

Abstract
The programmed death-ligand 1 (PD-L1) is a major checkpoint protein that helps cancer cells evade the immune system. A non-invasive imaging agent with rapid clearance rate would be an ideal tool to predict and monitor the efficacy of anti-PD-L1 therapy. The aim of this research was to engineer a subnanomolar, high-affinity fibronectin type 3 domain (FN3)-based small binder targeted against human PD-L1 (hPD-L1) present on tumor cells. A naive yeast G4 library containing the FN3 gene with three binding loop sequences was used to isolate high-affinity binders targeted to purified full-length hPD-L1. The selected binder clones displayed several mutations in the loop regions of the FN3 domain. One unique clone (FN3hPD-L1-01) with a 6x His-tag at the C-terminus had a protein yield of >5 mg/L and a protein mass of 12 kDa. In vitro binding assays on six different human cancer cell lines (MDA-MB-231, DLD1, U87, 293 T, Raji and Jurkat) and murine CT26 colon carcinoma cells stably expressing hPD-L1 showed that CT26/hPD-L1 cells had the highest expression of hPD-L1 in both basal and IFN-γ-induced states, with a binding affinity of 2.38 ± 0.26 nM for FN3hPD-L1-01. The binding ability of FN3hPD-L1-01 was further confirmed by immunofluorescence staining on ex vivo CT26/hPD-L1 tumors sections. The FN3hPD-L1-01 binder represents a novel, small, high-affinity binder for imaging hPD-L1 expression on tumor cells and would aid in earlier imaging of tumors. Future clinical validation studies of the labeled FN3hPD-L1 binder(s) have the potential to monitor immune checkpoint inhibitors therapy and predict responders.
Keywords: Fibronectin domain 3 (FN3)-based binder, immune checkpoint, yeast display, human programmed death ligand 1 (hPD-L1)
Introduction
Harnessing the body’s immune response against tumors is proving to be a promising approach in cancer research. Since William Coley’s accidental discovery of complete remissions in a sarcoma patient with erysipelas infection (Coley, 1910), there has been prevalent interest in using immunotherapy to treat cancer. Additional studies spearheaded several immune therapies using various immune checkpoints such as CTLA-4 (cytotoxic T-lymphocyte-associated protein 4 or CD152), PD-1 (programmed cell death protein 1 or CD279) and its ligand PD-L1 (programmed death-ligand 1/B7-H1/CD274).
The immune system plays a major role in regulating and suppressing cancer. Two components of the immune system exist: innate and adaptive immune systems. The innate system (including natural killer cells, mast cells, eosinophils, basophils, macrophages, neutrophils and dendritic cells) provides an immediate, first line of defense from pathogens (e.g. bacterial, viral, fungal, parasites, worms etc.). The adaptive system (including T and B lymphocytes) is also responsible for specific and long-term memory responses (Charles et al., 2001). Both systems complement each other in combating infections. Most of the work related to immunotherapy and immune blocking strategies have been focused on lymphocytes, due to their ability to recognize and kill tumor cells with specific antigens expressed on cell surfaces (Allison, 2015; Pardoll, 2012). This lymphocyte activity is highly controlled by a complex sequence of mechanisms, including stimulatory, co-stimulatory and inhibitory molecules. In this complex system, the central regulator of T-lymphocyte function is the T-cell receptor (TCR), the balance between positive and negative signaling input deeply shapes the response that lymphocytes mount upon exposure to reactive peptides/major histocompatibility complexes (MHC) (Maute, 2015). Consequently, cancer cells develop somatic mutations that distinguish from non-cancerous cells. These mutations can be recognized by the TCRs and trigger the endogenous antitumor response by the immune system. To escape from this attack, tumor cells express inhibitory molecule of PD-L1. This led to active research on PD1: PD-L1 pathway blocking molecules for cancer immunotherapy (Pardoll, 2012).
Many antibodies targeting or blocking these inhibitors have been recently approved by the Food and Drug Administration (FDA) due to their promising therapeutic effects: anti-CTLA4: YERVOY®/Ipilimumab (Yervoy, Bristol-Meyers Squibb) and Tremelimumab® (Pfizer), anti-PD-1: Pembrulizumab® (Merck) and anti-PD-L1: Nivolumab® (Bristol-Myers Squibb). PD-1 is expressed on the surface of CD4+ and CD8+ cells as well as activated T cells, B cells, NK cells and APCs (Keir et al., 2008; Lim et al., 2016). On the other hand, PD-L1 is expressed on several tumor cells—melanoma (Sunshine et al., 2017; Taube et al., 2014), breast cancer (Latchman et al., 2001), NSCLC (Taube et al., 2014; Champiat et al., 2014) and binds to the PD-1 receptor on T cells, thereby inhibiting T-cell killing of tumors. In addition, PD-L1 inhibits T-cell proliferation, cytokine production and cell adhesion (Ghiotto et al., 2010), thereby evading tumorigenesis. Antibodies targeting PD-L1 such as Nivolumab® (Bristol-Myers Squibb) were shown to reduce tumor margins in 30% of patients with Stage III-IV advanced melanoma (Long et al., 2017).
Following the success of Nivolumab®, PD-L1 antibodies were developed by Astrazeneca (Durvalumab®) and Genetech/Roche (Atezolizumab®) for clinical use for the treatment of locally advanced or metastatic urothelial carcinoma. As therapeutic agents, full-length antibodies are not limited by their low immunogenicity and long-serum half-life. However, as imaging agents, their large size (~150 kDa) and longer clearance times from circulation may preclude their utility for early time points and repetitive imaging (Zhan et al., 2016). The FDA recently approved two IHC tests (PD-L1 IHC 22C3 pharmDx and PD-L1 IHC 28-8 pharmDx) for the detection of PD-L1, but this requires the patients to undergo invasive biopsies and limits the detection of PD-L1 due to its heterogenic expression on tumors (Chatterjee et al., 2017). Moreover, in a study on non-small cell lung patient samples, only 40, 38, 18 and 30% of PD-L1 expression was detected using clones 22C3, SP263, SP142 and E1L3N, respectively (Kim et al., 2017). For these reasons, non-invasive imaging agents with faster clearance (<12 hours (Hackel et al., 2012; Natarajan et al., 2013)) would be an ideal tool for the prediction and monitoring of anti-PD-L1 therapy.
Low molecular weight protein scaffolds (6–20 KDa) such as monobodies, nanobodies, knottins, antibody fragments and fibronectin domains (Lipovšek, 2011) have been explored for cancer detection. In particular, the 10th type III domain of the human fibronectin (FN3) (Fig. 2A) has a low molecular weight of ~ 11 kDa, with a rapid clearance rate (<12 hours vs. ~ 72 hours in the case of full-length antibodies), high thermostability (Tm = 86°C), and demonstrates specific in vivo targeting with excellent tumor-to-background ratios (Hackel et al., 2012). The FN3 domain can be engineered by inducing mutations in three loops to facilitate binding to hPD-L1. In the current work, we present the selection, production of a small, non-invasive high-affinity FN3-based hPD-L1 binder that could likely be translated as an imaging agent for detection of hPD-L1 expression in cancers.
