

Abstract

The study of G-quadruplexes (G4s) in a cellular context has demonstrated links between these nucleic acid secondary structures, gene expression, and DNA replication. Ligands that bind to the G4 structure therefore present an excellent opportunity for influencing gene expression through the targeting of a nucleic acid structure rather than sequence. Here, we explore cyclic peptides as an alternative class of G4 ligands. Specifically, we describe the development of de novo G4-binding bicyclic peptides selected by phage display. Selected bicyclic peptides display submicromolar affinity to G4 structures and high selectivity over double helix DNA. Molecular simulations of the bicyclic peptide–G4 complexes corroborate the experimental binding strengths and reveal molecular insights into G4 recognition by bicyclic peptides via the precise positioning of amino acid side chains, a binding mechanism reminiscent of endogenous G4-binding proteins. Overall, our results demonstrate that selection of (bi)cyclic peptides unlocks a valuable chemical space for targeting nucleic acid structures.
Introduction
G-quadruplexes (G4s) are noncanonical nucleic acid secondary structures that consist of stacks of Hoogsteen-bonded guanine tetrads. The resulting quadruple helix is stabilized by cations (K+ > Na+ > Li+ in order of stability), and the structural characteristics of G4s have been extensively characterized in vitro(1) and in silico.(2,3) Numerous studies have shown that G4s are important for replication, transcription, and translation events in living cells.4 Consequently, targeting G4s with synthetic molecules represents an opportunity for influencing gene expression, with a view toward further elucidation of biological mechanisms and potential novel methods of therapeutic intervention.5
Nucleic acids are generally targeted with specificity through sequence recognition (e.g., RNAi/CRISPR or engineered transcription factors). In contrast, the globular structure of the G4 enables specific binding through recognition of its three-dimensional structure. For example, a number of small molecule ligands utilize a combination of flat, heteroaromatic moieties that are thought to stack on G4 tetrads via π–π interactions with pendant positive charges for electrostatic interaction with the phosphate backbone.6 However, endogenous G4-binding proteins appear to achieve G4 recognition in a more complex fashion and utilize a greater number of different interactions. In particular, the crystal structure of the G4 helicase DHX36 revealed that its picomolar affinity to G4s derives in part from hydrophobic interactions between the tetrad and amino acid residues (e.g., Ile65, Tyr69, and Ala70) positioned by the enzyme’s tertiary structure.7
We reasoned that such a prestructured binding modality, inspired by the potent G4 recognition properties of DHX36, can also be achieved by cyclic peptides. Cyclic peptides mimic preorganized configurations and modes-of-action of protein epitopes,8,9 allowing them to target well-defined binding pockets and disrupt protein–protein interactions. Additionally, cyclic peptides are genetically encodable and lend themselves to selection strategies that allow the identification of high-affinity binders from vast libraries.10 As a result, an increasing number of compounds from de novo cyclic peptides are entering or undergoing clincial trials.11
Here we report the use of phage display for the selection of bicyclic peptides as ligands displaying high affinity and selectivity for DNA G4s.12 Using energy landscape exploration and molecular dynamics, we find that these G4-binding peptides derive their affinities from precise positioning of amino acid residues, in a mode that is akin to endogenous G4-binding proteins.
Results
Establishing Phage Display of Bicyclic Peptide G-Quadruplex Ligands
To exploit genetically encoded cyclic peptides for targeting nucleic acids, it is necessary to ensure that the genetic barcode of each peptide does not interact with the nucleic acid target. Phage display is an ideal solution, as the phage coat provides a physical barrier between the peptide and its encoding gene. We opted for the phage display of bicyclic peptides established by Heinis and co-workers, as the double-ring structure results in more rigid structures and confers higher proteolytic stability than monocyclic peptides.9,12 In brief, bicyclic peptide libraries are accessed through in situ chemical modification of linear, cysteine-rich peptides displayed on phage coats with 1,3,5-tris(bromomethyl)benzene (TBMB) via efficient trinucleophilic substitution (Figure 1). These libraries are screened based on their affinity to biotinylated G4-forming oligonucleotides and captured with streptavidin magnetic beads. Weak binders are removed by washing, and enriched bicyclic peptides are eluted. Reinfection of E. coli amplifies selected phages and permits identification by sequencing or subsequent rounds of selection.
