

Abstract
Rationale
Significant progress has revealed transcriptional inputs that underlie regulation of artery and vein endothelial cell fates. However, little is known concerning genome-wide regulation of this process. Therefore, such studies are warranted to address this gap.
Objective
To identify and characterize artery- and vein-specific endothelial enhancers in the human genome, thereby gaining insights into mechanisms by which blood vessel identity is regulated.
Methods and Results
Using ChIP-seq for markers of active chromatin in human arterial and venous endothelial cells, we identified several thousand artery- and vein-specific regulatory elements. Computational analysis revealed that NR2F2 sites were over-represented in vein-specific enhancers, suggesting a direct role in promoting vein identity. Subsequent integration of ChIP-seq datasets with RNA-seq revealed that NR2F2 regulated three distinct aspects related to arteriovenous identity. First, consistent with previous genetic observations, NR2F2 directly activated enhancer elements flanking cell cycle genes to drive their expression. Second, NR2F2 was essential to directly activate vein-specific enhancers and their associated genes. Our genomic approach further revealed that NR2F2 acts with ERG at many of these sites to drive vein-specific gene expression. Finally, NR2F2 directly repressed only a small number of artery enhancers in venous cells to prevent their activation, including a distal element upstream of the artery-specific transcription factor, HEY2. In arterial endothelial cells, this enhancer was normally bound by ERG, which was also required for arterial HEY2 expression. By contrast, in venous endothelial cells NR2F2 was bound to this site, together with ERG, and prevented its activation.
Conclusions
By leveraging a genome-wide approach, we revealed mechanistic insights into how NR2F2 functions in multiple roles to maintain venous identity. Importantly, characterization of its role at a crucial artery enhancer upstream of HEY2 established a novel mechanism by which artery-specific expression can be achieved.
Keywords: Artery, vein, enhancer, differentiation, endothelial, vascular biology, gene expression/regulation, genomics
Subject Terms: Cellular Reprograming, Cell Signaling/Signal Transduction, Epigenetics, Gene Expression and Regulation, Vascular Biology
Graphical Abstract
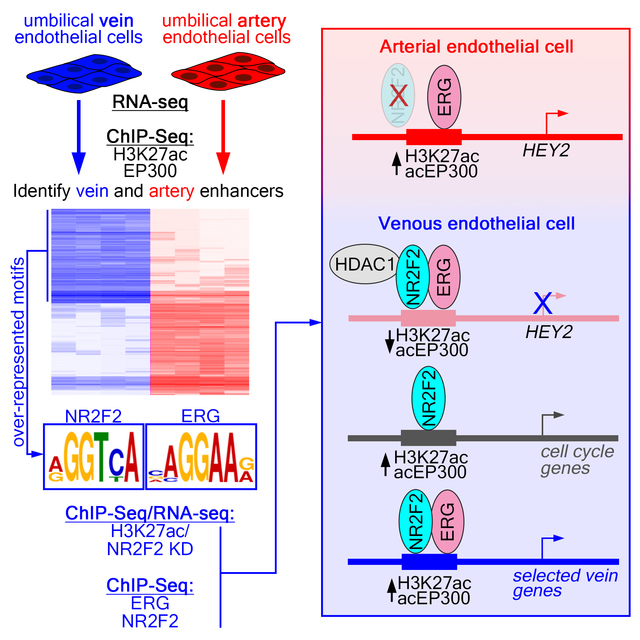
INTRODUCTION
Specification of arterial and venous (arteriovenous) endothelial cell fates is an essential step during the development of the circulatory system 1. While differences between these blood vessel types were long thought to be the result of physiological determinants, work over the past twenty years has shown that arteriovenous identity is established through genetic pathways acting prior to circulation. Additionally, defects in these pathways lead to abnormal vascular morphogenesis and patterning2, 3. Developmental studies in multiple model organisms demonstrate that arteriovenous identities are specified prior to blood vessel formation and are largely fixed by adulthood4–6, suggesting that re-programming these fates may be challenging in an established circulatory system. Indeed, limited arteriovenous plasticity has been noted in pathological settings in humans7. Notably, coronary artery bypass grafts using saphenous veins are more likely to exhibit stenosis and graft failure than those using radial arteries 8. Similarly, venous grafts subjected to arterial flow in animal models show loss of vein identity, but fail to induce artery gene expression9. Thus, a better understanding of the signals that determine artery and vein identity may allow manipulation of these fates in the adult circulatory system and lead to improved clinical outcomes.
A feature of cellular differentiation is lineage-specific gene expression governed by cell type-specific transcription factors. Accordingly, a number of transcription factors have been implicated in arteriovenous specification. Two related Forkhead transcription factors, Foxc1 and Foxc2, are expressed in endothelial cells and their combined deficiency leads to defects in vascular development in mouse10. Foxc proteins can interact directly with cis regulatory elements at the artery-specific Dll4 and Hey2 genes and drive their expression10. Similarly, the Ets transcription factor ERG binds to an artery-specific intronic enhancer of Dll4 to drive its expression11. Accordingly, mice lacking Erg show reduced artery Dll4 expression. Interestingly, a broad range of endothelial enhancers bear composite Forkhead:Ets motifs, which are capable of driving robust endothelial reporter expression12, although most of these are not artery-restricted. Sox family transcription factors have also been implicated in artery differentiation. Sox17 is expressed specifically in arterial endothelial cells in mouse13, while Sox7 and Sox18 exhibit similar patterns in zebrafish14. Sox17 can bind upstream of multiple artery-expressed genes and regulate their expression13. Thus, members of the Ets, Sox, and Forkhead transcription factor families have all been implicated in artery differentiation to directly promote expression of artery-specific genes. However, a genome-wide view of putative artery enhancers and genes acted upon by these upstream transcription factors in arterial endothelial cells is lacking.
While many factors have been implicated in artery differentiation, less is known about vein specification. The orphan nuclear receptor, Nr2f2 (also known as Coup-TFII), is expressed specifically in venous endothelial cells and is required for vascular morphogenesis during embryonic development 15. Mice lacking Nr2f2 show ectopic expression of artery markers in venous endothelial cells, while exogenous transgene-driven Nr2f2 suppresses artery gene expression16. Cell culture studies suggest that Nr2f2 binds to cis elements at the Hey2 and Foxc1 loci17, inhibiting their expression to block artery differentiation. More recently, a bone morphogenetic protein (BMP) signaling pathway has been implicated in venous differentiation. Loss of the BMP receptor Bmpr1a, or the downstream effector Smad4, in mice results in loss of vein-specific gene expression18. Moreover, Smad1/5 binding at the vein-specific Ephb4 locus was stimulated by Bmp9 leading to upregulation of Ephb4 expression. A similar binding site at the Nr2f2 locus suggests that a Bmp9/Bmpr1a/Smad pathway acts upstream of Nr2f2 during vein specification.
