

Abstract
Decades ago, Rap1, a small GTPase very similar to Ras, was observed to suppress oncogenic Ras phenotype, reverting its transformation. The proposed reason, persisting since, has been competition between Ras and Rap1 for a common target. Yet, none was found. There was also Rap1’s puzzling suppression of Raf-1 versus activation of BRAF. Reemerging interest in Rap1 envisages capturing its Ras suppression action by inhibitors. Here, we review the literature and resolve the enigma. In vivo oncogenic Ras exists in isoform-distinct nanoclusters. The presence of Rap1 within the nanoclusters reduces the number of the clustered oncogenic Ras molecules, thus suppressing Raf-1 activation and mitogen-activated protein kinase (MAPK) signaling. Nanoclustering suggests that Rap1 suppression is Ras isoform dependent. Altogether, a potent Rap1-like inhibitor appears unlikely.
Introduction
Decades ago, a paper published what was to become a pioneering and baffling finding: the discovery of the Rap1 protein (identical to Krev-1), with a high sequence similarity (53% identical) to Ras [1]. Rap1 has two isoforms, Rap1A and Rap1B, that differ in only a few amino acids. The work was intriguing, since the authors observed Rap1 to suppress oncogenic Ras phenotype, or as they put it, revert Ras transformation. They puzzled over the mechanism and suggested that Rap1 may antagonize Ras proteins, either by competing for a common target or by functioning as a G protein in transducing an inhibitory growth signal, opposing the Ras growth-promoting signal in fibroblasts. The question of why, or how, Rap1 suppresses Ras oncogenic signaling lingered in the decades that followed. A few years later, Rap1G12V expressed at physiological levels was observed to interfere with signal transduction from Ras to MAPKs in vivo [2]. Activated Rap1 inhibited extracellular signal-regulated kinase (ERK)s’ – but not Ras – activation, ruling out the involvement of nucleotide exchange factors. Nonetheless, in all experiments using conditional expression of Rap1G12V, MAPK inhibition was incomplete. As in the first publication, the authors suggested two possible explanations; in the first, Rap1G12V–GTP antagonizes Ras by competing for a Ras effector. In the second, Rap1G12V sends a downstream signal, antagonizing the MAPK pathway. The authors favored the first model since Ras and Rap1 proteins share a cluster of identical amino acids (residues 32–44 in HRas) in the effector-binding region. In further support of this rational, they cited Rap1–GTP potent inhibition of the interaction between Ras and p120 GTPase-activating protein (GAP) and that overexpressed Rap1 was unable to activate Raf-1 [2]. Altogether, this and subsequent publications (e.g., [3–7]; see also the references that follow) led to the still persistent view that Rap1 is a Ras antagonist, acting by competing with Ras and assembling into unproductive complexes with Ras effectors. However, more recently, biochemical, cellular, and developmental observations revealed that Rap1 has Ras-independent functions; primary among these is the control of cell adhesion-related events [4,7–29]. Such critical cellular functions are further ratified by Rap1 conservation across species, raising the question: is Rap1’s raison d’être indeed Ras suppression, and if a different mechanism is involved, as this appears to suggest, the question reemerges: what is it?
The quest for the underlying mechanism becomes particularly important under the premise that the apparent Rap1 suppressor action can be exploited in Ras drug discovery. Multiple experiments testify to Rap1 interfering with Ras downstream MAPK signaling, underscoring its significance. To date, the paradigm lingers: Rap1 attenuates, or abates Ras transforming activity through competitive binding to a common, though still unidentified, target. Later, we suggest another explanation: the presence of Rap1 in a Ras nanocluster reduces the effective local concentration of oncogenic Ras molecules in the nanocluster, thus Raf activation and MAPK signaling (Figure 1, Key Figure). As we noted recently, such a simple and straightforward explanation also holds for several other oncogenic Ras-related apparent puzzles, such as how wild-type Ras suppresses the oncogenicity of its oncogenic mutant variant [30,31]. It is also in line with earlier Rap1 observations [32,33]. Those findings suggest that the strength of the interaction of the Raf cysteine-rich domain (CRD) is a critical determinant in Raf’s activation. Thus, our rational simply stipulates that the presence of Rap1 ‘dilutes’ the effective concentration of the oncogenic Ras in the nanocluster. If Rap1 is active, it will activate BRAF – but not Raf-1; if inactive, which is the expected state in vivo, neither will, even though Raf-1 CRD whose Rap1 binding is nucleotide independent, will still bind. Altogether, oncogenic Ras signaling will shrink, albeit not entirely, in line with the experimental data [2]. Ras signaling through BRAF will go; via Raf-1 it will not.
Excellent reviews on Rap1 functional roles have been published over the years (these include [5–7,16–20,34–36]). Thus, here we refer the reader to these rather than review them again. Instead, we prefer to focus on the apparent puzzling Rap1 observations and how they can be understood, and also the implications of our simple explanation.
Key Figure
Ras and Rap1 Signaling Pathways
Figure 1.
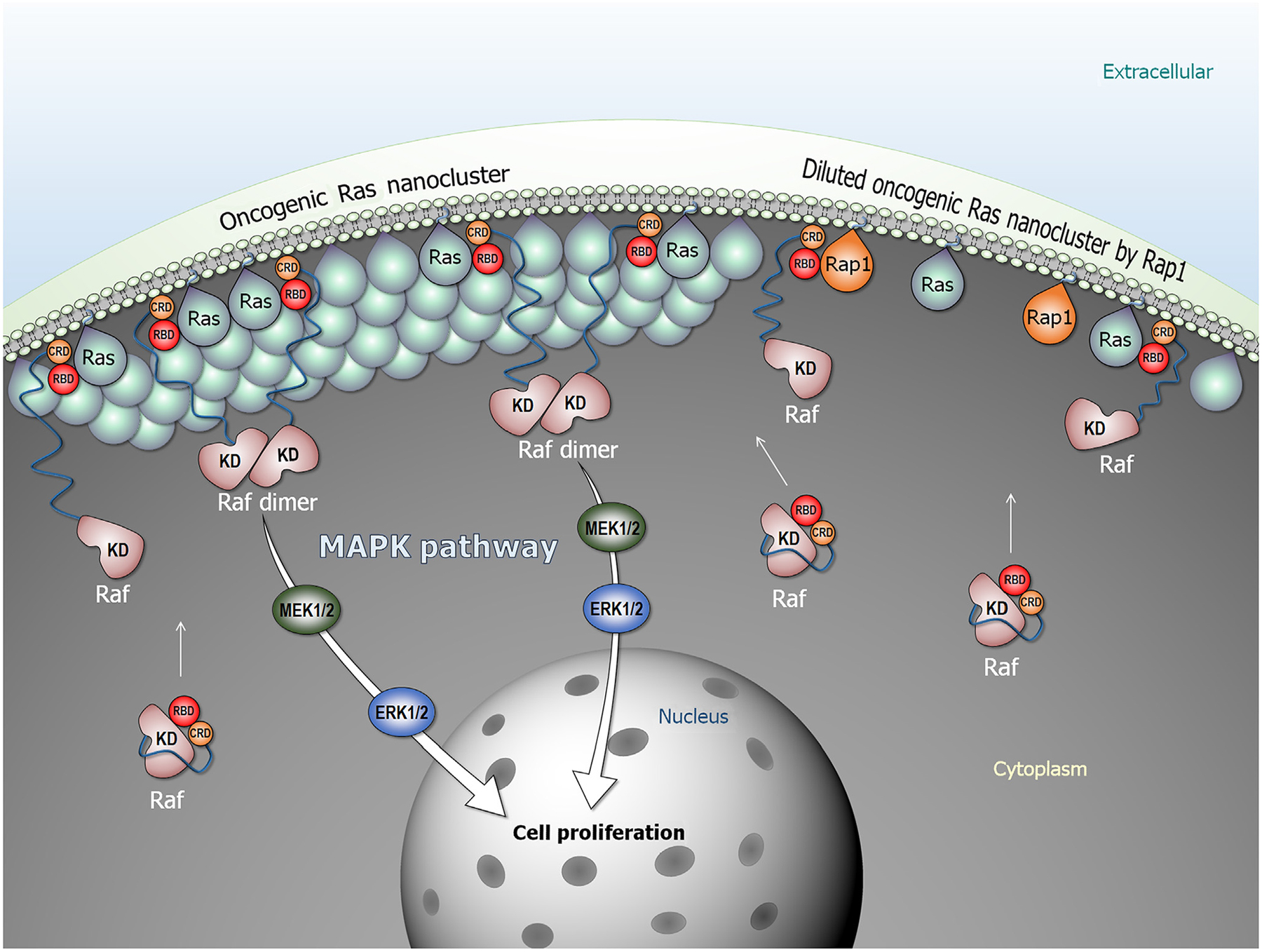
Oncogenic Ras forms nanoclusters and promotes Raf dimerization, activation, and signaling in the mitogen-activated protein kinase (MAPK) pathway (Raf/MEK/ERK). In the cytosol, Raf is autoinhibited, but the high-affinity Ras–(RBD) Ras-binding domain interaction releases the autoinhibition. Raf is activated through side-by-side dimerization. Rap1 dilutes oncogenic Ras nanoclusters, suppressing Raf’s activation. Abbreviations: CRD, cysteine-rich domain; KD, kinase domain.