Figure 1.

Phage display of bicyclic
peptides12 for affinity selection of G4
ligands. Bacteriophages express libraries of peptides with cysteines
at defined positions on their coat proteins (A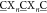 G, X = any of the 20 canonical amino acids, n = 3 or 4 in this study). These Cys-rich libraries are
bicyclized quantitatively by 1,3,5-tris(bromomethyl)benzene in situ. G4 ligands are selected from this bicyclic peptide
library (around 108 – 109 members) by
presenting biotinylated G4 targets for capture.
G, X = any of the 20 canonical amino acids, n = 3 or 4 in this study). These Cys-rich libraries are
bicyclized quantitatively by 1,3,5-tris(bromomethyl)benzene in situ. G4 ligands are selected from this bicyclic peptide
library (around 108 – 109 members) by
presenting biotinylated G4 targets for capture.
Initially we chose the G4-forming sequence from the human telomere
(hTelo) as a G4 of significant biological interest.13 This biotinylated sequence was incubated with phages displaying
a 4 × 4 bicyclic peptide library (A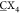
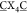 G, X = any of the 20 canonical amino acids). After
three successive rounds of selection we observed a substantial increase
in the phage titer alongside a significant reduction in the library
complexity (Figure S1), both indicative
of a successful selection. By next generation sequencing, we identified
b-ACGS CPISVCG (b-G4pep1, Figure 2a) as the most enriched sequence. We confirmed synthetic
b-G4pep1 binds a panel of common DNA G4 structures (Figure S4) by FRET melting14 and
fluorescence quenching assays,15 while
the linear precursor exhibited negligible binding (Figure S2). While these results demonstrate that G4-binding
bicyclic peptides can be selected from diverse libraries by phage
display, b-G4pep1 only displayed a ΔTm = 6 K at 5 μM and a modest binding affinity for the ckit-1
G4 DNA with Kd = 13 μM. However, b-G4pep1 does not
feature any cationic or aromatic residues, suggesting a distinct mode
of G4 recognition from the majority of small molecule G4 ligands.
G, X = any of the 20 canonical amino acids). After
three successive rounds of selection we observed a substantial increase
in the phage titer alongside a significant reduction in the library
complexity (Figure S1), both indicative
of a successful selection. By next generation sequencing, we identified
b-ACGS CPISVCG (b-G4pep1, Figure 2a) as the most enriched sequence. We confirmed synthetic
b-G4pep1 binds a panel of common DNA G4 structures (Figure S4) by FRET melting14 and
fluorescence quenching assays,15 while
the linear precursor exhibited negligible binding (Figure S2). While these results demonstrate that G4-binding
bicyclic peptides can be selected from diverse libraries by phage
display, b-G4pep1 only displayed a ΔTm = 6 K at 5 μM and a modest binding affinity for the ckit-1
G4 DNA with Kd = 13 μM. However, b-G4pep1 does not
feature any cationic or aromatic residues, suggesting a distinct mode
of G4 recognition from the majority of small molecule G4 ligands.
Figure 2.

(a) Schematic structures of three G4 bicyclic peptide ligands elicited by phage display, exhibiting both common G4 small molecule binding motifs and hydrophobic amino acids. (b) Next-generation sequencing permits motif analysis of the selection process; the optimal amino acids at each position in the final selected library are identified. Enriched amino acid functionalities include positive charges (position 8) and aromatic rings (9) but also hydrophobic residues (5) and proline (3). (c) ckit-1 G4 melting temperature increase (ΔTm) as measured by FRET melting. ΔTm demonstrates progressive improvement of G4 bicyclic peptides from b-G4pep1 to b-G4pep3. At the same time, no significant ΔTm is observed for a short double-stranded DNA. (d) Apparent Kd values measured by fluorescence quench equilibrium binding assay for b-G4pep1 and b-G4pep3; the quoted Kd is for ckit-1.