Fig. 5.
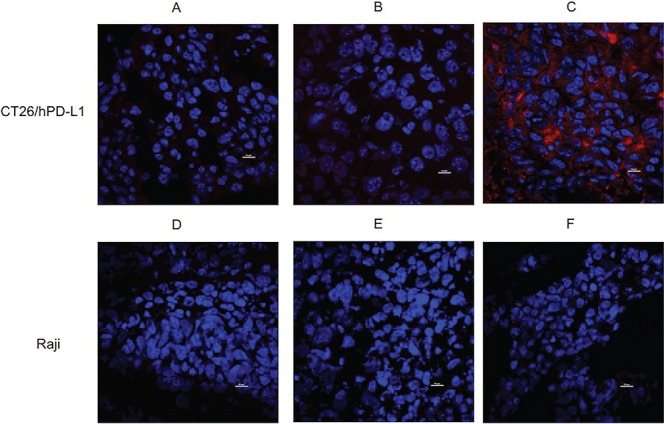
Specificity of FN3hPD-L1-01 binder for in vivo detection of hPD-L1 in tumors. (A) Tumor sections from mice bearing CT26/hPD-L1 xenografts were stained with Alexa Fluor 647® 6XHisTag antibody. (B) Tumor sections from mice bearing CT26/hPD-L1 xenografts and injected with 1 mg of FN3hPD-L1-01 binder 24 hours after injection with 100 μg Tecentriq® via tail vein and were stained with Alexa Fluor 647® 6XHisTag antibody. (C) Tumor sections from mice bearing CT26/hPD-L1 xenografts and injected with 1 mg binder via tail vein were stained with Alexa Fluor 647® 6XHisTag antibody (membrane staining). (D) Tumor sections from mice bearing Raji xenografts were stained with Alexa Fluor 647® 6XHisTag antibody. (E) Tumor sections from mice bearing Raji xenografts and injected with 1 mg of FN3hPD-L1-01 binder 24 hours after injection with 100 μg Tecentriq® via tail vein. (F) Tumor sections from mice bearing Raji xenografts and injected with 1 mg binder via tail vein. All sections were stained for the nuclei using DAPI (nuclei staining, center). Image acquisition was performed at 60× magnification using an intravital microscope. Scale bar = 10 μm.
Materials and Methods
hPD-L1 protein biotinylation
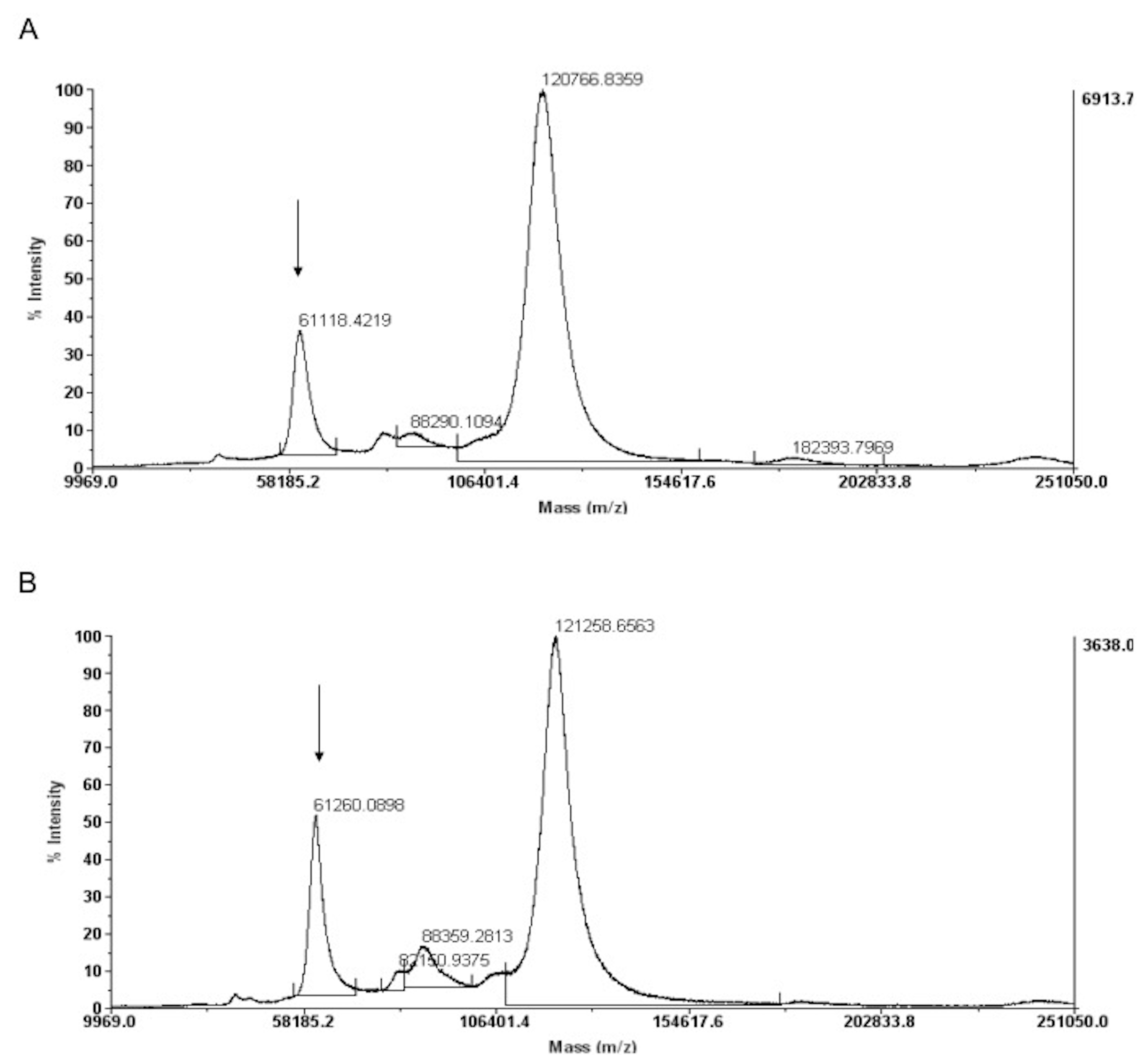
Recombinant human (rh) B7-H1/Fc chimera was purchased from Sino Biologicals (Cat. No 10084-H02H, Beijing, China) and biotinylated using EZ-Link™ NHS-PEG4-Biotinylation Kit (Thermo Fisher Scientific, Waltham, MA). Biotinylation was confirmed by Matrix-Assisted Laser Desorption/Ionization (MALDI) analysis (Fig. S1).