A Nanoclusters-Centric View Can Explain Puzzling Ras Observations
Ras signaling plays a critical role in cancer initiation and cell proliferation [37]. To signal, Ras needs to be GTP bound and membrane anchored [31,38–47]. Attachment to the membrane is required for Ras activation and nanocluster formation [43]. However, nanoclustering is not essential for all oncogenic Ras pathways: it is needed for Raf’s activation and MAPK signaling but not for PI3Kα activation and signaling through the PI3Kα/Akt/mTOR pathway [30,31]. This is because Raf’s activation requires side-to-side dimerization (homo- or hetero-) of two kinase domains [48–57] (Figure 2). As the figure shows, the two domains are contributed by two Raf molecules, each is bound to active Ras through its Ras-binding domain (RBD) (Figure 1). For the two kinase domains to interact efficiently, they need to be nearby, a requirement that is fulfilled when the Ras molecules are spatially adjacent in the nanocluster (or exist as dimers, oligomers). To activate Raf efficiently, the population of active Ras in the nanocluster should be of sufficient size. If sparse, the probability of dimerization of two Raf kinase domains decreases. This explains the necessity of Ras anchorage in the membrane. In solution, the chance of Ras molecules, thus Raf kinase domains, getting into proximity is low. Thus, if there are other small GTPases in the nanocluster that can bind – but do not activate – Raf, the outcome is essentially tantamount to ‘Ras suppression.’ Later, we suggest that Rap1 falls into this rubric. Nanoclusters are not needed for activation of PI3Kα, where the kinase domain from the p110 subunit exists as a dimer with the regulatory p85 subunit. p85 covers the active site, and activation involves a conformational change that exposes it at the membrane to its PIP2 lipid substrate [58,59].
Figure 2. Comparison between BRAF and Raf-1.
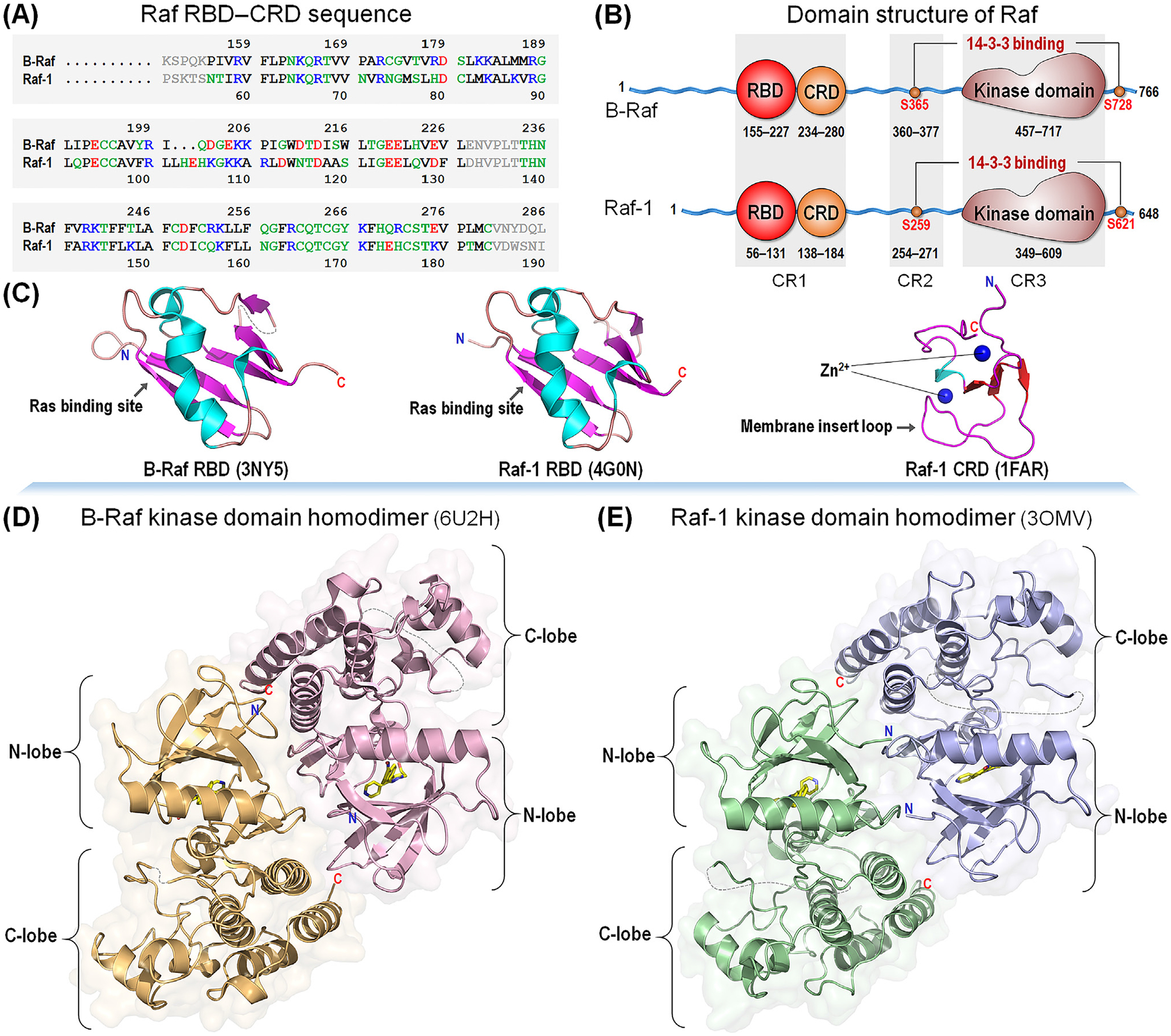
(A) Sequences of conserved region (CR)1 and (B) domain structures of BRAF and Raf-1. In the sequence, hydrophobic, polar/glycine, positively charged, and negatively charged residues are colored black, green, blue, and red, respectively. Gray denotes the unstructured loop region. All Raf kinases share three conserved regions; CR1 involves the Ras-binding domain (RBD) and cysteine-rich domain (CRD), CR2 contains the Ser/Thr-rich region at the flexible linker, and CR3 is the kinase domain. (C) Crystal structures of BRAF RBD (PDB: 3NY5) and Raf-1 RBD (PDB: 4G0N), and solution structure of Raf-1 CRD (PDB: 1FAR). (D) Crystal structures of the kinase domain homodimer for BRAF (PDB: 6U2H) and (E) Raf-1 (PDB: 3OMV). Raf domain structures are highly homologous among the isoforms.
