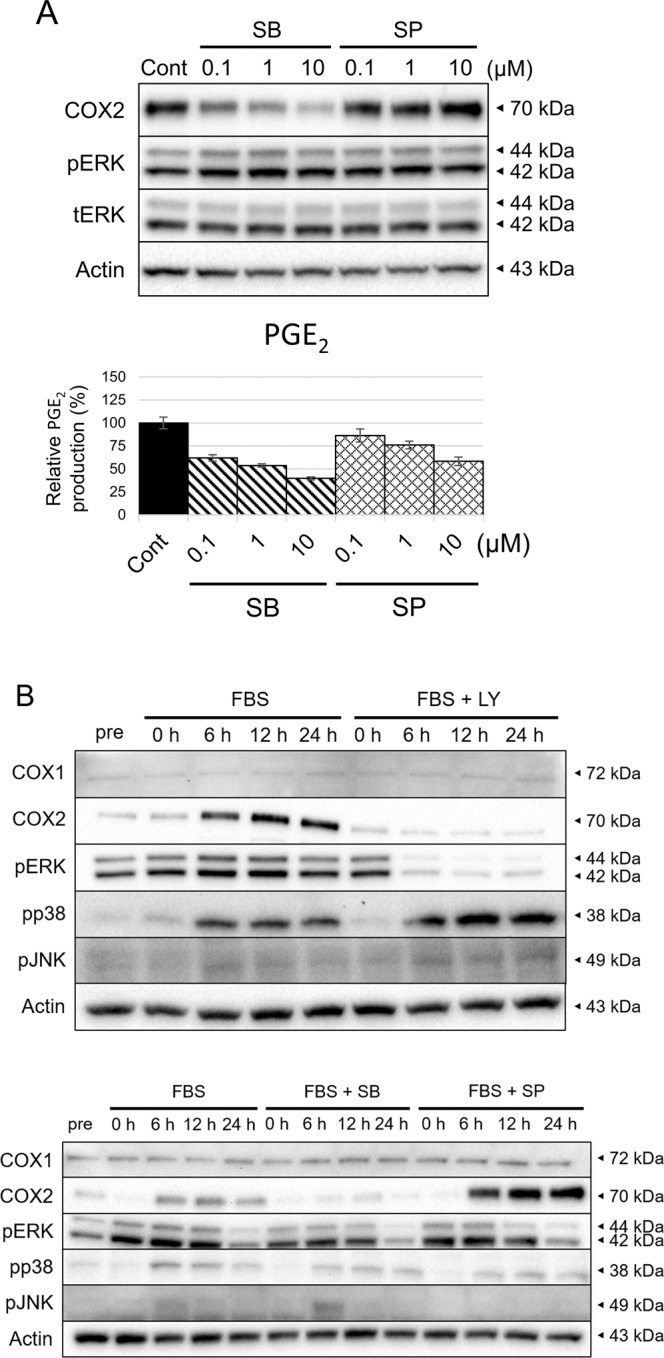
Contribution of p38 and JNK MAPK pathway to COX2 expression and PGE2 production in BRAFV595E cUC. Protein levels in whole cell lysates were detected by Western blotting with Actin as loading control. (A) cUC cells (Sora) were treated with vehicle (Cont), SB239068(SB) and SP600125 (SP) for 12 h at indicated dose. Amount of PGE2 in culture medium was measured by enzyme-linked immunosorbent assay and normalised to cell number. Bar graph represents % control of PGE2 production. (B) cUC cells (Sora) were treated with foetal bovine serum (FBS) and LY3009120 (LY) at 1 μM, SB239068 (SB) at 10 μM or SP600125 (SP) at 10 μM after 24 h-starvation. Whole cell lysates were corrected before (pre) and after treatment for indicated time.
To further explore the mechanism of action and relationship of the three MAPK pathways regarding control over cellular PGE2 production, starvation and refeeding experiments were performed in combination with inhibition of each pathway. As the p38 and JNK pathways are activated by external stimuli33, cells were refed with FBS after starvation to mimic them. Under starvation (“pre”), p38 and JNK were not activated while clear ERK phosphorylation was observed possibly because of the activating mutation in BRAF gene (Fig. 4B). It was also noted that the expression level of COX2 was low under starvation. Upon refeeding (“FBS”), COX2 expression was induced after 6 h and phosphorylation of p38 and JNK was also observed. When ERK MAPK pathway inhibitor was added with FBS (“FBS + LY”); ERK activation and COX2 induction were inhibited. No impact on COX1 and phosphorylated p38 and JNK was observed. Addition of p38 inhibitor at the same time with FBS (“FBS + SB”) also inhibited the induction of COX2 without affecting phosphorylation of ERK and JNK and expression of COX1. The ATP-competitive p38 inhibitor used in this study (SB239063) inhibited phosphorylation of downstream molecules by p38 without inhibiting the phosphorylation of p38 itself34,35. When refeeding was conducted with the JNK inhibitor (“FBS + SP”), phosphorylation of JNK was eradicated and the overexpression of COX2 was observed. There were no changes in COX1 expression and phosphorylation of ERK and p38.
