

Abstract
Background
A new endemic disease has spread across Wuhan City, China, in December 2019. Within few weeks, the World Health Organization (WHO) announced a novel coronavirus designated as coronavirus disease 2019 (COVID-19). In late January 2020, WHO declared the outbreak of a “public-health emergency of international concern” due to the rapid and increasing spread of the disease worldwide. Currently, there is no vaccine or approved treatment for this emerging infection; thus, the objective of this study is to design a multiepitope peptide vaccine against COVID-19 using an immunoinformatics approach.
Method
Several techniques facilitating the combination of the immunoinformatics approach and comparative genomic approach were used in order to determine the potential peptides for designing the T-cell epitope-based peptide vaccine using the envelope protein of 2019-nCoV as a target.
Results
Extensive mutations, insertion, and deletion were discovered with comparative sequencing in the COVID-19 strain. Additionally, ten peptides binding to MHC class I and MHC class II were found to be promising candidates for vaccine design with adequate world population coverage of 88.5% and 99.99%, respectively.
Conclusion
The T-cell epitope-based peptide vaccine was designed for COVID-19 using the envelope protein as an immunogenic target. Nevertheless, the proposed vaccine rapidly needs to be validated clinically in order to ensure its safety and immunogenic profile to help stop this epidemic before it leads to devastating global outbreaks.
1. Introduction
Coronaviruses (CoV) are a large family of zoonotic viruses that cause illness ranging from the common cold to more severe diseases such as Middle East Respiratory Syndrome (MERS-CoV) and Severe Acute Respiratory Syndrome (SARS-CoV). In the last decades, six strains of coronaviruses were identified; however, in December 2019, a new strain has spread across Wuhan City, China [1, 2]. It was designated as coronavirus disease 2019 (COVID-19) by the World Health Organization (WHO) [3]. In late January 2020, WHO declared the outbreak a global pandemic with cases in more than 45 countries where the COVID-19 was spreading fast outside China, most significantly in South Korea, Italy, and Iran with over 2,924 deaths and 85,212 cases confirmed while 39,537 recovered on 29 February 2020, 06:05 AM (GMT).
COVID-19 is a positive-sense single-stranded RNA virus (+ssRNA). Its RNA sequence is approximately 30,000 bases in length [4]. It belongs to the subgenus Sarbecovirus and genus Betacoronavirus within the family Coronaviridae. The corona envelope (E) protein is a small, integral membrane protein involved in several aspects of the virus' life cycle, such as pathogenesis, envelope formation, assembly, and budding, alongside with its interactions with both other CoV proteins (M, N, and S) and host cell proteins (release of infectious particles after budding) [5–9].
The infected person is characterized with fever, upper or lower respiratory tract symptoms, diarrhea, lymphopenia, thrombocytopenia, and increased C-reactive protein and lactate dehydrogenase levels or combination of all these within 3-6 days after exposure. Further molecular diagnosis can be made by real-time PCR for genes encoding the internal RNA-dependent RNA polymerase and Spike's receptor binding domain, which can be confirmed by Sanger sequencing and full genome analysis by NGS, multiplex nucleic acid amplification, and microarray-based assays [10–14].
A phylogenetic tree of the mutation history of a family of viruses is possible to reconstruct with a sufficient number of sequenced genomes. The phylogenetic analysis indicates that COVID-19 likely originated from bats [15]. It also showed that it is highly related with at most seven mutations relative to a common ancestor [16].
The sequence of COVID-19 RBD, together with its RBM that contacts receptor angiotensin-converting enzyme 2 (ACE2), was found similar to that of SARS coronavirus. In January 2020, a group of scientists demonstrated that ACE2 could act as the receptor for COVID-19 [17–21]. However, COVID-19 differs from other previous strains in having several critical residues at the 2019-nCoV receptor-binding motif (particularly Gln493) which provide advantageous interactions with human ACE2 [15]. This difference in affinity possibly explains why the novel coronavirus is more contagious than other viruses.
At present, there is no vaccine or approved treatment for humans, but Chinese traditional medicines, such as ShuFengJieDu capsules and Lianhuaqingwen capsules, could be possible treatments for COVID-19. However, there are no clinical trials approving the safety and efficacy for these drugs [22].
The main concept within all the immunizations is the ability of the vaccine to initiate an immune response in a faster mode than the pathogen itself. Although traditional vaccines, which depend on biochemical trials, induced potent neutralizing and protective responses in the immunized animals, they can be costly, allergenic, and time-consuming and require in vitro culture of pathogenic viruses leading to serious concern of safety [23, 24]. Thus, the need for safe and efficacious vaccines is highly recommended.
Peptide-based vaccines do not need in vitro culture making them biologically safe, and their selectivity allows accurate activation of immune responses [25, 26]. The core mechanism of the peptide vaccines is built on the chemical method to synthesize the recognized B-cell and T-cell epitopes that are immunodominant and can induce specific immune responses. A B-cell epitope of a target molecule can be linked with a T-cell epitope to make it immunogenic. The T-cell epitopes are short peptide fragments (8-20 amino acids), whereas the B-cell epitopes can be proteins [27, 28]. Therefore, in this study, we aimed to design a peptide-based vaccine to predict epitopes from the corona envelope (E) protein using immunoinformatics analysis [29–34]. Rapid further studies are recommended to prove the efficiency of the predicted epitopes as a peptide vaccine against this emerging infection.
2. Materials and Methods
The workflow summarizing the procedures for the epitope-based peptide vaccine prediction is shown in Figure 1.
Figure 1.
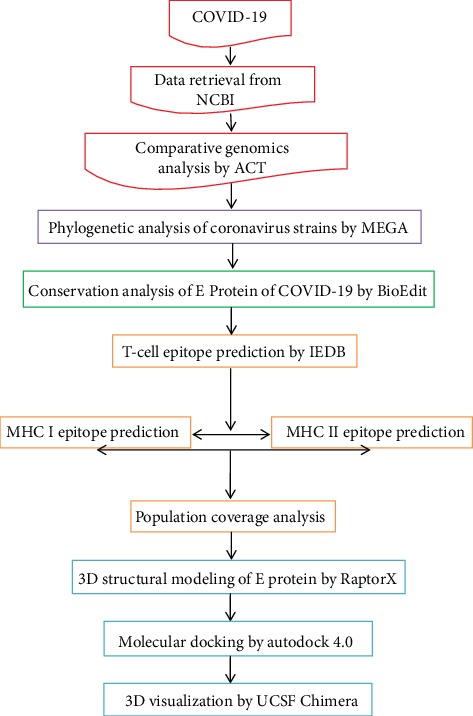
Descriptive workflow for the epitope-based peptide vaccine prediction.
2.1. Data Retrieval
Full GenBank files of the complete genomes and annotation of COVID-19 (NC_04551), SARS-CoV (FJ211859), MESA-CoV (NC_019843), HCoV-HKU1 (AY884001), HCoV-OC43 (KF923903), HCoV-NL63 (NC_005831), and HCoV-229E (KY983587) were retrieved from the National Center for Biotechnology Information (NCBI), while the FASTA format of the envelope (E) protein (YP_009724392.1), spike (S) protein (YP_009724390.1), nucleocapsid (N) protein (YP_009724397.2), and membrane (M) protein (YP_009724393.1) of 2019-nCoV and the envelope (E) protein of two Chinese and two American sequences (YP009724392.1, QHQ71975.1, QHO60596.1, and QHN73797.1) were obtained from the NCBI (https://www.ncbi.nlm.nih.gov/).