Despite advances in identifying transcriptional inputs that govern arteriovenous expression, past studies have focused on selected candidate target genes and their flanking enhancer elements. As such, a genome-wide view of enhancer dynamics in arterial and vein endothelial cells is lacking. The advent of deep sequencing technologies has enabled straightforward implementation of genome-wide approaches and these have begun to be applied to better understand control of endothelial gene expression. Notably, endothelial chromatin dynamics in response to Vascular endothelial growth factor A (Vegfa) have been extensively characterized and new tools to investigate similar mechanisms in vivo have been developed19, 20. Furthermore, the large-scale Encyclopedia of DNA elements (ENCODE) project includes datasets from several primary human endothelial lines21, which have been used to provide insight into enhancer variants relevant to cardiovascular disease22, 23. However, none of these efforts have yielded appropriate datasets to investigate mechanisms that impact arteriovenous identity. To address this knowledge gap, we have applied a genome-wide approach using primary human endothelial cells to identify and characterize artery and vein enhancers in the human genome. We then leverage the resulting datasets to provide new insights into how NR2F2 directly affects multiple aspects of arteriovenous gene regulation.
METHODS
Due to space limitations, a detailed description of all methods is provided in the Online Data supplement.
Raw and processed datasets are publicly available at the Gene Expression Omnibus (GEO; GSE128382) or can be found in the Online Data supplement.
RESULTS
Identification of artery and vein enhancers in the human genome
To gain insights into genome-wide control of arteriovenous gene expression, we identified and characterized all active cis regulatory elements in artery and vein endothelial cells. For this purpose, we isolated human umbilical artery and vein endothelial cells (HUAEC and HUVEC, respectively) and used them prior to their third passage (P3). HUAEC and HUVEC expressed canonical endothelial cell markers (e.g. CDH5, ERG, ETS1, and FLI1), compared to a non-endothelial cell type (HeLa), but not markers for blood cell lineages (CSF1R, SPI1; Online Figure IA). By RNA-seq, HUAEC and HUVEC exhibited expression of conserved identity markers, including arterial expression of DLL4, EFNB2, HEY2, and NOTCH4 and venous expression of EMCN, EPHB4 and NR2F2 (Figure 1A, Online Figure IB, Online Table I). HUAEC also exhibit higher levels of Notch activation than HUVEC, as assessed using the Notch-responsive TP1 reporter 24 (Online Figure IC) and expression of a previously identified artery-specific transcription factor signature 25 (Online Figure ID). Thus, HUAEC and HUVEC maintain their identities under our isolation and culture conditions.
Figure 1. Genome-wide identification of artery and vein enhancers.

A, Volcano plot of HUAEC/HUVEC RNAseq. Red and blue indicates significantly-enriched HUAEC- and HUVEC-specific genes, respectively (log2 fold change >= 1, or <=−1, padj <=0.01). Grey indicates genes that are not significantly changed. Selected artery- and vein-specific genes are noted. B, Binding affinity heat map showing normalized read density (score) of replicate acEP300 and H3K27ac ChIP-seq samples from HUAEC and HUVEC. Only features with FDR<0.01 are shown. C, Pie charts showing distribution of HUAEC, HUVEC and common elements at inter/intragenic regions, transcriptional start sites (TSS) or heterochromatin/centromeric regions. D, Proportion of indicated elements concordant to DNAseI hypersensitivity I sites (DHS) from human microvascular endothelial cells (HMVEC) or T helper cells (Th1). E,F, Plots showing relationship between fold-enrichment in HUAEC or HUVEC and number of flanking E, artery-specific, or F, vein-specific enhancers. r and P values from Pearson Correlation are shown. H, I, Selected over-represented sites in H, vein-specific or I, artery-specific enhancers. Also see Online Tables V and VI.
To identify active cis regulatory elements, we performed ChIP-seq on HUAEC and HUVEC for histone 3, acetylated at lysine 27 (H3K27ac) and the acetylated form of the EP300 histone acetyl transferase (acEP300), both of which associate with active promoters and enhancers 26, 27. We then used these data to identify regulatory elements that were artery- or vein-specific. For this purpose, we compared the level of H3K27ac and acEP300 occupancy at all active regulatory elements in HUAEC and HUVEC (see Detailed Methods section in the Online Supplement for a description of enhancer identification, differential analysis, and statistical thresholds). An element was defined as “artery-” or “vein-enriched” if it displayed a log2 fold change in read density of greater than 1 or less then −1 (FDR<0.01), respectively, from replicate H3K27ac and acEP300 ChIP-seq samples. This analysis yielded 2609 artery- and 1933 vein-enriched regulatory elements (Figure 1B, Online Table II). An additional forty thousand elements were occupied to a similar level in both cell types and are referred to hereafter as “common” (Online Table III). Most artery- and vein-enriched elements localized to inter- or intragenic regions with approximately fifteen percent at transcriptional start sites (TSS; +/−2.5 kb), while common elements were more equally distributed (Figure 1C). A small fraction of elements mapped to ENCODE-annotated heterochromatin or centromeric regions (Figure 1C) and were not subsequently considered. The majority of all element types (artery, vein, and common) are largely concordant with DNase I hypersensitivity sites (DHS) in primary human microvascular endothelial cells (HMVEC), which would be expected to include both arterial and venous cell types, but less so in non-endothelial cells (lymphoid, Th1, Figure 1D). Accordingly, enhancer elements are located preferentially near genes associated with angiogenesis or endothelial cell functions (Online Table IV). We further find that the number of flanking artery- or vein-enriched, but not common, enhancers strongly correlates with the degree of HUAEC- or HUVEC-enriched expression, respectively, of adjacent genes (Figure 1E,F, Online Figure IE). Together, these findings suggest that most of these artery- and vein-specific regulatory elements are endothelial enhancers that regulate arteriovenous gene expression. As an accessible resource, we provide annotation files for easy visualization of these elements on the human genome (hg19) using the UCSC Genome browser (Online Data Files I and II). We also provide a single file that summarizes and integrates the results from all of the ChIP-seq data presented in this work (Online Table III).
Coordinately expressed genes often possess common cis regulatory sequences responsible for their shared expression patterns. Interestingly, a consensus site for the vein-enriched transcription factor, NR2F2, was over-represented in HUVEC-, but not HUAEC-specific, elements (Figure 1H, Online Table V). Vein-enriched elements also showed a prevalence of binding sites for ETS factors, such as ERG, and Forkhead proteins (Figure 1H, Online Table V), such as Foxc1/2, despite their implicated roles in artery differentiation 28. These sites were also over-represented in HUAEC-specific elements (Figure 1I, Online Table VI) consistent with comparable levels of transcripts encoding both ETS and Forkhead proteins in HUAEC and HUVEC (Online Figure IF, G). Despite robust detection of individual Fox and ETS motifs, unbiased analysis failed to identify the previously described composite FOX:ETS motif 12 as over-represented. Furthermore, we could not identify enrichment of an obvious HUAEC-specific cis regulatory element expected to bind known factors, including those that would bind the previously described artery signature transcription factors 25.
