

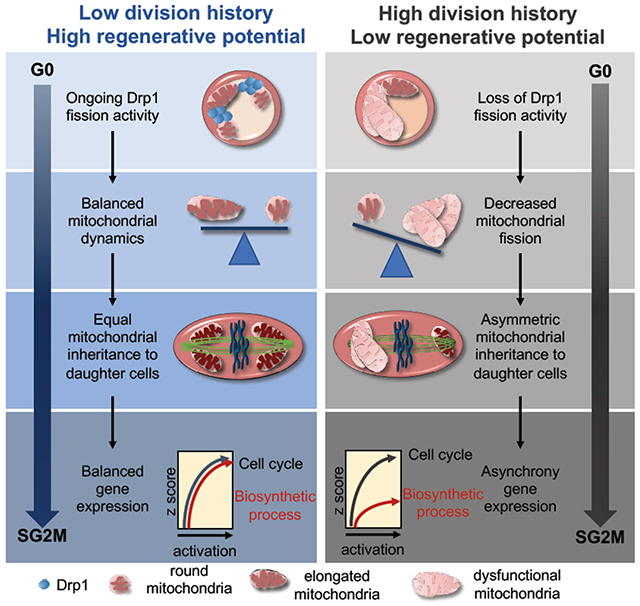
Summary
The metabolic requirements of hematopoietic stem cell (HSC) change with their cell cycle activity. However, the underlying role of mitochondria remain ill-defined. Here, we found that after mitochondrial activation with replication, HSC irreversibly remodel the mitochondrial network and this network is not repaired after HSC re-entry into quiescence, contrary to hematopoietic progenitors. HSC keep and accumulate dysfunctional mitochondria through asymmetric segregation during active division. Mechanistically, mitochondria aggregate and depolarize after stress due to loss of activity of mitochondrial fission regulator Drp1 onto mitochondria. Genetic and pharmacological studies indicate that inactivation of Drp1 causes a loss in HSC regenerative potential while maintaining HSC quiescence. Molecularly, HSC carrying dysfunctional mitochondria can re-enter quiescence but fail to synchronize the transcriptional control of core cell cycle and metabolic components in subsequent division. Thus, the loss of fidelity of mitochondrial morphology and segregation provides one type of HSC divisional memory and drives HSC attrition.
Graphical Abstract
eTOC blurb
Hinge et al show that mitochondria are permanently remodeled after HSC division despite re-entry into quiescence. HSCs keep dysfunctional mitochondria through asymmetric segregation during mitosis, which does not prevent reversible HSC quiescence and cell cycle progression but drives their functional decline via asynchrony in cell cycle and biosynthetic gene expression.
Introduction
Adult hematopoietic stem cells sustain the production of mature blood and immune cells. They are endowed with high regenerative potential and can self-renew for a limited number of divisions. (Qiu et al., 2014; Wilson A, 2008). In order to prevent excessive cell division and premature exhaustion, HSCs are maintained in a quiescent and low metabolic state (Hsu and Qu, 2013; Takubo and Suda, 2012; Vannini N, 2016). HSCs have low mitochondrial metabolic activity, with low membrane potential [MMP], low oxidative phosphorylation (OXPHOS), and low mitochondrial ROS (mtROS) production. Sustained ROS production (Ito et al., 2012; Ito et al., 2006; Simsek et al., 2010; Takubo et al., 2013) or sustained mitochondrial activation (Ho et al., 2017; Chen et al., 2008) prevent HSC quiescence, and alter HSC activity.
HSC self-renewal and regenerative potential inherently require HSCs to exit quiescence and produce daughter cells that will either maintain stem cell features or commit to differentiation. HSC cell cycle entry is accompanied by mitochondrial activation that is critical to achieve cell division (Ho et al., 2017; Ito and Suda, 2014; Luchsinger LL, 2016; Maryanovich M, 2015; Yu WM, 2013; Umemoto et al., 2018). Mitochondrial activity is equally important for HSC self-renewal (Ito et al., 2012; Ito K, 2016; Maryanovich M, 2015). Mitochondrial morphology controlled by the mitochondrial fusion regulator MFN2 is critical to maintain a pool of lymphoid-biased HSC (Luchsinger LL, 2016). Here, we show that HSCs use mitochondria as a natural checkpoint to track their divisional history and limit their self-renewal activity.
Results
Mitochondria permanently remodel after HSC replication under regenerative and homeostatic conditions.