As in the case of Rap1, decades ago another puzzling observation was made: wild-type HRas can suppress cancer [60,61]. Subsequent work clarified that this holds for KRas as well: wild-type KRas can inhibit lung cancer. Additional crucial information was obtained subsequently [62]: wild-type NRas can suppress the malignant phenotype in the presence or absence of its mutant, and importantly, this is particularly the case for KRas [63]. Wild-type KRas inhibited cancer initiation and progression in cell lines expressing KRas, with stronger effect than for NRas [62]. Follow-up experiments confirmed these results [64,65] and provided further crucial data: wild-type Ras proteins can suppress their mutant isoforms [66]; however, it is still unclear exactly how. It was hypothesized that alternative pathways, or complex feedback loops, for the mutants and wild type could be involved. Further work extended these seemingly perplexing data [67–69]. Explaining these tantalizing observations was challenging but deemed essential if we are to harness them toward Ras drug discovery [66].
So, why does wild-type Ras inhibit its oncogenic variants, and why KRas more so than NRas? A Ras nanoclustering-centric outlook can help in resolving this question [30] (Figure 1). It can explain why wild-type KRas can inhibit its oncogenic variant, by simply considering that the presence of inactive wild-type Ras diminishes the concentration of the oncogenic isoform in the nanocluster, thus ‘suppressing’ oncogenic Ras. It also explains why suppression is stronger for KRas than for NRas by simply considering their relative abundance. In the cell line in which the experiments were conducted KRas is more abundant than NRas. It can also explain why a wild-type Ras isoform does not suppress a mutant isoform of a different type [65,70]. This is because different Ras isoforms favor distinct membrane compositions, a preference which is dictated largely by their hypervariable region (HVR) membrane-binding domains [31]. KRas favors acidic disordered membranes; HRas and NRas prefer neutral ones. Consequently, they are unlikely to share clusters.
A nanocluster-centric view can provide a simple explanation as to why wild-type Ras suppresses its oncogenic mutant: wild-type Ras typically exists in an inactive state. To get activated it requires a signal from receptor tyrosine kinase (RTK). The concentration, or population, of the nanocluster is mixed, containing oncogenic (constitutively active) and inactive Ras molecules, thus depressing Raf kinase domain dimerization and activation, and MAPK signaling. Here, we suggest that the presence of Rap1 in oncogenic Ras nanoclusters lowers the concentration of active Ras molecules (Figure 1), thus weakening MAPK signaling, in much the same way the wild-type inactive Ras tampers signaling by its oncogenic isoform. This scenario rests on three notions. First, Ras nanoclustering plays a critical role in Raf activation, an increasingly accepted view in the Ras community, supported by experiments. Second, Rap1 (Rap1A) nestles in KRas nanoclusters. Even though to date there is no direct experimental data, considering the experimental evidence that HRas, NRas, and KRas do not share the same nanocluster due to the distinct chemical properties of their respective HVR membrane-attaching domains [71–73], it is reasonable that Rap1, that shares KRas HVR properties, will join its nanocluster. Third, Rap1 and Ras interact with BRAF and Raf-1 differently. Even though there is some experimental evidence in support of this, further confirmation and conformational details are essential. Our ongoing modeling and simulations are exploring this notion.
Consistent with this idea, we found that in the Cancer Genome Atlas (TCGA), pancreatic cancer and lung adenocarcinoma oncogenic RAS mutations co-occur with a shallow deletion of RAP1A. Importantly, shallow deletions of RAP1A are associated with its decreased expression in both cancer types (www.cbioportal.org) (Figure 3). These genetic data suggest that decreased Rap1 expression might favor development and progression of KRAS mutated tumors.
Figure 3. Decreased Rap1 Expression is Associated with KRas Mutation.
