

Abstract
Sex differences in redox signaling in the kidney present new challenges and opportunities for understanding the physiology and pathophysiology of the kidney. This review will focus on reactive oxygen species, immune-related signaling pathways and endothelin-1 as potential mediators of sex-differences in redox homeostasis in the kidney. Additionally, this review will highlight male-female differences in redox signaling in several major cardiovascular and renal disorders namely acute kidney injury, diabetic nephropathy, kidney stone disease and salt-sensitive hypertension. Furthermore, we will discuss the contribution of redox signaling in the pathogenesis of postmenopausal hypertension and preeclampsia.
Keywords: Sex-differences, Redox signaling, Endothelin-1, ROS, Inflammation, Kidney
1. Introduction
1.1. Kidney disease in males and females
Kidney disease is a public health problem that is associated with high morbidity and mortality. Worldwide, more than 850 million humans suffer from kidney disease [1]. In the United States, 2.4% of all adults are diagnosed with kidney disease [2]. Women are largely protected from the development of kidney disease, compared to age-matched men [3,4]. However, this female protection is lost when women reach menopause. Despite the protection of the female population against the initiation of kidney disease, the progression of multiple nephropathies displays worst outcomes amongst women, in comparison to men. Data points to estradiol (E2) as a central player in the renal protection elicited in females before menopause [5,6]. Ample evidence implicates that the mechanisms involved in the initiation and progression of kidney disease are sex dependent. The development of kidney disease is multi-factorial. Compelling evidence points to imbalances in redox signaling as a major player in the etiology of multiple nephropathies [[7], [8], [9]].
We herein provide an overview of sex-related differences in redox signaling pathways within the kidney, with emphasis on the contribution of reactive oxygen species and immune-related signaling pathways. Given our expertise in the field of endothelin-1, we highlight the role of this peptide in mediating sex-differences in the renal redox status and renal disease pathophysiology. Moreover, we also discuss sex-related differences in redox signaling that may contribute to the pathophysiology of kidney diseases, including acute kidney injury, diabetic nephropathy, kidney stones, salt-sensitive hypertension, postmenopausal hypertension and preeclampsia.
1.2. Biological factors underlying sex differences in renal physiology and pathophysiology
1.2.1. Sex chromosomes
Male-female differences in the sex chromosomal complement, XX vs. XY, have been poorly studied in the context of kidney diseases. There are some genes on the Y-chromosome that do not have an alternative on the X-chromosome. Mostly, one of two X-chromosomes is silenced in each cell in females. However, X-associated gene dosage may vary due to incomplete silencing of one X-chromosome in females [10,11]. More in-depth studies focusing on the impact of male-female differences in sex chromosomal complement on renal physiology and pathophysiology are needed.
1.2.2. Sex steroids
A large body of evidence suggests that sex steroidal hormones are major contributors to sex-differences in the kidney. For example, studies have shown that E2 and testosterone are capable of regulating different aspects of renal function [6,12,13]. Testosterone may be a predisposing factor for kidney disease [[14], [15], [16]], while on the other hand, E2 has been shown to exert renoprotective effects in females [6,16]. However, the impact of sex steroids on renal tubular function and renal hemodynamic is not fully understood.
1.2.3. Sex steroid receptors in the kidney
Male and female sex steroids are known to exert their actions mainly via activation of nuclear receptors, which typically facilitate genomic effects. Classic estrogen receptors are classified as two steroidal receptor subtypes; estrogen receptor ERα and ERβ. These receptors function as other steroid hormones in stimulating gene expression, although in recent years, evidence for membrane-associated estrogen receptors has been reported in multiple organs including the kidney [[17], [18], [19]]. Androgenic and estrogenic receptor expression have been localized to multiple sites in the renal tubule [20]. It was shown that androgen receptor mRNA is localized in the proximal tubule and the cortical collecting duct in the rat isolated renal tubule [21]. Using radiolabeled E2, Davidoff et al. demonstrated that labelling resides in proximal tubules cells from the S1 and S2 segments of juxtamedullary nephrons [22]. Radioactivity was also evident in the collecting duct cells in the inner medullary zone [22]. Estrogen receptor activation has been shown to elicit nephroprotection [[23], [24], [25]]. However, the mechanisms underlying estrogen receptor-mediated renal protection is not completely defined. Of note, activation of ERα, ERβ and G protein-coupled estrogen receptor, GPER, has been shown to exert protective effects by modulating redox signaling pathways [23,[26], [27], [28]].
2. Redox biology
2.1. Reactive oxygen species
Redox homeostasis is a continuous balance of processes within the cell that can either protect cells, stimulate their growth or promote their death in response to internal or external stimuli. Redox homeostasis is critical to normal physiological function and is maintained by feedback mechanisms involving reactive oxygen species (ROS)/reactive nitrogen species (RNS) and antioxidant signaling. Imbalance of this system can lead to increased oxidative stress, reduced antioxidant signaling, cell death, inflammation and tissue injury (Fig. 1).
Fig. 1.
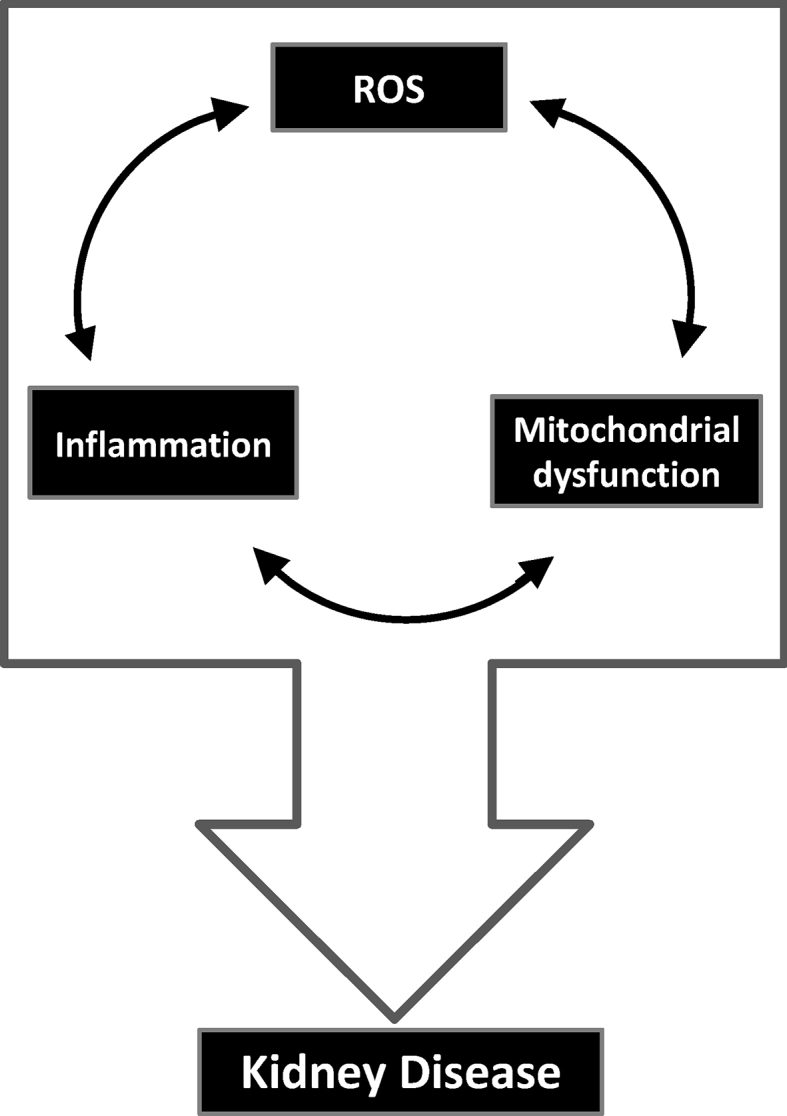
Overlapping role for reactive oxygen generation (ROS) generation, mitochondrial dysfunction and inflammation in kidney disease pathogenesis.
ROS and RNS are by-products of various enzymatic reactions throughout several cellular compartments (i.e. cytoplasm, peroxisome, endoplasmic reticulum and mitochondria) and serve as signaling molecules [29]. The proximal oxidant involved in aerobic metabolism is superoxide and is generated by the single electron reduction of molecular oxygen. It is poorly diffusible across the cellular membranes and is transported by anion channels. A number of different enzymes produce superoxide such as lipoxygenases, xanthine oxidases, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases and cyclooxygenases [30]. Of these, NADPH oxidases (NOX) are one of the most abundant sources of ROS in the cytoplasm [31]. They typically are dormant within membranes of resting cells and become activated following exposure to inflammatory or microbial stimuli. NADPH oxidases produce superoxide through electron exchange by gp91phox, the electron transferase [31]. NADPH oxidases exist in several isoforms, have heterodimeric subunits and are well known for their role in phagocytosis [31]. Importantly, these proteins are vital in promoting tissue repair and cell proliferation. Another well-known source of superoxide is the coupling of electron transport during oxidative phosphorylation within the mitochondria, which are essential for cellular metabolism [32]. Dysfunctional metabolism can lead to the overproduction of ROS and can cause further mitochondrial injury. Mitochondrial ROS production occurs at several sites in the respiratory chain, including complexes I and III and other major sites involving the low molecular weight redox shuttle CoQ [32,33].
Superoxide is highly reactive and can quickly transform into other ROS/RNS within the cytosol and mitochondria. Some of these reactive species include hydroxyl radical, peroxynitrite and hydrogen peroxide [30]. Hydroxyl radical is generated by Fenton reaction of peroxide with Fe2+, is highly reactive with cellular compartments and can produce additional free radicals [30]. Specifically, it can damage proteins and DNA and initiate lipid peroxidation and oxidized lipid products. These products can, in turn, promote cell injury, inflammation and fibrosis. Peroxynitrite is a derivative of superoxide and nitric oxide (NO) and a member of the RNS family. It is one of the most potent oxidants and causes NO to be depleted when it is formed [34]. NO is produced by three isoforms of nitric oxide synthase (NOS) including NOS1 (also known as neuronal NOS or nNOS), NOS2 (also known as inducible or iNOS) and NOS3 (also known as endothelial NOS or eNOS). All NOS isoforms use l-arginine, oxygen and other co-factors to produce NO [35], which is critical for exerting cytoprotective and cytotoxic properties in the cell. Hydrogen peroxide is a non-radical species formed by the rapid dismutation of superoxide. It is the least reactive ROS molecule and can easily cross the cell membranes to influence other nearby cells using its signaling properties [36].
To combat oxidative stress and regulate redox homeostasis, the cell stimulates the transcription and expression of several key antioxidants such as superoxide dismutases (SODs), glutathione peroxidase, catalase, thioredoxin, peroxiredoxin and glutathione transferase to modulate oxidative stress [33,37]. Of these antioxidants, both SODs and glutathione are the most abundant. SODs are found throughout the cell and exist in three different isoforms: copper-zinc-containing SOD (CuSOD), manganese-containing SOD (MnSOD) and the extracellular SOD (ecSOD). Glutathione is a small thiol located in the cytosol and is synthesized by the cell to protect against hydrogen peroxide-mediated oxidative damage and toxicity. It plays an important role in regulating cell death within the mitochondria and cell division in the nucleus [38]. Taken together, it is critical for all of these chemical processes to be properly orchestrated and balanced in order to maintain cell viability, activity and function.
2.2. Redox biology and sex differences in the kidney
Oxidative stress is associated with aging, cancer, neurodegenerative diseases and cardiovascular diseases such as hypertension and atherosclerosis [[39], [40], [41]]. A number of animal and human studies characterizing the role of ROS/RNS in kidney injury and disease progression have been reported [[42], [43], [44]]. Both sex and oxidative stress have been described to impact renal blood flow [45,46].
Primary sources of oxidative stress in the kidney include NADPH oxidases, which are expressed in a regional and cell specific manner. Specifically, NOX1, NOX2, NOX4 and NOX5 have been reported to be expressed in the kidney [47]. NOX4 is the most abundant based on mRNA levels and is located primarily in the renal cortex [31]. It actively produces hydrogen peroxide; whereas, the other NOXs primarily generate superoxide [47]. NOX4 has been shown to stimulate acute kidney injury by inducing ROS [48] and to induce diabetic nephropathy in female mice [49].
Another prominent source of oxidative stress in the kidney is the mitochondria. Proximal tubule cells are involved in reabsorption of inorganic solutes, sugars and amino acids and are enriched with mitochondria which makes them highly vulnerable to oxidative stress. Ischemia/reperfusion (I/R) has been thoroughly investigated and has been determined to cause significant injury to renal proximal tubules [50]. Renal I/R has been shown to increase ROS generation, mitochondrial damage and cell death in both renal proximal tubules and kidneys [43,51,52]. Sex differences have also been reported in renal I/R, where female rats have reduced I/R-induced injury and cell death compared to male rats [44,53].
An additional factor that contributes to renal ROS generation are infiltrating immune cells which will be discussed further below. More investigations are needed to delineate sex differences in the role of ROS/RNS under normal and pathological conditions of the kidney.
3. Mediators of sex differences in redox homeostasis in the kidney
3.1. ROS & mitochondria
Mitochondria are multifunctional organelles responsible for controlling energy production, cell growth and death [54,55]. They are at the center of cell metabolism and directly influence fatty acid oxidation, energy production and oxidant generation (i.e. superoxide) via the coupling of electron transport, oxidative phosphorylation and proton pumping across the inner mitochondrial membrane. Superoxide serves as a signaling molecule and is rapidly converted to hydrogen peroxide within the mitochondria [33,55]. In cases where superoxide is not detoxified, additional ROS and RNS are generated. Low levels of NO and superoxide are essential for cytokine and chemokine synthesis and leukocyte adhesion and migration [56]. However, excessive amounts of superoxide and NO can be toxic and cause damage to mitochondrial proteins and DNA. Ultimately, this can reduce mitochondrial efficiency and induce further ROS generation and pro-inflammatory signaling (Fig. 1).