2.2. The Artemis Comparison Tool (ACT)
ACT is an in silico analysis software for visualization of comparisons between complete genome sequences and associated annotations [35]. It is also applied to identify regions of similarity, rearrangements, and insertions at any level from base pair differences to the whole genome (https://www.sanger.ac.uk/science/tools/artemis-comparison-tool-act).
2.3. VaxiJen Server
It is the first server for alignment-independent prediction of protective antigens. It allows antigen classification solely based on the physicochemical properties of proteins without recourse to sequence alignment. It predicts the probability of the antigenicity of one or multiple proteins based on auto cross covariance (ACC) transformation of protein sequence. Structural CoV-2019 proteins (N, S, E, and M) were analyzed by VaxiJen with threshold of 0.4 [36] (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html).
2.4. BioEdit
It is a software package proposed to stream a distinct program that can run nearly any sequence operation as well as a few basic alignment investigations. The sequences of the E protein retrieved from UniProt were run in BioEdit to determine the conserved sites through ClustalW in the application settings [37].
2.5. The Molecular Evolutionary Genetics Analysis (MEGA)
MEGA (version 10.1.6) is software for the comparative analysis of molecular sequences. It is used for pairwise and multiple sequence alignment alongside construction and analysis of phylogenetic trees and evolutionary relationships. The gap penalty was 15 for opening and 6.66 for extending the gap for both pairwise and multiple sequence alignment. Bootstrapping of 300 was used in construction of the maximum like hood phylogenetic tree [38, 39] (https://www.megasoftware.net).
2.6. Prediction of T-Cell Epitopes
IEDB tools were used to predict the conserved sequences (10-mer sequence) from HLA class I and class II T-cell epitopes by using an Artificial Neural Network (ANN) approach [40–42]. The Artificial Neural Network (ANN) version 2.2 was chosen as the prediction method as it depends on the median inhibitory concentration (IC50) [40, 43–45]. For the binding analysis, all the alleles were carefully chosen, and the length was set at 10 before prediction was done. Analysis of epitopes binding to the MHC class I and II molecules was assessed by the IEDB MHC prediction server at http://tools.iedb.org/mhci/ and http://tools.iedb.org/mhcii/, respectively. All conserved immunodominant peptides binding to the MHC I and II molecules at scores equal or less than 100 median inhibitory concentrations (IC50) and 1000, respectively, were selected for further analysis while epitopes with IC50 greater than 100 were eliminated [46].
2.7. Population Coverage Analysis
Population coverage for each epitope was carefully determined by the IEDB population coverage calculation tool. Due to the diverse binding sites of epitopes with different HLA alleles, the most promising epitope candidates were calculated for population coverage against the population of the whole world, China, and Europe to get and ensure a universal vaccine [47, 48] (http://tools.iedb.org/population/).
2.8. Tertiary Structure (3D) Modeling
The reference sequence of the E protein that has been retrieved from GenBank was used as an input in RaptorX to predict the 3D structure of the E protein [49, 50]; the visualization of the obtained 3D protein structure was performed in UCSF Chimera (version1.8) [51].
2.9. In Silico Molecular Docking
2.9.1. Ligand Preparation
In order to estimate the binding affinities between the epitopes and the molecular structure of MHC I and MHC II, in silico molecular docking was used. Sequences of proposed epitopes were selected from the COVID-19 reference sequence using UCSF Chimera 1.10 and saved as a PDB file. The obtained files were then optimized and energy minimized. The HLA-A∗02:01 was selected as the macromolecule for docking. Its crystal structure (4UQ3) was downloaded from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do), which was in a complex with an azobenzene-containing peptide [52].
All water molecules and heteroatoms in the retrieved target file 4UQ3 were then removed. The target structure was further optimized and energy minimized using Swiss PDB Viewer V.4.1.0 software [53].
2.9.2. Molecular Docking
Molecular docking was performed using AutoDock 4.0 software, based on the Lamarckian genetic algorithm, which combines energy evaluation through grids of affinity potential to find the suitable binding position for a ligand on a given protein [54, 55] Polar hydrogen atoms were added to the protein targets, and Kollman united atomic charges were computed. The target's grid map was calculated and set to 60 × 60 × 60 points with grid spacing of 0.375 Ǻ. The grid box was then allocated properly in the target to include the active residue in the center. The genetic algorithm and its run were set to 100. The docking algorithms were set to default. Finally, results were retrieved as binding energies and poses that showed the lowest binding energies visualized using UCSF Chimera.
3. Results
3.1. The Artemis Comparison Tool
The reference sequence of the envelope protein was aligned with the HCoV-HKU1 reference protein using the Artemis Comparison Tool as illustrated in (Figure 2).
Figure 2.
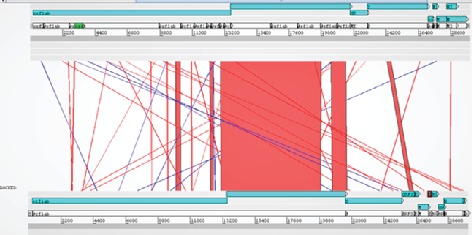
Artemis analysis of the envelope protein displaying 3 windows. The upper window represents the HCoV-HKU1 reference sequence, and its genes are highlighted in blue starting from orflab gene and ending with N gene. The middle window describes the similarities and the difference between the two genomes. Red lines indicate a match between genes from the two genomes; blue lines indicate inversion which represents the same sequences in the two genomes, but they are organized in the opposite direction. The lower window represents COVID-19 and its genes starting from orflab gene and ending with N gene.
3.2. VaxiJen Server
The mutated proteins were tested for antigenicity using VaxiJen software, where the envelope protein was found as the best immunogenic target in Table 1.
Table 1.
VaxiJen overall prediction of probable COVID-19 antigen.
| Protein | Result | VaxiJen prediction |
|---|---|---|
| Protein E | 0.6025 | Probable antigen |
| Protein M | 0.5102 | Probable antigen |
| Protein S | 0.4646 | Probable antigen |
| Protein N | 0.5059 | Probable antigen |
3.3. BioEdit
Sequence alignment of the COVID-19 envelope protein was done using BioEdit software which shows total conservation across four sequences which were retrieved from China and the USA (Figure 3).
Figure 3.

Sequence alignment of the envelope protein of COVID-19 using BioEdit software (total conservation through the 4 strains: 2 from China and 2 from the USA).
3.4. The Molecular Evolutionary Genetics Analysis
To study the evolutionary relationship between all the seven strains of coronavirus, a multiple sequence alignment (MSA) was performed using ClustalW by MEGA software. This alignment was used to construct the maximum likelihood phylogenetic tree as seen in Figure 4.
Figure 4.