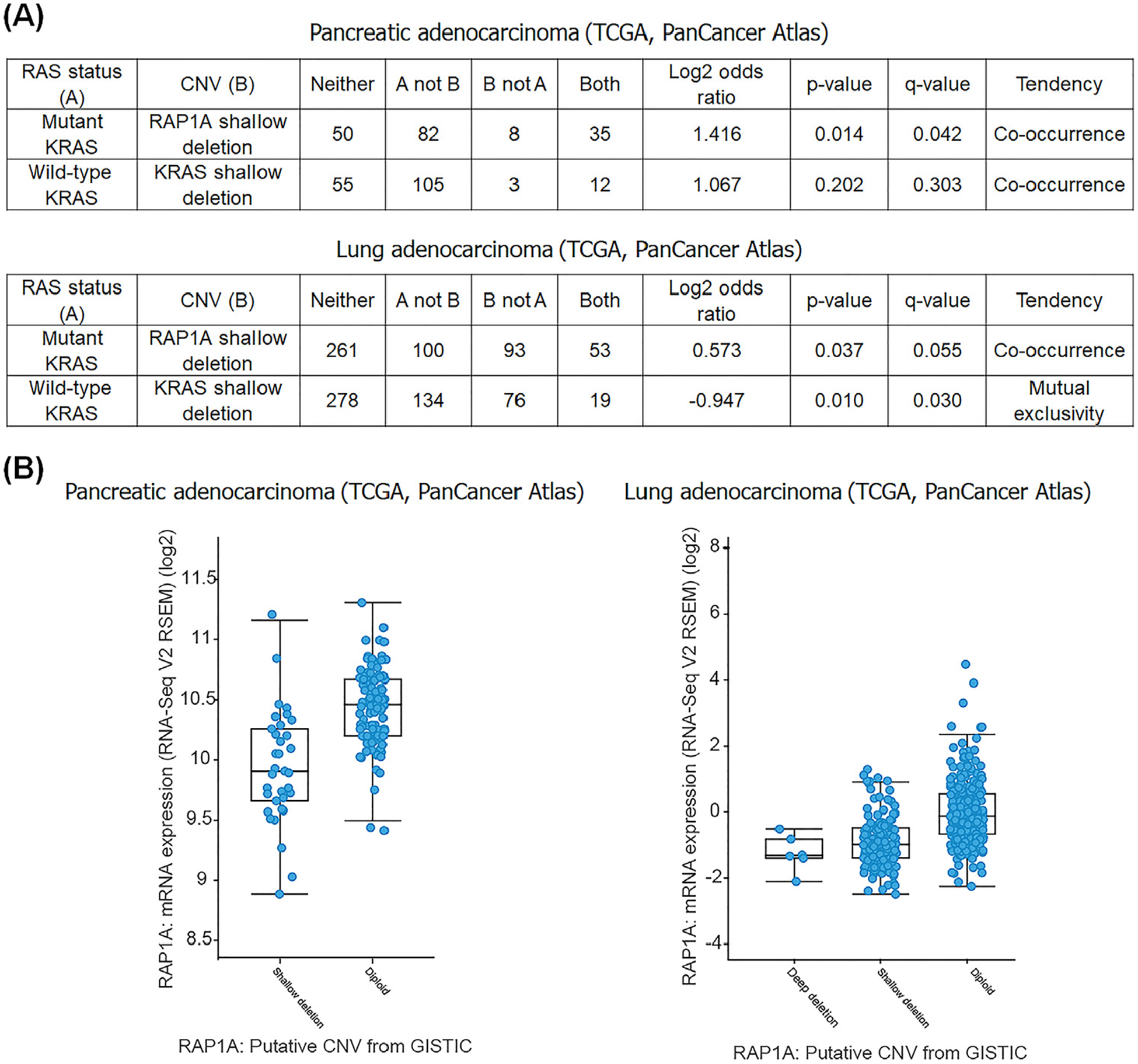
(A) Shallow deletion of RAP1A and KRAS mutation co-occur in pancreatic and lung cancers. The distribution of KRAS somatic mutations and RAP1A copy number variations (CNV) in the Cancer Genome Atlas (TCGA) pancreatic and lung cancers obtained from cBioPortal. cBioPortal was queried over all completed tumors from the PanCancer Atlas datasets using the Onco Query Language (OQL), ‘RAP1A:HETLOSS; KRAS: G12, G13, K61.’ (B) Shallow deletion of RAP1A leads to decreased mRNA in pancreatic and lung cancers. The plots are generated by cBioPortal (www.cbioportal.org) [97,98]. Abbreviation: GISTIC, Genomic Identification of Significant Targets in Cancer.
Rap1–Raf Interaction: Why the Difference between Raf-1 and BRAF and How Does It Relate to Rap1 Depressing Oncogenic Ras Signaling?
Why is it experimentally observed that Ras suppression is incomplete? This is because a certain extent of signaling will go through. Later, we discuss the apparent perplexing observations relating to the interactions of Rap1 with Raf. Early experiments indicated that Rap1 tampers with Raf-1 activation but not with BRAF’s.
HRas Activates Raf Via a Shift in the Equilibrium toward Kinase Domain-Exposed States
Raf is autoinhibited [48,74]. In the autoinhibited state the kinase domain is unable to establish a side-to-side dimer, which is an absolute requirement for wild-type Raf autophosphorylation. Occlusion of the kinase domain is accomplished by the RBD and CRD domains [75,76], with involvement of the linker connecting CRD with the kinase domain [the serine/threonine-rich conserved region (CR)2] [77] (Figure 2). Active, GTP-bound Ras activates Raf through its high-affinity binding to its RBD. This shifts the equilibrium toward the open, kinase domain-exposed state [74,78]. Likewise, binding of active Rap1 to Raf’s RBD is essential for Rap1-dependent Raf activation. The data on Rap1–Raf interactions via two Raf domains [32,33] were not entirely unexpected, given the earlier observations. Those observations showed that Raf-1 contains two distinct HRas-interaction regions, CRD and RBD, each recognizes distinct Ras sites [79–82], and only active Ras can bind and activate Raf [83]. Arguably, shifting the equilibrium in favor of conformations where both domains are away from the kinase domain would relieve the autoinhibition, thus facilitating the kinase domain dimerization. Those works were, however, confined to Raf-1. By contrast, the Rap1 works compared the interactions of Rap1 with Raf-1 with those of Rap1–BRAF and their consequences. Surprisingly, they discovered that whereas active Rap1 activates BRAF, it does not activate Raf-1.
Rap1 Activation Scenarios of Raf-1 Versus BRAF
Rap1 has an identical effector-binding domain to that of Ras (Figure 4A). However, whereas Raf-1 can bind active or inactive Rap1, active Rap1 does not activate Raf-1 and in vivo it even antagonizes some Ras functions [32,33]. To explore this puzzling observation, HRas protein residues, N26G and V45E, were replaced by those in Rap1. The mutations abolished Raf-1 CRD binding and activation, suggesting that HRasN26G/V45E impaired binding to both CRD and RBD. On the contrary, Rap1, with those residues, binds Raf-1 CRD more strongly than HRas does, even though it still does not activate it, indicating that those residues are not responsible for the Rap1–Raf-1 CRD interaction. E31K, another Rap1-mimic substitution, strengthened (tenfold) Raf-1 CRD binding; however, HRasE31K was incapable of Raf-1 activation. The failure of the HRasE31K mutant to activate Raf-1 led the authors to propose that K31 of Rap1 may play a role in the loss of activation. This hypothesis was confirmed by structural studies that observed that Raf-1 RBD binds HRas about 100-fold tighter than Rap1 does, and importantly, the lower Rap1 affinity was attributed to electrostatic repulsion by the interaction of K84 of Raf-1 RBD and K31 of Rap1 (which is Glu in HRas and can form a salt bridge) [84–86] (Figure 4B).
Figure 4. Comparison between Rap1 and Ras.