Modulating Phage Enrichment to Elicit More Potent Ligands
To enhance the selection of high-affinity G4-binders we made a number of improvements to our initial selection. First, given the known structural heterogeneity of the folded hTelo G4,16 we next chose the G4-forming sequence from the KIT promoter (ckit-1), known to form a unique, well-defined structure in which a loop folds back into the helix to form part of the middle tetrad.17 Since we observed deletions in the first loop in the previous experiment, we reduced the bicyclic peptide library ring size from 4 × 4 to 3 × 3 (ACX3CX3CG). Finally, we applied more stringent selection conditions by adding genomic DNA (salmon sperm DNA, 10 to 100-fold excess) as a competitor. The most enriched bicyclic peptide from this protocol was b-ACPPICIKFCG (b-G4pep2, Figure 2a), which exhibited stronger G4-binding properties (ΔTm = 20 K at 5 μM, Kd [ckit-1] = 1.0 μM, Figure 2c/d) compared to b-G4pep1. Furthermore, we derived the optimal amino acids at each position by scaling each peptide by its frequency in the library (Figure 2b). The resulting bicyclic peptide ACPRLCRRFCG (b-G4pep3, Figure 2a) exhibits further enhanced G4-binding properties (ΔTm = 34 K at 5 μM, Kd [ckit −1] = 630 nM, Figure 2c/d).
Enriched amino acid functionalities in these bicyclic peptides include positive charges and aromatic rings that often constitute small molecule G4 ligands, but bicyclic peptides additionally feature hydrophobic residues and proline. High selectivity for G4 structures over double helix DNA is maintained; both b-G4pep2 and b-G4pep3 do not exhibit any significant ΔTm for dsDNA up to 50 μM of ligand, and there is negligible effect on ΔTm for ckit-1 when a short double helix DNA is added at 100-fold molar excess (Figure S5). Again, all corresponding linear precursor peptides exhibit significantly reduced G4-binding properties (Figure S2), suggestive that the rigidity and structure conferred through bicyclization plays an important role in the G4-binding properties of these bicyclic peptides.
Computational Modeling of Bicyclic Peptide-G4 Complexes
To gain further insight into the recognition of G4s by bicyclic peptides, we studied G4-bicyclic peptide complexes computationally. First, we explored the energy landscapes for the bicyclic peptides alone using discrete path sampling (DPS).18,19 As expected, bicyclic peptides are structurally rigid and exhibit a relatively low number of local minima, with structural variations largely confined to side chain conformations. This rigidity is also observed in the calculated radii of gyration and solvent-accessible surface areas (S17 and S18). These observations are expected for a peptide bicyclized by TBMB and agree with previous structural studies of bicyclic peptides.9,12
A number of structural features are worth noting. In particular, the compact structure of b-G4pep1 (Figure 3a) arises from its high propensity to form α-helices, a feature shared with a number of other bicyclic peptides modeled in this study (SI Table S2). The prolines in all these bicyclic peptides permit sharp-turn motifs, while b-G4pep3 (Figure 3b/c) notably also utilizes its N-terminal arginine (Arg4) residue to facilitate hydrogen bonding with the backbone on the opposite side of the bicyclic peptide. This preorganization allows the remaining positive charges to be distributed in a belt-like configuration around the mesitylene core.
Figure 3.

Lowest energy structures for b-G4pep1 and b-G4pep3 (two perspectives); mesitylene core—orange, N-terminus—blue. (a) b-G4pep1 forms an α-helix (green) in the second loop between Cys5 and Cys10, facilitated by Pro6 reducing overall strain. (b) b-G4pep3 possesses a compact, elongated structure with a belt of positive charges around the center (green). (c) b-G4pep3 structure is stabilized by interactions between Arg4 and the backbone of the second loop.