This concludes that the three MAPK pathways independently regulate PGE2 production in the BRAF mutant cUC cells, not influencing each other. Moreover, ERK and p38 pathways mediate PGE2 production through COX2 expression. It seemed that activation of ERK MAPK itself does not induce COX2 overexpression, rather it maintains COX2 expression induced by other stimuli. Conversely, JNK may induce PGE2 production in COX1/2-independent manner.
Discussion
The COX2/PGE2 axis plays a crucial role in tumour development. However, mechanisms of the aberrant expression of COX2/PGE2 in cUC with BRAFV595E mutation remain to be elucidated. Influenced by our previous study which has found aberrant PGE2 production in BRAF mutant cUC cells, the series of in vitro and in silico analyses, as well as evaluation of the clinical samples from the patients, were performed to address this question. As a result, it was suggested that the activation of ERK MAPK pathway leads to upregulation of COX2/PGE2 in BRAFV595E mutant cUC cells. In addition, the involvement of the ERK MAPK pathway in the regulation of COX2 expression was observed in the clinical samples of cUC tissues. Furthermore, it was suggested that the p38 and JNK pathways contribute to PGE2 production independently of the ERK pathway in BRAFV595E cUC cells in a COX2-dependent and COX1/COX2-independent manner, respectively. ERK MAPK activation seems to maintain, rather than induce, COX2 expression induced by other stimuli. Further investigation thus is warranted for validation. Conclusively, our data revealed major mechanisms of PGE2 regulation in BRAFV595E cUC cells. The results offer a new direction of anti-inflammatory strategy in clinical management of cUC as well as may explain the association relationship oncogenic mutation and tumour-associated inflammation.
Emerging evidences suggest that activation of ERK MAPK pathway in human cancer contributes to the regulation of COX2/PGE2 axis. A recent study suggested that COX2/PGE2 expression depends on the activation of the pathway in a BRAF mutant melanoma cell line established from a genetically engineered mouse model36. It has been suggested that ERK signalling activated by hepatocyte growth factor leads to increased COX2 expression in human colorectal cancer and non-small cell lung cancer37,38. In the present study, by pharmacological targeting of the multiple components in the pathway, it was shown that the ERK pathway plays a significant role in regulation of COX2 expression and PGE2 production in BRAF mutant cUC cells (Fig. 5). Expression of phospho-ERK was also correlated with COX2 expression in clinical samples. Simultaneously, the results of this study suggest that the ERK pathway is major, but not an only factor that regulates COX2/PGE2 axis in cUC. Although most cUC cell lines used in this study produced PGE2, as a unique characteristic of cUC13,39, not all with activated ERK signalling showed overexpression of COX2. This observation suggested that there is an obvious heterogeneity among cUC cells in terms of regulation of COX2/PGE2 axis. Mechanistically, activation of ERK may not induce, rather maintain COX2 expression induced by other stimuli. As shown in Fig. 4B, under starvation, cells showed weak COX2 expression despite clear phosphorylation of ERK which possibly constitutively activated by mutated BRAF. When COX2 was strongly induced by refeeding with FBS, inhibition of the ERK MAPK pathway resulted in significant reduction of COX2/PGE2. Further, overexpression of COX2 and overproduction of PGE2 seemed to be caused by more than one pathway in cUC cells.
Figure 5.
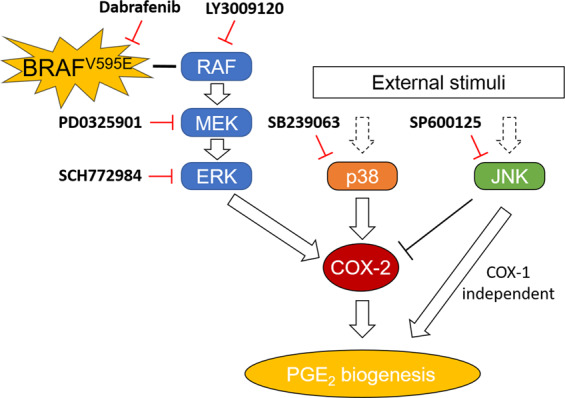
Schematic diagram of the involvement of ERK, p38 and JNK MAPK pathways in the regulation of COX2/PGE2 axis in BRAFV595E mutant cUC.