Cell culture
CT26 (murine colon carcinoma), Raji (Burkitt’s lymphoma from a human lymphoblast) and DLD-1 (human colorectal adenocarcinoma) cell were gifts from Dr. Irving L. Weissman lab (Stanford University, Stanford, CA) that also generated genetic variations of CT26 expressing human PD-L1 (CT26/hPD-L1) (Maute, 2015). Raji, MDA-MB-231 (human breast adenocarcinoma), U87 (human glioblastoma), 293 T (human embryonic kidney cells transformed with the large T antigen) and Jurkat (human T cell leukemia) were obtained from American Type Culture Collection (ATCC, Manassas, VA). CT26/hPD-L1, Raji, DLD1 and Jurkat cells were cultured in RPMI-1640 media supplemented with 10% (vol/vol) fetal bovine serum (FBS) and 1% (vol/vol) pencillin-streptomycin (P/S). MDA-MB-231, U87 and 293 T were grown in DMEM media supplemented with 10% (vol/vol) FBS and 1% (vol/vol) P/S. All cell lines were grown at 37°C with 5% CO2 in a humidified incubator. Cell lines were induced using 0–40 ng/ml interferon-gamma (IFN-γ, R&D Systems, Minneapolis, MN) for 24 hours to induce the expression of hPD-L1. All cell culture reagents were purchased from Thermo Fisher Scientific Inc. (Waltham, MA) unless otherwise stated.
FN3 yeast display construct and libraries
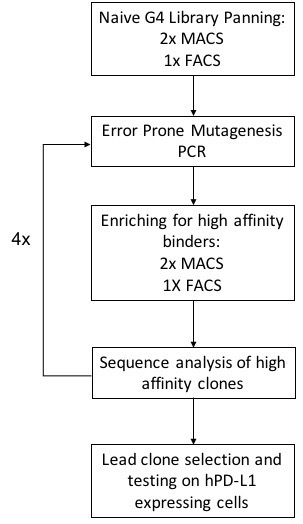
To further enhance affinity maturation and FN3 binder screening, the biotinylated hPD-L1 antigen was mixed with streptavidin-coated magnetic Dynabeads (Thermo Fisher Scientific, Waltham, MA) and incubated with the EBY100 strain of the Saccharomyces cerevisiae yeast surface displayed FN3 G4 library at room temperature for 90 minutes (Hackel et al., 2010). Two selection schemes (sequentially; Fig. S2) were used to isolate high-affinity binders: (i) MACS selection, using a biotinylated human PD-L1 antigen immobilized on a streptavidin-coated magnetic bead; and (ii) FACS sorting with a streptavidin (SA) Alexa Fluor 647® mixed with the biotinylated hPD-L1 antigen. Under MACS selection, the library was pre-cleared with 33 pmol biotinylated Fc domain alone, followed by a positive selection with the full-length hPD-L1 antigen-SA fusion. After every two rounds of MACS enrichment, the highest affinity binders (top 2% of the population) were sorted by FACS (Fig. S2).
Yeast cells were grown in S(D)CAA + P/S (6.7 g/L yeast nitrogen base, 5 g/L bacto casamino acids, 13.7 g/L sodium citrate dihydrate, 8.4 g/L anhydrous citric acid, 20 g/L dextrose, 100 kU/L penicillin and 0.1 g/L streptomycin) at 30°C, 250 rpm. 2 × 108 cells/ml were induced for protein expression in S(G) CAA (0.1 M sodium phosphate, pH 6.0, 6.7 g/L yeast nitrogen base, 5 g/L bacto casamino acids, 19 g/L galactose, 1 g/L glucose, 100 kU/L penicillin and 0.1 g/L streptomycin) at 30°C, 250 rpm for 16 hours. 2.5 × 108 induced cells were used for each round of affinity maturation. Each round of affinity maturation comprised of two rounds of magnetic selections and one round of fluorescence-based sorting, followed by an error-prone mutagenesis polymerase chain reaction (PCR) to induce mutations in the three loops (BC, DE and FG) of the fibronectin and reintroduced into the yeast by electroporation. The new diverse population was panned again using streptavidin-coated magnetic Dynabeads. Four cycles of selection and mutation were performed for selection of highest binding clones (Natarajan et al., 2013; Chao et al., 2006).
FN3hPD-L1 binder production in bacteria
The genomic DNA from each of the yeast clones was extracted using Zymoprep™ Yeast Plasmid Miniprep II (Zymo Research, Irvine, CA) and transformed into bacterial pETH Vector containing 6x His-tags using the SHuffle® T7 Express competent Escherichia coli (New England Biolabs, Ipswich, MA). Individual clones from the bacteria were sequenced using DNA sequencing services by Sequetech Corporation (Mountain View, CA). Unique clones were selected and grown in 1 L LB medium to 0.8–1.0 OD600. The cultures were induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 30°C, 250 rpm for 4 hours. Cells were pelleted, frozen, thawed and re-suspended in lysis buffer (50 mM NaPO4 pH 8.0 0.5 M NaCl (Thermo Fisher Scientific, Waltham, MA), 5% glycerol, 5 mM CHAPS, 25 mM imidazole and 1X EDTA-free protease inhibitors (Sigma Aldrich, St. Louis, MO). Lysed bacterial cells were sonicated on ice four times at 60 W and 60% amplitude and centrifuged for insoluble fraction at 12 000 g, 4°C for 5 minutes. The FN3hPD-L1 binders were purified by fast protein liquid chromatography (FPLC) and reverse-phase high-performance liquid chromatography (HPLC), using a Histrap FF column (GE Healthcare, Uppsala, Sweden) and a C4 semi-preparative column, respectively. Protein mass was verified by mass spectrometry and SDS page gel (Figs S1 and S3). The purified binder was produced in bacteria and used for staining of cell surface hPD-L1 in CT26/hPD-L1 cells by FACS analyses and in tumor sections, as well as injection into mice (see below).
Flow cytometry assays
All flow cytometry experiments were performed using FACS Aria III (BD Biosciences, San Jose, CA, USA), and the data were analyzed using FlowJo. FACS was carried out for (i) affinity maturation; (ii) determining cell-surface hPD-L1 expression on mammalian cell lines (CT26/hPD-L1, Raji, DLD1, Jurkat, 293 T, U87, MDA-MB-231); and (iii) binder characterization and titration on yeast and mammalian cells.
For affinity maturation studies, yeast cells in single-cell suspension were stained with chicken anti-Myc (A21281, Thermo Fisher Scientific, Waltham, MA) and goat anti-chicken Alexa Fluor 555® (A21437, Thermo Fisher Scientific, Waltham, MA) for 40 minutes at 4°C. Cells were washed twice with PBS containing 0.1% w/v BSA (PBSA) and re-suspended in PBSA for analyses and sorting.
For binder characterization and titration studies, yeast cells were incubated with a hPD-L1: streptavidin (Streptavidin Alexa Fluor 647® conjugate (S21371), Thermo Fisher Scientific, Waltham, MA) complex for 1 hour at 4°C. Cells were washed with PBSA and stained with 1:100 chicken anti-Myc and 1:100 goat anti-chicken Alexa Fluor 555® for 40 minutes at 4°C. Cells were washed twice and re-suspended in PBSA for flow analyses.
For blocking studies, CT26/hPD-L1 cells induced with 10 ng/ml IFN-γ were incubated with 150 pM–100 nM of the commercial hPD-L1 antibody E1L3N (Cell Signaling Technology, Danvers, MA) for 2 hours and then incubated with 100 nM of the FN3hPD-L1-01binder for 90 minutes at 4°C. Cells were washed with PBSA and stained with 1:300 α-His Tag Ab (MCA1396A647, Bio-Rad, Raleigh, NC) for 1 hour at 4°C. Cells were washed twice and re-suspended in PBSA and analyzed by flow cytometry.