Mitochondria are important for synthesizing steroid hormones and a crosstalk occurs between the two for normal physiological function. Specifically, mitochondria produce pregnenolone, a sex hormone that serves as a precursor for testosterone, progesterone, cortisol, estrogen and other hormones [57]. A decline in circulating sex hormones has been shown to enhance aging in mice and humans [58] and to be associated with increased coronary heart disease risk and diabetes [59,60]. Evidence indicates mitochondrial dysfunction contributes to aging, heart disease, diabetes and other pathologies [[61], [62], [63]]. Sex hormones can directly impact mitochondrial function by binding to nuclear receptors. Estrogen directly modulates genes that are important for mitochondrial function such as nuclear respiratory factor-1 and peroxisome proliferator-activated receptor gamma coactivator 1 [64,65]. These two genes are critical for mitochondrial respiration and mitochondrial biogenesis, a process that stimulates additional mitochondria in response to stress. It has been determined that mitochondrial activity, ROS generation and antioxidant activity are different in mitochondria from the heart and brain of male and female mice [66]. Sex-specific alterations have also been observed in renal mitochondria from rats in response to perinatal iron deficiency [67,68]. Additionally, women have also been reported to have superior intrinsic mitochondrial respiration in their skeletal muscle compared to men [69]. Thus, it appears that sex hormones can directly impact mitochondria and may contribute to different mechanisms occurring in both physiological and pathophysiological conditions. Further investigations about the role of sex hormones in the kidney are needed and could help explain the differences observed in the progression of kidney disease between the sexes.
3.2. Inflammation
Renal disease and injury can cause excessive ROS generation from renal endothelial cells and mesangial cells. This subsequently leads to activation of the innate and adaptive immune response and the release of pro-inflammatory cytokines and chemokines including chemokine (C–C motif) ligand 5 (CCL5 or RANTES), intercellular adhesion molecule-1 (ICAM-1), colony stimulating factor-1 (CSF-1) and monocyte chemoattractant protein-1 (MCP-1) [70]. These signaling molecules recruit leukocytes to the kidney and this causes an enhanced pro-oxidant and pro-inflammatory cellular environment. Two key leukocyte types involved in this process are macrophages and neutrophils. These cells are critical to innate immunity and play an integral role in resolving inflammation. Once present in the kidney, macrophages and neutrophils release additional cytokines such as tumor necrosis factor-ɑ (TNF-ɑ) and interleukin-1β (IL-1β) (pro-inflammatory cytokines) and elicit a respiratory burst [71,72]. The respiratory burst is a process where ROS (i.e. superoxide and hydrogen peroxide) are produced once large quantities of oxygen are consumed. Mitochondria play an important role in regulating inflammation within monocytes and macrophages using ROS signaling [29,[73], [74], [75]]; whereas, NADPH oxidases predominately play a role in generating ROS in neutrophils to promote defense against inflammation and infection [76].
Monocytes are derived from myeloid cells and can differentiate into macrophages once they enter into tissue [77]. Chronic over-production of mitochondrial ROS by the electron transport chain can cause oxidative stress [29] through reverse electron transport which occurs when electrons flow backwards through complex I [78]. Reverse electron transport is critical for macrophage metabolic reprograming and activation [79]. Importantly, elevated ROS production can negatively impact monocyte and macrophage function in atherosclerosis, diabetes, acute and chronic kidney disease [62,75,80,81].
Neutrophils are the most abundant leukocyte in the circulation and are a part of both innate and adaptive immunity. The sources of reactive species in neutrophils are due to NADPH oxidases as described above. Additionally, these cells have a limited number of mitochondria that do not respire at elevated rates. However, neutrophil mitochondria have been shown to participate in apoptosis [82]. It has been reported that neutrophil cell counts are higher in women compared to men [83] and their morphology is different among the two cohorts [84]. However, postmenopausal women tend to have lower neutrophil cell counts [85]. A recent study by Pace et al. showed that sex directly impacted biosynthesis of prostaglandins and leukotrienes, which are lipid mediators of inflammation in humans and mice [86]. This suggests that sex hormones may diminish the impact of the immune system. Damage to circulating monocytes, macrophages or neutrophils by oxidative stress could impact other cells and tissues and create a pro-oxidant and pro-inflammatory milieu throughout the body. As a result, these responses can cause deleterious outcomes such as fibrosis and tissue injury, two of the hallmarks of kidney disease. A better understanding of the effects of sex on the different components of the redox pathways in the kidney will undoubtedly lead to new therapeutic approaches for kidney disease.
3.3. Endothelin-1 signaling in the kidney
Endothelin-1 (ET-1) is the most potent vasoactive factor produced by the human body. It consists of a 21 amino acid-long sequence that acts through activation of two G protein-coupled receptors: the ETA and ETB receptors. Although ET-1 was first described as a product of endothelial cells [87], we now know that other cell types, for instance renal tubular epithelial cells [88], cardiomyocytes [89] and immune cells [90] also synthesize ET-1. Nonetheless, the kidney is the organ that contains the highest concentration of ET-1, especially in the inner medullary collecting duct [91]. Activation of ETA and ETB receptors triggers very different physiological and pathophysiological actions. Within the kidney, overactivation of the ETA receptor results in renal hypertrophy, inflammation and fibrosis [92,93]. On the other hand, activation of the ETB receptor is associated with clearance of ET-1 from the plasma, regulation of sodium and water excretion and release of vasodilators such as NO and prostaglandins [88]. The oftentimes opposite actions of these two receptors may be related to their different location within the kidney. Within the renal vasculature, ETA receptors are prominently located in the vascular smooth muscle cells in the afferent arterioles, while the vascular endothelial and smooth muscle cells of the efferent arterioles are enriched in ETB receptors. The vasa recta capillaries also contain ETB receptors, whereas ETA receptors are expressed in the pericytes. Glomeruli predominantly express ETA receptors, especially in mesangial cells and podocytes. The renal tubules, on the other hand, majorly express ETB receptors, although there is a small presence of ETA receptors in proximal and distal tubule [88,94].
Cardiovascular diseases like heart failure, vascular disease, hypertension, renal disease and diabetic nephropathy are associated with an upregulation of the ET-1 system, and in particular, ETA receptor activity [95]. Within the kidney, dysfunction and over-activation of the ET-1 system results in renal disease, probably due to an unbalanced ET-1 receptor activity (Fig. 2). For instance, proteinuria and podocyte damage have been attributed to elevated ET-1 levels and exaggerated activation of ETA receptors has been described in different renal diseases associated with hypertension [96], sickle cell disease [97] and diabetes [96]. Similarly, overexpression of ET-1 in transgenic animals leads to progressive kidney damage [98]. ET-1 also plays a critical role in the development of tubulointerstitial damage [99], as well as the tubular injury associated with ischemia reperfusion, sepsis-induced acute kidney injury [100] or polycystic kidney disease [99]. Importantly, the use of specific antagonists against ET-1 receptors retards the development of these renal diseases, emphasizing the significant role of the ET-1 system in the progression of renal disease.
Fig. 2.
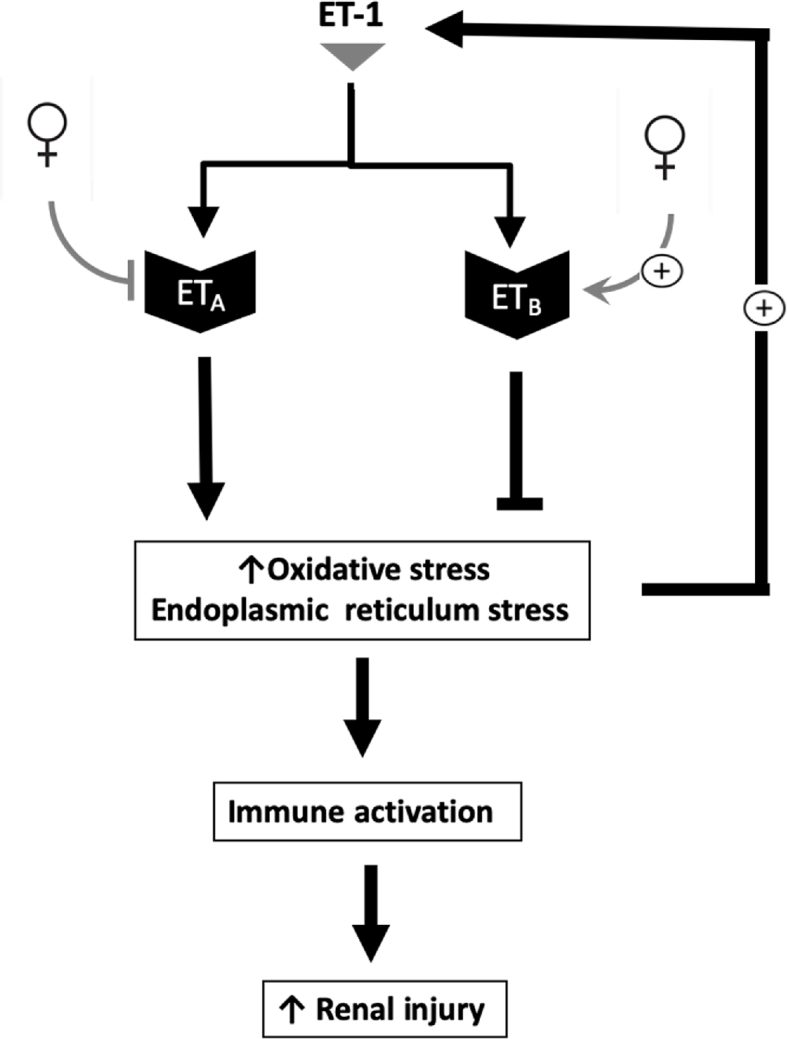
Proposed scheme for the impact of the female sex on the crosstalk between endothelin-1 and oxidative stress in the kidney.
3.3.1. Crosstalk between ET-1 and oxidative stress
Activation of the ET-1 signaling system generates ROS via the NADPH oxidase [101]. Notably, the role of ROS is well-recognized as an activator of the immune system activation and it plays a central role in kidney-related disease, including hypertension. A dialog between ET-1 and oxidative stress is evident, as depicted in Fig. 2. As ET-1 can evoke oxidative stress, ET-1 levels increase in response to oxidative stress [102,103]. It has also been shown that oxygen-derived free radicals generated by xanthine oxidase stimulate the mRNA expression of preproET-1 and big ET-1 in cultured human umbilical vein endothelial cells via activation of the preproET-1 promoter [103]. An earlier study also revealed an upregulation of prepro-ET-1 mRNA in endothelial cells from ovariectomized pigs, compared to gonadally-intact males or females, suggesting that ovarian hormones regulate prepro-ET-1 at the transcriptional level [104]. In addition, overactivation of the ET-1 system evokes the endoplasmic reticulum stress response renally and extra-renally [105,106]. In particular, overstimulation of the ETA receptor leads to renal endoplasmic stress and apoptosis, whereas activation of the ETB receptor has protective effects [106]. The bi-directional relationship between ET-1 and redox signaling pathways in the kidney is not completely understood.
3.3.2. Sex differences in the renal ET-1 system
ET-1 is among the putative mediators of sex difference in the development of cardiovascular and kidney disease. There are differences in the renal expression of ET-1 receptors between the sexes, with males presenting an increased expression of ETA receptor in the inner medullary collecting duct compared to females [107] as illustrated in Fig. 2. This observation suggests that males have an exaggerated ETA receptor activation in this part of the nephron, and, thus are more sensitive to kidney damage in response to an insult such as increased salt consumption. It was also demonstrated that when ET-1 is infused into the renal inner medulla, female rats do not decrease the blood flow in the renal medullary region as much as male rats do [108], which could be one of the mechanisms by which males develop exaggerated kidney damage.
Sex hormones regulate the ET-1 system, although mechanistic studies in this field remain scarce. In the setting of ischemic acute renal injury, E2 was shown to decrease the production of ET-1 in the kidney and, therefore, prevents the development of the associated renal injury [109]. Others also reported that the increased ETA expression observed in the kidney during mouse pregnancy is mediated by progesterone [110]. Ovariectomy increases the expression of ETA and ETB receptors in the inner medulla of the kidney [111]. This effect was reversed by supplementation of ovariectomized (OVX) rats with E2 [111]. Additionally, renal cortical ETA and ETB receptor expression is attenuated by ovariectomy and this reduction was prevented in E2-treated OVX rats [111]. On the other hand, the interaction between androgens and the ET-1 system is controversial in the literature. Some studies suggest that androgens stimulate the production of ET-1 in the kidney, inducing vasoconstriction and leading to renal injury and fibrosis [112]; these effects seemed to be attenuated after castration [113]. More research is needed in order to get a better understanding of the relationship between sex hormones and the renal ET-1 system.
4. Redox homeostasis during acute and chronic kidney disease
Redox homeostasis has been shown to be altered in a variety of renal diseases in both experimental animal models and clinical trials. In this section, we discuss the role of sex and/or sex hormones and imbalances in renal redox homeostasis signaling pathways in the pathogenesis of acute kidney injury, diabetic nephropathy, kidney stone disease, salt sensitivity, pre-eclampsia and postmenopausal hypertension.