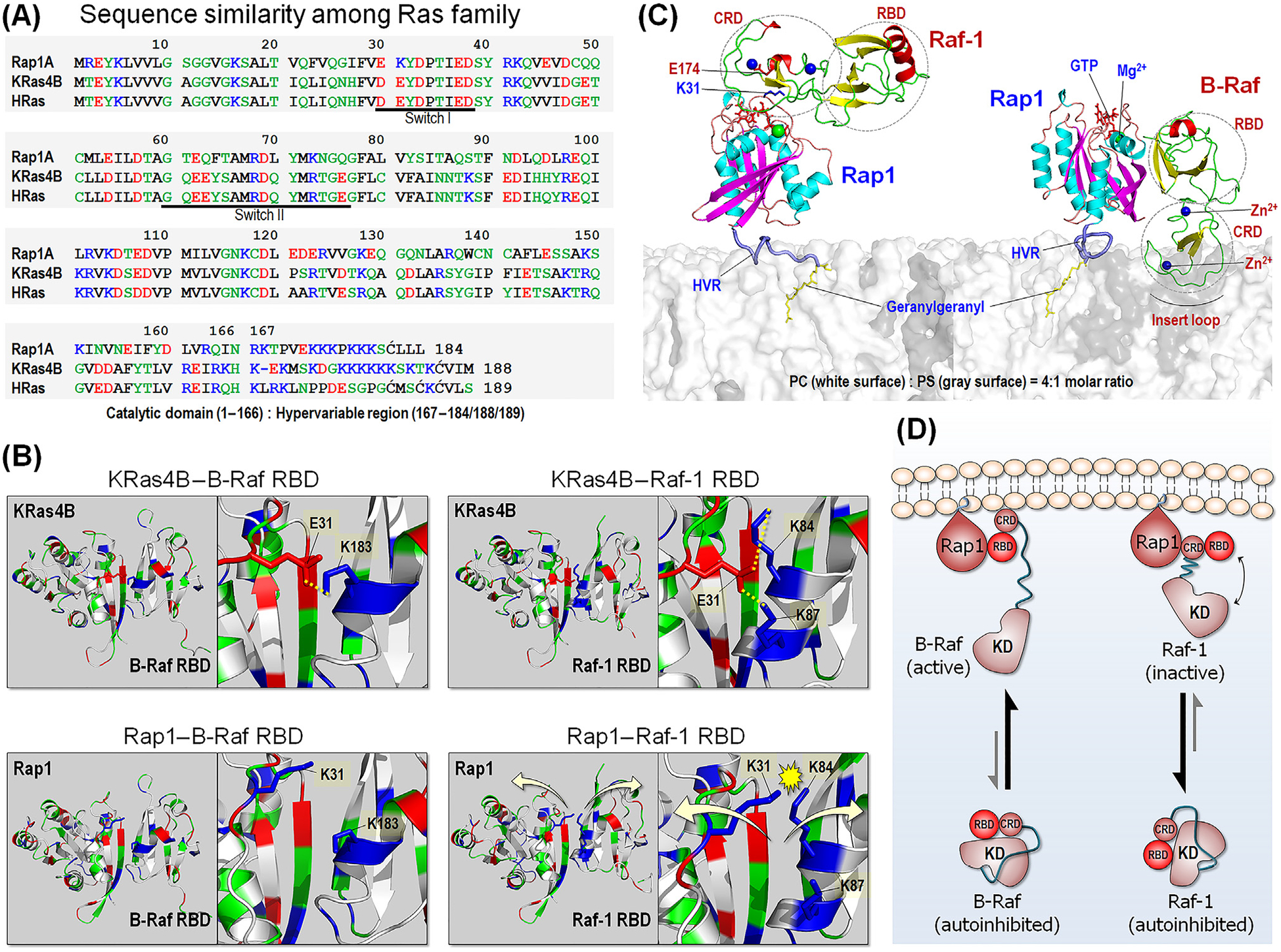
(A) Sequence similarity among Ras family, including Rap1, KRas4B, and HRas. In the sequence, hydrophobic, polar/glycine, positively charged, and negatively charged residues are colored black, green, blue, and red, respectively. (B) Model structures of KRas4B catalytic domain interacting with BRAF Ras-binding domain (RBD) (upper left) and Raf-1 RBD (upper right), based on our previous studies [87]. In addition to the strong β-sheet interaction, Ras adds stability to the interaction with the RBDs through the salt bridges between E31 and K183 for BRAF, and between E31 and K84/K87 for Raf-1. Rap1 can interact with BRAF RBD due to distant repulsive force between K31 and K183 (lower left). However, in the interaction with Raf-1 RBD, Rap1 K31 electrostatically crashes with K84/K87, leading to dissociation. (C) Model structures of Rap1 interacting with Raf-1 conserved region (CR)1 (left) and BRAF CR1 at the anionic membrane. Rap1 prefers to bind Raf-1 cysteine-rich domain (CRD), while it binds to BRAF RBD. (D) Schematic diagram of Raf activation by Rap1. Abbreviations: KD, kinase domain; HVR, hypervariable region.
BRAF was also tested [33]. The experiments indicated that BRAF is activated by both HRas and Rap1. The authors concluded that Rap1 not only obstructs HRas-dependent activation of Raf; but also, fails to activate Raf-1 in contrast to BRAF. Their measurements of the strengths of the respective CRD interactions to Rap1 indicated that, different from Raf-1, BRAF’s CRD interacted only weakly, leading them to conclude that the interactions of the CRDs hold the clue to the differential activation of Raf-1 versus BRAF. Domain-shuffling between Raf-1 and BRAF confirmed their conclusion: with BRAF’s CRD, Raf-1 was activated by Rap1, whereas with Raf-1 CRD, BRAF was not. BRAF CRD double mutant BRAFK253E/M279T, that strengthened the interaction, was not activated by Rap1 either, but it was still activated by Ras. Importantly, CRD is a membrane-interacting domain; but all experiments were carried out in vitro; thus, in the absence of the cell membrane.
Why the Distinct Rap1 Activation Scenarios of Raf-1 versus BRAF?
In the case of Raf-1, the tight binding of CRD to Rap1 is expected to interfere with optimized interaction of its RBD to Rap1’s catalytic domain, thus retaining the largely inactive state (Figure 4C). The figure provides a structural model of Rap1 interaction with the CR1 of Raf-1, which is based on our modeling of KRas4B with Raf-1 RBD and CRD [87]. In the model of Rap1–Raf-1 CRD interaction, we observed formation of a possible salt bridge between K31 and E174, residues that are Rap1 and Raf-1 specific, respectively. We have also modeled Rap1 with BRAF, based on our previous KRas4B-Raf-1 RBD–CRD model [87]. Rap1 interacts with BRAF RBD (Figure 4B) and activates it as Ras does. In the autoinhibited states of BRAF and Raf-1, both RBD and CRD are bound to the kinase domain (Figure 4D). BRAF is activated by Rap1, with the equilibrium shifting toward the active BRAF state. The higher affinity BRAF RBD binding to the GTPase catalytic domain, facilitates the weaker binding of BRAF CRD to Rap1, relieving the autoinhibition. By contrast, the strong Raf-1 CRD interaction with Rap1 [32,33] prevents optimal Raf-1 RBD interactions, retaining Raf-1 in the inactive state. As to Ras [87,88], binding is expected to be isoform dependent, in line with membrane composition and HVR preference.
The membrane is absent in some early experiments but is present in others [89]. Notably, these also include solution NMR. The experiments showed that the Raf-1 CRD–HRas interactions are nucleotide independent but are enhanced by Ras farnesylation. An impaired Raf-1 CRD variant helped identify the key interacting residues in Ras-mediated activation of Raf-1. These residues, 149, 150, 151, 152, 158, and 159, form a hydrophobic patch on Raf-1 CRD. Mutant Raf-1 CRDF151Q/L149T abolishes the HRas–Raf-1 interaction, and consequently Raf-1 activation. Guided by this observation, our inspection of Rap1 surface suggests the presence of several hydrophobic patches with which CRD may form a strong association. To date, there is no direct measurement of the dissociation constant between Rap1 and Raf-1 CRD domains.
How Does the Difference between Raf-1 and BRAF Relate to Rap1 Depressing Oncogenic Ras Signaling?
Ras-dependent Raf activation requires that Ras molecules be spatially close to each other (Figure 1). If they are too sparse, the chance of Raf’s kinase domains to dimerize is low. This is what happens if Rap1 is present in the Ras nanocluster. If Rap1 is active, BRAF will be activated; if inactive, it will not be. For Raf-1, in neither case will it be. In the absence of incoming activation signals, Rap1 is likely to be inactive in the oncogenic Ras nanoclusters. The reduced number of oncogenic molecules in the nanocluster, diminishes – but not abolishes – Ras signaling.