For cell binding assays, CT26/hPD-L1 cells were incubated with the binder (30 pM–500 nM) for 90 minutes at 4°C and stained with 1:300 α-His Tag Ab for 1 hour at 4°C. Cells were washed twice and re-suspended in PBSA for flow analyses. Cells were gated with reference to a negative cell line, CT26 that does not express hPD-L1.
Animal studies
Animal studies were performed in compliance with the guidelines from the Administrative Panel on Laboratory Animal Care (APLAC) at Stanford University. NSG (NOD.Cg-PrkdcscidIl2rgtm1wjl/SzJ) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained in-house in an AAALAC-accredited facility. Six-week-old female mice were subcutaneously implanted with 5 × 106 CT26/hPD-L1 cells on the left shoulder and 5 × 106 Raji cells on the right shoulder. Cells were re-suspended in 1:1 solution of PBS and matrigel (Corning, Inc., Corning, NY). Two mice were used in each group for the following intravenous injections: (i) control with saline; (ii) 1 mg of the purified binder; and (iii) blocking study with 100 μg commercial hPD-L1 antibody Tecentriq® (Genentech, South San Francisco, CA) 24 hours prior to injection of 1 mg of the purified binder. Mice were euthanized on day 26 post tumor implantation as per APLAC guidelines.
Immunofluorescence staining of hPD-L1 expression in tumors
CT26/hPD-L1 and Raji tumors harvested from the mice were washed in PBS and embedded in Optimal Cutting Temperature compound (OCT) (Scigen Scientific Inc., Gardena, CA). The tissues were sliced to 7 μm thick sections on slides using a Leica CM1850 cryostat. The sections were then fixed and dehydrated with 4% formalin for 12 minutes, washed thrice for 5 minutes with PBS with 1% w/v bovine serum albumin (PBSA) and blocked overnight at 4°C with blocking buffer (37525, Thermo Fisher Scientific, Waltham, MA). Each slide was then washed thrice for 5 minutes with PBSA and incubated with 6x-His Tag Monoclonal Antibody (4E3D10H2/E3) conjugated with Alexa Fluor 647® (MA1-135-A647, Thermo Fisher Scientific, Waltham, MA) overnight at 4°C. After three washes with PBSA, the slides were incubated with NucBlue® Fixed Cell ReadyProbes® Reagent (R37606, Thermo Fisher Scientific, Waltham, MA) at room temperature for 2 minutes to stain for the nuclei and washed thrice with PBSA. The slides were mounted using Permafluor Mountant (Thermo Fisher Scientific, Waltham, MA) and imaged at 60X using an oil immersion objective with the Nikon A1 Plus Intravital Microscope (Tokyo, Japan).
Statistical analyses
All the binding and blocking study data presented in the manuscript were tested for statistical significance (R2) using One Site—total and non-specific binding in the Graphpad Prism software. Student t-test was used to determine differences between different cell binding and incubation conditions, with P < 0.05 as the cut-off for statistical significance. A P value <0.05 was deemed statistically significant.
Results
Engineering of FN3hPD-L1 binder
After four iteration rounds and affinity maturations (Fig. 1), the enriched yeast populations were sorted based on highest fluorescence (P3) at a hPD-L1 binding concentration of 5 nM (Fig. 1A), 0.25 nM (Fig. 1B), 0.125 nM (Fig. 1C) and 0.060 nM (Fig. 1D). Approximately 3 × 105 cells were sorted during each round. The binding affinities (Kd) of the sorted mixed populations at 5 nM, 0.25 nM, 0.125 nM and 0.060 nM hPD-L1 were 12.50 ± 1.95 nM (R2 = 0.95); 5.52 ± 0.63 nM (R2 = 0.89); 3.66 ± 1.01 nM (R2 = 0.92) and 2.82 ± 0.55 nM (R2 = 0.97), respectively (Fig. 1E and F). The estimated Kd values for yeasts expressing the binders (mixed populations) to purified hPD-L1 antigen decreased in Rounds 2–4, relative to the Round 1 of sorting (P < 0.05 in all cases). Clones were selected at the end of each sort to check for unique sequences. Sequence analyses of the 24 clones sorted at 60 pM revealed three unique clones (Fig. 2B). Binder titration was performed using the yeast clones that express FN3hPD-L1-01 to FN3hPD-L1-03 binder on the surface by FACS (Fig. 2C). The biotinylated antigen was mixed with streptavidin-Alexa647® conjugate, incubated with yeast expressing the binders and stained with Alexa 555®-conjugated antibodies that bind to the myc tag on the yeast. The binding affinities of these FN3hPD-L1 clones were within the range for the Kd of the mixed populations (Fig. 1D) sorted at 60 pM (2.82 ± 0.55 nM) from which these individual clones originated from (P > 0.05 in all cases). The best yeast clone (FN3hPD-L1-01) from the 60 pM mixed population binders demonstrated a binding affinity of 2.38 ± 0.26 nM, compared to that of FN3hPD-L1-02 (7.89 ± 2.36 nM) and FN3hPD-L1-03 (9.06 ± 4.08 nM), and was therefore used for protein expression in bacteria. The full-length amino acid sequence of the FN3hPD-L1-01 clone within the BC, DE and FG loops, along with the 6x His-tag at the C-terminus (Fig. 2D).
Fig. 1.
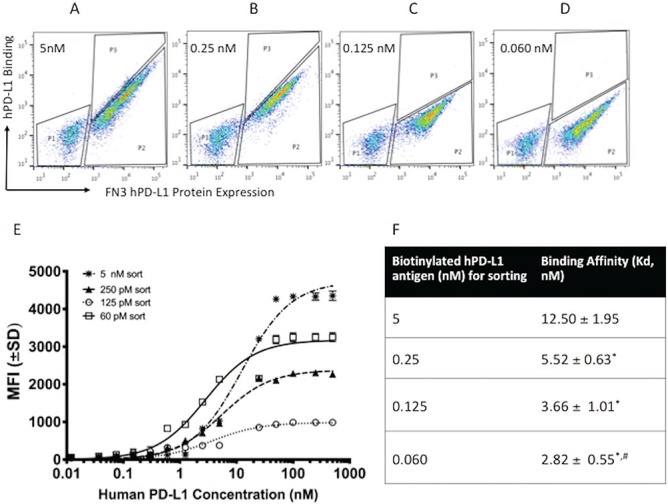
Enrichment of yeast populations for hPD-L1 binders after 4 rounds of affinity maturation. (A) Yeast cells were enriched with 5 nM of the biotinylated hPD-L1 antigen. Clones with high expression of hPD-L1 were sorted (P3 populations). (B) Sorted population from 5 nM were enriched further with 0.25 nM of the biotinylated hPD-L1 antigen. Populations within P3 gate (top 2% of total population) were sorted. (C) Sorted population from 0.25 nM were enriched further with 0.125 nM of the biotinylated hPD-L1 antigen. Populations within P3 gate (top 1% of total population) were sorted. (D) Sorted population from 0.125 nM were enriched further with 0.06 nM of the biotinylated hPD-L1 antigen. Populations within P3 gate (top 1% of total population) were sorted. (E) Binding dynamics of the yeast populations from each round of sorting at 5, 0.25, 0.125 and 0.06 nM at different concentrations of purified hPD-L1. The mean fluorescence intensity (MFI) values of each sample were measured by FACS. Error bars represent S.D. for duplicate samples. (F) Binding affinities (Kd) of the yeast from each round of sorting. *P < 0.05 relative to first round of sorting. #P < 0.05 relative to second round of sorting.