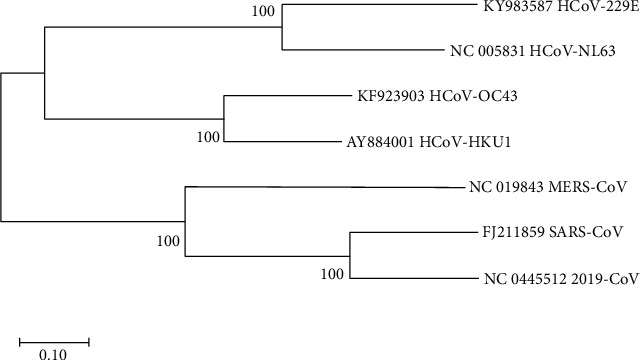
Maximum likelihood phylogenetic tree which describes the evolutionary relationship between the seven strains of coronavirus.
3.5. Prediction of T-Cell Epitopes and Population Coverage
The IEDB website was used to analyze the 2019-nCoV envelope protein for T-cell-related peptides. Results show ten MHC class I- and II-associated peptides with high population coverage (Tables 2 and 3; Figure 5). The most promising peptides were visualized using UCSF Chimera software (Figures 6(a) and 6(b)).
Table 2.
The most promising MHC class I-related peptides in the envelope protein-based vaccine of COVID-19 along with the predicted coverage of the world, China, Europe, and East Asia.
| Peptide | Alleles | Coverage | Combined coverage of 10 peptides |
|---|---|---|---|
| YVYSRVKNL | HLA-C∗14:02, HLA-C∗12:03, HLA-C∗07:01, HLA-C∗03:03, HLA-C∗06:02 | 50.02% | World: 88.5% |
| SLVKPSFYV | HLA-A∗02:06, HLA-A∗02:01, HLA-A∗68:02 | 42.53% | China: 78.17% |
| SVLLFLAFV | HLA-A∗02:06, HLA-A∗68:02, HLA-A∗02:01 | 42.53% | Europe: 92.94% |
| FLAFVVFLL | HLA-A∗02:01, HLA-A∗02:06 | 40.60% | East Asia: 80.78% |
| VLLFLAFVV | HLA-A∗02:01 | 39.08% | |
| RLCAYCCNI | HLA-A∗02:01 | 39.08% | |
| FVSEETGTL | HLA-C∗03:03, HLA-C∗12:03, HLA-A∗02:06, HLA-A∗68:02, HLA-B∗35:01 | 28.22% | |
| LTALRLCAY | HLA-A∗01:01, HLA-A∗30:02, HLA-B∗15:01 | 26.34% | |
| LVKPSFYVY | HLA-B∗15:01, HLA-A∗29:02, HLA-A∗30:02, HLA-B∗35:01 | 21.72% | |
| NIVNVSLVK | HLA-A∗68:01, HLA-A∗11:01 | 20.88% |
Table 3.
The most promising MHC class II-related peptides in the envelope protein-based vaccine of COVID-19 along with the predicted coverage of the world, China, Europe, and East Asia.
| Peptide sequence | Alleles | World coverage | Coverage/10 peptides |
|---|---|---|---|
| KPSFYVYSRVKNLNS | HLA-DPA1∗01:03, HLA-DPB1∗02:01, HLA-DPB1∗03:01, HLA-DPB1∗04:01, HLA-DPA1∗02:01, HLA-DPB1∗05:01, HLA-DPA1∗03:01, HLA-DPB1∗04:02, HLA-DPB1∗06:01, HLA-DPB1∗14:01, HLA-DPB1∗01:01, HLA-DQA1∗05:01, HLA-DQB1∗04:02, HLA-DQA1∗06:01, HLA-DQA1∗01:02, HLA-DQB1∗05:01, HLA-DQA1∗02:01, HLA-DRB1∗01:01, HLA-DRB1∗07:01, HLA-DRB1∗08:01, HLA-DRB1∗09:01, HLA-DRB1∗11:01, HLA-DRB4∗01:03, HLA-DRB1∗04:01, HLA-DRB1∗10:01, HLA-DRB1∗04:05, HLA-DRB1∗13:01, HLA-DRB1∗08:02, HLA-DRB1∗16:02, HLA-DRB1∗15:01, HLA-DRB3∗03:01, HLA-DRB5∗01:01, HLA-DRB3∗02:02, HLA-DRB1∗04:04, HLA-DRB1∗13:02 | 99.93% | World: 99.99% |
| VKPSFYVYSRVKNLN | HLA-DPA1∗01:03, HLA-DPB1∗02:01, HLA-DPB1∗04:01, HLA-DPB1∗03:01, HLA-DPA1∗02:01, HLA-DPB1∗05:01, HLA-DPA1∗03:01, HLA-DPB1∗04:02, HLA-DPB1∗06:01, HLA-DPB1∗01:01, HLA-DQA1∗05:01, HLA-DQB1∗04:02, HLA-DQA1∗06:01, HLA-DQA1∗01:02, HLA-DQB1∗05:01, HLA-DQA1∗02:01, HLA-DRB1∗07:01, HLA-DRB1∗08:01, HLA-DRB1∗01:01, HLA-DRB1∗09:01, HLA-DRB1∗11:01, HLA-DRB4∗01:03, HLA-DRB1∗15:01, HLA-DRB1∗13:01, HLA-DRB3∗03:01, HLA-DRB1∗10:01, HLA-DRB1∗16:02, HLA-DRB1∗08:02, HLA-DRB1∗04:05, HLA-DRB5∗01:01, HLA-DRB1∗13:02, HLA-DRB3∗02:02, HLA-DRB1∗04:01, HLA-DRB1∗04:04 | 99.92% | China: 99.96% |
| LVKPSFYVYSRVKNL | HLA-DPA1∗01:03, HLA-DPB1∗02:01, HLA-DPB1∗04:01, HLA-DPA1∗02:01, HLA-DPB1∗05:01, HLA-DPB1∗06:01, HLA-DPA1∗03:01, HLA-DPB1∗04:02, HLA-DPA1∗02:01, HLA-DPB1∗01:01, HLA-DQA1∗06:01, HLA-DQB1∗04:02, HLA-DQA1∗05:01, HLA-DQA1∗02:01, HLA-DQA1∗01:04, HLA-DQB1∗05:03, HLA-DQA1∗01:02, HLA-DQB1∗05:01, HLA-DRB1∗07:01, HLA-DRB1∗08:01, HLA-DRB1∗09:01, HLA-DRB1∗11:01, HLA-DRB4∗01:03, HLA-DRB3∗03:01, HLA-DRB1∗01:01, HLA-DRB1∗15:01, HLA-DRB1∗16:02, HLA-DRB1∗13:01, HLA-DRB1∗10:01, HLA-DRB1∗08:02, HLA-DRB5∗01:01, HLA-DRB1∗13:02, HLA-DRB1∗04:05, HLA-DRB3∗02:02, HLA-DRB1∗04:01 | 99.90% | Europe: 100.0% |