We then analyzed binding of the optimized bicyclic peptide structures to the solution structure of ckit-1 solved by NMR spectroscopy.17 Basin-hopping global optimization20−22 was used to explore the ability of the three selected peptides to interact with three potential binding sites in the ckit-1 G4: (1) the exposed 5′-tetrad, (2) the 3′-tetrad flanked by the AGGAG loop that folds back onto the central tetrad, and (3) the AGGAG loop without the tetrad (Figure 4). Next, molecular dynamics simulations were applied to calculate a measure of the interaction energy in the presence of explicit solvent and ions. This energy, defined as the sum of all nonbonded interactions between two molecules, is a proxy for the binding free energy and enables us to compare approximate relative binding strengths of different peptides rapidly. These simulations revealed that bicyclic peptides b-G4pep1-3 can productively interact with the 3′-tetrad and the pocket created by the AGGAG loop and that complex formation does not entail significant changes in either the G4 or the peptide structure. Furthermore, the calculated interaction energies for all complexes reflect the trends in biophysical data (Figure 5).
Figure 4.

Schematic representation and structure of ckit-1 showing the binding sites used for docking; G** represents the incomplete tetrad corners filled by Gs at the 3′ end. Red site (1): the 5′-tetrad is openly accessible with potential interaction partners corresponding to C11 and T12. The final AGGAG loop (nucleotides 16 to 20) in ckit-1 folds back to complete the middle and 3′ tetrad, presenting binding sites with the 3′-tetrad (blue site, 2) and without the tetrad (green site, 3).
Figure 5.

Calculated interaction energies in the molecular dynamics simulations for b-G4pep1 to b-G4pep3 with ckit-1 in explicit solvent, alongside experimental Kds. The plots show the evolution of all trajectories (in color) and the moving average value (black). The interaction for b-G4pep3 with the binding site between the 3′-tetrad and the AGGAG loop is the strongest.
G4-bpep3 achieves G4 binding in two ways. The linear interaction energy between b-G4pep3 is dominated by the Coulombic term and derives from the belt-like architecture of positive charges interacting with the AGGAG loop. In addition, leucine and the N-terminus stack against the 3′ -tetrad providing additional hydrophobic and Coulombic interactions (Figure 6a/b). The structures of ckit-1 and b-G4pep3 are not significantly perturbed upon complexation, and the preorganization of b-G4pep3 by Arg4 is maintained. b-G4pep1 in contrast lacks positive charges, with the exception of its N-terminal amine, and hydrogen bonds between serine residues and the phosphate backbone, rather than the corresponding Coulombic interactions with arginines in the case of b-G4pep3, can explain its lower binding strength (Figure 6c/d). The intermediate-strength binder b-G4pep2 displays fewer Coulombic interactions (one lysine rather than two arginines) but is additionally strengthened by a π-stacking interaction between the tetrad and phenylalanine (Figure S3). Overall, our calculations demonstrate that G4 recognition by these bicyclic peptides relies on preorganization of the overall peptide structure and the precise positioning of amino acid side-chains to facilitate key binding interactions. A similar binding mechanism is likely utilized by a cyclized peptide derived from RHAU/DHX3623 and in cyclic peptide ligands of the mi-21R miRNA.24
Figure 6.

(a) Low-energy structure of the b-G4pep3:ckit-1 complex, highlighting the arginine belt in orange. (b) Detail of complex, showing interactions between Arg7/8 belt (green) and the phosphate groups of G18/A19 in the AGGAG loop (red), with stacking on 3′-tetrad (yellow) through Leu5 and the N-terminus (behind Leu5). Arg4 preorganization is maintained. (c) Low-energy structure of b-G4pep1:ckit-1 complex, highlighting Ser4/7 in green. (d) Detail of b-G4pep1 showing weaker interactions of Ser4/7 and Ile7 with the loops.
Conclusions
We have demonstrated the generation of (bi)cyclic peptide G4 ligands via phage display to yield a water-soluble ligand with respectable G4-targeting properties (b-G4pep3: ΔTm = 34 K at 5 μM, Kd [ckit-1] = 630 nM, ΔTm ∼ 0 for dsDNA up to 50 μM). Molecular modeling reveals that b-G4pep3 shows a high degree of protein-like structural organization, and this arrangement of amino acids facilitates molecular recognition of ckit-1 via tetrad stacking and Coulombic interactions with the AGGAG loop. Overall, this study exemplifies the potential of de novo cyclic peptides as agents for targeting nucleic acid secondary structures.