We analyzed mitochondrial activity using tetramethylrhodamine-ethyl ester (TMRE) dyes to assess mitochondrial membrane potential (MMP) and mitoSOX redR dye for mitochondrial ROS (mtROS) detection in primary-bone-marrow HSCs (lineage-c-Kit+Sca1+CD150+CD48−; SLAM, Figure S1A) from naïve animals (pre-transplantation; NT), and after HSC replication following transplantation (post-transplantation; T) or 5-Fluorouracil (5FU) myeloablation. As previously reported, mitochondrial activity was low in SLAMs compared to progenitors (multipotent progenitors LSK-CD48+ (MPP) and committed progenitors as lineage-c-Kit+Sca1− (CP)) and increased with acute activation, both in vitro and in vivo (Figure S1B) (Ho et al., 2017; Simsek et al., 2010). However, T-SLAM mitochondria exhibited lower TMRE and mitotracker green staining, and sustained mtROS production (Figure 1A–D). Mitochondrial parameters were unchanged in progenitor populations after transplantation (Figure 1E–G). ATP production remained unchanged (Figure 1H). HSCs also exhibited depolarized mitochondria after 5FU-induced myeloablation that persisted up to 5 months after 5FU treatment (Figure S1B,C). Mitochondrial content was evaluated by immunostaining of Tom20, an outer mitochondrial membrane protein, instead of mitotracker-green, which reportedly cannot evaluate mitochondrial mass in HSC (de Almeida MJ, 2017; Norddahl et al., 2011). Tom20 levels in SLAMs were relatively high and remained unchanged in T-SLAMs, suggesting that their lower MMP was not due to lower mitochondrial mass (Figure 1I and S1D). To examine mitochondrial morphology we performed high resolution Z-stack imaging and 3D reconstruction analyses using HSCs from a mouse-transgenic-reporter system expressing mitochondrial-targeted-Dendra2 (mito-Dendra2), an eGFP fluorescent protein (Figure S1E). Although mito-Dendra2 levels were comparable in NT SLAMs and T-SLAMs (Figure 1J), mitochondrial organization was different. The mitochondrial network was well dispersed across the cell volume in NT SLAMs. In contrast, mitochondria were organized in a more compact network in T-SLAMs, which was restricted to one part of the cell (Figure 1K). Mitochondrial distribution was highly polarized in T-SLAMs compared to NT SLAMs (Figure 1L–M). Importantly, electron microscopy (Kumar and Filippi, 2016) showed that NT SLAMs had small and round mitochondria with defined cristae. In contrast, T-SLAMs had elongated and swollen mitochondria with dysregulated cristae (Figure 1N–P). The compact mitochondrial organization in T-SLAMs was observed both in TMRE high and low populations (Figure S1F). Mitochondrial organization was similar between NT and T LK cells (Figure S1G,H). To determine if abnormal mitochondria in T-SLAMs were indicative of dysfunctional mitochondria, we tested the ability of SLAMs to maintain MMP in response to mitochondrial electron transport chain complex I or ATPase inhibitors, rotenone and oligomycin, respectively. Under these conditions, cells with healthy mitochondria should maintain MMP (Martinez-Reyes et al., 2016). NT SLAMs did maintain MMP after rotenone or oligomycin treatments. In contrast, MMP remained lower in T-SLAMs after activation in culture and collapsed in a subpopulation of T-SLAMs that were treated with rotenone or oligomycin (Figure 1Q and S1I,J). Hence, following replication, HSCs carry mitochondria which differ both morphologically as well in their respond to mitochondrial stress.
Figure. 1. HSC keep dysfunctional mitochondria after replication.