| PSFYVYSRVKNLNSS | HLA-DPA1∗01:03, HLA-DPB1∗02:01, HLA-DPB1∗03:01, HLA-DPB1∗04:01, HLA-DPA1∗03:01, HLA-DPB1∗04:02, HLA-DPA1∗02:01, HLA-DPB1∗05:01, HLA-DPB1∗06:01, HLA-DQA1∗05:01, HLA-DQB1∗04:02, HLA-DQA1∗01:02, HLA-DQB1∗05:01, HLA-DQA1∗06:01, HLA-DRB1∗01:01, HLA-DRB1∗08:01, HLA-DRB1∗04:01, HLA-DRB1∗11:01, HLA-DRB1∗09:01, HLA-DRB1∗07:01, HLA-DRB4∗01:03, HLA-DRB1∗04:05, HLA-DRB1∗10:01, HLA-DRB1∗13:01, HLA-DRB1∗08:02, HLA-DRB1∗16:02, HLA-DRB1∗15:01, HLA-DRB3∗03:01, HLA-DRB3∗02:02, HLA-DRB1∗04:04, HLA-DRB5∗01:01, HLA-DRB1∗13:02 | 99.86% | East Asia:99.91% |
| NIVNVSLVKPSFYVY | HLA-DPA1∗01:03, HLA-DPB1∗02:01, HLA-DPB1∗04:01, HLA-DPB1∗06:01, HLA-DPA1∗02:01, HLA-DPB1∗01:01, HLA-DQA1∗01:02, HLA-DQB1∗05:01, HLA-DQA1∗05:01, HLA-DQB1∗04:02, HLA-DQA1∗02:01, HLA-DQB1∗03:01, HLA-DQB1∗03:03, HLA-DQB1∗03:03, HLA-DQB1∗04:02, HLA-DRB1∗12:01, HLA-DRB1∗01:01, HLA-DRB5∗01:01, HLA-DRB3∗03:01, HLA-DRB1∗13:01, HLA-DRB1∗07:01, HLA-DRB1∗15:01, HLA-DRB4∗01:03, HLA-DRB1∗04:04, HLA-DRB1∗08:02, HLA-DRB1∗09:01, HLA-DRB1∗13:02, HLA-DRB1∗11:01, HLA-DRB1∗04:05, HLA-DRB1∗10:01 | 99.77% | |
| LLVTLAILTALRLCA | HLA-DPA1∗01:03, HLA-DPB1∗02:01, HLA-DPB1∗06:01, HLA-DPA1∗03:01, HLA-DPB1∗04:02, HLA-DQA1∗01:02, HLA-DQB1∗05:01, HLA-DQA1∗02:01, HLA-DQB1∗03:01, HLA-DQB1∗03:03, HLA-DQA1∗05:01, HLA-DQB1∗04:02, HLA-DQA1∗06:01, HLA-DQA1∗01:03, HLA-DQB1∗06:03, HLA-DRB4∗01:03, HLA-DRB1∗01:01, HLA-DRB1∗13:01, HLA-DRB1∗04:04, HLA-DRB5∗01:01, HLA-DRB3∗03:01, HLA-DRB1∗10:01, HLA-DRB1∗15:01, HLA-DRB1∗07:01, HLA-DRB1∗11:01, HLA-DRB1∗08:01, HLA-DRB1∗12:01, HLA-DRB1∗03:01, HLA-DRB4∗01:01, HLA-DRB1∗16:02, HLA-DRB1∗08:02 | 99.72% | |
| SFYVYSRVKNLNSSR | HLA-DPA1∗01:03, HLA-DPB1∗03:01, HLA-DPB1∗02:01, HLA-DPA1∗03:01, HLA-DPB1∗04:02, HLA-DPA1∗02:01, HLA-DPB1∗05:01, HLA-DPB1∗06:01, HLA-DQA1∗05:01, HLA-DQB1∗04:02, HLA-DQA1∗01:02, HLA-DQB1∗05:01, HLA-DRB1∗04:01, HLA-DRB1∗01:01, HLA-DRB1∗11:01, HLA-DRB1∗08:01, HLA-DRB1∗07:01, HLA-DRB1∗09:01, HLA-DRB4∗01:03, HLA-DRB1∗10:01, HLA-DRB1∗13:01, HLA-DRB1∗04:05, HLA-DRB1∗08:02, HLA-DRB1∗16:02, HLA-DRB3∗02:02, HLA-DRB3∗03:01, HLA-DRB1∗04:04, HLA-DRB1∗15:01, HLA-DRB5∗01:01, HLA-DRB1∗13:02 | 99.72% | |
| LVTLAILTALRLCAY | HLA-DPA1∗01:03, HLA-DPB1∗02:01, HLA-DPB1∗06:01, HLA-DPA1∗03:01, HLA-DPB1∗04:02, HLA-DQA1∗01:02, HLA-DQB1∗05:01, HLA-DQA1∗02:01, HLA-DQB1∗03:01, HLA-DQB1∗03:03, HLA-DQA1∗05:01, HLA-DQB1∗04:02, HLA-DQB1∗06:02, HLA-DQA1∗02:01, HLA-DQA1∗06:01, HLA-DRB4∗01:03, HLA-DRB1∗01:01, HLA-DRB1∗13:01, HLA-DRB1∗04:04, HLA-DRB1∗12:01, HLA-DRB1∗10:01, HLA-DRB5∗01:01, HLA-DRB1∗15:01, HLA-DRB1∗11:01, HLA-DRB3∗03:01, HLA-DRB1∗03:01, HLA-DRB1∗08:01, HLA-DRB1∗07:01, HLA-DRB4∗01:01, HLA-DRB1∗16:02, HLA-DRB1∗04:02, HLA-DRB1∗08:02 | 99.69% | |
| VTLAILTALRLCAYC | HLA-DPA1∗01:03, HLA-DPB1∗06:01, HLA-DPA1∗03:01, HLA-DPB1∗04:02, HLA-DQA1∗02:01, HLA-DQB1∗03:01, HLA-DQA1∗01:02, HLA-DQB1∗05:01, HLA-DQB1∗06:02, HLA-DQA1∗05:01, HLA-DQB1∗04:02, HLA-DQB1∗03:03, HLA-DQA1∗06:01, HLA-DQB1∗04:02, HLA-DRB1∗01:01, HLA-DRB4∗01:03, HLA-DRB1∗13:01, HLA-DRB1∗04:04, HLA-DRB1∗12:01, HLA-DRB1∗10:01, HLA-DRB5∗01:01, HLA-DRB1∗15:01, HLA-DRB1∗11:01, HLA-DRB1∗03:01, HLA-DRB3∗03:01, HLA-DRB1∗08:01, HLA-DRB1∗07:01, HLA-DRB4∗01:01, HLA-DRB1∗04:02 | 99.56% | |
| CNIVNVSLVKPSFYV | HLA-DPA1∗01:03, HLA-DPB1∗06:01, HLA-DPB1∗04:02, HLA-DQA1∗01:02, HLA-DQB1∗05:01, HLA-DQA1∗05:01, HLA-DQB1∗04:02, HLA-DQA1∗02:01, HLA-DQB1∗03:01, HLA-DQB1∗03:03, HLA-DQA1∗01:03, HLA-DQB1∗06:03, HLA-DRB3∗03:01, HLA-DRB1∗12:01, HLA-DRB5∗01:01, HLA-DRB1∗01:01, HLA-DRB1∗07:01, HLA-DRB4∗01:03, HLA-DRB1∗13:01, HLA-DRB1∗15:01, HLA-DRB1∗08:02, HLA-DRB1∗04:04, HLA-DRB1∗09:01, HLA-DRB1∗13:02, HLA-DRB1∗11:01, HLA-DRB1∗04:05, HLA-DRB4∗01:01, HLA-DRB1∗10:01 | 99.53% |
Figure 5.