NR2F2 directly activates cell cycle genes and vein-specific enhancers in HUVEC
Previous studies suggest that NR2F2 controls arteriovenous differentiation by directly repressing artery gene expression and regulating the cell cycle 16, 17, 29. However, our finding of over-represented NR2F2 sites in vein-specific enhancers suggested a more direct role in activating a vein gene program. To gain mechanistic insights into these multiple possible roles, we first identified all NR2F2-regulated transcripts in HUVEC. RNA-seq following NR2F2 knockdown in HUVEC (Online Table I, Online Figure IIA) revealed more than 1000 transcripts that were significantly upregulated (log2FC>=1, adjP<=0.01), including the artery-specific transcription factor, HEY2, a known repressed target (Figure 2A, B). Strikingly, many more transcripts were downregulated (1821, log2FC<=1, padj<=0.01) consistent with a role for NR2F2 in actively driving gene activation in HUVEC. GO term analysis revealed an overwhelming association of all regulated genes with the cell cycle and most of these genes were downregulated by NR2F2 knockdown (Figure 2A; Online Figure IIB, Online Table VII). Further comparison of NR2F2-regulated genes with HUAEC and HUVEC transcriptomes (see above) revealed that a proportion of all artery- or vein-restricted genes were significantly regulated by NR2F2 (Figure 2B, C, Online Table I). Thus, our results are consistent with NR2F2 controlling multiple aspects of arteriovenous differentiation.
Figure 2. NR2F2 directly induces cell cycle and vein-specific genes.

A-C, Scatterplots of regulated genes following NR2F2 knockdown in HUVEC. Red and blue dots indicate genes significantly up and down regulated (log2 fold change >= 1, or <=−1, padj <=0.01), respectively. X- and Y-axes are log10 of mapped read counts of triplicate samples normalized by Median Ratio Normalization. The same scatter plot is shown at different magnification in each panel. Grey dots indicate genes that are not significantly changed. A, Green dots indicate cell-cycle genes that are significantly regulated. B, Green dots indicate all artery-specific genes. C, Green dots indicate all vein-specific genes. D, E, NR2F2 and H3K27ac ChIP-seq tracks at the D, RACGAP1, and E, XRCC3 loci. Boxes indicate regions bound by NR2F2 and showing significantly decreased H3K27ac (log2FC<=−1, FDR<0.05) following NR2F2 knockdown in triplicate samples. F, Heatmap of H3K27ac reads mapped to vein-specific elements significantly regulated (log2 fold change >= 1, or <=−1, FDR<=0.05; zscore = average of normalized read depth, n=3) by NR2F2 knockdown in HUVEC. NR2F2 bound elements are indicated, as are closest down-regulated genes following NR2F2 knockdown. Asterisk for CLU indicates that it falls just under the fold-change threshold for being an NR2F2-regulated gene (log2FC=−0.92, padj<0.01).
Nuclear receptors related to NR2F2 can typically act in a complex that leads to acetylation or deacetylation of histones to influence transcriptional changes 30. Using this characteristic as a guide, we took a two-pronged approach to identify direct functional NR2F2 target sites flanking the regulated genes identified above. First, we identified genomic regulatory elements bound by NR2F2 using ChIP-seq in HUVEC. From replicate samples, we identified 5108 sites, the majority of which exhibited a GGTCA half-site or direct repeat consistent with known NR2F2 or related nuclear receptor binding sites (Online Data File III, Online Figure IIIA, B; ref. 31). Next, we profiled genome-wide H3K27ac occupancy following knockdown of NR2F2 in HUVEC to identify elements with increased or reduced occupancy. Together, these data allowed us to identify all NR2F2 binding sites, along with those that were being directly regulated in a positive or negative manner by NR2F2 (see Online Tables I and III).
As noted above, cell cycle genes comprise a large proportion of NR2F2-regulated transcripts (Figure 2A). Accordingly, NR2F2 binding sites are found flanking these genes at a significantly higher rate than all genes (Online Figure IIIC). However, only 16 of 136 NR2F2-regulated cell cycle genes with flanking NR2F2 binding sites showed concomitant decrease in H3K27ac occupancy at these same sites following knockdown (Online Table I; 3 vein-specific elements and 13 common elements). These downregulated genes included RACGAP1, which shows robust NR2F2 binding at the promoter along with NR2F2-dependent H3K27ac at this same site (Figure 2D, Online Tables I, III) and XRCC3, which possesses an intronic enhancer that shows similar behavior (Figure 2E). These results indicate that NR2F2 can directly control the expression of cell cycle genes, but may act as a canonical activator on only a select group of these targets.
We next investigated whether NR2F2 directly induced vein-specific cis elements and similarly regulated flanking genes. NR2F2 ChIP-seq revealed that one-fifth of vein-specific elements are bound by NR2F2 consistent with our computational analysis (Online Figure IIID; Online Table V). Furthermore, 219 of 1933 vein-specific elements exhibited significantly reduced H3K27ac occupancy following NR2F2 knockdown (log2 fold change <=−1, FDR<=0.05 in triplicate ChIP-seq experiments; Online Table III), while 82 were up-regulated (Figure 2F). Intersection of these datasets revealed 76 of downregulated sites were also bound by NR2F2 suggesting that they were direct targets and only 4 of the 82 upregulated sites were likewise bound (Figure 2F). Further comparison to RNA-seq datasets revealed that only 6 vein-specific genes reduced by NR2F2 knockdown possessed a flanking vein-specific enhancer that was both bound by NR2F2 and displayed a similar decrease in H3K72ac occupancy (Figure 2F, Online Table I). CLU is a vein-specific gene that possessed flanking vein-specific enhancers that were similarly regulated by NR2F2. However, CLU was not initially detected since its expression in response to NR2F2 knockdown fell just below our threshold (log2 fold change=−0.92, adj.P<0.01; Online Figure IV). In any case, NR2F2 is essential to directly activate selected vein-specific cis regulatory elements in HUVEC and to drive expression of adjacent vein-specific genes.