Would Rap1 Suppress All Ras Isoforms?
Experimentally, Rap1A inhibits HRas activation of Raf-1. However, those early experiments were carried out in the absence of the cell membrane. Distinct Ras isoforms favor different membrane compositions and assemble in distinct nanoclusters. Since the HVR of Rap1A is highly positively charged, we expect that in the membrane it colocalizes with and suppresses KRas (KRas4A or KRas4B). By contrast, the HVR of Rap1B is fairly neutral; thus, we expect it to colocalize with and suppress NRas or HRas.
Concluding Remarks
So why does Rap1 interfere with Ras downstream MAPK signaling and revert its transformation? This baffling observation has stymied Ras biology for decades. Possible explanations were proposed early on [1], advanced again a few years later [2], and have been unchallenged since. These argued that Rap1 competes with a Ras target. Even though the similarity in the effector-binding domain lends support to such a hypothesis, no candidate target protein has been identified in the decades since. In addition, this hypothesis cannot explain why, if the two effector-binding surfaces are so similar, and the relative expression levels had no clear effects, there would be such an outcome. The same arguments would apply to interference by inhibiting a pathway protein. Tissue specificity is not an argument here either, since, if that was the case, Rap1 would not depress Ras. Here we take it up from a structural standpoint. This permits us to propose a coherent view that is consistent with available data, albeit one that still needs direct testing (see Outstanding Questions). In essence, we suggest that the key rests with small GTPases organized in the membrane in nanoclusters, an organization which is required for activating those effectors that need to dimerize in order to get activated, like Raf or Rassf5/NORE1A [90], although not for other pathways, like PI3K/Akt that do not [30].
Outstanding Questions.
Experiments discovered that active Rap1 interacts with BRAF RBD and activates it as Ras does, but it does not activate Raf-1. Structural determination of the Rap1–BRAF RBD would address the essential question of whether those early results are relevant to oncogenic cell signaling, as we suggest here.
Experimentally, in the absence of the cell membrane, Rap1 (Rap1A) inhibits activation of Raf-1 by HRas. However, the hypervariable region (HVR) of Rap1A is KRas-like, thus, it can colocalize and suppress KRas, and Rap1B can colocalize with and suppress NRas or HRas, whose HVRs it resembles. Thus, do these isoform distinction scenarios hold in vivo? This requires experimental verification.
RAP1A loss of heterozygosity is associated with KRAS-mutated lung and pancreatic tumors. Functional studies should address the question of whether decreased RAP1A expression triggers KRAS-mediated transformation.
Are there additional small GTPases that may interfere with oncogenic Ras signaling?
A nanocluster organization implies that Ras molecules are assembled in the membrane at favored membrane locations (such as certain lipid composition [91] and membrane geometric features [92]). Its aim is signaling efficiency. In the nanocluster, dynamic Ras molecules are spatially proximal. However, the presence of Rap1 within the cluster ‘dilutes’ active Ras content, making it sparser, thus, essentially ‘suppressing’ Raf’s activation. If Rap1 is inactive, which is likely to be the case in oncogenic Ras clusters since no incoming cue, neither Raf-1 nor BRAF, will be activated. If Rap1 is active, Raf-1 still wouldn’t be, but BRAF would. Notably, the Rap1–Raf data comes from in vitro experiments [32,33]. These should be repeated in the presence of the membrane to capture the in vivo environment.
The search is on for new drug targets and novelty is often the key [93,94]. Mechanism complexity is, however, often difficult to untangle and overlooked factors can be at play [95,96]. Nanoclustering is a fundamental organization in the cell. However, it is often not considered in explaining in vivo phenomena. We hope that this work as well as an earlier one, which similarly showed how the presence can explain puzzling observations [30], will help with making strides in this direction. However, the underlying assumption, that Rap1’s suppression action on oncogenic Ras can be captured by inhibitors, does not appear as a viable model for Ras inhibition.
Highlights.
Rap1, a small GTPase very similar to Ras, suppresses oncogenic Ras pheno-type; albeit not completely. The initial hypothesis was that Ras and Rap1 compete for a common target, but none has been found.
One reported function of Rap1 is to modulate the activity of Raf kinases, thus MAPK signaling. Based on published observations, we propose that Rap1 suppresses MAPK signaling via Raf-1 but not via BRAF. We suggest that Rap1 high-affinity binding to Raf-1’s cysteine-rich domain (CRD) disfavors Rap1 interaction with Raf-1’s Ras-binding domain (RBD). BRAF’s CRD has lower affinity to Rap1; its RBD can bind to Rap1.
The presence of active Rap1 in oncogenic Ras nanoclusters reduces (dilutes) the number of the oncogenic Ras molecules in the cluster, thus suppressing Ras activation of Raf-1 but not of BRAF.
We suggest that Rap1 suppression of Ras is likely to be Ras and Rap1 isoform specific.
Acknowledgments
This project has been funded in whole, or in part, with federal funds from the National Cancer Institute, National Institutes of Health (NIH), under contract HHSN261200800001E, and H2020 European Research Council (ub-RASDisease). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services; nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This research was supported (in part) by the Intramural Research Program of NIH, National Cancer Institute, Center for Cancer Research.