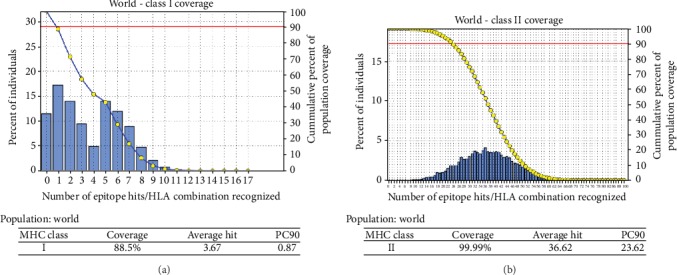
Schematic diagrams (a) and (b) showing world population coverage of the envelope protein of COVID-19 binding to the MHC class I and MHC class II molecules, respectively.
Figure 6.
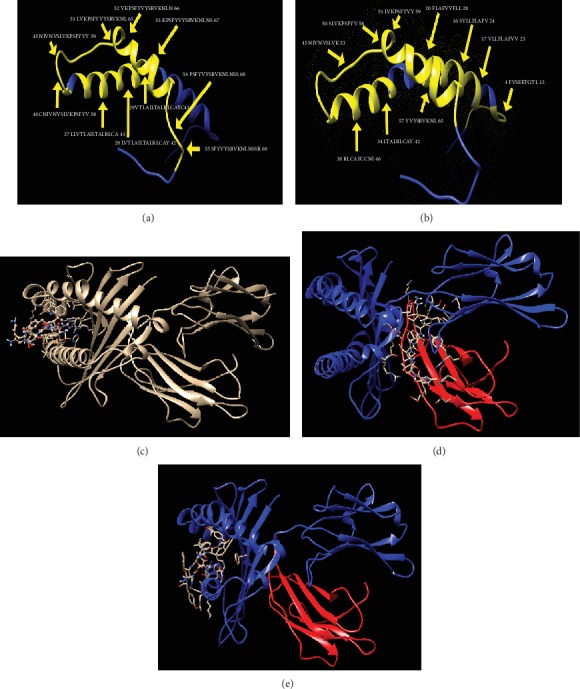
3D structures visualized by UCSF Chimera: (a) and (b) show the most promising peptides in the envelope protein of COVID-19 (yellow colored) binding to MHC class I and MHC class II, respectively, while (c), (d), and (e) show the molecular docking of the YVYSRVKNL, LAILTALRL, and SLVKPSFYV peptides of coronavirus docked in HLA-A∗02:01, respectively.
4. Discussion
Designing a novel vaccine is very crucial to defend against the rapid endless global burden of diseases [56–59]. In the last few decades, biotechnology has advanced rapidly, alongside with the understanding of immunology which assisted the rise of new approaches towards rational vaccine design [60]. Peptide-based vaccines are designed to elicit immunity particular pathogens by selectively stimulating antigen-specific B- and T-cells [25]. Applying the advanced bioinformatics tools and databases, various peptide-based vaccines could be designed where the peptides act as ligands [61–63]. This approach has been used frequently in Saint Louis encephalitis virus [64], dengue virus [65], and Chikungunya virus [66] proposing promising peptides for designing vaccines.
The COVID-19 is an RNA virus which tends to mutate more commonly than the DNA viruses [67]. These mutations lie on the surface of the protein, which makes COVID-19 more superior than other previous strains by inducing its sustainability leaving the immune system in a blind spot [68].
In our present work, different peptides were proposed for designing a vaccine against COVID-19 (Figure 1). In the beginning, the whole genome of COVID-19 was analyzed by a comparative genomic approach to determine the potential antigenic target [69]. The Artemis Comparison Tool (ACT) was used to analyze human coronavirus (HCoV-HKU1) reference sequence vs. Wuhan-Hu-1 COVID-19. Results obtained (Figure 2) revealed extensive mutations among the tested genomes. New genes (ORF8 and ORF6) were found inserted in COVID-19 which were absent in HCoV-HKU1 that might be acquired by the horizontal gene transmission [70]. The high rate of mutation between the two genomes was observed in the region from 20,000 bp to the end of the sequence. This region encodes the four major structural proteins in coronavirus which are the envelope (E) protein, nucleocapsid (N) protein, membrane (M) protein, and spike (S) protein, all of which are required to produce a structurally complete virus [71, 72].
These conserved antigenic sites were revealed in previous studies through sequence alignment between MERS-CoV and bat coronavirus [73] and analyzed in SARS-CoV [74].
The four proteins were then analyzed by VaxiJen software to test the probability of antigenic proteins. Protein E was found to be the most antigenic gene with the highest probability as shown in Table 1. A literature survey confirmed this result in which protein E was investigated in Severe Acute Respiratory Syndrome (SARS) in 2003 and, more recently, Middle-East Respiratory Syndrome (MERS) [71]. Furthermore, the conservation of this protein against the seven strains was tested and confirmed through the use of the BioEdit package tool (Figure 3).
Phylogenetic analysis is a very powerful tool for determining the evolutionary relationship between strains. Multiple sequence alignment (MSA) was performed using ClustalW for the seven strains of coronavirus, which are COVID-19 (NC_04551), SARS-CoV (FJ211859), MESA-CoV (NC_019843), HCoV-HKU1 (AY884001), HCoV-OC43 (KF923903), HCoV-NL63 (NC_005831), and HCoV-229E (KY983587). The maximum likelihood phylogenetic tree revealed that COVID-19 is found in the same clade of SARS-CoV; thus, the two strains are highly related to each other (Figure 4).
The immune response of T-cells is considered a long-lasting response compared to B-cells, where the antigen can easily escape the antibody memory response [75]. Vaccines that effectively generate cell-mediated responses are needed to provide protection against the invading pathogen. Moreover, the CD8+ and CD4+ T-cell responses play a major role in antiviral immunity [76]. Thus, designing a vaccine against T-cells is much more important.
Choosing protein E as the antigenic site, the binding affinity to MHC molecules was then evaluated. The protein reference sequence was submitted to the IEDB MHC predication tool. 21 peptides were found to bind MHC class I with different affinities (Table 1), from which ten peptides were selected for vaccine design based on the number of alleles and world population percentage (Table 2; Figure 5). Analysis in the IEDB MHC II binding prediction tool resulted in prediction of 61 peptides (Table 2), from which ten peptides were selected for vaccine design based on the number of alleles and world population percentage (Table 3; Figure 5). Unfortunately, IEDB did not give any result for B-cell epitopes; this might be due to the length of the COVID-19 (75 amino acids).