Methods
Phage Display of Bicyclic Peptides and G4 Ligand Selection
All the methods are described in full in the Supporting Information. In brief, production of bicyclic peptide-bearing phage largely follows protocols described by Rentero-Rebollo and Heinis.25 Starting and evolved peptide libraries were sequenced using next generation sequencing (Illumina MiSeq). G4-bicyclic peptide interactions were biophysically characterized using FRET melting14 and fluorescence-quench equilibrium binding.15
Computational Methods
We employed the computational potential energy landscape framework26 and molecular dynamics simulations. For each bicyclic peptide, we explored the energy landscapes and extracted a detailed picture of the structural variation within the low energy conformational ensemble. We then employed the lowest energy structures selectively docked to ckit-1 (PDB id: 2O3M)17 to generate structures for bound states. Basin-hopping global optimization20−22 and molecular dynamics simulations were then used to provide a better understanding of the structural basis for the binding in these favored structures. Details of the simulation methods and protocols are provided in the Supporting Information, and more information can be found in various reviews.26,27
Acknowledgments
The authors thank C. Heinis (EPFL Lausanne) for providing the phage display vectors, by kind permission of F.X. Schmid. KCL was supported by Jesus College, Cambridge (Embiricos Scholarship) and the Herchel Smith studentship. The Balasubramanian group receives programme funding (C9681/A18618) and core funding (C14303/A17197) from Cancer Research UK, and an Investigator Award from the Wellcome Trust (099232/Z/12/Z). KR and DJW received funding from the EPSRC (EP/N035003/1), and KR also received funding from the Cambridge Philosophical Society. Finally, we thank Chris Lowe for comments on the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c01879.
Further G4-bicyclic peptide biophysics, selection trends, characterization of synthesized bicyclic peptides, and experimental methodology. Description of computational tools employed; MD protocols; structural analysis of the bicyclic peptide simulations, analysis for the binding simulations, and structural representations for predicted low energy structures. (PDF)
The authors declare the following competing financial interest(s): SB is a founder, advisor and shareholder of Cambridge Epigenetix Ltd.
Supplementary Material
References
- Bochman M. L.; Paeschke K.; Zakian V. A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- S̆poner J.; Bussi G.; Stadlbauer P.; Kührová P.; Banás̆ P.; Islam B.; Haider S.; Neidle S.; Otyepka M. Folding of guanine quadruplex molecules–funnel-like mechanism or kinetic partitioning? An overview from MD simulation studies. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 1246–1263. 10.1016/j.bbagen.2016.12.008. [DOI] [PubMed] [Google Scholar]
- Salsbury A.; L J. Molecular dynamics simulations of the c-kit1 promoter G-quadruplex: Importance of electronic polarization on stability and cooperative ion binding. J. Phys. Chem. B 2019, 123, 148–159. 10.1021/acs.jpcb.8b11026. [DOI] [PubMed] [Google Scholar]
- Hänsel-Hertsch R.; Di Antonio M.; Balasubramanian S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. 10.1038/nrm.2017.3. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S.; Hurley L. H.; Neidle S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy?. Nat. Rev. Drug Discovery 2011, 10, 261–275. 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel J.; Adhikari S.; Balasubramanian S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. 10.1016/j.trechm.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. C.; Tippana R.; Demeshkina N. A.; Murat P.; Balasubramanian S.; Myong S.; Ferré-D’Amaré A. R. Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature 2018, 558, 465–469. 10.1038/s41586-018-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov A. A.; Yin Y.; Suga H. Macrocyclic peptides as drug candidates: Recent progress and remaining challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. 10.1021/jacs.8b13178. [DOI] [PubMed] [Google Scholar]