4.1. Acute kidney injury
Acute kidney injury (AKI) is a complex clinical syndrome that involves a sudden onset loss of kidney function and causes significant morbidity and mortality [114]. Emerging data reveal that mitochondrial dysfunction and oxidative stress are major players in the genesis of the deleterious cellular events in AKI, suggesting that intervening within these pathways may offer potential therapeutic targets for AKI [[115], [116], [117]]. Given the hypoxia and ischemia evident in renal injury, the role of ROS in AKI pathogenesis has been extensively examined. Enhanced ROS generation during the reperfusion stage triggers mitochondrial DNA damage, apoptosis and lipid peroxidation [118,119]. Earlier studies revealed that treatment with superoxide dismutase prior to renal ischemia prevents increased mitochondrial lipid peroxidation in the renal cortex [119]. The potential role for antioxidants in the management of AKI has been previously investigated [120].
Extensive evidence suggests that the female sex is protective against AKI in ischemic animal models [[121], [122], [123]]. In contrast, other studies have shown that the female sex is a predisposing risk factor in drug-induced AKI [[124], [125], [126]]. Despite the vast amount of literature highlighting gender-related differences in AKI incidence, progression and outcomes, the underlying mechanisms remain to be identified. Studies demonstrate that E2 elicits protective actions in ischemic renal injury [121,122,127], while testosterone plays a central role in predisposing males to ischemic renal injury [122]. Additional studies focused on identifying the contribution of sex hormonal receptors and sex chromosomes in AKI initiation and progression are required.
Despite our current knowledge that mitochondrial bioenergetics and oxidative stress play critical roles in AKI pathophysiology, the contribution of mitochondrial dysfunction to gender differences in AKI pathophysiology is not fully defined. Of note, sex differences in mitochondrial number, morphology, biogenesis and bioenergetics occur in renal [67,68] and non-renal tissues [66,69,128]. Pan and Sheikh-Hamad have recently comprehensively reviewed current advances in sex-specific alteration in mitochondrial function that relates to AKI [116]. Overall, evidence suggests differences exists between males and females in mitochondrial biology and ROS generation. Further studies are required to determine the contribution of mitochondrial dysfunction and ROS in male-female differences in AKI.
4.2. Diabetic nephropathy
Diabetes is a multifactorial metabolic disease that impacts approximately 9.4% of the U.S. population and is steadily becoming more prevalent worldwide [129]. The high prevalence of this disease is due to sedentary lifestyles, poor diet intake and increased metabolic diseases such as obesity and cardiovascular disease. Diabetes elicits systemic effects and accelerates the development of secondary complications such as high blood pressure, stroke, neuropathy retinopathy and diabetic nephropathy (DN) through excessive ROS production [130,131]. About 30–40% of diabetic patients develop kidney disease and about one third of them progress to DN [132], which is the leading cause of end-stage kidney disease. Because of this, DN is associated with substantial healthcare costs [133]. Key clinical identifiers of DN progression include albuminuria and renal mesangial expansion in the glomeruli, tubular hypertrophy, interstitial fibrosis and immune cell infiltration [134].
Sex differences play a critical role in the progression of DN in clinical and pre-clinical studies. Men with diabetes have been reported to have a higher risk for DN compared to women with diabetes [135]. However, diabetic women over 60 years of age tend to have a higher risk for DN compared to men [136]. These differences are thought to be associated with reduced endogenous E2 levels [137]. Sex differences have also been observed in DN progression to chronic kidney disease (CKD) and end stage renal disease. In general, women have a higher incidence for CKD compared to men; however, CKD progression is worse in men [6]. The reasons for these sex differences are not fully elucidated but appear to be influenced by sex hormones. These clinical studies amongst many others suggest sex hormones play a vital role in the progression of DN and are elegantly reviewed elsewhere [138,139].
Pre-clinical models have provided some additional evidence of sex differences during DN. Streptozotocin induction (STZ model) and Akita mice are widely accepted type 1 diabetes animal models and the db/db mouse model, Zucker diabetic fatty rats and Wistar fatty rats are common type 2 diabetes models [140]. A large proportion of research in this area has consisted of using male mice rather than females to understand DN pathogenesis and for drug development [141]. Female rodents are not typically included in many of these studies because they are especially resistant to the development of hyperglycemia, as well as the associated diabetic kidney disease. Female sex hormones are thought to be protective against DN, although the mechanisms involved in this protection are far from being understood. Interestingly, a recent study determined that male and female mice that lack nitric oxide synthase 3 develop diabetes equally and this model may be useful to study DN in both sexes [142].
ROS generated from NOXs and the mitochondria are elevated during diabetes and contribute to inflammation and disease progression [143]. Studies have investigated whether targeting such sources of ROS during diabetes could be useful to ameliorate the associated renal injury. A study by Zheng et al. showed that oxidative stress in the kidneys of animals with type 1 diabetes induced via STZ can be attenuated by dietary compounds through activation of the anti-oxidant actions of nuclear factor E2-related factor 2 (Nrf2) [144]. Additionally, replacement of E2 in ovariectomized diabetic rats induced with STZ reduced oxidative stress in the kidney by preserving glutathione redox cycling [145]. Other research groups using animal models of type 2 diabetes determined that reducing oxidative stress by antioxidants confers protection against DN [146,147].
Although sex differences are known to be involved in the processes contributing to DN, additional pre-clinical models and larger, prospective, randomized clinical studies examining both sexes are essential to better understand the progression of DN in both genders. Another topic of interest is to understand why renoprotective effects are reduced in diabetic women compared to women diagnosed with other renal diseases. Identifying the cause of such differences could be useful to design therapeutic targets.
4.3. Kidney stone disease
Kidney stones are hard, mineral deposits that form in the urinary tract due to elevated urine supersaturation which leads to urinary crystal formation, retention and growth. Kidney stones affect 1 in 11 people in the United States [148]. Kidney stones are more prevalent in men (10.6%) compared to women (7.1%) [148] and the reason why this occurs is not well defined, although E2 has been suggested to be protective against kidney stones in women [149]. However, recent data show women are starting to experience kidney stones more frequently and this may be a result of reduced fluid intake and increased dietary changes and obesity [150,151]. Tundo et al. recently reported this gender disparity is diminishing primarily in adults under fifty in the United States [152]. However, the incidence of kidney stones continues to be higher in postmenopausal women [153].
The most common type of kidney stone is found in men and women and is composed of calcium oxalate (CaOx) crystals. Stones primarily comprised of calcium phosphate (>50%) occur more frequently in women than men [154]. Irrespective of stone composition, approximately 50% of stone formers experience another stone episode within 5 years [155]. The etiology of kidney stones and the mechanisms leading to recurring stones are not well established. Lifestyle factors, genetics and high oxalate diets are all contributors to stone pathogenesis [148,[156], [157], [158]]. Stone formers also are at risk for developing chronic kidney disease, diabetes and cardiovascular diseases [[159], [160], [161], [162]]. Gillen et al. found women with kidney stones have elevated hypertension compared to male stone formers and healthy females [163].
Several experimental and clinical studies have suggested oxidative stress and inflammation are integral factors in kidney stone pathogenesis [[164], [165], [166], [167], [168], [169], [170], [171], [172], [173], [174], [175]]. This is important to highlight because both oxidative stress and inflammation stimulate metabolic dysfunction and cell death. Renal cells exposed to CaOx crystals or oxalate have increased superoxide generation from NADPH oxidases [176] and also induce expression of MCP-1 (chemokine that recruits monocytes), Interleukin-2R (IL-2Rβ) receptor (receptor for pleiotropic cytokine, IL-2) and IL-6 (pro-inflammatory cytokine) [173,[177], [178], [179]]. CaOx crystals have also been shown to increase IL-1β secretion and alter mitochondrial protein levels in immune cells [172,180]. It was recently reported that oxalate disrupts cellular energetics and redox status in a human monocytic cell line and primary monocytes from healthy subjects [181]. Patel et al. also found that CaOx crystals suppressed cellular energetics and reduced glutathione levels and MnSOD protein levels in monocytes [181]. Khan et al. examined the impact of renal crystal deposition on oxidative stress and inflammation in male rats fed a hydroxyproline diet [182]. The authors determined that the presence of renal crystals caused superoxide generation via NADPH oxidase and upregulation of crystallization modulators, such as osteopontin, in renal tissue. These findings were reversed when animals were fed apocynin, a NAPDH oxidase inhibitor. Of note, the androgen receptor has been found to promote crystal formation by inducing NADPH oxidases and oxalate synthesis [183]. Taguchi et al. used colony stimulating factor-1-deficient mice to determine anti-inflammatory macrophages play an important role in renal crystal formation during hyperoxaluria [184]. A study by Fan et al. used both male and female stone forming rats to examine the role of sex hormones [185]. They found that implanting rats with E2 mitigated crystal deposition after ethylene glycol exposure and suggested that E2 decreased urinary oxalate. Another study suggested that E2 exerts protective effects against CaOx crystal injury in renal cells by altering the cellular proteome [24]. These findings suggest E2 may be an important factor in stone disease.
As indicated earlier, men tend to form kidney stones at a higher rate than women. A potential reason why this occurs may be due to the interaction between sex hormones and immune system. It has been well established that immune response differs among men and women [186]. Further, immunity can be impacted by obesity and diet, which are both associated with stone formation and may be impacted by gender [187]. Obesity and diet could reduce the ability of leukocytes to respond to crystal growth in the kidney. Clinical studies have reported patients with kidney stones have increased pro-inflammatory cytokines and chemokines in their serum and urine [[169], [170], [171]]. Williams et al. demonstrated that monocytes from CaOx stone formers have reduced cellular energetics compared to healthy subjects [188]. It was recently established that CaOx crystals differentiates human monocytes into pro-inflammatory macrophages [189]. Pro-inflammatory gene expression has been shown to be elevated in papillary tissue from Randall plaques of CaOx stone formers and was suggested to contribute to oxidative stress and renal injury [190]. It would be interesting to examine such responses in men versus women and to compare responses in stone formers compared to healthy individuals.
Taken together, understanding the interplay between oxidative stress and inflammation in kidney stone disease is necessary to develop strategies to prevent stone formation and to reduce the development of secondary complications in patients. Importantly, evaluating how sex hormones influence the immune system and inflammatory responses leading to this pathology warrants further investigation.
4.4. Salt sensitivity
The kidney plays a fundamental role in the regulation of blood pressure and sodium homeostasis through the pressure-natriuresis mechanism. Multiple studies revealed that many renal regulatory mechanisms that contribute to the maintenance of blood pressure and sodium balance are sex specific. Compelling evidence indicates that oxidative stress is a major contributor to the development of multiple forms of hypertension [191]. In addition, activation of the immune system is involved in the development of hypertension [192]. Some individuals can efficiently excrete a high salt intake, while others cannot do so effectively without an increase in blood pressure. The latter are referred to as salt sensitive subjects [193]. The kidneys are equipped with an enormous capacity to handle dietary salt challenges, however dysfunction of the kidney to maintain sodium balance will lead to the development of salt-sensitive hypertension.
Sexual dimorphism exists in the renal response to high salt. Female rats exhibit a more rapid natriuretic response to an acute intraperitoneal saline load [194]. Similarly, the urinary excretion of salt was higher in women than men injected with an intravenous saline load [195]. These studies suggest that the female kidney has an enhanced capacity to handle salt challenges compared to males.
Improving our understanding of the impact of male and female sex steroids on renal sodium handling and renal hemodynamics is critical to better understand sex differences in salt sensitivity. Depletion of ovarian hormones by ovariectomy potentiated salt-induced elevation in blood pressure in Dahl salt sensitive female rats [196]. Castration resulted in a more pronounced decline in the hypertensive response to salt loading in male Dahl salt-sensitive rats [197]. These studies suggest an inhibitory role for ovarian hormones and a facilitatory role for androgenic hormones in salt sensitivity in this animal model. In female rats treated with salt and deoxycorticosterone (DOCA), there was an increase in oxidative stress level in the kidney [198]. Renal malondialdehyde (oxidant) level was elevated in DOCA-salt treated rats [198]. Treatment with E2 resulted in a decrease in malondialdehyde level in the kidney from female rats [198]. GPER, which mediated rapid estrogenic signaling, has been shown to elicit cardiovascular and renal protective effects in salt-induced complications. Lindsey et al. have shown that GPER activation attenuated renal tubular oxidative stress in salt-sensitive mRen2 female rats [23], a transgenic rat model expressing the murine Ren2 gene which quickly develops hypertension. Urinary excretion of the lipid oxidation marker 8-isoprostane was declined by chronic GPER activation in salt-loaded mRen2 female rats [23]. Studying sex hormonal and receptor-related differences in the renal response to salt challenges is fundamental to understanding the mechanisms triggering salt sensitivity in males and females.
ET-1 is a well-established natriuretic factor that inhibits tubular reabsorption of sodium. ET-1 natriuretic responses are mediated mainly via activation of ETB receptors and lack of functional ETB receptors results in salt sensitive hypertension [199]. ETA receptors contribute to ET-1 facilitatory effects on sodium excretions in female rats only [108]. Despite the large body of evidence pointing to sex differences in ET-1 tubular effects, sex does not impact afferent arteriolar responsiveness to ET receptor activation [200]. Given that the mechanisms that regulate renal sodium handling are different based on the sex, the therapeutic targets for salt sensitivity should be tailored based on the patient's gender.
4.5. Preeclampsia
Preeclampsia is a complication of pregnancy that is characterized by persistent hypertension, edema and proteinuria. The mechanisms involved in the initiation and progression of preeclampsia are complex and involves many factors. It has been extensively shown in the literature that this pregnancy complication is linked to placental oxidative and nitrosative stresses [[201], [202], [203]]. In preeclampsia, oxidative stress markers are increased and antioxidant levels are decreased in the maternal plasma [[204], [205], [206]]. Poor perfusion of the fetal placenta elevates lipid peroxidation byproducts, thromboxane and cytokines, which contribute to the endothelial dysfunction observed in preeclampsia [202,203]. The endothelium of the maternal kidney is highly affected [202]. The endothelial-podocyte interplay may be involved in the deterioration of the filtration barrier that contributes to proteinuria in preeclamptic patients [207].