Fig. 2.
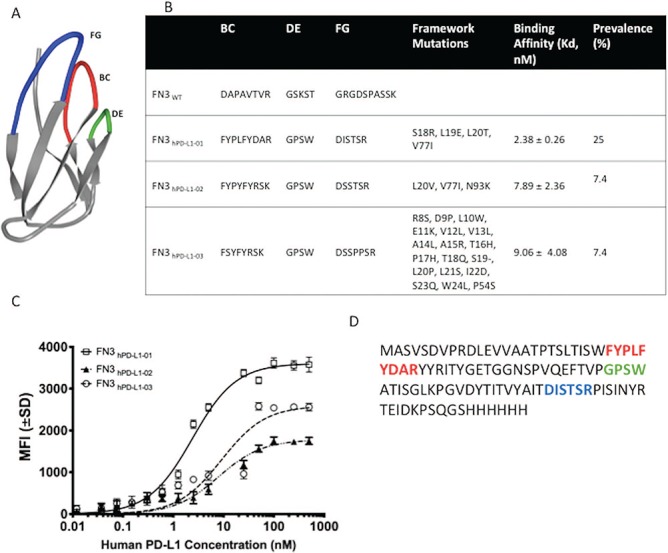
Engineering and development of a high-affinity binder for human PD-L1. (A) NMR structure of wild-type fibronectin domain (PDB ID: 1TTG) showing the engineered loops—BC (top middle), DE (top right) and FG (top left). (B) The diverse sequences of the framework and loops—BC, DE and FG between the three unique clones (FN3hPD-L1-01 to FN3hPD-L1-03) isolated from the mixed binder populations enriched at 60 pM (Fig. 1D) along with their binding affinities (Kd) are shown. (C) Binding dynamics of each yeast clone at different concentrations of purified hPD-L1. Error bars represent S.D. for duplicate samples. (D) Full-length amino acid sequence of the FN3hPD-L1-01 clone within the BC, DE and FG loops, along with the 6x His-tag at the C-terminus. All subsequent experiments were performed using the FN3hPD-L1-01 clone.
Production of soluble FN3hPD-L1-01 binder
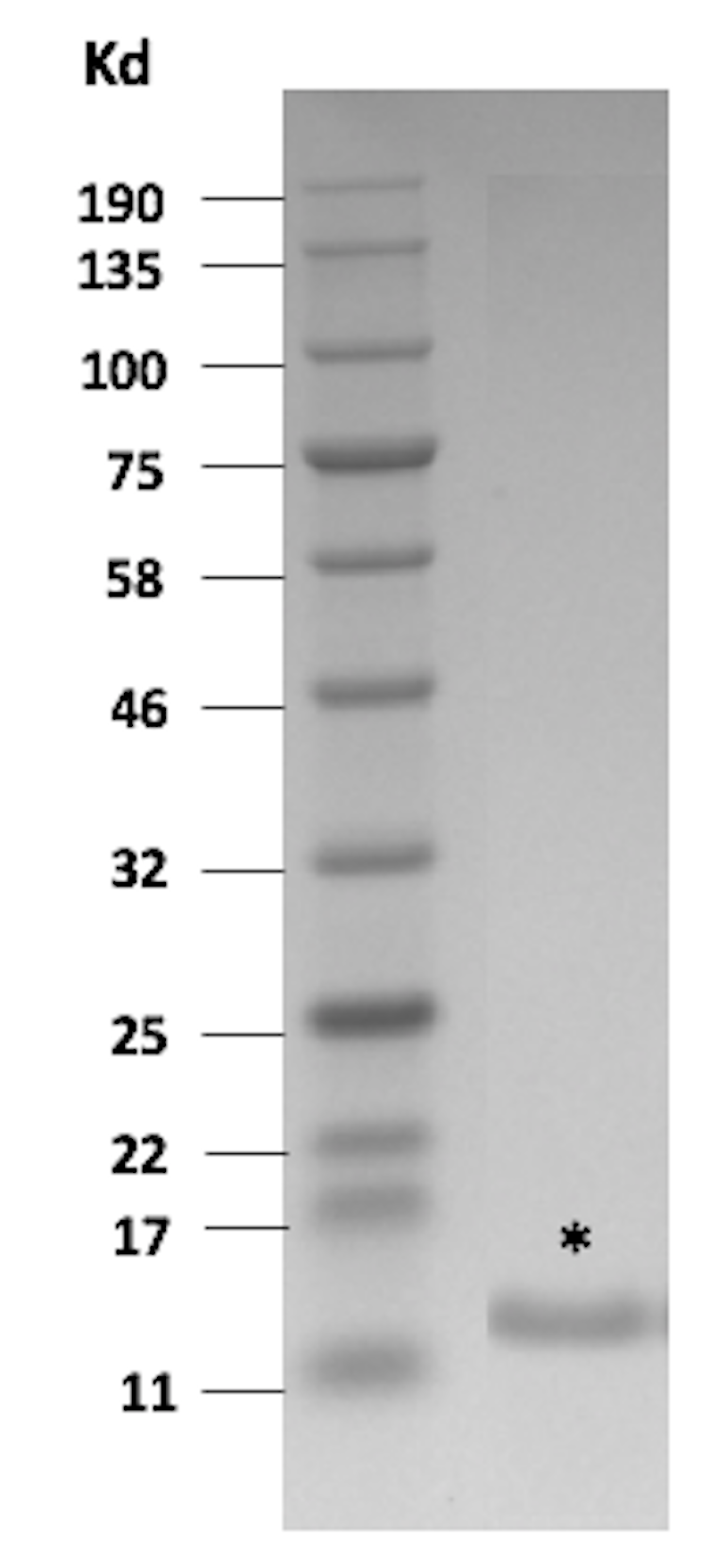
Genomic DNA was isolated from the yeast expressing FN3hPD-L1-01, and the insert was released by restriction enzymes digest, subcloned into bacterial pETH vector and transformed into bacteria to facilitate protein purification by taking advantage of the 6x His-tag at the C-terminus. Transformed individual bacterial clones were analyzed by DNA sequencing, and one unique clone was selected, expressed and purified by fast protein liquid chromatography (FPLC). The eluted protein was lyophilized and then re-suspended in DMSO. The approximate yield of purified protein was ~ 5 mg/L. Mass spectrometry measured a protein mass of 11 807 Da. SDS PAGE also indicated a clear band at 11–12 kDa (Fig. S3). This purified FN3hPD-L1-01 binder was used for all subsequent experiments.