References
- 1.Kitayama H et al. (1989) A ras-related gene with transformation suppressor activity. Cell 56, 77–84 [DOI] [PubMed] [Google Scholar]
- 2.Cook SJ et al. (1993) RapV12 antagonizes Ras-dependent activation of ERK1 and ERK2 by LPA and EGF in Rat-1 fibro-blasts. EMBO J. 12, 3475–3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sprang SR (1995) How Ras works: structure of a Rap–Raf complex. Structure 3, 641–643 [DOI] [PubMed] [Google Scholar]
- 4.Zwartkruis FJ and Bos JL (1999) Ras and Rap1: two highly related small GTPases with distinct function. Exp. Cell Res 253, 157–165 [DOI] [PubMed] [Google Scholar]
- 5.Shah S et al. (2019) Ras and Rap1: a tale of two GTPases. Semin. Cancer Biol 54, 29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bos JL (1998) All in the family? New insights and questions regarding interconnectivity of Ras, Rap1 and Ral. EMBO J. 17, 6776–6782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caron E (2003) Cellular functions of the Rap1 GTP-binding protein: a pattern emerges. J. Cell Sci 116, 435–440 [DOI] [PubMed] [Google Scholar]
- 8.Takahashi M et al. (2017) Phosphorylation of Rap1 by cAMP-dependent protein kinase (PKA) creates a binding site for KSR to sustain ERK activation by cAMP. J. Biol. Chem 292, 1449–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang YC et al. (2019) Molecular basis for autoinhibition of RIAM regulated by FAK in integrin activation. Proc. Natl. Acad. Sci. U. S. A 116, 3524–3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeiller C et al. (2012) Ras and Rap1 govern spatiotemporal dynamic of activated ERK in pituitary living cells. Cell. Signal 24, 2237–2248 [DOI] [PubMed] [Google Scholar]
- 11.Li Y et al. (2016) Protein kinase A-independent Ras protein activation cooperates with Rap1 protein to mediate activation of the extracellular signal-regulated kinases (ERK) by cAMP. J. Biol. Chem 291, 21584–21595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su W et al. (2015) Rap1 and its effector RIAM are required for lymphocyte trafficking. Blood 126, 2695–2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu W et al. (2018) Small GTPase Rap1A/B is required for lymphatic development and adrenomedullin-induced stabilization of lymphatic endothelial junctions. Arterioscler. Thromb. Vasc. Biol 38, 2410–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.York RD et al. (1998) Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 392, 622–626 [DOI] [PubMed] [Google Scholar]
- 15.Raaijmakers JH and Bos JL (2009) Specificity in Ras and Rap signaling. J. Biol. Chem 284, 10995–10999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bos JL et al. (2001) Rap1 signalling: adhering to new models. Nat. Rev. Mol. Cell Biol 2, 369–377 [DOI] [PubMed] [Google Scholar]
- 17.Knox AL and Brown NH (2002) Rap1 GTPase regulation of adherens junction positioning and cell adhesion. Science 295, 1285–1288 [DOI] [PubMed] [Google Scholar]
- 18.Wittchen ES et al. (2011) Isoform-specific differences between Rap1A and Rap1B GTPases in the formation of endothelial cell junctions. Small GTPases 2, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeyaraj SC et al. (2011) Rap1 GTPases: an emerging role in the cardiovasculature. Life Sci. 88, 645–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bos JL (2005) Linking Rap to cell adhesion. Curr. Opin. Cell Biol 17, 123–128 [DOI] [PubMed] [Google Scholar]
- 21.Jeon TJ et al. (2007) Rap1 controls cell adhesion and cell motility through the regulation of myosin II. J. Cell Biol 176, 1021–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arthur WT et al. (2004) Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J. Cell Biol 167, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itoh M et al. (2007) Rap1 integrates tissue polarity, lumen formation, and tumorigenic potential in human breast epithelial cells. Cancer Res. 67, 4759–4766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lakshmikanthan S et al. (2011) Rap1 promotes VEGFR2 activation and angiogenesis by a mechanism involving integrin αvβ3. Blood 118, 2015–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailey CL et al. (2009) Activation of Rap1 promotes prostate cancer metastasis. Cancer Res. 69, 4962–4968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wittchen ES et al. (2013) Rap1 GTPase activation and barrier enhancement in RPE inhibits choroidal neovascularization in vivo. PLoS One 8, e73070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goto M et al. (2010) Rap1 stabilizes β-catenin and enhances β-catenin-dependent transcription and invasion in squamous cell carcinoma of the head and neck. Clin. Cancer Res 16, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu L et al. (2016) Rap1A promotes ovarian cancer metastasis via activation of ERK/p38 and notch signaling. Cancer Med. 5, 3544–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jordan JD et al. (1999) Modulation of rap activity by direct interaction of Gαo with Rap1 GTPase-activating protein. J. Biol. Chem 274, 21507–21510 [DOI] [PubMed] [Google Scholar]
- 30.Nussinov R et al. (2019) Is nanoclustering essential for all oncogenic KRas pathways? Can it explain why wild-type KRas can inhibit its oncogenic variant? Semin. Cancer Biol 54, 114–120 [DOI] [PubMed] [Google Scholar]
- 31.Nussinov R et al. (2018) Oncogenic Ras isoforms signaling specificity at the membrane. Cancer Res. 78, 593–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu CD et al. (1997) Coassociation of Rap1A and Ha-Ras with Raf-1 N-terminal region interferes with Ras-dependent activation of Raf-1. J. Biol. Chem 272, 11702–11705 [DOI] [PubMed] [Google Scholar]
- 33.Okada T et al. (1999) The strength of interaction at the Raf cysteine-rich domain is a critical determinant of response of Raf to Ras family small GTPases. Mol. Cell. Biol 19, 6057–6064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stork PJ (2005) Directing NGF’s actions: it’s a Rap. Nat. Cell Biol 7, 338–339 [DOI] [PubMed] [Google Scholar]
- 35.Stork PJ (2003) Does Rap1 deserve a bad Rap? Trends Biochem. Sci 28, 267–275 [DOI] [PubMed] [Google Scholar]
- 36.Hattori M and Minato N (2003) Rap1 GTPase: functions, regulation, and malignancy. J. Biochem 134, 479–484 [DOI] [PubMed] [Google Scholar]
- 37.Stout MC et al. (2014) Analyzing Ras-associated cell proliferation signaling. Methods Mol. Biol 1170, 393–409 [DOI] [PubMed] [Google Scholar]
- 38.Boriack-Sjodin PA et al. (1998) The structural basis of the activation of Ras by Sos. Nature 394, 337–343 [DOI] [PubMed] [Google Scholar]
- 39.Cherfils J and Zeghouf M (2013) Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev 93, 269–309 [DOI] [PubMed] [Google Scholar]
- 40.Hancock JF et al. (1989) All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57, 1167–1177 [DOI] [PubMed] [Google Scholar]
- 41.Jang H et al. (2016) The higher level of complexity of K-Ras4B activation at the membrane. FASEB J. 30, 1643–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmick M et al. (2014) KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 157, 459–471 [DOI] [PubMed] [Google Scholar]
- 43.Zhou Y and Hancock JF (2015) Ras nanoclusters: versatile lipid-based signaling platforms. Biochim. Biophys. Acta 1853, 841–849 [DOI] [PubMed] [Google Scholar]
- 44.Nussinov R et al. (2019) Oncogenic KRas mobility in the membrane and signaling response. Semin. Cancer Biol 54, 109–113 [DOI] [PubMed] [Google Scholar]
- 45.Muratcioglu S et al. (2017) PDEδ binding to Ras isoforms provides a route to proper membrane localization. J. Phys. Chem. B 121, 5917–5927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jang H et al. (2016) Membrane-associated Ras dimers are isoform-specific: K-Ras dimers differ from H-Ras dimers. Biochem. J 473, 1719–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bandaru P et al. (2019) The interdependent activation of Sonof-Sevenless and Ras. Cold Spring Harb. Perspect. Med 9, a031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rezaei Adariani S et al. (2018) Structural snapshots of RAF kinase interactions. Biochem. Soc. Trans 46, 1393–1406 [DOI] [PubMed] [Google Scholar]
- 49.Tsai CJ and Nussinov R (2018) Allosteric activation of RAF in the MAPK signaling pathway. Curr. Opin. Struct. Biol 53, 100–106 [DOI] [PubMed] [Google Scholar]