Patients suffering from renal disease are at a higher risk for developing hypertensive disorders during pregnancy [208]. Serum creatinine levels were correlated with the risk for preeclampsia [208]. Preeclampsia predisposes women to AKI with high risk of maternal and fetal morbidity [207,209]. Preeclampsia-induced renal injury involves antiangiogenetic factors such as soluble fms-like tyrosine kinase-1 [207]. The contribution of renal redox mechanisms to the pathogenesis of preeclampsia is still an area of active investigation.
Data suggest that ET-1 contributes to the progression of preeclampsia. Multiple human studies showed that circulating levels of ET-1 are elevated in preeclampsia, compared to normal pregnancy [210]. In addition, preeclampsia elevates ET-1 level in the umbilical vein [211]. Both placental and renal ET-1 levels are increased in experimental models of preeclampsia [212]. Besides, ET-1 receptor signaling is altered in preeclampsia. Animal studies showed that ETA receptor antagonism completely abolished sFlt-1-induced hypertension in pregnancy [213]. Previously preeclamptic women exhibit ETB receptor dysfunction in their cutaneous vessels postpartum [214].
Importantly, it has been shown that ET-1 evokes oxidative stress in preeclampsia (Fig. 3) [215,216]. In human placenta, ET-1 increased malondialdehyde (oxidant) and decreased glutathione, glutathione disulfide and ascorbic acid (antioxidants) [217]. ET-1-induced oxidative stress can act as a positive feedback loop to generate more ET-1 [216]. ET-1-induced imbalance between the oxidant and antioxidant forces in preeclampsia setting provides a potential target for therapeutic intervention.
Fig. 3.
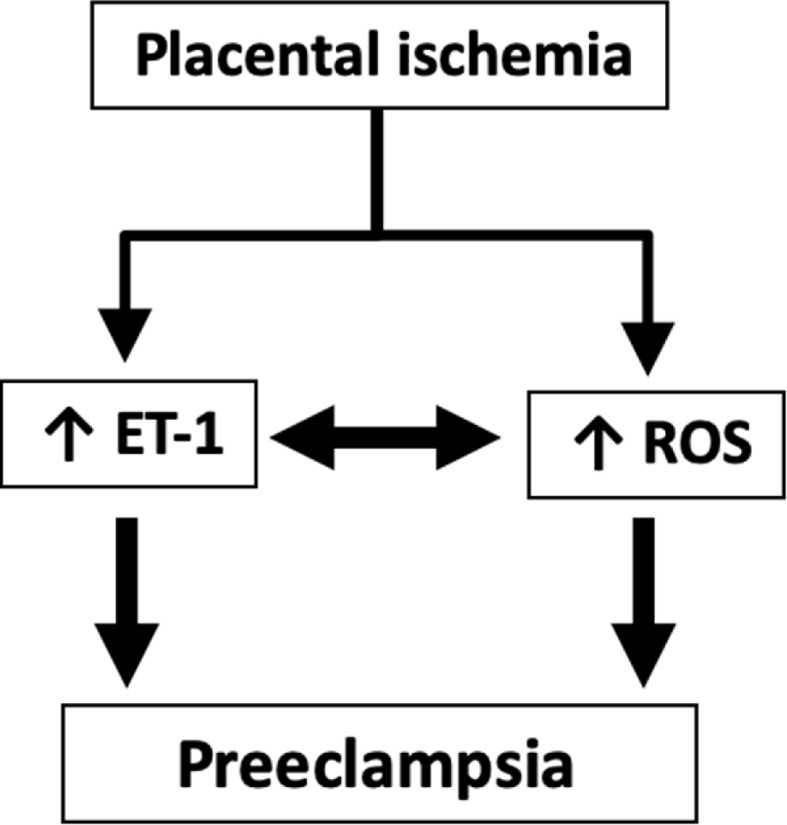
Proposed scheme for the involvement of endothelin-1 (ET-1) and reactive oxygen generation (ROS) in the development of preeclampsia.
Recent data suggest that placental endoplasmic reticulum stress contributes to the pathogenesis of hypertensive pregnancy [218]. Moreover, ET-1 has been shown to induce endoplasmic reticulum stress in preeclampsia [105]. ET-1 acts via ETB receptor to activate phospholipase C pathway and facilitate the release of calcium from endoplasmic reticulum, thereby triggering endoplasmic reticulum stress [105]. Collectively, ET-1-induced oxidative and endoplasmic reticulum stress may present the link between the maternal syndrome of preeclampsia and renal injury.
4.6. Postmenopausal hypertension
Premenopausal women have lower incidence of hypertension that men at similar age. In postmenopausal women, blood pressure rises to levels that are higher than those in age-matched men. It has been also shown that the prevalence of salt sensitivity increases after menopause [219]. While the lack of E2 as a contributing factor in the development of postmenopausal hypertension seems likely, this is most likely not the only component underlying the pathogenesis of postmenopausal hypertension.
In postmenopausal women, the circulating levels of oxidative stress markers increase as menopause proceeds [220,221]. The longer the time elapsed after menopause, the lower the plasma levels of glutathione peroxidase [220]. Chronic treatment of post-cycling female spontaneously hypertensive rats with the antioxidant vitamins, E and C, reduces hypertension [222], suggesting that oxidative stress contributes to the development of postmenopausal hypertension. Of note, the urinary excretion of 8-iso-prostaglandin F (2α), a major isoprostane and a biomarker for oxidative stress, was lower in premenopausal women compared with postmenopausal women [221].
Studies suggest that ET-1 contributes to the progression of postmenopausal hypertension. Plasma ET-1 levels increase after menopause and may contribute to hypertension and cardiovascular disease in postmenopausal women [223,224]. In comparison to young women, ETB-mediated vasodilatory function is attenuated in postmenopausal women [225]. In aged female spontaneously hypertensive, a model of postmenopausal hypertension, ETA receptor antagonism induced an additional decrease in blood pressure, compared to the combined antihypertensive effect of angiotensin converting enzyme inhibitor and arachidonic acid metabolism inhibition [226]. Ovariectomy, a surgical model of menopause, and hormone replacement have been shown to regulate ETA and ETB receptors mRNA expression in renal cortical and medullary tissues [111].
In response to high salt, ET-1 is released within the kidney to prevent tubular reabsorption of sodium. Infusion of a salt load to the renal medulla evokes a natriuretic response that is mediated via ETA and ETB receptors in male and OVX female rats [227,228]. In contrast, the natriuretic response to medullary salt loading in intact female rats is ET-1 independent [227]. These studies highlight that the ET-1 signaling system in the medulla of the kidney has a more important contribution to salt excretion in OVX rats, in comparison to intact female animals. Altogether, data from human and animal studies point to the ET-1 and redox signaling pathways as potential therapeutic targets to manage hypertension in postmenopausal women.
5. Future directions
Despite the release of the NIH Revitalization Act which requires the inclusion of women and minorities in NIH-funded research in 1993 [229], women remain under-represented in clinical trials [230]. Even when women are included, data are often not presented based on gender [231], causing a gap in knowledge about gender disparities in renal disease. The National Institute of Diabetes and Digestive and Kidney Diseases Workshop on “Sex and the Kidneys” have recently highlighted current understanding and research opportunities for the renal research community to advance our understanding of the sex differences in the kidney in clinical, basic and translational research [232]. Better understanding the contribution of the sex to renal function in basic science research will guide future clinical research to study gender-related differences in kidney disease pathophysiology.
As a result of the current review, it is clear that the crosstalk between the sex steroids and redox homeostasis in the development and progression of renal pathologies needs to be further investigated. Future studies of the mechanisms underlying male-female differences in renal disease will reveal potential therapeutic targets and facilitate the development of therapies that are tailored based on the gender and the hormonal status of patients.
Author contributions
TM, CDM and EYG wrote the manuscript. All authors participated in editing the manuscript and approved the final version to be submitted.
Sources of funding
This project was supported by the National Institute of Health (K01DK106284 to TM, K01HL145324 to CDM and K99DK119413 to EYG).
Declaration of competing interest
The authors have declared that no conflict of interest exists. Dr. Gohar is also affiliated with the Department of Pharmacology and Toxicology, Faculty of Pharmacy, Alexandria University, Egypt.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101489.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Press Release from ASN, ERA-EDTA and ISN The hidden epidemic: worldwide, over 850 million people suffer from kidney diseases. 2018. https://renal.org/press-release-asn-era-edta-isn-hidden-epidemic-worldwide-850-million-people-suffer-kidney-diseases/ Press Release. Retrieved from. June 29.
- 2.CDC/National center for health statistics. Kidney disease. 2013. https://www.cdc.gov/nchs/fastats/kidney-disease.htm February 18.
- 3.Carrero J.J., Hecking M., Chesnaye N.C., Jager K.J. Sex and gender disparities in the epidemiology and outcomes of chronic kidney disease. Nat. Rev. Nephrol. 2018;14(3):151–164. doi: 10.1038/nrneph.2017.181. [DOI] [PubMed] [Google Scholar]
- 4.Neugarten J., Golestaneh L. Influence of sex on the progression of chronic kidney disease. Mayo Clin. Proc. 2019;94(7):1339–1356. doi: 10.1016/j.mayocp.2018.12.024. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki H., Kondo K. Chronic kidney disease in postmenopausal women. Hypertens. Res. : official journal of the Japanese Society of Hypertension. 2012;35(2):142–147. doi: 10.1038/hr.2011.155. [DOI] [PubMed] [Google Scholar]
- 6.Valdivielso J.M., Jacobs-Cacha C., Soler M.J. Sex hormones and their influence on chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2019;28(1):1–9. doi: 10.1097/MNH.0000000000000463. [DOI] [PubMed] [Google Scholar]
- 7.Krata N., Zagozdzon R., Foroncewicz B., Mucha K. Oxidative stress in kidney diseases: the cause or the consequence? Arch. Immunol. Ther. Exp. 2018;66(3):211–220. doi: 10.1007/s00005-017-0496-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamma G., Valenti G. Evaluating the oxidative stress in renal diseases: what is the role for S-glutathionylation? Antioxidants Redox Signal. 2016;25(3):147–164. doi: 10.1089/ars.2016.6656. [DOI] [PubMed] [Google Scholar]
- 9.Lever J.M., Boddu R., George J.F., Agarwal A. Heme oxygenase-1 in kidney health and disease. Antioxidants Redox Signal. 2016;25(3):165–183. doi: 10.1089/ars.2016.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Posynick B.J., Brown C.J. Escape from X-chromosome inactivation: an evolutionary perspective. Front Cell Dev Biol. 2019;7:241. doi: 10.3389/fcell.2019.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galupa R., Heard E. X-chromosome inactivation: a crossroads between chromosome architecture and gene regulation. Annu. Rev. Genet. 2018;52:535–566. doi: 10.1146/annurev-genet-120116-024611. [DOI] [PubMed] [Google Scholar]
- 12.Khalil R., Kim N.R., Jardi F., Vanderschueren D., Claessens F., Decallonne B. Sex steroids and the kidney: role in renal calcium and phosphate handling. Mol. Cell. Endocrinol. 2018;465:61–72. doi: 10.1016/j.mce.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Arias-Loza P.A., Muehlfelder M., Elmore S.A., Maronpot R., Hu K., Blode H. Differential effects of 17beta-estradiol and of synthetic progestins on aldosterone-salt-induced kidney disease. Toxicol. Pathol. 2009;37(7):969–982. doi: 10.1177/0192623309350475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filler G., Ramsaroop A., Stein R., Grant C., Marants R., So A. Is testosterone detrimental to renal function? Kidney Int Rep. 2016;1(4):306–310. doi: 10.1016/j.ekir.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDonald S.P., Craig J.C. Australian, and New Zealand Paediatric Nephrology A. Long-term survival of children with end-stage renal disease. N. Engl. J. Med. 2004;350(26):2654–2662. doi: 10.1056/NEJMoa031643. [DOI] [PubMed] [Google Scholar]
- 16.Silbiger S.R. Raging hormones: gender and renal disease. Kidney Int. 2011;79(4):382–384. doi: 10.1038/ki.2010.474. [DOI] [PubMed] [Google Scholar]
- 17.Prabhushankar R., Krueger C., Manrique C. Membrane estrogen receptors: their role in blood pressure regulation and cardiovascular disease. Curr. Hypertens. Rep. 2014;16(1):408. doi: 10.1007/s11906-013-0408-6. [DOI] [PubMed] [Google Scholar]
- 18.Hutson D.D., Gurrala R., Ogola B.O., Zimmerman M.A., Mostany R., Satou R. Estrogen receptor profiles across tissues from male and female Rattus norvegicus. Biol. Sex Differ. 2019;10(1):4. doi: 10.1186/s13293-019-0219-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bjornstrom L., Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. (Baltimore, Md) 2005;19(4):833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 20.Sabolic I., Asif A.R., Budach W.E., Wanke C., Bahn A., Burckhardt G. Gender differences in kidney function. Pflügers Archiv. 2007;455(3):397–429. doi: 10.1007/s00424-007-0308-1. [DOI] [PubMed] [Google Scholar]
- 21.Boulkroun S., Le Moellic C., Blot-Chabaud M., Farman N., Courtois-Coutry N. Expression of androgen receptor and androgen regulation of NDRG2 in the rat renal collecting duct. Pflügers Archiv. 2005;451(2):388–394. doi: 10.1007/s00424-005-1410-x. [DOI] [PubMed] [Google Scholar]
- 22.Davidoff M., Caffier H., Schiebler T.H. Steroid hormone binding receptors in the rat kidney. Histochemistry. 1980;69(1):39–48. doi: 10.1007/BF00508365. [DOI] [PubMed] [Google Scholar]
- 23.Lindsey S.H., Yamaleyeva L.M., Brosnihan K.B., Gallagher P.E., Chappell M.C. Estrogen receptor GPR30 reduces oxidative stress and proteinuria in the salt-sensitive female mRen2. Lewis rat. Hypertension. 2011;58(4):665–671. doi: 10.1161/HYPERTENSIONAHA.111.175174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peerapen P., Thongboonkerd V. Protective cellular mechanism of estrogen against kidney stone formation: a proteomics approach and functional validation. Proteomics. 2019;19(19) doi: 10.1002/pmic.201900095. [DOI] [PubMed] [Google Scholar]