Detection of cell-surface hPD-L1 expression in cancer cells using FN3hPD-L1-01
To determine the optimal concentration of IFN-γ for induction of hPD-L1 expression, CT26/hPD-L1 cells were induced with 2.5, 5, 10, 20 and 40 ng/ml of IFN-γ for 24 hours. The highest expression of hPD-L1 was observed when the cells were induced with 10 ng/ml of IFN-γ (Fig. 3A). As shown in Fig. 3B, CT26/hPD-L1 had the highest PD-L1 expression at the basal level, since it was genetically engineered to express hPD-L1. None of the other six cell lines show basal expression of hPD-L1 (Soliman et al., 2014). When induced with 10 ng/ml IFN-γ, CT26/hPD-L1, MDA-MB-231, DLD1 and U87 were shown to express elevated levels of hPD-L1, with CT26/hPD-L1 having the highest inducible expression (P < 0.001 relative to non-induced cells). 293 T, Raji and Jurkat had no expression of hPD-L1 at both their constitutive and induced states (P > 0.05 relative to non-induced cells).
Fig. 3.
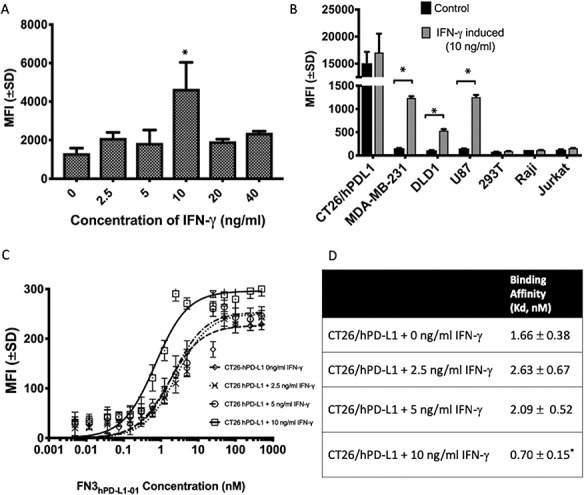
Detection of cell-surface hPD-L1 in CT26/hPD-L1 cells using the FN3hPD-L1-01 binder. (A) Dose response of IFN-γ for induction of hPD-L1 expression on CT26/hPD-L1 cells as measured by mean fluorescence intensity (MFI). Highest expression of hPD-L1 was achieved at 10 ng/ml of IFN-γ. Error bars represent S.E.M from duplicate samples. *P < 0.05 relative cells without induction. (B) Expression of hPD-L1 in different human cancer cell lines upon induction with 10 ng/ml IFN-γ. Except for CT26/hPD-L1, the basal levels of hPD-L1 expression across six cell lines were low. When induced with IFN-γ, CT26/hPD-L1 showed the highest expression of hPD-L1 when compared to other cell lines. MDA-MB-231, U87 and DLD-1 showed IFN-γ-induced expression of hPD-L1. However, 293 T, Raji and Jurkat did not show any expression of hPD-L1 even with IFN-γ induction. Error bars represent S.D. from duplicate samples. *P < 0.05. (C) Binding dynamics of FN3hPD-L1-01 in CT26/hPD-L1 cells induced with different concentrations of IFN-γ. Error bars represent S.D. from duplicate samples. (D) Binding affinity (Kd) decreased as the expression of hPD-L1 increases with IFN-γ.*P < 0.05 relative to non-induced CT26/hPD-L1 cells.
Cell binding assays to determine the affinity FN3hPD-L1-01 to hPD-L1
To determine the correlation between expression of hPD-L1 with staining of the binder, CT26/hPD-L1 cells were induced with three concentrations (2.5, 5 and 10 ng/ml) of IFN-γ, followed by incubation with several concentrations of the binder (15 pM to 500 nM). Cells were gated with reference to the negative CT26 cell line that does not express hPD-L1. The MFI (mean fluorescence intensity) from the binding of FN3hPD-L1-01 to CT26/hPDL1 cells increased with the concentrations of IFN-γ, as shown by the upward shift in the binding curves in Fig. 3C. The Kd values for binding of FN3hPD-L1-01 to CT26/hPD-L1 decreased relative to non-induced cells (P < 0.05), as the expression of hPD-L1 increased upon induction with 10 ng/ml of IFN- γ (P < 0.05, Fig. 3A). All subsequent experiments were done using CT26/hPD-L1 using 10 ng/ml of IFN-γ.
To determine the specificity of the staining of cell-surface hPD-L1 with our binder, blocking studies were performed using a commercial hPD-L1 antibody (E1L3N). CT26/hPD-L1 cells were incubated with several concentrations of the commercial antibody prior to staining with purified of FN3hPD-L1-01 binder (100 nM) that was subsequently detected using the Alexa Fluor 647®-conjugated anti-6xHisTag antibody. The IC50 for inhibition of FN3hPD-L1-01 binding was 51.26 ± 10.12 nM (R2 = 0.95, Fig. 4).
Fig. 4.
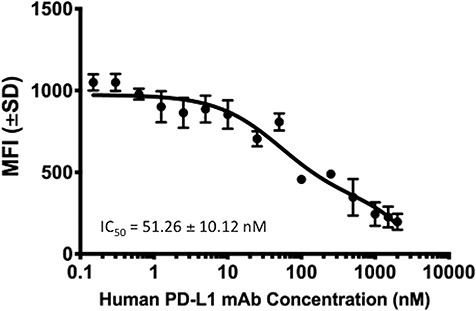
Determination of the specificity of FN3hPD-L1-01 binding using a commercial hPD-L1 antibody. CT26/hPD-L1 cells induced with 10 ng/ml IFN-γ were incubated with different concentrations of E1L3N, prior to incubation with FN3hPD-L1-01. The mean fluorescence intensity (MFI) values of each sample were measured by FACS. Error bars represent S.D from duplicate samples.
In vivo detection of human PD-L1 in tumor xenografts using FN3hPD-L1-01
To determine if FN3hPD-L1-01 can be used to detect hPD-L1 in tumors, mice bearing CT26/hPD-L1 and Raji xenografts were injected with the binder for 1 hour prior to tumor excision. To determine the specificity of the targeting, mice were injected with the commercial hPD-L1 antibody Tecentriq® prior to injection of FN3hPD-L1-01. Compared to mice injected with the FN3hPD-L1-01 binder (Fig. 5C), minimal cell-surface hPD-L1 staining was seen on tumors blocked with Tecentriq® (Fig. 5B) or injected with saline (Fig. 5A). Minimal hPD-L1 staining was observed in Raji tumor sections under all 3 conditions. To confirm the presence of hPD-L1 in CT26/hPD-L1 tumors, CT26/hPD-L1 and Raji tumor sections were stained with two concentrations of the binder (0.3 and 1.25 nM) ex vivo, followed by detection with Alexa Fluor 647® 6XHisTag antibody. Clear cell-surface staining on CT26/hPD-L1 tumor section was seen at 1.25 nM (Fig. 6A) but not in Raji cells (Fig. 6C).
Fig. 6.