- Angelini A.; Cendron L.; Chen S.; Touati J.; Winter G.; Zanotti G.; Heinis C. Bicyclic peptide inhibitor reveals large contact interface with a protease target. ACS Chem. Biol. 2012, 7, 817–821. 10.1021/cb200478t. [DOI] [PubMed] [Google Scholar]
- Davis A. M.; Plowright A. T.; Valeur E. Directing evolution: The next revolution in drug discovery?. Nat. Rev. Drug Discovery 2017, 16, 681–698. 10.1038/nrd.2017.146. [DOI] [PubMed] [Google Scholar]
- Zorzi A.; Deyle K.; Heinis C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. 10.1016/j.cbpa.2017.02.006. [DOI] [PubMed] [Google Scholar]
- Heinis C.; Rutherford T.; Freund S.; Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- Rhodes D.; Lipps H. J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. 10.1093/nar/gkv862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cian A.; Guittat L.; Kaiser M.; Saccà B.; Amrane S.; Bourdoncle A.; Alberti P.; Teulade-Fichou M.-P.; Lacroix L.; Mergny J.-L. Fluorescence-based melting assays for studying quadruplex ligands. Methods 2007, 42, 183–195. 10.1016/j.ymeth.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Le D. D.; Antonio M. D.; Chan L. K. M.; Balasubramanian S. G-quadruplex ligands exhibit differential G-tetrad selectivity. Chem. Commun. 2015, 51, 8048–8050. 10.1039/C5CC02252E. [DOI] [PubMed] [Google Scholar]
- Lee J. Y.; Okumus B.; Kim D. S.; Ha T. Extreme conformational diversity in human telomeric DNA. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 18938–18943. 10.1073/pnas.0506144102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan A. T.; Kuryavyi V.; Burge S.; Neidle S.; Patel D. J. Structure of an unprecedented G-quadruplex scaffold in the human c-kit promoter. J. Am. Chem. Soc. 2007, 129, 4386–4392. 10.1021/ja068739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales D. J. Discrete path sampling. Mol. Phys. 2002, 100, 3285–3305. 10.1080/00268970210162691. [DOI] [Google Scholar]
- Wales D. J. Some further applications of discrete path sampling to cluster isomerization. Mol. Phys. 2004, 102, 891–908. 10.1080/00268970410001703363. [DOI] [Google Scholar]
- Li Z.; Scheraga H. A. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 6611–6615. 10.1073/pnas.84.19.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Scheraga H. A. Structure and free-energy of complex thermodynamic systems. J. Mol. Struct.: THEOCHEM 1988, 48, 333–352. 10.1016/0166-1280(88)80133-7. [DOI] [Google Scholar]
- Wales D. J.; Doye J. P. K. Global optimization by basin-hopping and the lowest energy structures of Lennard-Jones clusters containing up to 110 atoms. J. Phys. Chem. A 1997, 101, 5111–5116. 10.1021/jp970984n. [DOI] [Google Scholar]
- Ngo K. H.; Yang R.; Das P.; Nguyen G. K. T.; Lim K. W.; Tam J. P.; Wu B.; Phan A. T. Cyclization of a G4-specific peptide enhances its stability and G-quadruplex binding affinity. Chem. Commun. 2020, 56, 1082–1084. 10.1039/C9CC06748E. [DOI] [PubMed] [Google Scholar]
- Shortridge M. D.; Walker M. J.; Pavelitz T.; Chen Y.; Yang W.; Varani G. A macrocyclic peptide ligand binds the oncogenic microRNA-21 precursor and suppresses Dicer processing. ACS Chem. Biol. 2017, 12, 1611–1620. 10.1021/acschembio.7b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentero Rebollo I.; Heinis C. Phage selection of bicyclic peptides. Methods 2013, 60, 46–54. 10.1016/j.ymeth.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Joseph J. A.; Röder K.; Chakraborty D.; Mantell R. G.; Wales D. J. Exploring biomolecular energy landscapes. Chem. Commun. 2017, 53, 6974–6988. 10.1039/C7CC02413D. [DOI] [PubMed] [Google Scholar]
- Röder K.; Joseph J. A.; Husic B. E.; Wales D. J. Energy landscapes for proteins: From single funnels to multifunctional systems. Adv. Theory Simul. 2019, 2, 1800175. 10.1002/adts.201800175. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.