- 25.Gong W., Song J., Chen X., Li S., Yu J., Xia W. Estrogen-related receptor-alpha mediates puromycin aminonucleoside-induced mesangial cell apoptosis and inflammatory injury. Am. J. Physiol. Ren. Physiol. 2019;316(5):F906–F913. doi: 10.1152/ajprenal.00507.2018. [DOI] [PubMed] [Google Scholar]
- 26.Khan M., Ullah R., Rehman S.U., Shah S.A., Saeed K., Muhammad T. 17beta-Estradiol modulates SIRT1 and halts oxidative stress-mediated cognitive impairment in a male aging mouse model. Cells. 2019;8(8) doi: 10.3390/cells8080928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kowalska K., Habrowska-Gorczynska D.E., Urbanek K.A., Dominska K., Sakowicz A., Piastowska-Ciesielska A.W. Estrogen receptor beta plays a protective role in zearalenone-induced oxidative stress in normal prostate epithelial cells. Ecotoxicol. Environ. Saf. 2019;172:504–513. doi: 10.1016/j.ecoenv.2019.01.115. [DOI] [PubMed] [Google Scholar]
- 28.Ogola B.O., Zimmerman M.A., Sure V.N., Gentry K.M., Duong J.L., Clark G.L. G protein-coupled estrogen receptor protects from angiotensin II-induced increases in pulse pressure and oxidative stress. Front. Endocrinol. 2019;10:586. doi: 10.3389/fendo.2019.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheng Y., Abreu I.A., Cabelli D.E., Maroney M.J., Miller A.F., Teixeira M. Superoxide dismutases and superoxide reductases. Chem. Rev. 2014;114(7):3854–3918. doi: 10.1021/cr4005296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 32.Brand M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016;100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Speckmann B., Steinbrenner H., Grune T., Klotz L.O. Peroxynitrite: from interception to signaling. Arch. Biochem. Biophys. 2016;595:153–160. doi: 10.1016/j.abb.2015.06.022. [DOI] [PubMed] [Google Scholar]
- 35.Forstermann U., Sessa W.C. Nitric oxide synthases: regulation and function. Eur. Heart J. 2012;33(7):829–837. doi: 10.1093/eurheartj/ehr304. 37a-37d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bienert G.P., Schjoerring J.K., Jahn T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta. 2006;1758(8):994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Birben E., Sahiner U.M., Sackesen C., Erzurum S., Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5(1):9–19. doi: 10.1097/WOX.0b013e3182439613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Forman H.J., Zhang H., Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol. Aspect. Med. 2009;30(1–2):1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stocker R., Keaney J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004;84(4):1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 40.Kudryavtseva A.V., Krasnov G.S., Dmitriev A.A., Alekseev B.Y., Kardymon O.L., Sadritdinova A.F. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget. 2016;7(29):44879–44905. doi: 10.18632/oncotarget.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pham-Huy L.A., He H., Pham-Huy C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008;4(2):89–96. [PMC free article] [PubMed] [Google Scholar]
- 42.Miranda-Diaz A.G., Pazarin-Villasenor L., Yanowsky-Escatell F.G., Andrade-Sierra J. Oxidative stress in diabetic nephropathy with early chronic kidney disease. J Diabetes Res. 2016;2016:7047238. doi: 10.1155/2016/7047238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitchell T., Saba H., Laakman J., Parajuli N., MacMillan-Crow L.A. Role of mitochondrial-derived oxidants in renal tubular cell cold-storage injury. Free Radic. Biol. Med. 2010;49(8):1273–1282. doi: 10.1016/j.freeradbiomed.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park K.M., Kim J.I., Ahn Y., Bonventre A.J., Bonventre J.V. Testosterone is responsible for enhanced susceptibility of males to ischemic renal injury. J. Biol. Chem. 2004;279(50):52282–52292. doi: 10.1074/jbc.M407629200. [DOI] [PubMed] [Google Scholar]
- 45.Steinhausen M., Ballantyne D., Fretschner M., Parekh N. Sex differences in autoregulation of juxtamedullary glomerular blood flow in hydronephrotic rats. Am. J. Physiol. 1990;258(4 Pt 2):F863–F869. doi: 10.1152/ajprenal.1990.258.4.F863. [DOI] [PubMed] [Google Scholar]
- 46.Hamza S.M., Dyck J.R. Systemic and renal oxidative stress in the pathogenesis of hypertension: modulation of long-term control of arterial blood pressure by resveratrol. Front. Physiol. 2014;5:292. doi: 10.3389/fphys.2014.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajaram R.D., Dissard R., Jaquet V., de Seigneux S. Potential benefits and harms of NADPH oxidase type 4 in the kidneys and cardiovascular system. Nephrol. Dial. Transplant. 2019;34(4):567–576. doi: 10.1093/ndt/gfy161. [DOI] [PubMed] [Google Scholar]
- 48.Meng X.M., Ren G.L., Gao L., Yang Q., Li H.D., Wu W.F. NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation. Lab. Invest. J. Tech. Methods Pathol. 2018;98(1):63–78. doi: 10.1038/labinvest.2017.120. [DOI] [PubMed] [Google Scholar]
- 49.Gorin Y., Block K., Hernandez J., Bhandari B., Wagner B., Barnes J.L. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J. Biol. Chem. 2005;280(47):39616–39626. doi: 10.1074/jbc.M502412200. [DOI] [PubMed] [Google Scholar]
- 50.Malek M., Nematbakhsh M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Ren. Inj. Prev. 2015;4(2):20–27. doi: 10.12861/jrip.2015.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wen Y., Liu Y.R., Tang T.T., Pan M.M., Xu S.C., Ma K.L. mROS-TXNIP axis activates NLRP3 inflammasome to mediate renal injury during ischemic AKI. Int. J. Biochem. Cell Biol. 2018;98:43–53. doi: 10.1016/j.biocel.2018.02.015. [DOI] [PubMed] [Google Scholar]
- 52.Saba H., Batinic-Haberle I., Munusamy S., Mitchell T., Lichti C., Megyesi J. Manganese porphyrin reduces renal injury and mitochondrial damage during ischemia/reperfusion. Free Radic. Biol. Med. 2007;42(10):1571–1578. doi: 10.1016/j.freeradbiomed.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fekete A., Vannay A., Ver A., Vasarhelyi B., Muller V., Ouyang N. Sex differences in the alterations of Na(+), K(+)-ATPase following ischaemia-reperfusion injury in the rat kidney. J. Physiol. 2004;555(Pt 2):471–480. doi: 10.1113/jphysiol.2003.054825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blake R., Trounce I.A. Mitochondrial dysfunction and complications associated with diabetes. Biochim. Biophys. Acta. 2013;1840(4):1404–1412. doi: 10.1016/j.bbagen.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 55.Hill B.G., Benavides G.A., Lancaster J.R., Jr., Ballinger S., Dell'Italia L., Jianhua Z. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012;393(12):1485–1512. doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guzik T.J., Korbut R., Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J. Physiol. Pharmacol. 2003;54(4):469–487. [PubMed] [Google Scholar]
- 57.Ventura-Clapier R., Moulin M., Piquereau J., Lemaire C., Mericskay M., Veksler V. Mitochondria: a central target for sex differences in pathologies. Clin. Sci. (Lond). 2017;131(9):803–822. doi: 10.1042/CS20160485. [DOI] [PubMed] [Google Scholar]
- 58.Velarde M.C. Mitochondrial and sex steroid hormone crosstalk during aging. Longev. Heal. 2014;3(1):2. doi: 10.1186/2046-2395-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matthews K.A. Interactive effects of behavior and reproductive hormones on sex differences in risk for coronary heart disease. Health Psychol. 1989;8(4):373–387. [PubMed] [Google Scholar]
- 60.Muka T., Nano J., Jaspers L., Meun C., Bramer W.M., Hofman A. Associations of steroid sex hormones and sex hormone-binding globulin with the risk of type 2 diabetes in women: a population-based cohort study and meta-analysis. Diabetes. 2017;66(3):577–586. doi: 10.2337/db16-0473. [DOI] [PubMed] [Google Scholar]
- 61.Sun N., Youle R.J., Finkel T. The mitochondrial basis of aging. Mol. Cell. 2016;61(5):654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ballinger S.W. Mitochondrial dysfunction in cardiovascular disease. Free Radic. Biol. Med. 2005;38(10):1278–1295. doi: 10.1016/j.freeradbiomed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 63.Sivitz W.I., Yorek M.A. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxidants Redox Signal. 2010;12(4):537–577. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mattingly K.A., Ivanova M.M., Riggs K.A., Wickramasinghe N.S., Barch M.J., Klinge C.M. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol. Endocrinol. 2008;22(3):609–622. doi: 10.1210/me.2007-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tcherepanova I., Puigserver P., Norris J.D., Spiegelman B.M., McDonnell D.P. Modulation of estrogen receptor-alpha transcriptional activity by the coactivator PGC-1. J. Biol. Chem. 2000;275(21):16302–16308. doi: 10.1074/jbc.M001364200. [DOI] [PubMed] [Google Scholar]
- 66.Khalifa A.R., Abdel-Rahman E.A., Mahmoud A.M., Ali M.H., Noureldin M., Saber S.H. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiological reports. 2017;5(6) doi: 10.14814/phy2.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Woodman A.G., Mah R., Keddie D.L., Noble R.M.N., Holody C.D., Panahi S. Perinatal iron deficiency and a high salt diet cause long-term kidney mitochondrial dysfunction and oxidative stress. Cardiovasc. Res. 2020;116(1):183–192. doi: 10.1093/cvr/cvz029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Woodman A.G., Mah R., Keddie D., Noble R.M.N., Panahi S., Gragasin F.S. Prenatal iron deficiency causes sex-dependent mitochondrial dysfunction and oxidative stress in fetal rat kidneys and liver. Faseb. J. : official publication of the Federation of American Societies for Experimental Biology. 2018;32(6):3254–3263. doi: 10.1096/fj.201701080R. [DOI] [PubMed] [Google Scholar]
- 69.Cardinale D.A., Larsen F.J., Schiffer T.A., Morales-Alamo D., Ekblom B., Calbet J.A.L. Superior intrinsic mitochondrial respiration in women than in men. Front. Physiol. 2018;9:1133. doi: 10.3389/fphys.2018.01133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schlondorff D., Nelson P.J., Luckow B., Banas B. Chemokines and renal disease. Kidney Int. 1997;51(3):610–621. doi: 10.1038/ki.1997.90. [DOI] [PubMed] [Google Scholar]
- 71.El-Benna J., Hurtado-Nedelec M., Marzaioli V., Marie J.C., Gougerot-Pocidalo M.A., Dang P.M. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol. Rev. 2016;273(1):180–193. doi: 10.1111/imr.12447. [DOI] [PubMed] [Google Scholar]
- 72.Iles K.E., Forman H.J. Macrophage signaling and respiratory burst. Immunol. Res. 2002;26(1–3):95–105. doi: 10.1385/IR:26:1-3:095. [DOI] [PubMed] [Google Scholar]
- 73.West A.P., Shadel G.S., Ghosh S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011;11(6):389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lopez-Armada M.J., Riveiro-Naveira R.R., Vaamonde-Garcia C., Valcarcel-Ares M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion. 2013;13(2):106–118. doi: 10.1016/j.mito.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 75.Ravi S., Mitchell T., Kramer P.A., Chacko B., Darley-Usmar V.M. Mitochondria in monocytes and macrophages-implications for translational and basic research. Int. J. Biochem. Cell Biol. 2014;53:202–207. doi: 10.1016/j.biocel.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baehner R.L., Gilman N., Karnovsky M.L. Respiration and glucose oxidation in human and Guinea pig leukocytes: comparative studies. J. Clin. Invest. 1970;49(4):692–700. doi: 10.1172/JCI106281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Geissmann F., Manz M.G., Jung S., Sieweke M.H., Merad M., Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327(5966):656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murphy M.P. Understanding and preventing mitochondrial oxidative damage. Biochem. Soc. Trans. 2016;44(5):1219–1226. doi: 10.1042/BST20160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scialo F., Fernandez-Ayala D.J., Sanz A. Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease. Front. Physiol. 2017;8:428. doi: 10.3389/fphys.2017.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lowell B.B., Shulman G.I. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 81.Cao Q., Wang Y., Harris D.C. Pathogenic and protective role of macrophages in kidney disease. Am. J. Physiol. Ren. Physiol. 2013;305(1):F3–F11. doi: 10.1152/ajprenal.00122.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maianski N.A., Geissler J., Srinivasula S.M., Alnemri E.S., Roos D., Kuijpers T.W. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. 2004;11(2):143–153. doi: 10.1038/sj.cdd.4401320. [DOI] [PubMed] [Google Scholar]