Possible candidates of such pathways were p38 and JNK pathways. p38 and JNK are members of the MAPK family and activated by environmental stress, including ultraviolet exposure, oxidative stress or inflammatory cytokines33. These pathways have been suggested to induce COX2 expression in inflammatory processes, such as rheumatoid arthritis or Parkinson’s disease, and in human cancer-promoting inflammation40–42. In this study, activation of the p38 pathway was involved in COX2 regulation and indicated to be essential to induce COX2 expression in the BRAFV595E cUC cells based on the starvation and refeeding study. Addition of FBS induced phosphorylation of p38. Then, inhibition of p38 by the specific inhibitor decreased the COX2 level without affecting the COX1 expression and other MAPK pathways. Interestingly, induction of COX2 was almost completely inhibited by either of p38 or ERK MAPK inhibition. ERK MAPK inhibition did not interfere with p38 MAPK pathway. In addition, activation of ERK MAPK did not induce COX2 expression as discussed above (“pre” state when cells are under starvation). One hypothesis for this observation is a cooperative regulation of COX2 by the two pathways. Souza et al. reported that the ERK MAPK pathway stabilises COX2 mRNA induced by the p38 MAPK pathway in human Barrett’s-associated adenocarcinoma treated with the low pH environment43. In our cell line characterization, overproduction of PGE2 were found mostly in the cell lines with both p38 and ERK activation (TCCUB, Sora, Nene, and NMTCC). This hypothesis needs to be further examined in cUC by simultaneous inhibition of both pathways with pharmacological or, more preferentially, genetic approaches. The importance of the p38 pathway in regulation of COX2 also needs to be validated in clinical samples.
Conversely, JNK inhibition upregulated COX2 at the protein level despite a significant decrease in PGE2 production. Refeeding with FBS induced phosphorylation of JNK, which was counteracted by JNK inhibitor. The drug did not have clear impact on either of ERK, p38 or COX1. These results indicated that JNK promotes PGE2 production in the cUC cells, independent of ERK and p38, which involves regulation of neither of COX1 and COX2. A previous report indicated that the JNK pathway can regulate PGE2 production in a COX-independent manner. In the paper, JNK pathway activated by TNF-α was involved in the gene expression of one of the PGE2 synthases, mPGES-1, in gingival fibroblasts44. The exact mechanism of action of JNK pathway in cellular PGE2 production can be further explored by a combination of a specific JNK activator33 and an unbiased comprehensive approach such as transcriptome analysis and drug screening. Simultaneous inhibition of three MAPK pathways using specific RNA interference will also clarify the association between the pathways.
The heterogeneity observed in cUC cell lines and clinical samples could be explained by two hypotheses. One is that complexed regulation of PGE2 production by the three different MAPK pathways. It seemed that activation of one of them is not sufficient to induce and maintain the eicosanoid production at an increased level. Another hypothesis is that there can be multiple pathways to trigger activation of ERK MAPK pathway. In cUC clinical samples, the association between BRAF genotype and COX2 expression was not clear, partly because some wild-type tumours showed increased COX2 expression. There is a similar observation in human medicine. Although COX2 plays a critical role in BRAF mutant human colorectal cancers, expression of COX2 is not associated with the presence of BRAF mutation45. This may be because there is another major driver mutation of KRAS in human CRC, which also leads to constitutive activation of ERK MAPK pathway. In cUC, overexpression of HER2 has been recently reported and this can contribute to activation of the ERK MAPK pathway46,47. Therefore, a future comprehensive study will need to assess BRAF genotype, HER2 expression, activation of the MAPK pathways and COX1/2 expression simultaneously. From this study, since BRAF mutation was associated with pERK IHS score and pERK IHS score was clearly associated with COX2 expression, we believe that BRAF mutation would contribute to the overexpression of COX2 in cUC although the direct causal relationship of them should be further examined. In a recent study, the association between BRAF mutation and COX2 expression was suggested in cUC tissues in specific dog breeds48.