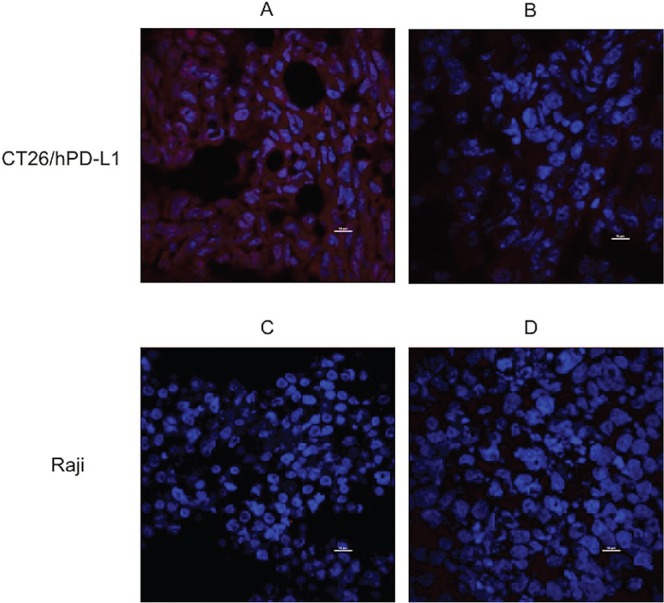
Specificity of FN3hPD-L1-01 binder for ex vivo detection of hPD-L1 in tumors. (A) CT26/hPD-L1 tumor section stained with 1.25 nM of the FN3hPD-L1-01 binder and Alexa Fluor 647® 6XHisTag antibody (membrane staining). (B) CT26/hPD-L1 tumor sections stained with 0.3 nM of the FN3hPD-L1-01 binder and Alexa Fluor 647® 6XHisTag antibody. (C) Raji tumor section stained with 1.25 nM of the FN3hPD-L1-01 binder and Alexa Fluor 647® 6XHisTag antibody. (D) Raji tumor section stained with 0.3 nM of the FN3hPD-L1-01 binder and Alexa Fluor 647® 6XHisTag antibody. All sections were stained for the nuclei using DAPI (nuclei staining, center). Image acquisition was performed at 60× using an intravital microscope. Scale bar = 10 μm.
Discussion
The hPD-L1 checkpoint protein is expressed on several types of cancers (Sunshine et al., 2017; Taube et al., 2014; Latchman et al., 2001; Champiat et al., 2014). We developed a novel, subnanomolar hPD-L1 binder based on a 11 kDa FN3 scaffold. Even though several hPD-L1 targeting full-length antibodies have shown remarkable therapeutic efficacies (Long et al., 2017), the slow clearance from the bloodstream (Warram et al., 2014) and non-target tissues of full-length antibodies may limit their utility as molecular imaging agents (Haylock et al., 2017). The engineered small protein binder, FN3hPD-L1-01, is only 11.8 kDa and hence is expected to have a rapid clearance from background tissues of 12–24 hours based on previous studies with FN3-based binders (Hackel et al., 2012; Natarajan et al., 2013).
Engineering and selection of hPD-L1 binders
FN3 has been shown as a useful platform for engineering of small protein imaging agents (Natarajan et al., 2013; Lipovšek, 2011; Bloom and Calabro, 2009; Koide et al., 1998). The three solvent-exposed loops (BC, DE and FG, Fig. 2A) within the FN3 domain are ideal regions to induce diversity and thus improve binding to target interfaces (Hackel et al., 2010; Hackel et al., 2008). The FN3 domain is structurally similar to the antibody variable domain (Hackel et al., 2012; Lipovšek, 2011; Hackel et al., 2010) and can be produced in bacteria at a high yield. Modifications of the amino acid sequences within the three loops of the FN3 were previously shown to enhance the binding activity and stability of the scaffold (Natarajan et al., 2013; Lipovšek, 2011; Hackel et al., 2010). Furthermore, it is less than one-tenth the size of a full-length antibody and is expected to demonstrate a rapid clearance from blood and background tissues effectively based on our previous experience with FN3-based binders (Hackel et al., 2012; Natarajan et al., 2013).
There was a 4-fold improvement in binding affinity of the yeast expressing the mixed populations of binders between Rounds 1 and 4, and a 2-fold improvement in binding affinity between Rounds 2 and 4 (P < 0.05 for both cases), validating our strategy for selection of higher affinity clones through multiple rounds of sorting (Fig. 1F). Yeast populations with the highest affinity to biotinylated hPD-L1 antigen at 60 pM demonstrated a Kd of 2.82 ± 0.55 nM (Fig. 1E). We understood that with each round of sorting with lower concentrations of the purified hPD-L1, we were enriching for binders with higher affinities with the potential loss of diversity of binders that have lower affinities. However, our goal is to identify clones with the highest affinity to hPD-L1 that could be developed as imaging agents through multiple rounds of sorting. In future binder development studies, we will significantly increase the number of clones used for sequencing with each round of sorting and perform binding studies on the monoclones during each round of sorting to circumvent the loss of diversity in high-affinity binders.
The best yeast clone (FN3hPD-L1-01) isolated from the 60-pM mixed population binders demonstrated a binding affinity of 2.38 ± 0.26 nM and was expressed in bacteria for all subsequent experiments. Having affinity in the sub-nanomolar range suggests that FN3hPD-L1-01 has the potential for pre-clinical imaging applications for hPD-L1.
In vitro and in vivo specificity of FN3hPD-L1-01 for detection of cell-surface hPD-L1 in cancer
We tested the in vitro efficacy of FN3hPD-L1-01 in detection of cell-surface hPD-L1 in different cancer cell lines, including CT26 cells that were engineered to overexpress hPD-L1 (Fig. 3B). CT26/hPD-L1 cells exhibited the highest expression of PD-L1 both in basal and IFN-γ-induced states and were used in all subsequent experiments. The optimal concentration of IFN-γ for maximum induction of cell-surface expression of hPD-L1 in CT26/hPD-L1 cells was 10 ng/ml.
To correlate the expression of the hPD-L1 in CT26/hPD-L1 cells with the binding affinity of FN3hPD-L1-01, we performed binding study using CT26/hPD-L1 cells induced with different concentrations of IFN-γ. We showed that as the levels of hPD-L1 increased (Fig. 3A), the mean fluorescence intensity increased (Fig. 3C) while the Kd decreased (Fig. 3D), thus validating hPD-L1 as a target for our FN3hPD-L1-01 binder. Blocking studies with a commercial antibody (E1L3N, Fig. 4) further confirmed that our binder is specific for detection of cell-surface CT26/hPD-L1.
In vivo targeting of FN3hPD-L1-01 to hPD-L1 expressing tumors
Our newly engineered binder shows high in vivo tumor targeting in CT26/hPD-L1 tumor xenografts, but not Raji tumor xenografts that do not express hPD-L1 (Fig. 5). Specificity of in vivo targeting of FN3hPD-L1-01 was further confirmed in mice that were pre-injected with the commercial hPD-L1 antibody Tecentriq® prior to FN3hPD-L1-01 injection (Fig. 5). Furthermore, ex vivo staining of CT26/hPD-L1 tumor sections with 1.25 and 0.3 nM of FN3hPD-L1-01 confirmed the subnanomolar affinity of the binder (Fig. 6).