- 83.Bain B.J., England J.M. Normal haematological values: sex difference in neutrophil count. Br. Med. J. 1975;1(5953):306–309. doi: 10.1136/bmj.1.5953.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davidson W.M., Smith D.R. A morphological sex difference in the polymorphonuclear neutrophil leucocytes. Br. Med. J. 1954;2(4878):6–7. doi: 10.1136/bmj.2.4878.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Y., Zhang Y., Zhao G., Chen C., Yang P., Ye S. Difference in leukocyte composition between women before and after menopausal age, and distinct sexual dimorphism. PloS One. 2016;11(9) doi: 10.1371/journal.pone.0162953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pace S., Rossi A., Krauth V., Dehm F., Troisi F., Bilancia R. Sex differences in prostaglandin biosynthesis in neutrophils during acute inflammation. Sci. Rep. 2017;7(1):3759. doi: 10.1038/s41598-017-03696-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yanagisawa M., Kurihara H., Kimura S., Tomobe Y., Kobayashi M., Mitsui Y. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 88.Kohan D.E., Inscho E.W., Wesson D., Pollock D.M. Physiology of endothelin and the kidney. Comprehensive Physiology. 2011;1(2):883–919. doi: 10.1002/cphy.c100039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tonnessen T., Giaid A., Saleh D., Naess P.A., Yanagisawa M., Christensen G. Increased in vivo expression and production of endothelin-1 by porcine cardiomyocytes subjected to ischemia. Circ. Res. 1995;76(5):767–772. doi: 10.1161/01.res.76.5.767. [DOI] [PubMed] [Google Scholar]
- 90.Elisa T., Antonio P., Giuseppe P., Alessandro B., Giuseppe A., Federico C. Endothelin receptors expressed by immune cells are involved in modulation of inflammation and in fibrosis: relevance to the pathogenesis of systemic sclerosis. Journal of immunology research. 2015;2015:147616. doi: 10.1155/2015/147616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kohan D.E. Endothelin synthesis by rabbit renal tubule cells. Am. J. Physiol. 1991;261(2 Pt 2):F221–F226. doi: 10.1152/ajprenal.1991.261.2.F221. [DOI] [PubMed] [Google Scholar]
- 92.Simonson M.S., Ismail-Beigi F. Endothelin-1 increases collagen accumulation in renal mesangial cells by stimulating a chemokine and cytokine autocrine signaling loop. J. Biol. Chem. 2011;286(13):11003–11008. doi: 10.1074/jbc.M110.190793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sorokin A., Kohan D.E. Physiology and pathology of endothelin-1 in renal mesangium. Am. J. Physiol. Ren. Physiol. 2003;285(4):F579–F589. doi: 10.1152/ajprenal.00019.2003. [DOI] [PubMed] [Google Scholar]
- 94.Wendel M., Knels L., Kummer W., Koch T. Distribution of endothelin receptor subtypes ETA and ETB in the rat kidney. J. Histochem. Cytochem. 2006;54(11):1193–1203. doi: 10.1369/jhc.5A6888.2006. [DOI] [PubMed] [Google Scholar]
- 95.Schonthal A.H. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Sci. Tech. Rep. 2012;2012:857516. doi: 10.6064/2012/857516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Barton M. Therapeutic potential of endothelin receptor antagonists for chronic proteinuric renal disease in humans. Biochim. Biophys. Acta. 2010;1802(12):1203–1213. doi: 10.1016/j.bbadis.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 97.Nath K.A., Hebbel R.P. Sickle cell disease: renal manifestations and mechanisms. Nat. Rev. Nephrol. 2015;11(3):161–171. doi: 10.1038/nrneph.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hocher B., Thone-Reineke C., Rohmeiss P., Schmager F., Slowinski T., Burst V. Endothelin-1 transgenic mice develop glomerulosclerosis, interstitial fibrosis, and renal cysts but not hypertension. J. Clin. Invest. 1997;99(6):1380–1389. doi: 10.1172/JCI119297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ong A.C., von Websky K., Hocher B. Endothelin and tubulointerstitial renal disease. Semin. Nephrol. 2015;35(2):197–207. doi: 10.1016/j.semnephrol.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 100.Heunisch F., von Einem G., Alter M., Weist A., Dschietzig T., Kretschmer A. Urinary ET-1 excretion after exposure to radio-contrast media in diabetic patients and patients with preexisting mild impaired renal function. Life Sci. 2014;118(2):440–445. doi: 10.1016/j.lfs.2013.12.233. [DOI] [PubMed] [Google Scholar]
- 101.Montezano A.C., Burger D., Paravicini T.M., Chignalia A.Z., Yusuf H., Almasri M. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ. Res. 2010;106(8):1363–1373. doi: 10.1161/CIRCRESAHA.109.216036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kahler J., Ewert A., Weckmuller J., Stobbe S., Mittmann C., Koster R. Oxidative stress increases endothelin-1 synthesis in human coronary artery smooth muscle cells. J. Cardiovasc. Pharmacol. 2001;38(1):49–57. doi: 10.1097/00005344-200107000-00006. [DOI] [PubMed] [Google Scholar]
- 103.Kahler J., Mendel S., Weckmuller J., Orzechowski H.D., Mittmann C., Koster R. Oxidative stress increases synthesis of big endothelin-1 by activation of the endothelin-1 promoter. J. Mol. Cell. Cardiol. 2000;32(8):1429–1437. doi: 10.1006/jmcc.2000.1178. [DOI] [PubMed] [Google Scholar]
- 104.Wang X., Barber D.A., Lewis D.A., McGregor C.G., Sieck G.C., Fitzpatrick L.A. Gender and transcriptional regulation of NO synthase and ET-1 in porcine aortic endothelial cells. Am. J. Physiol. 1997;273(4):H1962–H1967. doi: 10.1152/ajpheart.1997.273.4.H1962. [DOI] [PubMed] [Google Scholar]
- 105.Jain A., Olovsson M., Burton G.J., Yung H.W. Endothelin-1 induces endoplasmic reticulum stress by activating the PLC-IP(3) pathway: implications for placental pathophysiology in preeclampsia. Am. J. Pathol. 2012;180(6):2309–2320. doi: 10.1016/j.ajpath.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 106.De Miguel C., Hamrick W.C., Hobbs J.L., Pollock D.M., Carmines P.K., Pollock J.S. Endothelin receptor-specific control of endoplasmic reticulum stress and apoptosis in the kidney. Sci. Rep. 2017;7:43152. doi: 10.1038/srep43152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jin C., Speed J.S., Hyndman K.A., O'Connor P.M., Pollock D.M. Sex differences in ET-1 receptor expression and Ca2+ signaling in the IMCD. Am. J. Physiol. Ren. Physiol. 2013;305(8):F1099–F1104. doi: 10.1152/ajprenal.00400.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nakano D., Pollock D.M. Contribution of endothelin A receptors in endothelin 1-dependent natriuresis in female rats. Hypertension. 2009;53(2):324–330. doi: 10.1161/HYPERTENSIONAHA.108.123687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Takaoka M., Yuba M., Fujii T., Ohkita M., Matsumura Y. Clinical science; London, England: 1979. Oestrogen Protects against Ischaemic Acute Renal Failure in Rats by Suppressing Renal Endothelin-1 Overproduction. 2002;103 Suppl 48:434S-7S. [DOI] [PubMed] [Google Scholar]
- 110.Zhang Y., Knutsen G.R., Brown M.D., Ruest L.B. Control of endothelin-a receptor expression by progesterone is enhanced by synergy with Gata2. Mol. Endocrinol. (Baltimore, Md) 2013;27(6):892–908. doi: 10.1210/me.2012-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gohar E.Y., Yusuf C., Pollock D.M. Ovarian hormones modulate endothelin A and B receptor expression. Life Sci. 2016;159:148–152. doi: 10.1016/j.lfs.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dousdampanis P., Trigka K., Fourtounas C., Bargman J.M. Role of testosterone in the pathogenesis, progression, prognosis and comorbidity of men with chronic kidney disease. Ther. Apher. Dial. : official peer-reviewed journal of the International Society for Apheresis, the Japanese Society for Apheresis, the Japanese Society for Dialysis Therapy. 2014;18(3):220–230. doi: 10.1111/1744-9987.12101. [DOI] [PubMed] [Google Scholar]
- 113.Kalk P., Thone-Reineke C., Schwarz A., Godes M., Bauer C., Pfab T. Renal phenotype of ET-1 transgenic mice is modulated by androgens. Eur. J. Med. Res. 2009;14:55–58. doi: 10.1186/2047-783X-14-2-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gameiro J., Agapito Fonseca J., Jorge S., Lopes J.A. Acute kidney injury definition and diagnosis: a narrative review. J. Clin. Med. 2018;7(10) doi: 10.3390/jcm7100307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tomsa A.M., Alexa A.L., Junie M.L., Rachisan A.L., Ciumarnean L. Oxidative stress as a potential target in acute kidney injury. PeerJ. 2019;7 doi: 10.7717/peerj.8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pan J.S., Sheikh-Hamad D. Mitochondrial dysfunction in acute kidney injury and sex-specific implications. Med Res Arch. 2019;7(2) doi: 10.18103/mra.v7i2.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dennis J.M., Witting P.K. Protective role for antioxidants in acute kidney disease. Nutrients. 2017;9(7) doi: 10.3390/nu9070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Martin J.L., Gruszczyk A.V., Beach T.E., Murphy M.P., Saeb-Parsy K. Mitochondrial mechanisms and therapeutics in ischaemia reperfusion injury. Pediatr. Nephrol. 2019;34(7):1167–1174. doi: 10.1007/s00467-018-3984-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Paller M.S., Hoidal J.R., Ferris T.F. Oxygen free radicals in ischemic acute renal failure in the rat. J. Clin. Invest. 1984;74(4):1156–1164. doi: 10.1172/JCI111524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chatterjee P.K. Novel pharmacological approaches to the treatment of renal ischemia-reperfusion injury: a comprehensive review. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2007;376(1–2):1–43. doi: 10.1007/s00210-007-0183-5. [DOI] [PubMed] [Google Scholar]
- 121.Muller V., Losonczy G., Heemann U., Vannay A., Fekete A., Reusz G. Sexual dimorphism in renal ischemia-reperfusion injury in rats: possible role of endothelin. Kidney Int. 2002;62(4):1364–1371. doi: 10.1111/j.1523-1755.2002.kid590.x. [DOI] [PubMed] [Google Scholar]
- 122.Tanaka R., Tsutsui H., Ohkita M., Takaoka M., Yukimura T., Matsumura Y. Sex differences in ischemia/reperfusion-induced acute kidney injury are dependent on the renal sympathetic nervous system. Eur. J. Pharmacol. 2013;714(1–3):397–404. doi: 10.1016/j.ejphar.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 123.Ikeda M., Swide T., Vayl A., Lahm T., Anderson S., Hutchens M.P. Estrogen administered after cardiac arrest and cardiopulmonary resuscitation ameliorates acute kidney injury in a sex- and age-specific manner. Crit. Care. 2015;19:332. doi: 10.1186/s13054-015-1049-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sweileh W.M. Gender differences in aminoglycoside induced nephrotoxicity: a prospective, hospital-based study. Curr. Clin. Pharmacol. 2009;4(3):229–232. doi: 10.2174/157488409789375339. [DOI] [PubMed] [Google Scholar]
- 125.Latcha S., Jaimes E.A., Patil S., Glezerman I.G., Mehta S., Flombaum C.D. Long-Term renal outcomes after cisplatin treatment. Clin. J. Am. Soc. Nephrol. 2016;11(7):1173–1179. doi: 10.2215/CJN.08070715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mehran R., Aymong E.D., Nikolsky E., Lasic Z., Iakovou I., Fahy M. A simple risk score for prediction of contrast-induced nephropathy after percutaneous coronary intervention: development and initial validation. J. Am. Coll. Cardiol. 2004;44(7):1393–1399. doi: 10.1016/j.jacc.2004.06.068. [DOI] [PubMed] [Google Scholar]
- 127.Hutchens M.P., Nakano T., Kosaka Y., Dunlap J., Zhang W., Herson P.S. Estrogen is renoprotective via a nonreceptor-dependent mechanism after cardiac arrest in vivo. Anesthesiology. 2010;112(2):395–405. doi: 10.1097/ALN.0b013e3181c98da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zawada I., Masternak M.M., List E.O., Stout M.B., Berryman D.E., Lewinski A. Gene expression of key regulators of mitochondrial biogenesis is sex dependent in mice with growth hormone receptor deletion in liver. Aging (Albany NY) 2015;7(3):195–204. doi: 10.18632/aging.100733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Centers for Disease Control and Prevention, Atlanta G.A. U.S. Dept of Health and Human Services; 2017. Centers for Disease Control and Prevention. [Google Scholar]
- 130.Tiwari B.K., Pandey K.B., Abidi A.B., Rizvi S.I. Markers of oxidative stress during diabetes mellitus. J Biomark. 2013;2013:378790. doi: 10.1155/2013/378790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Deshpande A.D., Harris-Hayes M., Schootman M. Epidemiology of diabetes and diabetes-related complications. Phys. Ther. 2008;88(11):1254–1264. doi: 10.2522/ptj.20080020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.International Diabetes Federation . ninth ed. 2019. IDF Diabetes Atlas. Brussels, Belgium. [Google Scholar]
- 133.Foley R.N., Collins A.J. The growing economic burden of diabetic kidney disease. Curr. Diabetes Rep. 2009;9(6):460–465. doi: 10.1007/s11892-009-0075-9. [DOI] [PubMed] [Google Scholar]