One major hypothesis built primarily on in silico analysis, was that BRAFV595E mutation directly causes COX2 overexpression via upregulation of the ERK/MAPK pathway. In this study, we focused on the importance of ERK and other MAPK pathways in regulation of COX2/PGE2 in BRAF mutant cUC. The direct causal association between the mutation and COX2 expression should be further validated by gain-of-function and loss-of-function studies using canine cells. Activation in BRAF gene is known to induce aberrant activation of ERK MAPK pathways in mice, human and canine cells14,16,19,36. Moreover, in several human tumours, it has been suggested that the COX2/PGE2 axis is affected by oncogenic mutations including BRAFV600E 49,50. Considering the important role of the COX2/PGE2 axis in the pro-tumoural microenvironment and the unique vulnerability of cUC to anti-COX2 therapies, BRAFV595E cUC would serve as a valuable model to explore the possible association between oncogenic mutation and pro-tumoural microenvironment.
cUC is a unique tumour often managed with NSAID monotherapy, indicating the importance of the COX2/PGE2 axis in the development and progression of cUC9. Since no direct cytotoxicity was observed in our previous study13, the therapeutic effect of NSAIDs was considered to be indirect, possibly on tumour microenvironment. This hypothesis was further supported in this study by the fact that no correlation was observed between suppression of PGE2 production and cell growth. Although a causal relationship between BRAF mutation and PGE2 production is further to be determined, the findings of this study may suggest that driver mutation can render tumour cells malignant phenotypes through modulation of the microenvironment to a pro-tumoural niche rather than direct growth advantages.
In conclusion, our study revealed that activation of the ERK MAPK pathway mediated overexpression of COX2 and PGE2 production in BRAFV595E cUC cells. In addition, COX2 expression was cooperatively regulated by the p38 MAPK pathway. Oppositely, the JNK pathway was suggested to facilitate PGE2 production in a COX1- /COX2-independent manner (Fig. 5). By understanding how significantly and differently each MAPK pathway regulates PGE2 production in cUC, these findings may open up the possibility of PGE2-targeted therapy replacing the current standard treatment with NSAIDs, or COX inhibitors. The major hypothesis to be answered in future studies is direct causal relationship between the BRAF mutation and COX2 expression. This may help us to understand heterogeneity in COX2 expression in cUC and how tumour cells establish pro-tumoural environment surrounding them. As cUC has its uniqueness of the clinical vulnerability to the COX inhibitors, we believe that BRAFV595E cUC serve as a good model to study relationship between BRAF mutation and COX2/PGE2 axis, or driver mutation and pro-tumoural microenvironment, from a wider perspective.
Methods
Cell culture
Eight cUC cell lines, TCCUB, Sora, Love, Nene (originally established in our laboratory), NMTCC, LTCC, MCTCC and OMTCC (kindly provided by Hokkaido University) were used in this study13,51,52. Each cell line was maintained in RPMI-1640 supplemented with 10% heat-inactivated foetal bovine serum (FBS) and 5 mg/L gentamicin at 37 °C in a humidified atmosphere with 5% CO2.
Drugs
For in vitro drug screening, the SCADS inhibitor kits (kit 1, ver 3.3; kit 2, ver 2.0; kit 3, ver 1.6; kit 4, ver 2.3) were kindly provided by Molecular Profiling Committee, Grant-in-Aid for Scientific Research on Innovative Areas “Platform of Advanced Animal Model Support” from The Ministry of Education, Culture, Sports, Science and Technology, Japan (KAKENHI 16H06276). For the following experiment, dabrafenib (BRAF inhibitor), PD0325901 (MEK inhibitor) and SCH772984 (ERK inhibitor) were purchased from Selleck. LY3009120 (pan-RAF inhibitor) was purchased from Cayman Chemical. SB239063 (p38 inhibitor) and SP600125 (JNK inhibitor) were purchased from Sigma-Aldrich. These inhibitors were reconstituted in DMSO and stored at −20 °C or −80 °C.
PGE2 measurement
PGE2 concentration in culture supernatant was measured using enzyme-linked immunosorbent assay kit (Cayman Chemical) according to the manufacturer’s instructions. To normalise the amount of PGE2 to the cell density, direct cell count or sulforhodamine B (SRB) assay was performed53.
In vitro drug screening
Sora cells were seeded in a 96-well plate at a density of 11,000 cells per well. After 24-h incubation, the culture medium in each well was discarded, and fresh medium containing the drugs from the SCADS inhibitor kits was added at a final concentration of 10 µM. Following a 12-h incubation, the culture supernatants were collected. After normalising PGE2 production to cell density as mentioned above, percentage change in PGE2 production from control was calculated.