Future directions
Our binder FN3hPD-L1-01 binds to hPD-L1 expressing cancer cell lines in culture and in tumor sections, and it also targets hPD-L1 expressing tumors in vivo. Therefore, it is a prime candidate for future development as an imaging agent to predict and monitor immune checkpoint blockade therapies. For example, the binder can further be radiolabeled for positron emission tomography (PET) imaging and fluorescently labeled for endoscopy and guided surgery using hPD-L1 expression as a marker in human cancers (Natarajan et al., 2013; Chatterjee et al., 2017; Lwin et al., 2018). Further studies are warranted to determine the serum stability, thermostability and efficacies as an in vivo imaging agent.
Although we did not directly compare our lead binder to a full-length antibody as in vivo tumor imaging agents, the potential advantages of the FN3-based scaffold are well documented in the literature (Wu, 2014). The full-length antibodies have slower clearance from circulation and therefore may not be optimal in distinguishing imaging signals in the tumors from that of surrounding tissues at early time points. Other peptides may have similar behavior to our binder, but this would require a true head-to-head comparison. It is likely that the current binder would have advantages over other peptide binders because of the stability of other FN3-based binders that were evaluated in vivo (Natarajan et al., 2013; Kimura et al., 2009). Future studies comparing the detection sensitivity of FN3hPD-L1-01 vs. the commercial antibody E1L3N with different levels of hPD-L1 expression will be performed.
Conclusions
We have engineered and validated a subnanomolar FN3-based binder for in vivo tumor targeting and ex vivo detection of hPD-L1 expression in cancer. Imaging studies to assess the binder for detection of hPD-L1 in small animal tumor models are currently underway. Additionally, radiation dosimetry studies and other experiments needed for filing an exploratory IND (eIND) are also in progress. It will be important to learn the advantages and limitations of this binder for clinical translation in human volunteers and cancer patients using PET imaging.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors would like to acknowledge the support of Mr. Aaron Mayer, Ken Lau, Drs. Lingyun Xu, Mark Stolowitz for technical assistance with experiments, and Drs. Sharon Hori and Mirwais Wardak for advice on statistical analyses. The authors acknowledge the support of the Irv Weissman laboratory, the Stanford Shared FACS Facility and Stanford Center for Innovation in In-Vivo Imaging (Sci3).
Conflict of Interest
The authors declared no potential conflicts of interest with respect to the work presented in this manuscript.
Funding
The authors would like to thank the Ben & Catherine Ivy Foundation, the Canary Foundation and the National Cancer Institute (R01 CA201719) for their support in funding this research.
Author Contributions Statement
SR, AN, CTC and SSG performed study design, development and methodology; SR and PSP performed binder selection experiments, obtained and analyzed the data; AN performed the animal experiments, SR and CTC performed immunofluorescence staining and imaging of tumor sections, wrote the initial draft and revised the manuscript; all other authors reviewed and edited the manuscript. The study concept and overall study supervision was provided by SSG.
References
- Allison J.P. (2015) JAMA, 314, 1113–1114. [DOI] [PubMed] [Google Scholar]
- Bloom L. and Calabro V. (2009) Drug Discov. Today, 14, 949–955. [DOI] [PubMed] [Google Scholar]
- Champiat S., Ileana E., Giaccone G., Besse B., Mountzios G., Eggermont A., Soria J.C. (2014) J. Thorac. Oncol., 9, 144–153. [DOI] [PubMed] [Google Scholar]
- Chao G., Lau W.L., Hackel B.J., Sazinsky S.L., Lippow S.M., Wittrup K.D. (2006) Nat. Protoc., 1, 755–768. [DOI] [PubMed] [Google Scholar]
- Charles A., Janeway J., Travers P., Walport M., Shlomchik M.J. (2001) Immunobiology: The Immune System in Health and Disease, 5th edn. Garland Science, New York. [Google Scholar]
- Chatterjee S., Lesniak W.G., Nimmagadda S. (2017) Mol. Imaging, 16, 1536012117718459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S., Lesniak W.G.. et al. (2017) Biochem. Biophys. Res. Commun., 483, 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley W.B. (1910) Proc. R. Soc. Med., 3, 1–48. [PMC free article] [PubMed] [Google Scholar]
- Ghiotto M., Gauthier L., Serriari N., Pastor S., Truneh A., Nunès J.A., Olive D. (2010) Int. Immunol., 22, 651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackel B.J., Kapila A., Wittrup K.D. (2008) J. Mol. Biol., 381, 1238–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackel B.J., Kimura R.H., Gambhir S.S. (2012) Radiology, 263, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stability and CDR composition biases enrich binder functionality landscapes. (2010) J. Mol. Biol., 401, 84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haylock A.-K., Nilvebrant J., Mortensen A., Velikyan I., Nestor M., Oncotarget R.F. (2017) Oncotarget, 8, 65152–65170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir M.E., Butte M.J., Freeman G.J., Sharpe A.H. (2008) Annu. Rev. Immunol., 26, 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Kwon J.H., Park S.Y., Park E., Chung J.H. (2017) Oncotarget, 8, 98524–98532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R.H., Zhen C., Gambhir S.S., Cochran J.R. (2009) Cancer Res., 69, 2435–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide A., Bailey C.W., Huang X., Koide S. (1998) J. Mol. Biol., 284, 1141–1151. [DOI] [PubMed] [Google Scholar]
- Latchman Y., Wood C.R., Chernova T. et al. (2001) Nat. Immunol., 2, 261–268. [DOI] [PubMed] [Google Scholar]
- Lim T.S., Chew V., Sieow J.L., Goh S., Yeong J.P., Soon A.L., Ricciardi-Castagnoli P. (2016) Oncoimmunology, 5, e1085146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipovšek D. (2011) Protein Eng. Des. Sel., 24, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long G.V., Weber J.S., Larkin J.. et al. (2017) JAMA Oncol., 3, 1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lwin T.M., Hoffman R.M., Bouvet M. (2018) Expert. Rev. Anticancer. Ther., 18, 651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maute R.L. (2015) Proc. Natl. Acad. Sci. U. S. A., 112, E6506–E6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan A., Hackel B.J., Gambhir S.S. (2013) Clin. Cancer Res., 19, 6820–6829. [DOI] [PubMed] [Google Scholar]
- Pardoll D.M. (2012) Nat. Rev. Cancer, 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman H., Khalil F., Antonia S. (2014) PLoS One, 9, e88557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunshine J.C., Nguyen P.L., Kaunitz G.J.. et al. (2017) Clin. Cancer Res., 23, 4938–4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube J.M., Klein A., Brahmer J.R., Xu H., Pan X., Kim J.H., Chen L5 Pardoll D.M., Topalian S.L., Anders R.A. (2014) Clin. Cancer Res., 20, 5064–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warram J.M., Esther D.B., Anna G.S., Thomas K.C., Hyunki K., Rick G.P., Gooitzen M.V.D., Eben L.R. (2014) Cancer Metastasis Rev., 33, 809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A.M. (2014) Methods (San Diego, Calif.), 65, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan M.-M.,Hu X.Q., Liu X.X., Ruan B.F., Xu J., Liao C. (2016) Drug Discov. Today, 21, 1027–1036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.