NR2F2 and ERG induce vein-specific gene expression
In the course of analyzing our NR2F2 ChIP-seq data, we noticed enrichment for consensus ETS binding sites that were centered on, or adjacent to, NR2F2 half-sites (Figure 3A). Therefore, we performed ChIP-seq for ERG in HUVEC to determine if it was actually bound to these sites. We would note that the ERG antibody used in these studies also recognizes the highly related ETS factor, FLI1, and transcripts encoding these two factors are the most abundantly expressed of the ETS family members in HUVEC (Online Figure IF). Thus, they likely represent the bulk of ETS-bound sites in HUVEC. ERG ChIP-seq from replicate samples identified approximately 8,000 binding sites (Online Data File IV), which were likewise enriched for an ETS motif but much less so for the NR2F2 half-site (Figure 3B). ERG ChIP-seq reads, but not those from input, were detectable at most NR2F2 sites (Figure 3C) and more than half of significantly called NR2F2 sites in replicate samples overlapped with similarly called ERG sites (Figure 3D). These observations suggest that most NR2F2 binding sites were bound by both transcription factors in HUVEC. By contrast, a much smaller proportion of ERG sites are co-bound by NR2F2, making them more difficult to detect by only computational means (Figure 3B). This would be consistent with recent studies on genome-wide ERG binding in HUVEC that failed to identify a co-bound NR2F2 site from computational analysis 32. Consistent with our ChIP-seq analysis, co-immunoprecipitation of endogenous NR2F2 revealed an interaction with ERG in HUVEC (Figure 3E). We further noted NR2F2 and ERG binding together at shared sites in vein-enriched enhancers flanking FAM174B, ADAMTS18, and LHX6 (Figure 3F, G; data not shown). These particular elements also showed significantly reduced H3K27ac occupancy following NR2F2 knockdown, along with concomitant reduction of expression of the adjacent vein-specific genes (Figure 3F, G, Online Table I). ERG knockdown (Online Figure VA) likewise caused a significant downregulation of these genes, similar to NR2F2 loss, although knockdown of both ERG and NR2F2 did not further reduce expression (Figure 3H). Together, these observations suggest that ERG and NR2F2 act together in a complex to directly regulate the activation of selected vein enhancers and induce the expression of vein-specific genes.
Figure 3. NR2F2 and ERG are required for activation of vein enhancers and flanking vein genes.

A, B, Motif probability graphs using sequences from ChIP-seq output for A, NR2F2 and B, ERG. Consensus binding sequences identified in ChIP-seq-enriched fragments are shown; in both graphs red indicates ERG site, blue is NR2F2 half-site. C, Heatmap showing reads from ERG ChIP-seq mapped onto all NR2F2 sites in HUVEC. Zscore = average of normalized read depth, n=2. D, Venn diagram showing overlap of ERG and NR2F2 binding sites in HUVEC. Each set of sites was determined from duplicate ChIP-seq experiments. Note that total number of intersected elements is not equal to each individual dataset since single features from one set can intersect with multiple features from the other, in which case they are collapsed. E, immunoblot to detect ERG in lysates from HUVEC immunoprecipitated with IgG or NR2F2 antibody; HC is antibody heavy chain. F, G, Genomic regions flanking F, FAM174B and G, ADAMTS18. Tracks show read depth for H3K27ac from HUAEC and HUVEC, control and NR2F2 shRNA-expressing HUVEC, along with ChIP-seq of NR2F2 and ERG from HUVEC. Cyan boxes denote significant H3K27ac enrichment in HUVEC (HUAEC/HUVEC log2 FC<−1, FDR<0.05). Purple boxes are elements with significant reduction in H3K27ac occupancy following NR2F2 knockdown in triplicate samples (log2 fold change<−1, FDR<0.05). Dark blue boxes denote significant binding of ERG and NR2F2 from replicate samples. H, qRT-PCR for indicated transcripts in HUVEC expressing indicated shRNAs, n=3. Mean plus or minus S.E.M., 1-way Anova (ADAMTS18, P<1×10−4; FAM174B, P= 6×10−4; LHX6, P=4×10−4), Tukey’s post-hoc test.
NR2F2 and HDAC1 interact to repress a selected group of artery enhancers in HUVEC
Our findings demonstrate that NR2F2 can regulate cell cycle and vein-specific genes, in part by direct activation of flanking enhancers in HUVEC. Since NR2F2 has also been implicated in repressing artery gene expression, we further leveraged our genome-wide datasets to better characterize the mechanistic basis for its role in this context. Mapping and differential analysis of H3K27ac ChIP-seq data from HUVEC lacking NR2F2 revealed that only 15 out of 2609 artery-specific elements exhibited a significant decrease in H3K27ac occupancy and none of these bound to NR2F2 suggesting an indirect effect (Figure 4A, Online Table III). By contrast,139 artery elements exhibited a significant increase in H3K27ac occupancy following loss of NR2F2 (Figure 4A; logFC >=1, FDR<=0.05; considering both TSS and enhancers together; Online Table III), suggesting they are normally repressed by this factor in venous endothelial cells. However, only 15 of these sites were directly bound to NR2F2 in HUVEC (Figure 4A, Online Table III). Further integration of our RNA-seq and H3K27ac datasets indicated that these sites flanked only 3 artery-specific genes that were also upregulated by NR2F2 knockdown: ANTXR1, RASGRF2, and HEY2 (Figure 4A), the latter of which has previously been identified as a direct target of NR2F2 17. Thus, NR2F2 appears to act by directly repressing a highly selective group of artery-specific regulatory elements and their associated genes in venous endothelial cells.
Figure 4. NR2F2 represses artery-specific elements.

A, Heatmap of K27ac ChIPseq reads mapped onto artery-specific elements. Only elements with significant differences are shown (log2 fold change >= 1, or <=−1, FDR<=0.05; zscore = average of normalized read depth, n=3). NR2F2 bound elements are indicated, as are closest genes up-regulated by NR2F2 knockdown. B, C, qRT-PCR to detect indicated transcripts, Student’s t-test or Wilcoxon signed rank-sum test (B, EFNB2). B, HUVEC treated with DMSO or indicated concentration of TSA, n=3. C, HUVEC expressing control or HDAC1 shRNA, n=4. D, Immunoblot to detect HDAC1 in lysates from HUVEC immunoprecipitated with IgG or NR2F2 antibody. HC – denotes heavy chain of NR2F2 antibody. E, Heat map of read depth from single replicates of HDAC1 Cut&Run or NR2F2 ChIP-seq mapped onto NR2F2 and HDAC1 binding sites. F, Venn diagram indicating intersection of artery-specific elements, NR2F2 sites and HDAC1 sites.
In other contexts, NR2F2 interacts with histone deacetylase 1 (HDAC1) to facilitate gene repression 33, 34, raising the possibility of a similar role in negative regulation of artery genes in HUVEC. To determine if this was the case, we focused on the HEY2 locus. We first treated HUVEC with trichostatin A (TSA), a global HDAC inhibitor that causes global increase in H3K27ac levels (Online Figure VIA). TSA treatment causes significant upregulation of HEY2, but not EFNB2, which was unchanged, or DLL4, which, along with FLI1, a general endothelial marker, was downregulated (Figure 4B). Knockdown of HDAC1 in HUVEC likewise caused HEY2 upregulation, while also leading to the significant induction of EFNB2, but not DLL4 or FLI1 (Figure 4C; Online Figure VIB). EFNB2 upregulation in this case is likely secondary to HEY2 induction and may be due to the longer incubation time between knockdown and RNA isolation compared to TSA treatment (see Methods). Consistent with a physical interaction between HDAC1 and NR2F2 in HUVEC, immunoprecipitation of endogenous NR2F2 in HUVEC was capable of pulling down HDAC1 (Figure 4D). We next determined genome-wide occupancy of HDAC1 using the Cut&Run method 35, which identified 11528 bound sites in replicate experiments from HUVEC (Online Data File V). Similar to previous genome-wide profiling in non-endothelial cell types 36, HDAC1 binding was predominantly at promoter and enhancer elements in HUVEC (Online Figure VIC). Consistent with our immunoprecipitation results, visualization of mapped read density and intersection of significantly enriched NR2F2 and HDAC1 binding sites suggested that nearly one-third of all NR2F2 sites were co-bound by HDAC1 (Figure 4E, Online Figure VID). Importantly, 12 out of the 15 NR2F2-bound and -repressed artery elements identified above were bound by HDAC1 in HUVEC (Figure 4F), including the site at the HEY2 locus (see below). These results suggest that NR2F2 works together with HDAC1 at a select few loci to mediate repression of artery-specific enhancers in the HUVEC genome.