Growth inhibition
To compare with the changes in PGE2 production, data regarding cell growth inhibition after treatment with the inhibitors from the SCADS inhibitor kits were obtained from our previous study, where the detailed protocol is described52. Briefly, Sora cells were treated with the drugs from the SCADS inhibitor kits at 10 µM as described in In vitro drug screening section. After 72-h incubation, cell densities were determined by SRB assay.
Protein detection
cUC cells were seeded in serum-free medium and incubated for 24 h as in our previous report (Supplementary Table S3)52. After 24-h serum starvation, cUC cells were treated with final concentration of 10% FBS and ERK MAPK, p38 MAPK or JNK MAPK inhibitors as indicated in each figure. Following treatment for the indicated times, cells were lysed for 30 min on ice in RIPA buffer consisting of 50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 0.1% sodium dodecyl sulphate, 1% Triton-X, 10 mM NaF, 2 mM NaVO4 and complete protease inhibitor cocktail (Roche Diagnostics). After centrifugation at 12000 g, 4 °C for 20 min, protein concentrations were measured using bicinchoninic acid protein assay kit. Equal amounts (10 µg) of total protein were separated by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% skim milk in Tris-buffered saline containing 0.1% Tween (TBST) for 1 h at room temperature to avoid non-specific antibody binding. Further, the membranes were incubated overnight at 4 °C with each primary antibody (Supplementary Table S4). The membranes were then washed with TBST and incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody (1:10,000) from GE Healthcare for 1 h at room temperature. Protein signal was developed with a chemiluminescent system (Merck Millipore) and captured with an imaging system (BioRad Laboratories).
In silico BRAF gain-of-function analysis
Data search was performed using two public database (PubMed and GREIN) maintained by National Institute of Health, the United States23. The databases were accessed on 1/27/2020 and queries were performed with the term of “BRAF” and “transfect”. Four studies were identified with publicly available data (Supplementary Table S2)24–27. For the study by Becker et al., the fold change data was collected from the table in the original manuscript24. For the other studies, the normalised expression values were retrieved through GREIN. The expression values of the genes in the transfected cells were further normalised by the expression values in the parental cells and presented as the fold changes.
Immunohistochemistry
Formalin-fixed paraffin-embedded (FFPE) cUC tissues were retrospectively evaluated. Surgically resected cUC tissue samples (n = 43) were obtained from the archival collection of the Veterinary Medical Centre of the University of Tokyo (samples collected from 2009 to 2012 and 2015 to 2016) and Veterinary Medical Teaching Hospital of Nippon Veterinary and Life Science University (from 2003 to 2017). The clients from respective hospitals provided informed consent for the use of these samples for this study. cUC tissues of 4 μm thickness were deparaffinised in xylene and hydrated in graded alcohol. Antigen retrieval was performed for 10 min at 121 °C in citrate buffer (pH 6.0). The sections were then treated with 3% hydrogen peroxide for 30 min at room temperature. After blocking with 5% normal goat serum in TBST for 1 h at room temperature, the sections were incubated overnight with 1:100 of anti-COX2 mouse monoclonal antibody (BD Bioscience) or 1:400 of anti-phospho-ERK1/2 (Thr202/Tyr204) rabbit monoclonal antibody (Cell Signaling Technology) at 4 °C in a humidified chamber. Subsequently, the sections were rinsed with TBST and then incubated with EnVision polymer reagent for mouse (Dako) for 60 min at room temperature. After rinsing with TBST again, peroxidase reactions were developed for 3 min with 3,3′-diaminobenzidine (Dako). The sections were counterstained with haematoxylin.
BRAF genotyping
BRAF genotype of cUC tissue was determined as described in a previous study52. Briefly, gDNA was extracted from FFPE sections of cUC tissue using Qiaamp DNA FFPE Tissue Kit (Qiagen). BRAFV595E and wild-type BRAF gene were amplified with Taqman probe and signal detection was performed using QuantStudio 3D Digital PCR System (Thermo Fisher Scientific).
Immunohistochemical evaluation
For scoring COX2 and phospho-ERK (pERK) expression in cUC tissue, a semi-quantitative immunohistochemical score (IHS) system described in a previous report was used10,28–32. Briefly, the percentage of COX2- or pERK-positive tumour cells in the entire section and their signal intensity were evaluated when viewed at 100x and 200x magnification, respectively. Positivity was graded as 1 = <1%, 2 = 1%–9%, 3 = 10%–50%, 4 = >50% for COX2 and as 0 = <5%, 1 = 5%–25%, 2 = 26%–50%, 3 = 51%–75%, 4 = >75% for pERK, and intensity was graded as 0 = no staining, 1 = mild staining, 2 = moderate staining, 3 = strong staining for COX2 and pERK. IHS was obtained by multiplying positivity score and intensity score.