- 134.Lim A. Diabetic nephropathy - complications and treatment. Int. J. Nephrol. Renovascular Dis. 2014;7:361–381. doi: 10.2147/IJNRD.S40172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Abbate R., Mannucci E., Cioni G., Fatini C., Marcucci R. Diabetes and sex: from pathophysiology to personalized medicine. Intern Emerg Med. 2012;7(Suppl 3):S215–S219. doi: 10.1007/s11739-012-0804-y. [DOI] [PubMed] [Google Scholar]
- 136.Yu M.K., Lyles C.R., Bent-Shaw L.A., Young B.A., Pathways A. Risk factor, age and sex differences in chronic kidney disease prevalence in a diabetic cohort: the pathways study. Am. J. Nephrol. 2012;36(3):245–251. doi: 10.1159/000342210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Maric C. Sex, diabetes and the kidney. Am. J. Physiol. Ren. Physiol. 2009;296(4):F680–F688. doi: 10.1152/ajprenal.90505.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shepard B.D. Sex differences in diabetes and kidney disease: mechanisms and consequences. Am. J. Physiol. Ren. Physiol. 2019;317(2):F456–F462. doi: 10.1152/ajprenal.00249.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ahmed S.B., Ramesh S. Sex hormones in women with kidney disease. Nephrol. Dial. Transplant. 2016;31(11):1787–1795. doi: 10.1093/ndt/gfw084. [DOI] [PubMed] [Google Scholar]
- 140.Kitada M., Ogura Y., Koya D. Rodent models of diabetic nephropathy: their utility and limitations. Int. J. Nephrol. Renovascular Dis. 2016;9:279–290. doi: 10.2147/IJNRD.S103784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sembach F.E., Fink L.N., Johansen T., Boland B.B., Secher T., Thrane S.T. Impact of sex on diabetic nephropathy and the renal transcriptome in UNx db/db C57BLKS mice. Phys. Rep. 2019;7(24) doi: 10.14814/phy2.14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tian L., Nikolic-Paterson D.J., Tesch G.H. Establishing equivalent diabetes in male and female Nos3-deficient mice results in a comparable onset of diabetic kidney injury. Phys. Rep. 2019;7(18) doi: 10.14814/phy2.14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Newsholme P., Haber E.P., Hirabara S.M., Rebelato E.L., Procopio J., Morgan D. Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J. Physiol. 2007;583(Pt 1):9–24. doi: 10.1113/jphysiol.2007.135871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zheng H., Whitman S.A., Wu W., Wondrak G.T., Wong P.K., Fang D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes. 2011;60(11):3055–3066. doi: 10.2337/db11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ulas M., Cay M. 17β-Estradiol and vitamin E modulates oxidative stress-induced kidney toxicity in diabetic ovariectomized rat. Biol. Trace Elem. Res. 2011;144(1):821–831. doi: 10.1007/s12011-011-9025-x. [DOI] [PubMed] [Google Scholar]
- 146.Yiu W.H., Wong D.W., Wu H.J., Li R.X., Yam I., Chan L.Y. Kallistatin protects against diabetic nephropathy in db/db mice by suppressing AGE-RAGE-induced oxidative stress. Kidney Int. 2016;89(2):386–398. doi: 10.1038/ki.2015.331. [DOI] [PubMed] [Google Scholar]
- 147.Feng B., Yan X.F., Xue J.L., Xu L., Wang H. The protective effects of alpha-lipoic acid on kidneys in type 2 diabetic Goto-Kakisaki rats via reducing oxidative stress. Int. J. Mol. Sci. 2013;14(4):6746–6756. doi: 10.3390/ijms14046746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Scales C.D., Smith A.C., Hanley J.M., Saigal C.S., UDiA Project. Prevalence of kidney stones in the United States. Eur. Urol. 2012;62(1):160–165. doi: 10.1016/j.eururo.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Heller H.J., Sakhaee K., Moe O.W., Pak C.Y. Etiological role of estrogen status in renal stone formation. J. Urol. 2002;168(5):1923–1927. doi: 10.1016/S0022-5347(05)64264-4. [DOI] [PubMed] [Google Scholar]
- 150.Strope S.A., Wolf J.S., Jr., Hollenbeck B.K. Changes in gender distribution of urinary stone disease. Urology. 2010;75(3):543–546. doi: 10.1016/j.urology.2009.08.007. 6 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Scales C.D., Jr., Curtis L.H., Norris R.D., Springhart W.P., Sur R.L., Schulman K.A. Changing gender prevalence of stone disease. J. Urol. 2007;177(3):979–982. doi: 10.1016/j.juro.2006.10.069. [DOI] [PubMed] [Google Scholar]
- 152.Tundo G., Khaleel S., Pais V.M., Jr. Gender equivalence in the prevalence of nephrolithiasis among adults younger than 50 Years in the United States. J. Urol. 2018;200(6):1273–1277. doi: 10.1016/j.juro.2018.07.048. [DOI] [PubMed] [Google Scholar]
- 153.Prochaska M., Taylor E.N., Curhan G. Menopause and risk of kidney stones. J. Urol. 2018;200(4):823–828. doi: 10.1016/j.juro.2018.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lieske J.C., Rule A.D., Krambeck A.E., Williams J.C., Bergstralh E.J., Mehta R.A. Stone composition as a function of age and sex. Clin. J. Am. Soc. Nephrol. 2014;9(12):2141–2146. doi: 10.2215/CJN.05660614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fink H.A., Wilt T.J., Eidman K.E., Garimella P.S., MacDonald R., Rutks I.R. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann. Intern. Med. 2013;158(7):535–543. doi: 10.7326/0003-4819-158-7-201304020-00005. [DOI] [PubMed] [Google Scholar]
- 156.Goodman H.O., Brommage R., Assimos D.G., Holmes R.P. Genes in idiopathic calcium oxalate stone disease. World J. Urol. 1997;15(3):186–194. doi: 10.1007/BF02201856. [DOI] [PubMed] [Google Scholar]
- 157.Holmes R.P., Assimos D.G. The impact of dietary oxalate on kidney stone formation. Urol. Res. 2004;32(5):311–316. doi: 10.1007/s00240-004-0437-3. [DOI] [PubMed] [Google Scholar]
- 158.Mitchell T., Kumar P., Reddy T., Wood K.D., Knight J., Assimos D.G. Dietary oxalate and kidney stone formation. Am. J. Physiol. Ren. Physiol. 2019;316(3):F409–F413. doi: 10.1152/ajprenal.00373.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xu H., Zisman A.L., Coe F.L., Worcester E.M. Kidney stones: an update on current pharmacological management and future directions. Expet Opin. Pharmacother. 2013;14(4):435–447. doi: 10.1517/14656566.2013.775250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shoag J., Halpern J., Goldfarb D.S., Eisner B.H. Risk of chronic and end stage kidney disease in patients with nephrolithiasis. J. Urol. 2014;192(5):1440–1445. doi: 10.1016/j.juro.2014.05.117. [DOI] [PubMed] [Google Scholar]
- 161.Daudon M., Traxer O., Conort P., Lacour B., Jungers P. Type 2 diabetes increases the risk for uric acid stones. J. Am. Soc. Nephrol. 2006;17(7):2026–2033. doi: 10.1681/ASN.2006030262. [DOI] [PubMed] [Google Scholar]
- 162.Lange J.N., Mufarrij P.W., Wood K.D., Holmes R.P., Assimos D.G. The association of cardiovascular disease and metabolic syndrome with nephrolithiasis. Curr. Opin. Urol. 2012;22(2):154–159. doi: 10.1097/MOU.0b013e32834fc31f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gillen D.L., Coe F.L., Worcester E.M. Nephrolithiasis and increased blood pressure among females with high body mass index. Am. J. Kidney Dis. 2005;46(2):263–269. doi: 10.1053/j.ajkd.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 164.Khan S.R. Hyperoxaluria-induced oxidative stress and antioxidants for renal protection. Urol. Res. 2005;33(5):349–357. doi: 10.1007/s00240-005-0492-4. [DOI] [PubMed] [Google Scholar]
- 165.Tracy C.R., Henning J.R., Newton M.R., Aviram M., Bridget Zimmerman M. Oxidative stress and nephrolithiasis: a comparative pilot study evaluating the effect of pomegranate extract on stone risk factors and elevated oxidative stress levels of recurrent stone formers and controls. Urolithiasis. 2014;42(5):401–408. doi: 10.1007/s00240-014-0686-8. [DOI] [PubMed] [Google Scholar]
- 166.Boonla C., Hunapathed C., Bovornpadungkitti S., Poonpirome K., Tungsanga K., Sampatanukul P. Messenger RNA expression of monocyte chemoattractant protein-1 and interleukin-6 in stone-containing kidneys. BJU Int. 2008;101(9):1170–1177. doi: 10.1111/j.1464-410X.2008.07461.x. [DOI] [PubMed] [Google Scholar]
- 167.Joshi S., Wang W., Peck A.B., Khan S.R. Activation of the NLRP3 inflammasome in association with calcium oxalate crystal induced reactive oxygen species in kidneys. J. Urol. 2015;193(5):1684–1691. doi: 10.1016/j.juro.2014.11.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Okada A., Yasui T., Hamamoto S., Hirose M., Kubota Y., Itoh Y. Genome-wide analysis of genes related to kidney stone formation and elimination in the calcium oxalate nephrolithiasis model mouse: detection of stone-preventive factors and involvement of macrophage activity. J. Bone Miner. Res. 2009;24(5):908–924. doi: 10.1359/jbmr.081245. [DOI] [PubMed] [Google Scholar]
- 169.Shoag J., Eisner B.H. Relationship between C-reactive protein and kidney stone prevalence. J. Urol. 2014;191(2):372–375. doi: 10.1016/j.juro.2013.09.033. [DOI] [PubMed] [Google Scholar]
- 170.Rhee E., Santiago L., Park E., Lad P., Bellman G.C. Urinary IL-6 is elevated in patients with urolithiasis. J. Urol. 1998;160(6 Pt 1):2284–2288. doi: 10.1097/00005392-199812010-00101. [DOI] [PubMed] [Google Scholar]
- 171.Suen J.L., Liu C.C., Lin Y.S., Tsai Y.F., Juo S.H., Chou Y.H. Urinary chemokines/cytokines are elevated in patients with urolithiasis. Urol. Res. 2010;38(2):81–87. doi: 10.1007/s00240-010-0260-y. [DOI] [PubMed] [Google Scholar]
- 172.Mulay S.R., Kulkarni O.P., Rupanagudi K.V., Migliorini A., Darisipudi M.N., Vilaysane A. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1beta secretion. J. Clin. Invest. 2013;123(1):236–246. doi: 10.1172/JCI63679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Koul S., Khandrika L., Pshak T.J., Iguchi N., Pal M., Steffan J.J. Oxalate upregulates expression of IL-2Rbeta and activates IL-2R signaling in HK-2 cells, a line of human renal epithelial cells. Am. J. Physiol. Ren. Physiol. 2014;306(9):F1039–F1046. doi: 10.1152/ajprenal.00462.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Khan S.R. Crystal-induced inflammation of the kidneys: results from human studies, animal models, and tissue-culture studies. Clin. Exp. Nephrol. 2004;8(2):75–88. doi: 10.1007/s10157-004-0292-0. [DOI] [PubMed] [Google Scholar]
- 175.Lu X., Gao B., Wang Y., Liu Z., Yasui T., Liu P. Renal tubular epithelial cell injury, apoptosis and inflammation are involved in melamine-related kidney stone formation. Urol. Res. 2012;40(6):717–723. doi: 10.1007/s00240-012-0507-x. [DOI] [PubMed] [Google Scholar]
- 176.Khan A., Byer K., Khan S.R. Exposure of Madin-Darby canine kidney (MDCK) cells to oxalate and calcium oxalate crystals activates nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase. Urology. 2014;83(2):510 e1–7. doi: 10.1016/j.urology.2013.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Umekawa T., Chegini N., Khan S.R. Oxalate ions and calcium oxalate crystals stimulate MCP-1 expression by renal epithelial cells. Kidney Int. 2002;61(1):105–112. doi: 10.1046/j.1523-1755.2002.00106.x. [DOI] [PubMed] [Google Scholar]
- 178.Umekawa T., Tsuji H., Uemura H., Khan S.R. Superoxide from NADPH oxidase as second messenger for the expression of osteopontin and monocyte chemoattractant protein-1 in renal epithelial cells exposed to calcium oxalate crystals. BJU Int. 2009;104(1):115–120. doi: 10.1111/j.1464-410X.2009.08374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Huang M.Y., Chaturvedi L.S., Koul S., Koul H.K. Oxalate stimulates IL-6 production in HK-2 cells, a line of human renal proximal tubular epithelial cells. Kidney Int. 2005;68(2):497–503. doi: 10.1111/j.1523-1755.2005.00427.x. [DOI] [PubMed] [Google Scholar]
- 180.Singhto N., Sintiprungrat K., Sinchaikul S., Chen S.T., Thongboonkerd V. Proteome changes in human monocytes upon interaction with calcium oxalate monohydrate crystals. J. Proteome Res. 2010;9(8):3980–3988. doi: 10.1021/pr100174a. [DOI] [PubMed] [Google Scholar]
- 181.Patel M., Yarlagadda V., Adedoyin O., Saini V., Assimos D.G., Holmes R.P. Oxalate induces mitochondrial dysfunction and disrupts redox homeostasis in a human monocyte derived cell line. Redox Biol. 2018;15:207–215. doi: 10.1016/j.redox.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Khan S.R., Joshi S., Wang W., Peck A.B. Regulation of macromolecular modulators of urinary stone formation by reactive oxygen species: transcriptional study in an animal model of hyperoxaluria. Am. J. Physiol. Ren. Physiol. 2014;306(11):F1285–F1295. doi: 10.1152/ajprenal.00057.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Liang L., Li L., Tian J., Lee S.O., Dang Q., Huang C.K. Androgen receptor enhances kidney stone-CaOx crystal formation via modulation of oxalate biosynthesis & oxidative stress. Mol. Endocrinol. 2014;28(8):1291–1303. doi: 10.1210/me.2014-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Taguchi K., Okada A., Kitamura H., Yasui T., Naiki T., Hamamoto S. Colony-stimulating factor-1 signaling suppresses renal crystal formation. J. Am. Soc. Nephrol. 2014;25(8):1680–1697. doi: 10.1681/ASN.2013060675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Fan J., Chandhoke P.S., Grampsas S.A. Role of sex hormones in experimental calcium oxalate nephrolithiasis. J. Am. Soc. Nephrol. 1999;10(Suppl 14):S376–S380. [PubMed] [Google Scholar]