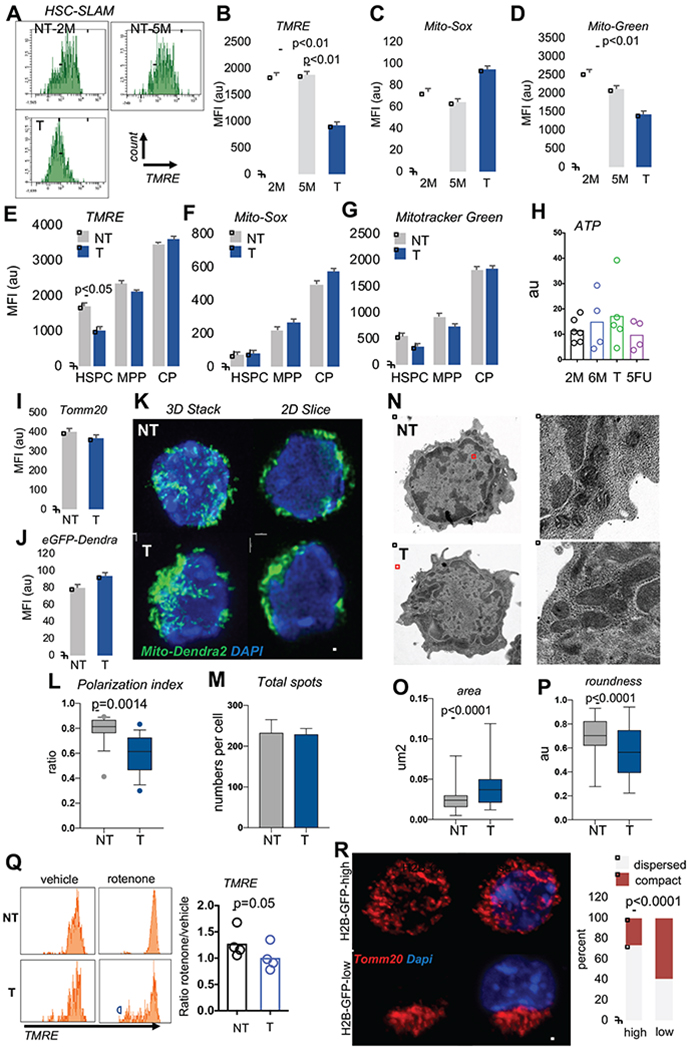
(A) Representative flow cytometry histogram of mitochondrial TMRE levels in 2 month(M)-old and 5 month-old non-transplanted [NT] mice, and transplanted [T] mice.
(B-D) quantification of mitochondrial parameters in 2M-old, and 5M-old NT SLAM and T SLAM, (mean±sem; n=7 mice).
(E-G) quantification of mitochondrial parameters, mean±SD, n=6 mice. (HSPC: LSK-CD48-; MPP: LSK-CD48+; CP:LK)
(H) quantification of ATP production in SLAMs from NT (2M, 6M), T and 14 days after 5FU (mean±SD, n=5).
(I) mitochondrial mass in NT and T SLAMs using Tom20 staining, mean fluorescent intensity (MFI) of Tom20 staining (mean±SD, n=203 in NT; 95 in T).
(J) Quantification of mito-Dendra2 MFI by flow cytometry mean±SD, n=6 mice.
(K) Representative IF images of mito-Dendra2 NT or T SLAM, shown as a 3D stack or one 2D slice view. scale bar 2μm.
(L-M) Mito-Drendra2 signals analyzed in Imaris using spot detection algorithm positioned relative to a reference point. (L) Polarization of mitochondrial spots per cell. (M) Total number of mito-Dendra2 spots per cell (n=24 NT and 20T, one representative experiment of 3)
(N-P) Mitochondrial analysis using transmission electron microscopy. (N) Representative images of NT and T SLAM. Zoom view of cell area highlighted in red is shown on the right images. (O) Area occupied by mitochondria. (P) quantification of roundness of mitochondria. (n=77 NT, 99 T mitochondria in 24NT and 19T cells).
(Q) Flow cytometry histogram and quantification of TMRE in NT and T HSPC after treatment with rotenone (1uM). (mean±s.e.m, paired student T-Test).
(R) Representative IF images of Tom20 staining in SLAMs from GFP-labeled H2B mice [high GFP and low GFP] isolated after 5 months of doxycycline removal and frequency of SLAMs with large mitochondrial clusters (n=90-100 cells; exact fisher test). All data are from 2-3 independent experiments. scale bar 2μm
To determine if mitochondrial morphology also changes in HSCs that have divided several times in vivo under homeostatic conditions, we used the doxycycline-inducible histone-2B [H2B]-GFP fusion label-retaining assay (Wilson A, 2008) and analyzed HSCs 5 months after doxycycline removal. HSCs with high GFP levels that have divided less number of times display more potency than those that have less GFP which have divided more times(Qiu et al., 2014; Wilson A, 2008). Five months after doxycycline removal (Figure S1K), mitochondria were well dispersed and homogeneously distributed in H2B-GFP-label-retaining SLAMs, whereas a significant proportion of H2B-GFP-low SLAMs had larger and more compact mitochondria (Figure 1R). Hence, once HSCs have divided and returned to quiescence under homeostatic or regenerative conditions, mitochondria do not completely return to their initial network organization or functional state.