Statistical analysis
Statistical analysis was performed using R software (https://www.R-project.org/). To determine enriched pathways revealed in drug screening, all of the 331 compounds were divided into 98 groups according to their targeting pathways, and p value for each pathway was calculated using hypergeometric distribution. Multiple comparison was adjusted by Benjamini–Hochberg method and the FDR cut-off was set as 0.1. To test the effect of ERK MAPK inhibition on PGE2 production, Dunnett’s multiple comparison test was used. Difference in the IHS score depending on the BRAF genotype or pERK IHS score (pERK_high; IHS ≥ 6 vs pERK_low; IHS < 6) was calculated using Wilcoxon rank sum test. All values are shown as the mean value ± standard deviation. p values <0.05 were considered statistically significant.
Supplementary information
Acknowledgements
We thank Molecular Profiling Committee, Grant-in-Aid for Scientific Research on Innovative Areas “Platform of Advanced Animal Model Support” from the Ministry of Education, Culture, Sports, Science and Technology, Japan, which is supported by KAKENHI 16H06276 for providing the drug screening kits. Also, we are grateful to Yuki Hoshino (Iwate University) for providing the cell lines (NMTCC, LTCC, MCTCC and OMTCC) and Masaki Michishita (Nippon Veterinary and Life Science University) for providing the FFPE cUC tissues. This work was supported by JSPS KAKENHI Grant Number 16K20968.
Author contributions
R.Y., K.S., R.N. and T.N. designed this study. R.Y. performed the experiments. S.E. and M.S. contributed to the data analysis. R.Y. and K.S. prepared the figures and wrote this manuscript. K.S., R.N., H.S., Y.E., N.F., R.N. and T.N. supervised this study.
Data availability
The data obtained during the current study will be available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
10/28/2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
Supplementary information
is available for this paper at 10.1038/s41598-020-64832-5.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Wang D, Dubois RN. Eicosanoids and cancer. Nat. Rev. Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DuBois RN, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073. [PubMed] [Google Scholar]
- 4.Petkova DK, et al. Overexpression of cyclooxygenase-2 in non-small cell lung cancer. Respir. Med. 2004;98:164–172. doi: 10.1016/j.rmed.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Agrawal U, Kumari N, Vasudeva P, Mohanty NK, Saxena S. Overexpression of COX2 indicates poor survival in urothelial bladder cancer. Ann. Diagn. Pathol. 2018;34:50–55. doi: 10.1016/j.anndiagpath.2018.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Eberhart CE, et al. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 7.Kim HS, et al. COX2 overexpression is a prognostic marker for Stage III breast cancer. Breast Cancer Res. Treat. 2012;132:51–59. doi: 10.1007/s10549-011-1521-3. [DOI] [PubMed] [Google Scholar]
- 8.Greenhough A, et al. The COX-2/PGE2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377–386. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]
- 9.Fulkerson CM, Knapp DW. Management of transitional cell carcinoma of the urinary bladder in dogs: A review. Vet. J. 2015;205:217–225. doi: 10.1016/j.tvjl.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 10.Khan KNM, Knapp DW, Denicola DB, Harris RK. Expression of cyclooxygenase-2 in transitional cell carcinoma of the urinary bladder in dogs. Am. J. Vet. Res. 2000;61:478–481. doi: 10.2460/ajvr.2000.61.478. [DOI] [PubMed] [Google Scholar]
- 11.Knottenbelt C, Mellor D, Nixon C, Thompson H, Argyle DJ. Cohort study of COX-1 and COX-2 expression in canine rectal and bladder tumours. J. Small Anim. Pract. 2006;47:196–200. doi: 10.1111/j.1748-5827.2006.00094.x. [DOI] [PubMed] [Google Scholar]
- 12.Sledge DG, Patrick DJ, Fitzgerald SD, Xie Y, Kiupel M. Differences in Expression of Uroplakin III, Cytokeratin 7, and Cyclooxygenase-2 in Canine Proliferative Urothelial Lesions of the Urinary Bladder. Vet. Pathol. 2015;52:74–82. doi: 10.1177/0300985814522819. [DOI] [PubMed] [Google Scholar]