NR2F2 prevents ERG-mediated HEY2 expression in HUVEC
Previous studies using ChIP-qPCR suggested that NR2F2 bound near the HEY2 promoter to inhibit its expression 17. However, we failed to identify an NR2F2-bound site that was enriched over background at this location in our ChIP-seq data (Online Figure VII). Instead, we observed robust NR2F2 binding at a site 161 kb upstream of the HEY2 promoter (Online Figure VII). This site is located in an artery-specific element identified above (see Figure 4A) that shows significantly increased H3K37ac occupancy following NR2F2 knockdown in HUVEC (Figure 5A, Online Table III). We further noted HDAC1 binding together with NR2F2 at this site (Figure 5A). This enhancer, referred to hereafter as −161K, is in the vicinity of several elements that are specifically occupied by H3K27ac and acEP300 in HUAEC, but not at all in HUVEC (Figure 5B, data not shown) indicative of their artery-specific activity. Most of these also exhibit increased H3K27ac following NR2F2 knockdown in HUVEC (Figure 5B, Online Table III). Thus, NR2F2 appears to regulate HEY2 expression through binding of the −161K enhancer leading to repression of this and flanking elements in HUVEC.
Figure 5. NR2F2 prevents ERG-mediated expression of HEY2 in HUVEC.

A, Genomic tracks at the HEY2 −161K enhancer showing read depth for H3K27ac, HDAC1 and NR2F2 ChIP-seq in HUVEC. B, Genomic region flanking HEY2. Red boxes denote enhancers with significant H3K27ac enrichment in HUAEC (HUAEC/HUVEC log2 FC>1, FDR<0.01). Purple boxes are elements with increased H3K27ac occupancy following NR2F2 knockdown (log2 fold change>1, FDR<0.05). Sequence of conserved element in the −161K enhancer shows putative ETS and NR2F2 sites. C, Genomic tracks at the −161K enhancer (same coordinates as in A) showing read depth for ERG ChIP-seq from HUVEC and ERG, NR2F2 and H3K27ac from HUAEC. D, Relative luminescence in reporter assays in indicated cell type with indicated reporters, n= 3. Mean plus or minus S.E.M., 1-way Anova (P<1×10−4), Tukey’s post-hoc test. E, Relative luminescence of −161K reporter in HUVECs expressing control or NR2F2 shRNA, n=3, Student’s t-test. F, G, HEY2 expression determined by qRT-PCR in HUAEC expressing F, control or ERG shRNA, or G, an EGFP or NR2F2 transgene driven by a constitutive promoter, n=3, Student’s t-test. H, HEY2 expression by qRT-PCR in HUVEC expressing indicated shRNAs, n=4, Mean plus or minus S.E.M., 1-way Anova (P=0.0237), Tukey’s post-hoc test.
The −161K enhancer bears a highly conserved core element that includes two NR2F2 binding sites, as expected from ChIP-seq (see Figure 5A, B). Interestingly, this element also contained several ETS binding sites and ChIP-seq revealed robust ERG binding in HUVEC (Figure 5B, C, Online Table III, Online Data File IV). ChIP-seq from HUAEC similarly shows ERG binding at this element in HUAEC along with high levels of H3K27ac, while NR2F2 is absent, consistent with its low level of expression in arterial endothelial cells (Figure 5C; see Online Figure IB). To interrogate the functional importance of these binding sites, we performed reporter assays using the −161K conserved core element in both HUAEC and HUVEC. In HUAEC, but not HUVEC, we observed a significant increase in −161K reporter activity over that associated with a basal promoter alone (Figure 5D). Consistent with the artery-specific H3K27ac signature for this enhancer (see Figure 5B), we observed significantly higher reporter levels in HUAEC compared to HUVEC (Figure 5D). Mutation of the NR2F2 binding sites, or knockdown of NR2F2 itself, caused a significant increase in reporter activity in HUVEC, to levels near that seen in HUAEC, while levels in HUAEC were largely unaffected (Figure 5D, E). By contrast, HUAEC −161K reporter activity was extinguished by mutating two out of the three ETS binding sites (Figure 5D).
Our ChIP-seq analysis and reporter assays suggested a model where ERG can drive HEY2 expression in part through activation of the −161K enhancer in HUAEC, while this effect is counteracted through NR2F2 binding and repression in HUVEC. Accordingly, knockdown of ERG, or over-expression of NR2F2, in HUAEC reduced HEY2 expression (Figure 5F, G, Online Figure VA, B). Finally, combined knockdown of ERG and NR2F2 in HUVEC eliminated HEY2 induction seen with loss of NR2F2 alone (Figure 5H). Together, these results suggest a model in which the −161K enhancer acts as a bimodal regulatory element depending on its cellular context: in venous endothelial cells, NR2F2 binds to prevent ERG-mediated activation of the enhancer and associated upregulation of HEY2. In arterial endothelial cells, where NR2F2 is absent, ERG is able to activate the −161K enhancer and drive HEY2 expression.
DISCUSSION
In this study, we provide a genome-level view of endothelial identity, with an emphasis on artery and vein differentiation. While an increasing number of studies have identified transcription factors and selected target enhancers relevant to arteriovenous differentiation, our study is among the first to begin to investigate artery and vein identity at a genomic scale. As such, the accompanying datasets will provide a valuable resource for ongoing genetic studies and serve as the starting point for new studies. Importantly, our work underscores the utility of this approach by providing unique mechanistic insights into how a known lineage-specific transcription factor, NR2F2, functions to maintain cell fate in mature endothelial cells.