- 50.Yuan J et al. (2018) The dimer-dependent catalytic activity of RAF family kinases is revealed through characterizing their oncogenic mutants. Oncogene 37, 5719–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lavoie H et al. (2018) MEK drives BRAF activation through allosteric control of KSR proteins. Nature 554, 549–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varga A et al. (2017) RAF1/BRAF dimerization integrates the signal from RAS to ERK and ROKα. Sci. Signal 10, eaai8482. [DOI] [PubMed] [Google Scholar]
- 53.Verkhivker GM (2016) Molecular dynamics simulations and modelling of the residue interaction networks in the BRAF kinase complexes with small molecule inhibitors: probing the allosteric effects of ligand-induced kinase dimerization and paradoxical activation. Mol. BioSyst 12, 3146–3165 [DOI] [PubMed] [Google Scholar]
- 54.Jambrina PG et al. (2016) Phosphorylation of RAF kinase dimers drives conformational changes that facilitate transactivation. Angew. Chem. Int. Ed. Engl 55, 983–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Freeman AK et al. (2013) The importance of Raf dimerization in cell signaling. Small GTPases 4, 180–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freeman AK et al. (2013) Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol. Cell 49, 751–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Udell CM et al. (2011) Mechanistic principles of RAF kinase signaling. Cell. Mol. Life Sci 68, 553–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang M et al. (2019) The mechanism of PI3Kα activation at the atomic level. Chem. Sci 10, 3671–3680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang M et al. (2019) The structural basis for Ras activation of PI3Kα lipid kinase. Phys. Chem. Chem. Phys 21, 12021–12028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spandidos A and Wilkie NM (1988) The normal human H-ras1 gene can act as an onco-suppressor. Br. J. Cancer Suppl 9, 67–71 [PMC free article] [PubMed] [Google Scholar]
- 61.Spandidos DA et al. (1990) Expression of the normal H-ras1 gene can suppress the transformed and tumorigenic phenotypes induced by mutant ras genes. Anticancer Res. 10, 1543–1554 [PubMed] [Google Scholar]
- 62.Diaz R et al. (2002) The N-ras proto-oncogene can suppress the malignant phenotype in the presence or absence of its oncogene. Cancer Res. 62, 4514–4518 [PubMed] [Google Scholar]
- 63.Zhang Z et al. (2001) Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat. Genet 29, 25–33 [DOI] [PubMed] [Google Scholar]
- 64.To MD et al. (2013) Interactions between wild-type and mutant Ras genes in lung and skin carcinogenesis. Oncogene 32, 4028–4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu J et al. (2013) Dominant role of oncogene dosage and absence of tumor suppressor activity in Nras-driven hematopoietic transformation. Cancer Discov. 3, 993–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou B et al. (2016) The role of wild type RAS isoforms in cancer. Semin. Cell Dev. Biol 58, 60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Staffas A et al. (2015) Wild-type KRAS inhibits oncogenic KRAS-induced T-ALL in mice. Leukemia 29, 1032–1040 [DOI] [PubMed] [Google Scholar]
- 68.Kong G et al. (2016) Loss of wild-type Kras promotes activation of all Ras isoforms in oncogenic Kras-induced leukemogenesis. Leukemia 30, 1542–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qiu W et al. (2011) Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget 2, 862–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matallanas D et al. (2011) Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol. Cell 44, 893–906 [DOI] [PubMed] [Google Scholar]
- 71.Henis YI et al. (2009) Ras acylation, compartmentalization and signaling nanoclusters (review). Mol. Membr. Biol 26, 80–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou Y et al. (2014) Signal integration by lipid-mediated spatial cross talk between Ras nanoclusters. Mol. Cell. Biol 34, 862–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nussinov R et al. (2015) Oligomerization and nanocluster organization render specificity. Biol. Rev. Camb. Philos. Soc 90, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nussinov R et al. (2018) Autoinhibition in Ras effectors Raf, PI3Kα, and RASSF5: a comprehensive review underscoring the challenges in pharmacological intervention. Biophys. Rev 10, 1263–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kondo Y et al. (2019) Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 366, 109–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park E et al. (2019) Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature 575, 545–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lavoie H and Therrien M (2015) Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol 16, 281–298 [DOI] [PubMed] [Google Scholar]
- 78.Nussinov R et al. (2019) Does Ras activate Raf and PI3K allosterically? Front. Oncol 9, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brtva TR et al. (1995) Two distinct Raf domains mediate interaction with Ras. J. Biol. Chem 270, 9809–9812 [DOI] [PubMed] [Google Scholar]
- 80.Drugan JK et al. (1996) Ras interaction with two distinct binding domains in Raf-1 may be required for Ras transformation. J. Biol. Chem 271, 233–237 [DOI] [PubMed] [Google Scholar]
- 81.Hu CD et al. (1995) Cysteine-rich region of Raf-1 interacts with activator domain of post-translationally modified Ha-Ras. J. Biol. Chem 270, 30274–30277 [DOI] [PubMed] [Google Scholar]
- 82.Morrison DK and Cutler RE (1997) The complexity of Raf-1 regulation. Curr. Opin. Cell Biol 9, 174–179 [DOI] [PubMed] [Google Scholar]
- 83.Moodie SA et al. (1993) Complexes of Ras. GTP with Raf-1 and mitogen-activated protein kinase. Science 260, 1658–1661 [DOI] [PubMed] [Google Scholar]
- 84.Herrmann C et al. (1996) Differential interaction of the Ras family GTP-binding proteins H-Ras, Rap1A, and R-Ras with the putative effector molecules Raf kinase and Ral-guanine nucleotide exchange factor. J. Biol. Chem 271, 6794–6800 [DOI] [PubMed] [Google Scholar]
- 85.Nassar N et al. (1996) Ras/Rap effector specificity determined by charge reversal. Nat. Struct. Biol 3, 723–729 [DOI] [PubMed] [Google Scholar]
- 86.Vetter IR et al. (1999) Structural and biochemical analysis of Ras-effector signaling via RalGDS. FEBS Lett. 451, 175–180 [DOI] [PubMed] [Google Scholar]
- 87.Li S et al. (2018) Raf-1 cysteine-rich domain increases the affinity of K-Ras/Raf at the membrane, promoting MAPK signaling. Structure 26, 513–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fischer A et al. (2007) B- and C-RAF display essential differences in their binding to Ras: the isotype-specific N terminus of B-RAF facilitates Ras binding. J. Biol. Chem 282, 26503–26516 [DOI] [PubMed] [Google Scholar]
- 89.Williams JG et al. (2000) Elucidation of binding determinants and functional consequences of Ras/Raf-cysteine-rich domain interactions. J. Biol. Chem 275, 22172–22179 [DOI] [PubMed] [Google Scholar]
- 90.Liao TJ et al. (2016) RASSF5: An MST activator and tumor suppressor in vivo but opposite in vitro. Curr. Opin. Struct. Biol 41, 217–224 [DOI] [PubMed] [Google Scholar]
- 91.Zhou Y and Hancock JF (2018) Electron microscopy combined with spatial analysis: quantitative mapping of the nano-assemblies of plasma membrane-associating proteins and lipids. Biophys. Rep 4, 320–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee Y et al. (2019) High-throughput single-particle tracking reveals nested membrane domains that dictate KRasG12D diffusion and trafficking. eLife 8, e46393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lou K et al. (2019) A bounty of new challenging targets in oncology for chemical discovery. Biochemistry 58, 3328–3330 [DOI] [PubMed] [Google Scholar]
- 94.Zhao Z et al. (2014) Exploration of type II binding mode: a privileged approach for kinase inhibitor focused drug discovery? ACS Chem. Biol 9, 1230–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y et al. (2013) Mutant N-RAS protects colorectal cancer cells from stress-induced apoptosis and contributes to cancer development and progression. Cancer Discov. 3, 294–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roskoski R Jr. (2019) Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol. Res 142, 151–168 [DOI] [PubMed] [Google Scholar]
- 97.Cerami E et al. (2012) The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gao J et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]