- 13.Yoshitake R, et al. Molecular investigation of the direct anti-tumour effects of nonsteroidal anti-inflammatory drugs in a panel of canine cancer cell lines. Vet. J. 2017;221:38–47. doi: 10.1016/j.tvjl.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Decker B, et al. Homologous Mutation to Human BRAF V600E Is Common in Naturally Occurring Canine Bladder Cancer–Evidence for a Relevant Model System and Urine-Based Diagnostic Test. Mol. Cancer Res. 2015;13:993–1002. doi: 10.1158/1541-7786.MCR-14-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mochizuki H, Kennedy K, Shapiro SG, Breen MB. BRAF mutations in canine cancers. PLoS One. 2015;10:1–9. doi: 10.1371/journal.pone.0129534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 17.Venderbosch S, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin. Cancer Res. 2014;20:5322–5330. doi: 10.1158/1078-0432.CCR-14-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimura, E. T. et al. High Prevalence of BRAF Mutations in Thyroid Cancer: Genetic Evidence for Constitutive Activation of the RET/PTC-RAS-BRAF Signaling Pathway in Papillary Thyroid Carcinoma. Cancer Research63, (2003). [PubMed]
- 19.Wan PTC, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 20.Chapman PB, et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hauschild A, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 22.Lake D, Corrêa SAL, Müller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cellular and Molecular Life Sciences. 2016;73:4397–4413. doi: 10.1007/s00018-016-2297-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahi, N. A, Najafabadi, M. F., Pilarczyk, M., Kouril, M. & Medvedovic, M. GREIN: An Interactive Web Platform for Re-analyzing GEO RNA-seq Data. Sci. Rep. 9, (2019). [DOI] [PMC free article] [PubMed]
- 24.Becker TM, et al. Mutant B-RAF-Mcl-1 survival signaling depends on the STAT3 transcription factor. Oncogene. 2014;33:1158–1166. doi: 10.1038/onc.2013.45. [DOI] [PubMed] [Google Scholar]
- 25.Flockhart RJ, et al. BRAFV600E remodels the melanocyte transcriptome and induces BANCR to regulate melanoma cell migration. Genome Res. 2012;22:1006–1014. doi: 10.1101/gr.140061.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim, Y. H. et al. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun. 8, (2017). [DOI] [PMC free article] [PubMed]
- 27.Pawlikowski JS, et al. Wnt signaling potentiates neèogenesis. Proc. Natl. Acad. Sci. USA. 2013;110:16009–16014. doi: 10.1073/pnas.1303491110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Queiroga FL, Alves A, Pires I, Lopes C. Expression of Cox-1 and Cox-2 in Canine Mammary Tumours. J. Comp. Pathol. 2007;136:177–185. doi: 10.1016/j.jcpa.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Belshaw Z, et al. COX-2 expression and outcome in canine nasal carcinomas treated with hypofractionated radiotherapy. Vet. Comp. Oncol. 2011;9:141–148. doi: 10.1111/j.1476-5829.2010.00243.x. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, et al. ERK expression and its correlation with STAT1 in esophageal squamous cell carcinoma. Oncotarget. 2017;8:45249–45258. doi: 10.18632/oncotarget.16902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma R, et al. AZ304, a novel dual BRAF inhibitor, exerts anti-tumour effects in colorectal cancer independently of BRAF genetic status /692/4028/67 /631/67 article. Br. J. Cancer. 2018;118:1453–1463. doi: 10.1038/s41416-018-0086-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao J, et al. Targeting of the MAPK and AKT pathways in conjunctival melanoma shows potential synergy. Oncotarget. 2017;8:58021–58036. doi: 10.18632/oncotarget.10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kyriakis JM, Avruch J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 34.Kumar S, Jiang MS, Adams JL, Lee JC. Pyridinylimidazole compound SB 203580 inhibits the activity but not the activation of p38 mitogen-activated protein kinase. Biochem. Biophys. Res. Commun. 1999;263:825–831. doi: 10.1006/bbrc.1999.1454. [DOI] [PubMed] [Google Scholar]
- 35.Griego SD, Weston CB, Adams JL, Tal-Singer R, Dillon SB. Role of p38 Mitogen-Activated Protein Kinase in Rhinovirus-Induced Cytokine Production by Bronchial Epithelial Cells. J. Immunol. 2000;165:5211–5220. doi: 10.4049/jimmunol.165.9.5211. [DOI] [PubMed] [Google Scholar]