Our initial impetus for these studies was to identify signature motifs responsible for driving artery expression. However, we were unable to identify such a pattern in HUAEC-specific regulatory elements. There are several possible reasons for this result. First, a basic signature motif may have avoided detection using the standard computational approach applied in our analysis. However, we readily detected an NR2F2 half-site in HUVEC-specific elements, supporting our ability to identify over-represented sites for a known lineage-specifying transcription factor. A second possibility is that an artery-specific motif is more complex than just a single cis regulatory sequence. A significant number of transcription factors have now been implicated in artery differentiation1, 25. It is possible that many of these act in a combinatorial manner through the action of separate binding sites in distinct enhancers flanking a particular artery gene. In this case, a more complex computational approach may be required to deconvolute such combinations of cis sequences in artery enhancers. Alternatively, with the large number of artery-enriched transcription factors, along with the potential combinatorial inputs and context dependence, it is possible that no single unifying cis regulatory signature exists to broadly drive artery gene expression. Finally, our studies focused on mature human endothelial cells where arteriovenous regulatory pathways may be predominantly involved in maintaining, rather than establishing, identity. As such, obvious regulatory motifs may be associated with developmentally active enhancers that are not similarly accessible in mature endothelial cells. Why this would be the case for arterial, but not venous motifs, at least one of which was readily detected in our analysis, is not clear. In any case, it will be worthwhile to apply similar genomic approaches in multiple contexts where arteriovenous identity is being actively programmed.
Instead of an obvious pro-artery signature, we identified a unique mechanism by which artery-restricted expression is achieved through active repression in venous endothelial cells (see Figure 6). In this case, ERG, which is robustly expressed in arterial and venous endothelial cells, provides a constitutive “on” signal in both cell types, but is limited for activation in HUVEC by NR2F2 (Figure 6). Interestingly, the −161K core element can still drive appreciable reporter expression in HUVEC, although it is clearly regulated by both ERG and NR2F2. This suggests that the surrounding chromatin landscape at the HEY2 locus likely plays a role in directing the activity of this regulatory element. Given the broad endothelial expression of numerous ETS transcription factors, along with a prevalence of ETS binding sites in artery enhancers, our observations suggest that this could be a general mechanism to prevent inappropriate activation of these elements in venous endothelial cells. However, only a small number of elements exhibited similar characteristics. Given the large number of ETS factors in endothelial cells, it is possible that related family members also play a role in regulating artery genes. Similarly, a number of other nuclear receptors, as well as other transcriptional repressors, may contribute to limiting inappropriate activation of artery enhancers bearing ETS-binding sites in venous endothelial cells.
Figure 6. Schematic showing mechanisms of gene regulation mediated by NR2F2.

Model of the multifunctional roles of NR2F2 in regulating artery and vein gene expression in endothelial cells. See Discussion text for details.
From genetic studies, NR2F2 is required for vein identity, where it has been largely thought to be a repressor of arterial endothelial cell fate 16. Our findings regarding the −161K HEY2 enhancer are consistent with that model. However, we find that the number of artery-specific targets regulated directly by NR2F2 is limited, suggesting a less broad repressor effect than predicted from genetic studies. Rather, NR2F2 likely mediates artery repression through targeting HEY2, which itself can partially induce artery marker gene expression when over-expressed in HUVEC37. More recent work in mouse suggests that Nr2f2 affects artery identity through cell cycle regulation29. Consistent with this work and earlier cell culture studies17, we find that several genes encoding positive regulators of the cell cycle are direct targets of NR2F2 in HUVEC (Figure 6). Interestingly, these genes and their associated target enhancers are not themselves vein-specific, suggesting that NR2F2 can modulate identity through more general regulators. In addition, we also find that NR2F2 directly induces expression of several vein-specific genes and does so with ERG to activate vein-specific enhancers adjacent to these genes (Figure 6). Thus, NR2F2 plays a multi-modal role in promoting vein identity. Our studies suggest that direct targets in these contexts appear surprisingly limited. However, we restricted our analysis to elements with direct binding, significant H3K27ac changes in response to NR2F2 loss, and were relatively near to NR2F2-regulated genes. Additional targets may be affected at longer distances, or through changes to enhancer elements that are not vein-specific. Our caveat noted above concerning cellular context is likewise applicable in this case.
Our results reveal an interesting paradox for NR2F2 function in venous endothelial cells. Namely, an NR2F2/ERG complex can yield a different transcriptional output depending on the target locus in the same cell type. It is not clear what governs this distinction, but there is precedent for related retinoid receptors acting in this manner. Classically, the function of a nuclear receptor as an activator or repressor depends on ligand binding 38. However, there are cases of liganded nuclear receptors acting as a repressor at particular loci 39. Distinct outputs can also be governed by target site orientation. While NR2F2 is an orphan nuclear receptor and may not require ligand binding, similar mechanisms may govern its differential transcriptional output. In addition, there are likely to be distinct complexes comprising NR2F2 and other co-regulators that govern activation or repression. Which complexes are formed may be determined by local chromatin architecture. The increasing availability of genome-scale endothelial datasets will be helpful in further elucidating these mechanisms.
The regulation of arterial and venous identity is clearly important for normal vascular morphogenesis and patterning during development1. However, maintenance and control of arteriovenous identity has also been implicated in a number of clinical settings7. As noted above, vascular grafts often apply a vein, which will carry arterial flow. In these cases, partial loss of vein identity is observed, but without proper induction of artery gene expression. Notably, venous grafts perform more poorly in coronary artery bypass grafts than arterial ones, suggesting that modulation of identity in these cases may be beneficial. Although numerous studies have successfully re-programmed non-endothelial cell types into functional endothelial cells, few studies to date have focused on generating only arterial endothelial cells. Furthermore, previous efforts to re-program HUVEC into arterial endothelial cells required a combination of eight transcription factors25. Our observations suggest that a combined approach that targets regulators such as NR2F2, which is important for directly repressing crucial artery enhancers, together with selected artery transcription factors may prove a more straightforward approach to program artery identity.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Artery and vein endothelial cells are molecularly distinct.
A number of transcription factors have been identified that are required for guiding artery and vein identity.
What New Information Does This Article Contribute?
Identifies and characterizes artery- and vein-specific endothelial enhancers in the human genome.
Reveals how NR2F2 contributes to venous endothelial identity through direct activation of both cell cycle and vein-specific genes, while directly repressing selected artery genes.
Demonstrates new mechanism by which artery-specific gene expression can be maintained.
Artery and vein endothelial cells are molecularly distinct and their identities are fixed during embryogenesis. This process contributes to subsequent functional and structural differences between these blood vessels. However, veins are often used in place of arteries in surgical procedures, such as coronary artery bypass grafts (CABG), where they perform more poorly in follow up studies. Therefore, a better understanding of how arteriovenous identities are controlled would be beneficial. In this work, we identified regulatory elements in the human genome responsible for controlling artery and vein gene expression. We then leveraged these data to gain insights into how arteriovenous identities are maintained in human endothelial cells. In particular, we find that the vein-specific transcription factor, NR2F2, directly controls multiple facets of vein identity. Together with the ERG transcription factor, NR2F2 directly activates vein-specific enhancers to turn on nearby vein-specific genes. At the same time, NR2F2 directly represses the expression of a crucial artery-specific transcription factor, HEY2, to maintain vein identity. Together, our studies provide new mechanistic insights into how endothelial identities are maintained in human endothelial cells. These studies provide the genomic framework to understand how artery and vein identity may be manipulated in a therapeutic setting.