It is well known that peptides recognized with a high number of HLA molecules are potentially inducing immune response. Based on the aforementioned results and taking into consideration the high binding affinity to both MHC class I and II, conservancy, and population coverage, three peptides are strongly proposed to formulate a new vaccine against COVID-19.
These findings were further confirmed by the results obtained for the molecular docking of the proposed peptides and HLA-A∗02:01. The formed complex between the MHC molecule and the three peptides (YVYSRVKNL, SLVKPSFYV, and LAILTALRL) has shown peptide amino- and carboxyl-termini forming one and three hydrogen bonds, respectively, at the two ends of a binding groove with MHC residues with the least binding energy -13.2 kcal/mol, -11 kcal/mol, and -11.3 kcal/mol, respectively (Figures 6(c)–6(e)).
Although both flu and anti-HIV drugs are used currently in China for treatment of COVID-19, chloroquine phosphate, an old drug for treatment of malaria, has recently been found to have apparent efficacy and acceptable safety against COVID-19 [77, 78]; nevertheless, more studies are required to standardize these therapies. In addition, there has been some success in the development of mouse models of MERS-CoV and SARS-CoV infection, and candidate vaccines where the envelope (E) protein is mutated or deleted have been described [79–85]. To the best of our knowledge, this is the first study to identify certain peptides in the envelope (E) protein as candidates for COVID-19. Accordingly, these epitopes were strongly recommended as promising epitope vaccine candidates against T-cells.
5. Conclusion
Extensive mutations, insertion, and deletion were discovered in the COVID-19 strain using the comparative sequencing. In addition, a number of the MHC class I- and II-related peptides were found to be promising candidates. Among which, the peptides YVYSRVKNL, SLVKPSFYV, and LAILTALRL show high potentiality for vaccine design with adequate world population coverage. The T-cell epitope-based peptide vaccine was designed for COVID-19 using the envelope protein as an immunogenic target; nevertheless, the proposed vaccine rapidly needs to be validated clinically ensuring its safety and immunogenic profile to help stop this epidemic before it leads to devastating global outbreaks.
Acknowledgments
The authors acknowledge the Deanship of Scientific Research at University of Bahri for the supportive cooperation.
Abbreviations
- WHO:
World Health Organization
- CoV:
Coronaviruses
- MERS-CoV:
Middle East Respiratory Syndrome
- SARS-CoV:
Severe Acute Respiratory Syndrome
- COVID-19:
Novel coronavirus
- HCoV-HKU1:
Human coronavirus HKU1
- HCoV-OC43:
Human coronavirus OC43
- HCoV-NL63:
Human coronavirus NL63
- HCoV-229E:
Human coronavirus 229E
- ACE2:
Angiotensin-converting enzyme 2
- RBM:
Receptor-binding motif
- RBD:
Receptor-binding domain
- vs.:
Versus
- ACT:
Artemis Comparison Tool
- ACC:
Auto cross covariance
- MEGA:
Molecular Evolutionary Genetics Analysis
- ANN:
Artificial Neural Network
- IEDB:
Immune Epitope Database
- IC50:
Median inhibitory concentrations
- MHC I:
Major histocompatibility complex class I
- MHC II:
Major histocompatibility complex class II
- PDB:
Protein database
- MSA:
Multiple sequence alignment.
Data Availability
All data underlying the results are available as part of the article, and no additional source data are required.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
The contributions of the authors involved in this study are as follows: MIA: conceptualization, formal analysis, investigation, methodology, validation, visualization, and writing (original draft); AHA: formal analysis, investigation, and methodology; MIM: methodology, writing (original draft), and writing (review and editing); NME: formal analysis, methodology, and visualization; NSM: conceptualization, resources, and writing (review and editing); SWS: visualization, validation, and writing (review and editing); and AMM: data curation, conceptualization, project administration, supervision, and writing (review and editing). All authors have read and approved the final manuscript.
References
- 1.Lu H., Stratton C. W., Tang Y. W. Outbreak of pneumonia of unknown etiology in Wuhan China: the mystery and the miracle. Journal of Medical Virology. 2020;92(4):401–402. doi: 10.1002/jmv.25678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hui D. S., I Azhar E., Madani T. A., et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health — The latest 2019 novel coronavirus outbreak in Wuhan, China. International Journal of Infectious Diseases. 2020;91:264–266. doi: 10.1016/j.ijid.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benvenuto D., Giovanetti M., Ciccozzi A., Spoto S., Angeletti S., Ciccozzi M. The 2019-new coronavirus epidemic: evidence for virus evolution. Journal of Medical Virology. 2020;92(4):455–459. doi: 10.1002/jmv.25688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.CoV2020. GISAID EpifluDB. Archived from the original on 12 January 2020. 2020.
- 5.Venkatagopalan P., Daskalova S. M., Lopez L. A., Dolezal K. A., Hogue B. G. Coronavirus envelope (E) protein remains at the site of assembly. Virology. 2015;478:75–85. doi: 10.1016/j.virol.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nieto-Torres J. L., DeDiego M. L., Álvarez E., et al. Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology. 2011;415(2):69–82. doi: 10.1016/j.virol.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curtis K. M., Yount B., Baric R. S. Heterologous gene expression from transmissible gastroenteritis virus replicon particles. Journal of Virology. 2002;76(3):1422–1434. doi: 10.1128/JVI.76.3.1422-1434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ortego J., Escors D., Laude H., Enjuanes L. Generation of a replication-competent, propagation-deficient virus vector based on the transmissible gastroenteritis coronavirus genome. Journal of Virology. 2002;76(22):11518–11529. doi: 10.1128/JVI.76.22.11518-11529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruch T. R., Machamer C. E. A single polar residue and distinct membrane topologies impact the function of the infectious bronchitis coronavirus E protein. PLoS Pathogens. 2012;8(5, article e1002674) doi: 10.1371/journal.ppat.1002674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan J. F.-W., Yuan S., Kok K.-H., et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395(10223):514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang N., Wang L., Deng X., et al. Recent advances in the detection of respiratory virus infection in humans. Journal of Medical Virology. 2020;92(4):408–417. doi: 10.1002/jmv.25674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahony J. B., Petrich A., Smieja M. Molecular diagnosis of respiratory virus infections. Critical reviews in clinical laboratory sciences. 2011;48(5-6):217–249. doi: 10.3109/10408363.2011.640976. [DOI] [PubMed] [Google Scholar]
- 13.Corman V. M., Landt O., Kaiser M., et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance. 2020;25(3) doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. Journal of Medical Virology. 2020;92(4):418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wan Y., Shang J., Graham R., Baric R. S., Li F. Receptor recognition by the Novel Coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS Coronavirus. Journal of Virology. 2020;94(7) doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bedford T., Neher R. Genomic Epidemiology of Novel Coronavirus (nCoV) Using Data Generated by Fudan University, China CDC, Chinese Academy of Medical Sciences, Chinese Academy of Sciences and the Thai National Institute of Health Shared via GISAID. 2020.