Loss of mitochondrial dynamism in HSCs after replication causes asymmetric segregation of mitochondria during HSC division.
To examine mitochondrial organization upon cell activation mito-Dendra2 SLAMs were activated in vitro for 24h, fixed and imaged. In NT activated SLAMs, the mitochondrial network was dispersed randomly through the cell body and extended into distinguishable mitochondrial filaments as well as smaller mitochondrial units or short segments. Some NT SLAMs exhibited a more connected network into long filaments than others but they were able to scatter in the 3D space (Figure 2A–E and Movie 1). However, in activated T-SLAMs mitochondria remained organized in a more compact network of intertwined filaments that failed to scatter (Figure 2A–E and Movie 2). Because HSCs are round cells and contain mitochondria dispersed in 3D space, an accurate measure of mitochondria length is not possible. Instead, we used Imaris surface building algorithm to cover the mitochondrial network into surface objects (Figure S2A). The number of surfaces and volume per surface provide information on distribution of the mitochondrial network. The total mitochondrial volume of surfaces per cell remained unchanged between the groups. However, the numbers of surfaces were lower in T-SLAMs than in NT SLAMs and the volume per surface was higher, indicative of a more compact mitochondrial network in T-SLAMs (Figure 2A,C). The observed frequency of cells with a compact mitochondrial organization was higher in T-SLAMs (Figure 2D). Consistently, the mitochondrial network was more polarized in T-SLAMs (Figure 2E).
Figure 2. Mitochondria are asymmetrically inherited during HSC division.
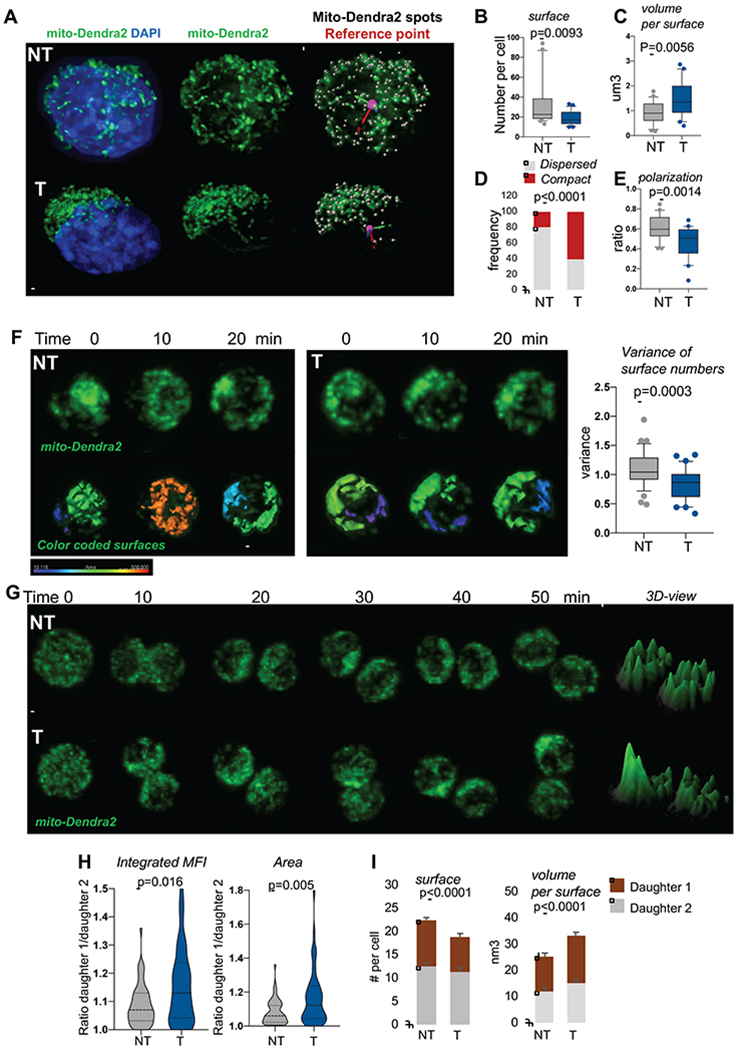
(A) Representative IF images of mitochondria in activated SLAMs, scale bar 1μm. Mitochondria in green, nucleus stained with DAPI in blue. Scale bar 2μm
(B-C) Mitochondrial network analyzed using Imaris surface building algorithm. (B) Number of separated mito-Dendra2 segments identified as surfaces per cell. (C) Average volume per mito-Dendra2 segments per cell, n=21 NT and 25 T.