- 36.Zelenay S, et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell. 2015;162:1257–1270. doi: 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore AE, et al. HGF/Met signalling promotes PGE2 biogenesis via regulation of COX-2 and 15-PGDH expression in colorectal cancer cells. Carcinogenesis. 2009;30:1796–1804. doi: 10.1093/carcin/bgp183. [DOI] [PubMed] [Google Scholar]
- 38.Siegfried JM, Gubish CT, Rothstein ME, de Oliveira PEQ, Stabile LP. Signaling Pathways Involved in Cyclooxygenase-2 Induction by Hepatocyte Growth Factor in Non Small-Cell Lung Cancer. Mol. Pharmacol. 2007;72:769–779. doi: 10.1124/mol.107.034215. [DOI] [PubMed] [Google Scholar]
- 39.Mohammed SI, et al. Prostaglandin E2 concentrations in naturally occurring canine cancer. Prostaglandins Leukot. Essent. Fat. Acids. 2001;64:1–4. doi: 10.1054/plef.2000.0231. [DOI] [PubMed] [Google Scholar]
- 40.Namba S, et al. ERK2 and JNK1 contribute to TNF-α-induced IL-8 expression in synovial fibroblasts. PLoS One. 2017;12:e0182923. doi: 10.1371/journal.pone.0182923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hunot S, et al. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA. 2004;101:665–670. doi: 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sung S, et al. Overexpression of cyclooxygenase-2 in NCI-H292 human alveolar epithelial carcinoma cells: Roles of p38 MAPK, ERK-1/2, and PI3K/PKB signaling proteins. J. Cell. Biochem. 2011;112:3015–3024. doi: 10.1002/jcb.23226. [DOI] [PubMed] [Google Scholar]
- 43.Souza RF, et al. Acid increases proliferation via ERK and p38 MAPK-mediated increases in cyclooxygenase-2 in Barrett’s adenocarcinoma cells. Am. J. Physiol. Liver Physiol. 2004;287:G743–G748. doi: 10.1152/ajpgi.00144.2004. [DOI] [PubMed] [Google Scholar]
- 44.Båge T, Lindberg J, Lundeberg J, Modéer T, Yucel-Lindberg T. Signal pathways JNK and NF-κB, identified by global gene expression profiling, are involved in regulation of TNFα-induced mPGES-1 and COX-2 expression in gingival fibroblasts. BMC Genomics. 2010;11:241. doi: 10.1186/1471-2164-11-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kosumi K, et al. Prognostic association of PTGS2 (COX-2) over-expression according to BRAF mutation status in colorectal cancer: Results from two prospective cohorts and CALGB 89803 (Alliance) trial. Eur. J. Cancer. 2019;111:82–93. doi: 10.1016/j.ejca.2019.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maeda S, et al. Comprehensive gene expression analysis of canine invasive urothelial bladder carcinoma by RNA-Seq. BMC Cancer. 2018;18:1–11. doi: 10.1186/s12885-018-4409-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsuboi M, et al. Assessment of HER2 Expression in Canine Urothelial Carcinoma of the Urinary Bladder. Vet. Pathol. 2019;56:369–376. doi: 10.1177/0300985818817024. [DOI] [PubMed] [Google Scholar]
- 48.Grassinger JM, Merz S, Aupperle-Lellbach H, Erhard H, Klopfleisch R. Correlation of BRAF variant V595E, breed, histological grade and cyclooxygenase-2 expression in canine transitional cell carcinomas. Vet. Sci. 2019;6:31. doi: 10.3390/vetsci6010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eisinger AL, et al. The adenomatous polyposis coli tumor suppressor gene regulates expression of cyclooxygenase-2 by a mechanism that involves retinoic acid. J. Biol. Chem. 2006;281:20474–20482. doi: 10.1074/jbc.M602859200. [DOI] [PubMed] [Google Scholar]
- 50.Wang X-Q, et al. Oncogenic K-Ras Regulates Proliferation and Cell Junctions in Lung Epithelial Cells through Induction of Cyclooxygenase-2 and Activation of Metalloproteinase-9. Mol. Biol. Cell. 2009;20:791–800. doi: 10.1091/mbc.E08-07-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakai K, et al. Anti-tumour effect of lapatinib in canine transitional cell carcinoma cell lines. Vet. Comp. Oncol. 2018;16:642–649. doi: 10.1111/vco.12434. [DOI] [PubMed] [Google Scholar]
- 52.Eto S, et al. Anti-tumor effects of the histone deacetylase inhibitor vorinostat on canine urothelial carcinoma cells. PLoS One. 2019;14:e0218382. doi: 10.1371/journal.pone.0218382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data obtained during the current study will be available from the corresponding author upon reasonable request.