- 17.Letko M., Munster V. Functional assessment of cell entry and receptor usage for lineage B β-coronaviruses, including 2019-nCoV. bioRxiv; 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Letko M., Marzi A., Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nature Microbiology. 2020;5(4):562–569. doi: 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El Sahly H. M. New England Journal of Medicine. Massachusetts, USA: 2020. Genomic characterization of the 2019 novel coronavirus. [Google Scholar]
- 20.Gralinski L. E., Menachery V. D. Return of the Coronavirus: 2019-nCoV. Viruses. 2020;12(2):p. 135. doi: 10.3390/v12020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu R., Zhao X., Li J., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu H. Drug treatment options for the 2019-new coronavirus (2019-nCoV) BioScience Trends. 2020;14(1):69–71. doi: 10.5582/bst.2020.01020. [DOI] [PubMed] [Google Scholar]
- 23.Lo Y. T., Pai T. W., Wu W. K., Chang H. T. Prediction of conformational epitopes with the use of a knowledge-based energy function and geometrically related neighboring residue characteristics. BMC Bioinformatics. 2013;14(S4) Supplement 4:p. S3. doi: 10.1186/1471-2105-14-S4-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W., Joshi M., Singhania S., Ramsey K., Murthy A. Peptide vaccine: progress and challenges. Vaccines. 2014;2(3):515–536. doi: 10.3390/vaccines2030515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell A. W., McCluskey J., Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nature Reviews Drug Discovery. 2007;6(5):404–414. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- 26.Dudek N. L., Perlmutter P., Aguilar M. I., Croft N. P., Purcell A. W. Epitope discovery and their use in peptide based vaccines. Current Pharmaceutical Design. 2010;16(28):3149–3157. doi: 10.2174/138161210793292447. [DOI] [PubMed] [Google Scholar]
- 27.Dermime S., Gilham D. E., Shaw D. M., et al. Vaccine and antibody-directed T cell tumour immunotherapy. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2004;1704(1):11–35. doi: 10.1016/j.bbcan.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Meloen R. H., Langeveld J. P. M., Schaaper W. M. M., Slootstra J. W. Synthetic peptide vaccines: unexpected fulfillment of discarded hope? Biologicals. 2001;29(3-4):233–236. doi: 10.1006/biol.2001.0298. [DOI] [PubMed] [Google Scholar]
- 29.Brusic V., Petrovsky N. Immunoinformatics and its relevance to understanding human immune disease. Expert Review of Clinical Immunology. 2014;1(1):145–157. doi: 10.1586/1744666X.1.1.145. [DOI] [PubMed] [Google Scholar]
- 30.Tomar N., De R. K. Immunoinformatics: a brief review. Methods in Molecular Biology. 2014;1184:23–55. doi: 10.1007/978-1-4939-1115-8_3. [DOI] [PubMed] [Google Scholar]
- 31.Khalili S., Jahangiri A., Borna H., Ahmadi Zanoos K., Amani J. Computational vaccinology and epitope vaccine design by immunoinformatics. Acta Microbiologica et Immunologica Hungarica. 2014;61(3):285–307. doi: 10.1556/AMicr.61.2014.3.4. [DOI] [PubMed] [Google Scholar]
- 32.Tomar N., De R. K. Immunoinformatics: an integrated scenario. Immunology. 2010;131(2):153–168. doi: 10.1111/j.1365-2567.2010.03330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hegde N. R., Gauthami S., Sampath Kumar H. M., Bayry J. The use of databases, data mining and immunoinformatics in vaccinology: where are we? Expert Opinion on Drug Discovery. 2017;13:117–130. doi: 10.1080/17460441.2018.1413088. [DOI] [PubMed] [Google Scholar]
- 34.Bahrami A. A., Payandeh Z., Khalili S., Zakeri A., Bandehpour M. Immunoinformatics:In SilicoApproaches and computational design of a multi-epitope, immunogenic protein. International Reviews of Immunology. 2019;38(6):307–322. doi: 10.1080/08830185.2019.1657426. [DOI] [PubMed] [Google Scholar]
- 35.Carver T. J., Rutherford K. M., Berriman M., Rajandream M. A., Barrell B. G., Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21(16):3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 36.Doytchinova I. A., Flower D. R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8(1):p. 4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oni O. O., Owoade A. A., Adeyefa C. A. O. Design and evaluation of primer pairs for efficient detection of avian rotavirus. Tropical Animal Health and Production. 2018;50(2):267–273. doi: 10.1007/s11250-017-1425-2. [DOI] [PubMed] [Google Scholar]
- 38.Stecher G., Tamura K., Kumar S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Molecular Biology and Evolution. 2020;37(4):1237–1239. doi: 10.1093/molbev/msz312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamura K., Dudley J., Nei M., Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution. 2007;24(8):1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 40.Nielsen M., Lundegaard C., Worning P., et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Science. 2003;12(5):1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lundegaard C., Lamberth K., Harndahl M., Buus S., Lund O., Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Research. 2008;36(suppl_2):W509–W512. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andreatta M., Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32(4):511–517. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buus S., Lauemoller S. L., Worning P., et al. Sensitive quantitative predictions of peptide-MHC binding by a 'Query by Committee' artificial neural network approach. Tissue Antigens. 2003;62(5):378–384. doi: 10.1034/j.1399-0039.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- 44.Lundegaard C., Lund O., Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics. 2008;24(11):1397–1398. doi: 10.1093/bioinformatics/btn128. [DOI] [PubMed] [Google Scholar]
- 45.Lundegaard C., Nielsen M., Lund O. The validity of predicted T-cell epitopes. Trends in Biotechnology. 2006;24(12):537–538. doi: 10.1016/j.tibtech.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 46.Southwood S., Sidney J., Kondo A., et al. Several common HLA-DR types share largely overlapping peptide binding repertoires. The Journal of Immunology. 1998;160(7):3363–3373. [PubMed] [Google Scholar]
- 47.Patronov A., Doytchinova I. T-cell epitope vaccine design by immunoinformatics. Open Biology. 2013;3(1, article 120139) doi: 10.1098/rsob.120139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bui H. H., Sidney J., Dinh K., Southwood S., Newman M. J., Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics. 2006;7(1):p. 153. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benson D. A., Cavanaugh M., Clark K., et al. GenBank. Nucleic Acids Research. 2017;45(D1):D37–d42. doi: 10.1093/nar/gkw1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang S., Li W., Liu S., Xu J. RaptorX-Property: a web server for protein structure property prediction. Nucleic Acids Research. 2016;44(W1):W430–W435. doi: 10.1093/nar/gkw306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pettersen E. F., Goddard T. D., Huang C. C., et al. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 52.Choo J. A., Thong S. Y., Yap J., et al. Bioorthogonal cleavage and exchange of major histocompatibility complex ligands by employing azobenzene-containing peptides. Angewandte Chemie (International ed in English). 2014;53(49):13390–13394. doi: 10.1002/anie.201406295. [DOI] [PubMed] [Google Scholar]