ACKNOWLEDGEMENTS
We would like to thank Tiffany Moore-Simas for facilitating human umbilical cord collection, as well as Ellen Kittler and members of the UMass Medical School Deep Sequencing Core for their valuable contributions. We thank Chinmay Trivedi and Tom Fazzio for helpful insights on the manuscript. We also thank Sarah De Val for providing laboratory space for several of the studies. We are grateful to Steve Henikoff for providing recombinant pA-MN protein. Plasmid 7TFC was a gift from Roel Nusse (Addgene plasmid # 24307) and plasmids psPAX2 (Addgene, # 12260) and pMD2.G (Addgene, #12259) were gifts from Didier Trono.
SOURCES OF FUNDING
This work was supported by NIH R35HL140017, an AHA Innovative Research Grant, and Leducq Foundation awards to N. D. L. and U01HG007910 from NHGRI and National Center for Advancing Translational Sciences grant UL1TR001453, which supported A. K. and O. Y.
Nonstandard Abbreviations And Acronyms
- acEP300
acetylated E1A binding protein p300
- ANTXR1
Anthrax toxin receptor 1
- BMP
bone morphogenetic protein
- Bmpr1a
bone morphogenetic protein receptor, type 1A
- Bmp9
bone morphogenetic protein 9
- CDH5
adherin 5
- ChIP-seq
chromatin immunoprecipitation and deep sequencing
- CLDN5
claudin 5
- CLU
Clusterin
- DHS
DNAse I hypersensitive site
- DLL4
Delta-like 4
- EFNB2
ephrin-B2
- ENCODE
Encyclopedia of DNA elements
- EphB4
Ephrin Type-B receptor 4
- ERG
ETS-related gene
- ETS
E26 transformation specific
- FAM174B
family with sequence similarity 174 member B
- FDR
false discovery rate
- FLI1
Fli-1 proto-oncogene, ETS transcription factor
- Foxc1/c2
Forkhead box C1/C2
- H3K27ac
histone 3, acetylated at lysine 27
- HDAC
histone deacetylase
- HEY2
hes related family bHLH transcription factor with YRPW motif 2
- HMVEC
human microvascular endothelial cells
- HUAEC
human umbilical arterial endothelial cells
- HUVEC
human umbilical venous endothelial cells
- LIM
homeobox 6
- LHX6
nuclear receptor subfamily 2, group F, member 2
- NR2F2
Notch-regulated ankyrin repeat protein
- NRARP
Rac GTPase Activating Protein 1
- RACGAP1Ras
Protein Specific Guanine Nucleotide Releasing Factor 2
- RASGRF2
recombination signal binding protein for immunoglobulin kappa J region
- RBPJ
RNA sequencing
- SMAD1/4/5
RNA-seq Smad family members 1/4/5
- Sox7/17/18
sex determining region Y-box 7/17/18
- TSA
Trichostatin A
- TSS
transcriptional start site
- Vegfa
vascular endothelial growth factor A
- XRCC3
X-Ray Repair Cross Complementing 3
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Fish JE and Wythe JD. The molecular regulation of arteriovenous specification and maintenance. Dev Dyn. 2015;244:391–409. [DOI] [PubMed] [Google Scholar]
- 2.Lawson ND, Scheer N, Pham VN, Kim CH, Chitnis AB, Campos-Ortega JA and Weinstein BM. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development. 2001;128:3675–83. [DOI] [PubMed] [Google Scholar]
- 3.Wang HU, Chen ZF and Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93:741–53. [DOI] [PubMed] [Google Scholar]
- 4.Moyon D, Pardanaud L, Yuan L, Breant C and Eichmann A. Plasticity of endothelial cells during arterial-venous differentiation in the avian embryo. Development. 2001;128:3359–70. [DOI] [PubMed] [Google Scholar]
- 5.Quillien A, Moore JC, Shin M, Siekmann AF, Smith T, Pan L, Moens CB, Parsons MJ and Lawson ND. Distinct Notch signaling outputs pattern the developing arterial system. Development. 2014;141:1544–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong TP, Childs S, Leu JP and Fishman MC. Gridlock signalling pathway fashions the first embryonic artery. Nature. 2001;414:216–20. [DOI] [PubMed] [Google Scholar]
- 7.Wolf K, Hu H, Isaji T and Dardik A. Molecular identity of arteries, veins, and lymphatics. J Vasc Surg. 2019;69:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaudino M, Benedetto U, Fremes S, Biondi-Zoccai G, Sedrakyan A, Puskas JD, Angelini GD, Buxton B, Frati G, Hare DL, Hayward P, Nasso G, Moat N, Peric M, Yoo KJ, Speziale G, Girardi LN, Taggart DP and Investigators R. Radial-Artery or Saphenous-Vein Grafts in Coronary-Artery Bypass Surgery. N Engl J Med. 2018;378:2069–2077. [DOI] [PubMed] [Google Scholar]
- 9.Kudo FA, Muto A, Maloney SP, Pimiento JM, Bergaya S, Fitzgerald TN, Westvik TS, Frattini JC, Breuer CK, Cha CH, Nishibe T, Tellides G, Sessa WC and Dardik A. Venous identity is lost but arterial identity is not gained during vein graft adaptation. Arterioscler Thromb Vasc Biol. 2007;27:1562–71. [DOI] [PubMed] [Google Scholar]
- 10.Kume T, Jiang H, Topczewska JM and Hogan BL. The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes & development. 2001;15:2470–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wythe JD, Dang LT, Devine WP, Boudreau E, Artap ST, He D, Schachterle W, Stainier DY, Oettgen P, Black BL, Bruneau BG and Fish JE. ETS factors regulate Vegf-dependent arterial specification. Dev Cell. 2013;26:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Val S, Chi NC, Meadows SM, Minovitsky S, Anderson JP, Harris IS, Ehlers ML, Agarwal P, Visel A, Xu SM, Pennacchio LA, Dubchak I, Krieg PA, Stainier DY and Black BL. Combinatorial regulation of endothelial gene expression by ets and forkhead transcription factors. Cell. 2008;135:1053–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corada M, Orsenigo F, Morini MF, Pitulescu ME, Bhat G, Nyqvist D, Breviario F, Conti V, Briot A, Iruela-Arispe ML, Adams RH and Dejana E. Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat Commun. 2013;4:2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cermenati S, Moleri S, Cimbro S, Corti P, Del Giacco L, Amodeo R, Dejana E, Koopman P, Cotelli F and Beltrame M. Sox18 and Sox7 play redundant roles in vascular development. Blood. 2008;111:2657–66. [DOI] [PubMed] [Google Scholar]
- 15.Pereira FA, Qiu Y, Zhou G, Tsai MJ and Tsai SY. The orphan nuclear receptor COUP-TFII is required for angiogenesis and heart development. Genes & development. 1999;13:1037–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.You LR, Lin FJ, Lee CT, DeMayo FJ, Tsai MJ and Tsai SY. Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature. 2005;435:98–104. [DOI] [PubMed] [Google Scholar]