- 186.Markle J.G., Fish E.N. SeXX matters in immunity. Trends Immunol. 2014;35(3):97–104. doi: 10.1016/j.it.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 187.Andersen C.J., Murphy K.E., Fernandez M.L. Impact of obesity and metabolic syndrome on immunity. Adv Nutr. 2016;7(1):66–75. doi: 10.3945/an.115.010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Williams J., Holmes R.P., Assimos D.G., Mitchell T. Monocyte mitochondrial function in calcium oxalate stone formers. Urology. 2016;93:224 e1–6. doi: 10.1016/j.urology.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Dominguez-Gutierrez P.R., Kusmartsev S., Canales B.K., Khan S.R. Calcium oxalate differentiates human monocytes into inflammatory M1 macrophages. Front. Immunol. 2018;9:1863. doi: 10.3389/fimmu.2018.01863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Taguchi K., Hamamoto S., Okada A., Unno R., Kamisawa H., Naiki T. Genome-Wide gene expression profiling of randall's plaques in calcium oxalate stone formers. J. Am. Soc. Nephrol. 2017;28(1):333–347. doi: 10.1681/ASN.2015111271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rubattu S., Forte M., Raffa S. Circulating leukocytes and oxidative stress in cardiovascular diseases: a state of the art. Oxid Med Cell Longev. 2019;2019:2650429. doi: 10.1155/2019/2650429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pai A.V., Maddox T., Sandberg K.T. Cells and hypertension: solved and unsolved mysteries regarding the female rat. Physiology. 2018;33(4):254–260. doi: 10.1152/physiol.00011.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Choi H.Y., Park H.C., Ha S.K. vol. 13. Electrolyte Blood Press; 2015. pp. 7–16. (Salt Sensitivity and Hypertension: A Paradigm Shift from Kidney Malfunction to Vascular Endothelial Dysfunction). 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Veiras L.C., Girardi A.C.C., Curry J., Pei L., Ralph D.L., Tran A. Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J. Am. Soc. Nephrol. : JASN (J. Am. Soc. Nephrol.) 2017;28(12):3504–3517. doi: 10.1681/ASN.2017030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Stachenfeld N.S., Splenser A.E., Calzone W.L., Taylor M.P., Keefe D.L. Sex differences in osmotic regulation of AVP and renal sodium handling. J. Appl. Physiol. 1985;91(4):1893–1901. doi: 10.1152/jappl.2001.91.4.1893. 2001. [DOI] [PubMed] [Google Scholar]
- 196.Sartori-Valinotti J.C., Venegas-Pont M.R., Lamarca B.B., Romero D.G., Yanes L.L., Racusen L.C. Rosiglitazone reduces blood pressure in female Dahl salt-sensitive rats. Steroids. 2010;75(11):794–799. doi: 10.1016/j.steroids.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yanes L.L., Sartori-Valinotti J.C., Iliescu R., Romero D.G., Racusen L.C., Zhang H. Testosterone-dependent hypertension and upregulation of intrarenal angiotensinogen in Dahl salt-sensitive rats. Am. J. Physiol. Ren. Physiol. 2009;296(4):F771–F779. doi: 10.1152/ajprenal.90389.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kafami M., Hosseini M., Niazmand S., Farrokhi E., Hajzadeh M.A., Nazemi S. The effects of estradiol and testosterone on renal tissues oxidative after central injection of angiotensin II in female doca - salt treated rats. Horm. Mol. Biol. Clin. Invest. 2018;37(3) doi: 10.1515/hmbci-2018-0044. [DOI] [PubMed] [Google Scholar]
- 199.Taylor T.A., Gariepy C.E., Pollock D.M., Pollock J.S. Gender differences in ET and NOS systems in ETB receptor-deficient rats: effect of a high salt diet. Hypertension. 2003;41(3 Pt 2):657–662. doi: 10.1161/01.HYP.0000048193.85814.78. [DOI] [PubMed] [Google Scholar]
- 200.Gohar E.Y., Cook A.K., Pollock D.M., Inscho E.W. Afferent arteriole responsiveness to endothelin receptor activation: does sex matter? Biol. Sex Differ. 2019;10(1):1. doi: 10.1186/s13293-018-0218-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lefevre G., Berkane N., Uzan S., Etienne J. [Pre-eclampsia and oxygenated free radicals] Ann. Biol. Clin. (Paris) 1997;55(5):443–450. [PubMed] [Google Scholar]
- 202.Abad C., Proverbio T., Pinero S., Botana D., Chiarello D.I., Marin R. Preeclampsia, placenta, oxidative stress, and PMCA. Hypertens. Pregnancy. 2012;31(4):427–441. doi: 10.3109/10641955.2012.690058. [DOI] [PubMed] [Google Scholar]
- 203.Aouache R., Biquard L., Vaiman D., Miralles F. Oxidative stress in preeclampsia and placental diseases. Int. J. Mol. Sci. 2018;19(5) doi: 10.3390/ijms19051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wisdom S.J., Wilson R., McKillop J.H., Walker J.J. Antioxidant systems in normal pregnancy and in pregnancy-induced hypertension. Am. J. Obstet. Gynecol. 1991;165(6 Pt 1):1701–1704. doi: 10.1016/0002-9378(91)90018-m. [DOI] [PubMed] [Google Scholar]
- 205.Mikhail M.S., Anyaegbunam A., Garfinkel D., Palan P.R., Basu J., Romney S.L. Preeclampsia and antioxidant nutrients: decreased plasma levels of reduced ascorbic acid, alpha-tocopherol, and beta-carotene in women with preeclampsia. Am. J. Obstet. Gynecol. 1994;171(1):150–157. doi: 10.1016/0002-9378(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 206.Aydin S., Benian A., Madazli R., Uludag S., Uzun H., Kaya S. Plasma malondialdehyde, superoxide dismutase, sE-selectin, fibronectin, endothelin-1 and nitric oxide levels in women with preeclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004;113(1):21–25. doi: 10.1016/S0301-2115(03)00368-3. [DOI] [PubMed] [Google Scholar]
- 207.Pagani F., Cantaluppi V. Renal injury during preclampsia: role of extracellular vesicles. Nephron. 2019;143(3):197–201. doi: 10.1159/000502456. [DOI] [PubMed] [Google Scholar]
- 208.Fassio F., Attini R., Masturzo B., Montersino B., Chatrenet A., Saulnier P. Risk of preeclampsia and adverse pregnancy outcomes after heterologous egg donation: hypothesizing a role for kidney function and comorbidity. J. Clin. Med. 2019;8(11) doi: 10.3390/jcm8111806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Conti-Ramsden F.I., Nathan H.L., De Greeff A., Hall D.R., Seed P.T., Chappell L.C. Pregnancy-related acute kidney injury in preeclampsia: risk factors and renal outcomes. Hypertension. 2019;74(5):1144–1151. doi: 10.1161/HYPERTENSIONAHA.119.13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lu Y.P., Hasan A.A., Zeng S., Hocher B. Plasma ET-1 concentrations are elevated in pregnant women with hypertension -Meta-Analysis of clinical studies. Kidney Blood Press. Res. 2017;42(4):654–663. doi: 10.1159/000482004. [DOI] [PubMed] [Google Scholar]
- 211.Mastrogiannis D.S., O'Brien W.F., Krammer J., Benoit R. Potential role of endothelin-1 in normal and hypertensive pregnancies. Am. J. Obstet. Gynecol. 1991;165(6 Pt 1):1711–1716. doi: 10.1016/0002-9378(91)90020-r. [DOI] [PubMed] [Google Scholar]
- 212.LaMarca B., Parrish M., Ray L.F., Murphy S.R., Roberts L., Glover P. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin-1. Hypertension. 2009;54(4):905–909. doi: 10.1161/HYPERTENSIONAHA.109.137935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Murphy S.R., LaMarca B.B., Cockrell K., Granger J.P. Role of endothelin in mediating soluble fms-like tyrosine kinase 1-induced hypertension in pregnant rats. Hypertension. 2010;55(2):394–398. doi: 10.1161/HYPERTENSIONAHA.109.141473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Stanhewicz A.E., Jandu S., Santhanam L., Alexander L.M. Alterations in endothelin type B receptor contribute to microvascular dysfunction in women who have had preeclampsia. Clinical science (London, England. 1979;131(23):2777–2789. doi: 10.1042/CS20171292. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Saleh L., Verdonk K., Visser W., van den Meiracker A.H., Danser A.H. The emerging role of endothelin-1 in the pathogenesis of pre-eclampsia. Ther Adv Cardiovasc Dis. 2016;10(5):282–293. doi: 10.1177/1753944715624853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Jain A. Endothelin-1: a key pathological factor in pre-eclampsia? Reprod. Biomed. Online. 2012;25(5):443–449. doi: 10.1016/j.rbmo.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 217.Fiore G., Florio P., Micheli L., Nencini C., Rossi M., Cerretani D. Endothelin-1 triggers placental oxidative stress pathways: putative role in preeclampsia. J. Clin. Endocrinol. Metabol. 2005;90(7):4205–4210. doi: 10.1210/jc.2004-1632. [DOI] [PubMed] [Google Scholar]
- 218.Yung H.W., Calabrese S., Hynx D., Hemmings B.A., Cetin I., Charnock-Jones D.S. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am. J. Pathol. 2008;173(2):451–462. doi: 10.2353/ajpath.2008.071193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kim J.M., Kim T.H., Lee H.H., Lee S.H., Wang T. Postmenopausal hypertension and sodium sensitivity. Journal of menopausal medicine. 2014;20(1):1–6. doi: 10.6118/jmm.2014.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Signorelli S.S., Neri S., Sciacchitano S., Di Pino L., Costa M.P., Pennisi G. Duration of menopause and behavior of malondialdehyde, lipids, lipoproteins and carotid wall artery intima-media thickness. Maturitas. 2001;39(1):39–42. doi: 10.1016/s0378-5122(01)00174-8. [DOI] [PubMed] [Google Scholar]
- 221.Helmersson J., Mattsson P., Basu S. Prostaglandin F(2alpha) metabolite and F(2)-isoprostane excretion rates in migraine. Clinical science (London, England. 1979;102(1):39–43. 2002. [PubMed] [Google Scholar]
- 222.Fortepiani L.A., Zhang H., Racusen L., Roberts L.J., 2nd, Reckelhoff J.F. Characterization of an animal model of postmenopausal hypertension in spontaneously hypertensive rats. Hypertension. 2003;41(3 Pt 2):640–645. doi: 10.1161/01.HYP.0000046924.94886.EF. [DOI] [PubMed] [Google Scholar]
- 223.Wilcox J.G., Hatch I.E., Gentzschein E., Stanczyk F.Z., Lobo R.A. Endothelin levels decrease after oral and nonoral estrogen in postmenopausal women with increased cardiovascular risk factors. Fertil. Steril. 1997;67(2):273–277. doi: 10.1016/S0015-0282(97)81910-3. [DOI] [PubMed] [Google Scholar]
- 224.Komatsumoto S., Nara M. [Changes in the level of endothelin-1 with aging] Nihon Ronen Igakkai Zasshi. 1995;32(10):664–669. doi: 10.3143/geriatrics.32.664. [DOI] [PubMed] [Google Scholar]
- 225.Wenner M.M., Sebzda K.N., Kuczmarski A.V., Pohlig R.T., Edwards D.G. ETB receptor contribution to vascular dysfunction in postmenopausal women. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017;313(1):R51–R57. doi: 10.1152/ajpregu.00410.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lima R., Yanes L.L., Davis D.D., Reckelhoff J.F. Roles played by 20-HETE, angiotensin II and endothelin in mediating the hypertension in aging female spontaneously hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013;304(3):R248–R251. doi: 10.1152/ajpregu.00380.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gohar E.Y., Kasztan M., Becker B.K., Speed J.S., Pollock D.M. Ovariectomy uncovers purinergic receptor activation of endothelin-dependent natriuresis. Am. J. Physiol. Ren. Physiol. 2017;313(2):F361–F369. doi: 10.1152/ajprenal.00098.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Gohar E.Y., Speed J.S., Kasztan M., Jin C., Pollock D.M. Activation of purinergic receptors (P2) in the renal medulla promotes endothelin-dependent natriuresis in male rats. Am. J. Physiol. Ren. Physiol. 2016;311(2):F260–F267. doi: 10.1152/ajprenal.00090.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.National Institutes of Health NIH guidelines on the inclusion of women and minorities as subjects in clinical research. Fed. Regist. 1994;59:14508–14513. [Google Scholar]
- 230.Scott P.E., Unger E.F., Jenkins M.R., Southworth M.R., McDowell T.Y., Geller R.J. Participation of women in clinical trials supporting FDA approval of cardiovascular drugs. J. Am. Coll. Cardiol. 2018;71(18):1960–1969. doi: 10.1016/j.jacc.2018.02.070. [DOI] [PubMed] [Google Scholar]
- 231.Geller S.E., Koch A., Pellettieri B., Carnes M. Inclusion, analysis, and reporting of sex and race/ethnicity in clinical trials: have we made progress? J Womens Health (Larchmt) 2011;20(3):315–320. doi: 10.1089/jwh.2010.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Bairey Merz C.N., Dember L.M., Ingelfinger J.R., Vinson A., Neugarten J., Sandberg K.L. Sex and the kidneys: current understanding and research opportunities. Nat. Rev. Nephrol. 2019;15(12):776–783. doi: 10.1038/s41581-019-0208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.