- 53.Guex N., Peitsch M. C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 54.Morris G. M., Huey R., Lindstrom W., et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morris G. M., Goodsell D. S., Halliday R. S., et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19(14):1639–1662. doi: 10.1002/(sici)1096-987x(19981115)19:14<1639::aid-jcc10>3.0.co;2-b. [DOI] [Google Scholar]
- 56.Marshall S. J. Developing countries face double burden of disease. Bulletin of the World Health Organization. 2004;82:p. 556. [PMC free article] [PubMed] [Google Scholar]
- 57.De Groot A. S., Rappuoli R. Genome-derived vaccines. Expert Review of Vaccines. 2014;3:59–76. doi: 10.1586/14760584.3.1.59. [DOI] [PubMed] [Google Scholar]
- 58.Korber B., LaBute M., Yusim K. Immunoinformatics comes of age. PLoS Computational Biology. 2006;2(6):p. e71. doi: 10.1371/journal.pcbi.0020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fauci A. S. Emerging and re-emerging infectious diseases: influenza as a prototype of the host-pathogen balancing act. Cell. 2006;124(4):665–670. doi: 10.1016/j.cell.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chaudhury S., Duncan E. H., Atre T., et al. Combining immunoprofiling with machine learning to assess the effects of adjuvant formulation on human vaccine-induced immunity. Human Vaccines & Immunotherapeutics. 2020;16(2):400–411. doi: 10.1080/21645515.2019.1654807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Usmani S. S., Kumar R., Bhalla S., Kumar V., Raghava G. P. S. In silico tools and databases for designing peptide-based vaccine and drugs. Therapeutic Proteins and Peptides. 2018;112:221–263. doi: 10.1016/bs.apcsb.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 62.Dhanda S. K., Usmani S. S., Agrawal P., Nagpal G., Gautam A., Raghava G. P. S. Novel in silico tools for designing peptide-based subunit vaccines and immunotherapeutics. Briefings in Bioinformatics. 2017;18(3):467–478. doi: 10.1093/bib/bbw025. [DOI] [PubMed] [Google Scholar]
- 63.Khazaei-Poul Y., Farhadi S., Ghani S., Ahmadizad S. A., Ranjbari J. Monocyclic peptides: types, synthesis and applications. Current Pharmaceutical Biotechnology. 2020;21 doi: 10.2174/1573412916666200120155104. [DOI] [PubMed] [Google Scholar]
- 64.Hasan M. A., Hossain M., Alam M. J. A computational assay to design an epitope-based peptide vaccine against Saint Louis encephalitis virus. Bioinformatics and Biology Insights. 2013;7:347–355. doi: 10.4137/BBI.S13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chakraborty S., Chakravorty R., Ahmed M., et al. A computational approach for identification of epitopes in dengue virus envelope protein: a step towards designing a universal dengue vaccine targeting endemic regions. In Silico Biology. 2010;10(5,6):235–246. doi: 10.3233/ISB-2010-0435. [DOI] [PubMed] [Google Scholar]
- 66.Qamar M. T. U., Bari A., Adeel M. M., et al. Peptide vaccine against Chikungunya virus: immuno-informatics combined with molecular docking approach. Journal of Translational Medicine. 2018;16(1):p. 298. doi: 10.1186/s12967-018-1672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Twiddy S. S., Holmes E. C., Rambaut A. Inferring the rate and time-scale of dengue virus evolution. Molecular Biology and Evolution. 2003;20(1):122–129. doi: 10.1093/molbev/msg010. [DOI] [PubMed] [Google Scholar]
- 68.Christie J. M., Chapel H., Chapman R. W., Rosenberg W. M. Immune selection and genetic sequence variation in core and envelope regions of hepatitis C virus. Hepatology. 1999;30(4):1037–1044. doi: 10.1002/hep.510300403. [DOI] [PubMed] [Google Scholar]
- 69.Hardison R. C. Comparative genomics. PLoS Biology. 2003;1(2):p. E58. doi: 10.1371/journal.pbio.0000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gilbert C., Chateigner A., Ernenwein L., et al. Population genomics supports baculoviruses as vectors of horizontal transfer of insect transposons. Nature Communications. 2014;5(1):p. 3348. doi: 10.1038/ncomms4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schoeman D., Fielding B. C. Coronavirus envelope protein: current knowledge. Virology Journal. 2019;16(1):p. 69. doi: 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mortola E., Roy P. Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Letters. 2004;576(1-2):174–178. doi: 10.1016/j.febslet.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sharmin R., Islam A. B. Conserved antigenic sites between MERS-CoV and Bat-coronavirus are revealed through sequence analysis. Source Code for Biology and Medicine. 2016;11(1):p. 3. doi: 10.1186/s13029-016-0049-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu J., Hu J., Wang J., et al. Genome organization of the SARS-CoV. Genomics Proteomics Bioinformatics. 2003;1(3):226–235. doi: 10.1016/S1672-0229(03)01028-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Black M., Trent A., Tirrell M., Olive C. Advances in the design and delivery of peptide subunit vaccines with a focus on toll-like receptor agonists. Expert Review of Vaccines. 2010;9(2):157–173. doi: 10.1586/erv.09.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sesardic D. Synthetic peptide vaccines. Journal of Medical Microbiology. 1993;39(4):241–242. doi: 10.1099/00222615-39-4-241. [DOI] [PubMed] [Google Scholar]
- 77.Gao J., Tian Z., Yang X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Bioscience trends. 2020;14(1):72–73. doi: 10.5582/bst.2020.01047. [DOI] [PubMed] [Google Scholar]
- 78.Arya R., Das A., Prashar V., Kumar M. Potential inhibitors against papain-like protease of novel coronavirus (COVID-19) from FDA approved drugs. 2020.
- 79.Westerbeck J. W., Machamer C. E. A Coronavirus E Protein Is Present in Two Distinct Pools with Different Effects on Assembly and the Secretory Pathway. Journal of Virology. 2015;89(18):9313–9323. doi: 10.1128/JVI.01237-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dediego M. L., Pewe L., Alvarez E., Rejas M. T., Perlman S., Enjuanes L. Pathogenicity of severe acute respiratory coronavirus deletion mutants in hACE-2 transgenic mice. Virology. 2008;376(2):379–389. doi: 10.1016/j.virol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Netland J., DeDiego M. L., Zhao J., et al. Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology. 2010;399(1):120–128. doi: 10.1016/j.virol.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McCray P. B., Jr., Pewe L., Wohlford-Lenane C., et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. Journal of Virology. 2007;81(2):813–821. doi: 10.1128/JVI.02012-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Regla-Nava J. A., Nieto-Torres J. L., Jimenez-Guardeño J. M., et al. Severe acute respiratory syndrome coronaviruses with mutations in the E protein are attenuated and promising vaccine candidates. Journal of Virology. 2015;89(7):3870–3887. doi: 10.1128/JVI.03566-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao J., Li K., Wohlford-Lenane C., et al. Rapid generation of a mouse model for Middle East respiratory syndrome. Proceedings of the National Academy of Sciences. 2014;111(13):4970–4975. doi: 10.1073/pnas.1323279111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo X., Deng Y., Chen H., et al. Systemic and mucosal immunity in mice elicited by a single immunization with human adenovirus type 5 or 41 vector-based vaccines carrying the spike protein of Middle East respiratory syndrome coronavirus. Immunology. 2015;145(4):476–484. doi: 10.1111/imm.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data underlying the results are available as part of the article, and no additional source data are required.