- 17.Chen X, Qin J, Cheng CM, Tsai MJ and Tsai SY. COUP-TFII is a major regulator of cell cycle and Notch signaling pathways. Mol Endocrinol. 2012;26:1268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neal A, Nornes S, Payne S, Wallace MD, Fritzsche M, Louphrasitthiphol P, Wilkinson RN, Chouliaras KM, Liu K, Plant K, Sholapurkar R, Ratnayaka I, Herzog W, Bond G, Chico T, Bou-Gharios G and De Val S. Venous identity requires BMP signalling through ALK3. Nat Commun. 2019;10:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang B, Day DS, Ho JW, Song L, Cao J, Christodoulou D, Seidman JG, Crawford GE, Park PJ and Pu WT. A dynamic H3K27ac signature identifies VEGFA-stimulated endothelial enhancers and requires EP300 activity. Genome Res. 2013;23:917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou P, Gu F, Zhang L, Akerberg BN, Ma Q, Li K, He A, Lin Z, Stevens SM, Zhou B and Pu WT. Mapping cell type-specific transcriptional enhancers using high affinity, lineage-specific Ep300 bioChIP-seq. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta RM, Hadaya J, Trehan A, Zekavat SM, Roselli C, Klarin D, Emdin CA, Hilvering CRE, Bianchi V, Mueller C, Khera AV, Ryan RJH, Engreitz JM, Issner R, Shoresh N, Epstein CB, de Laat W, Brown JD, Schnabel RB, Bernstein BE and Kathiresan S. A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression. Cell. 2017;170:522–533 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, Ren B, Fu XD, Topol EJ, Rosenfeld MG and Frazer KA. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato H, Sakai T, Tamura K, Minoguchi S, Shirayoshi Y, Hamada Y, Tsujimoto Y and Honjo T. Functional conservation of mouse Notch receptor family members. FEBS Lett. 1996;395:221–4. [DOI] [PubMed] [Google Scholar]
- 25.Aranguren XL, Agirre X, Beerens M, Coppiello G, Uriz M, Vandersmissen I, Benkheil M, Panadero J, Aguado N, Pascual-Montano A, Segura V, Prosper F and Luttun A. Unraveling a novel transcription factor code determining the human arterial-specific endothelial cell signature. Blood. 2013;122:3982–92. [DOI] [PubMed] [Google Scholar]
- 26.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA and Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, Afzal V, Ren B, Rubin EM and Pennacchio LA. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seo S, Fujita H, Nakano A, Kang M, Duarte A and Kume T. The forkhead transcription factors, Foxc1 and Foxc2, are required for arterial specification and lymphatic sprouting during vascular development. Dev Biol. 2006;294:458–70. [DOI] [PubMed] [Google Scholar]
- 29.Su T, Stanley G, Sinha R, D’Amato G, Das S, Rhee S, Chang AH, Poduri A, Raftrey B, Dinh TT, Roper WA, Li G, Quinn KE, Caron KM, Wu S, Miquerol L, Butcher EC, Weissman I, Quake S and Red-Horse K. Single-cell analysis of early progenitor cells that build coronary arteries. Nature. 2018;559:356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glass CK and Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes & development. 2000;14:121–41. [PubMed] [Google Scholar]
- 31.Cooney AJ, Leng X, Tsai SY, O’Malley BW and Tsai MJ. Multiple mechanisms of chicken ovalbumin upstream promoter transcription factor-dependent repression of transactivation by the vitamin D, thyroid hormone, and retinoic acid receptors. J Biol Chem. 1993;268:4152–60. [PubMed] [Google Scholar]
- 32.Kalna V, Yang Y, Peghaire CR, Frudd K, Hannah R, Shah AV, Osuna Almagro L, Boyle JJ, Gottgens B, Ferrer J, Randi AM and Birdsey GM. The Transcription Factor ERG Regulates Super-Enhancers Associated With an Endothelial-Specific Gene Expression Program. Circ Res. 2019;124:1337–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smirnov DA, Hou S and Ricciardi RP. Association of histone deacetylase with COUP-TF in tumorigenic Ad12-transformed cells and its potential role in shut-off of MHC class I transcription. Virology. 2000;268:319–28. [DOI] [PubMed] [Google Scholar]
- 34.Zhao B, Hou S and Ricciardi RP. Chromatin repression by COUP-TFII and HDAC dominates activation by NF-kappaB in regulating major histocompatibility complex class I transcription in adenovirus tumorigenic cells. Virology. 2003;306:68–76. [DOI] [PubMed] [Google Scholar]
- 35.Skene PJ and Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W and Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D and Brown PO. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci U S A. 2003;100:10623–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gudas LJ and Wagner JA. Retinoids regulate stem cell differentiation. J Cell Physiol. 2011;226:322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santos GM, Fairall L and Schwabe JW. Negative regulation by nuclear receptors: a plethora of mechanisms. Trends Endocrinol Metab. 2011;22:87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skene PJ, Henikoff JG and Henikoff S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc. 2018;13:1006–1019. [DOI] [PubMed] [Google Scholar]
- 41.Fuerer C and Nusse R. Lentiviral vectors to probe and manipulate the Wnt signaling pathway. PLoS One. 2010;5:e9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li B and Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kucukural A, Yukselen O, Ozata DM, Moore MJ and Garber M. DEBrowser: interactive differential expression analysis and visualization tool for count data. BMC Genomics. 2019;20:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Love MI, Huber W and Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maza E, Frasse P, Senin P, Bouzayen M and Zouine M. Comparison of normalization methods for differential gene expression analysis in RNA-Seq experiments: A matter of relative size of studied transcriptomes. Commun Integr Biol. 2013;6:e25849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benjamini Y and Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple hypothesis testing. J R Statistic Soc B. 1995;57:289–300. [Google Scholar]
- 48.Langmead B, Trapnell C, Pop M and Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W and Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quinlan AR. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics. 2014;47:11 12 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C, Caldas C and Carroll JS. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stark R and Brown GD. DiffBind: differential binding analysis of ChIP-Seq peak data. 2011. [Google Scholar]
- 53.Shao Z, Zhang Y, Yuan GC, Orkin SH and Waxman DJ. MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 2012;13:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khan A and Mathelier A. Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC bioinformatics. 2017;18:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma W, Noble WS and Bailey TL. Motif-based analysis of large nucleotide data sets using MEME-ChIP. Nat Protoc. 2014;9:1428–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Machanick P and Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu LJ, Gazin C, Lawson ND, Pages H, Lin SM, Lapointe DS and Green MR. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC bioinformatics. 2010;11:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramirez F, Dundar F, Diehl S, Gruning BA and Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ihaka R and Gentleman R. R: A language for analysis and graphics Journal of Computational and Graphical Statistics. 1996;5:299–314. [Google Scholar]
- 60.Royston P Algorithm AS 181: The W test for Normality. Applied Statistics. 1982;31:176–180. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.