(D) Quantification of cells observed with dispersed versus compact mitochondrial organization; n=57 for NT, 74 for T, fisher exact test.
(E) Quantification of polarization of mitochondria, n=23 NT and 24 T.
(F) NT and T mito-Dendra2 SLAM were imaged live during interphase for several minutes. Representative IF images and quantification of mitochondrial motility. Graph is variance of surfaces per time point, n=36. Scale bar 5μm
(G-I) NT and T mito-Dendra2 SLAM divisions were traced in vitro using live IF. (G) Representative images of mitochondria during mitosis, 3D representation of maximum intensity project using ImageJ. Scale bar 5μm (H) Integrated MFI and area occupied by mitochondria per daughter cell. Graph is ratio of each paired-daughter cell, n=55 NT and 44 T, Mann Whitney Test. (I) quantification of mitochondrial inheritance using Imaris surface building, numbers of surface per cell and average volumes per mitochondrial objects per cell are shown, mean±SD, n=30-35 cells.
Mitochondria are highly mobile and move throughout the cell body (Anesti and Scorrano, 2006). To examine this, mito-Dendra2 SLAMs were imaged live during HSC activation. Z-stack images were recorded every 10 minutes for several hours. The mitochondrial network was highly dynamic and motile in NT SLAMs since the organization of the network, assessed by number of surfaces per time point, changed every 10 minutes. However, the network remained unchanged over the course of several minutes in T-SLAMs such that the variance of number of surfaces calculated over one hour period was significantly lower (Figure 2F). The mitochondrial motility defects of T-SLAMs were not due to abnormal microtubules, as microtubule filaments extended across T-SLAMs cells similarly as NT SLAMs (Figure S2B), and thus may rather reflect impaired mitochondrial dynamism.
We next examined how mitochondria are inherited to the progeny of SLAM division using live imaging. Mitochondria were inherited homogeneously by daughter cells of most NT SLAM divisions since ratio of integrated MFI and area occupied by mitochondria in the daughter cells was less than 1.1 in 90% of SLAM divisions (Figure 2G–I). The mitochondrial organization remained quite similar in each daughter cell over several minutes following cytokinesis (Figure 2G–I). However, 40% of T-SLAM divisions were asymmetric with one daughter cell receiving more mitochondria, i.e. ratio of Integrated MFI more than 1.1 (Figure 2G–H), and/or had a more compact mitochondrial network than the other, i.e. ratio of area more than 1.1 (Figure 2G,H). Higher number of SLAMs also unequally distribute mitochondria after 5FU stress (not shown). Similarly, numbers of mitochondrial surfaces were lower in one daughter cell of T-SLAM division whereas the volume per surface was higher (Figure 2G,I). The differences between the 2 daughter cells were rarely more than 1.5 fold in this assay, as recently published (Loeffler et al., 2019). Loss of fidelity of mitochondria segregation and organization may prevent mitochondria from returning to homeostatic conditions.
Loss of Drp1 distribution onto mitochondria in HSC after replication.
The defects in mitochondrial functions seen in T-SLAM are reminiscent of defects in mitochondrial fission (Benard and Karbowski, 2009; Santel and Frank, 2008). Drp1, the main mitochondrial fission regulator, oligomerizes onto mitochondria to regulate mitochondrial division (Hatch et al., 2014). One way to measure Drp1 activity is thus assessing Drp1 distribution onto mitochondria (Adachi et al., 2016). We used Imaris spot detection algorithm to mark Drp1 signals, and quantified total numbers of Drp1 spots and numbers of Drp1 spots located within 0.3um of mitochondrial surfaces. We found numerous Drp1 spots onto mitochondria in NT SLAMs that were located at what appeared to be constriction sites, at the tip or between mitochondrial segments. The total number of Drp1 spots was significantly lower in T-SLAMs (Figure 3A,B). Further, the numbers of Drp1 spots close to mitochondria and at the tips of mitochondria were significantly lower in T-SLAMs (Figure 3A,C,D). In addition, the number of mitochondrial fission events was lower in T-SLAMs (Figure 3Avi,vii,E). This suggests that Drp1 failed to properly accumulate onto mitochondria in T-SLAMs, perhaps due to defective Drp1 expression. Quantification of Drp1 protein expression by flow cytometry confirmed reduced Drp1 expression in T-SLAMs (Figure S2C).
Figure 3. Impaired distribution of Drp1 on mitochondria in HSC after division. Mitochondrial remodeling drives HSC functional decline.
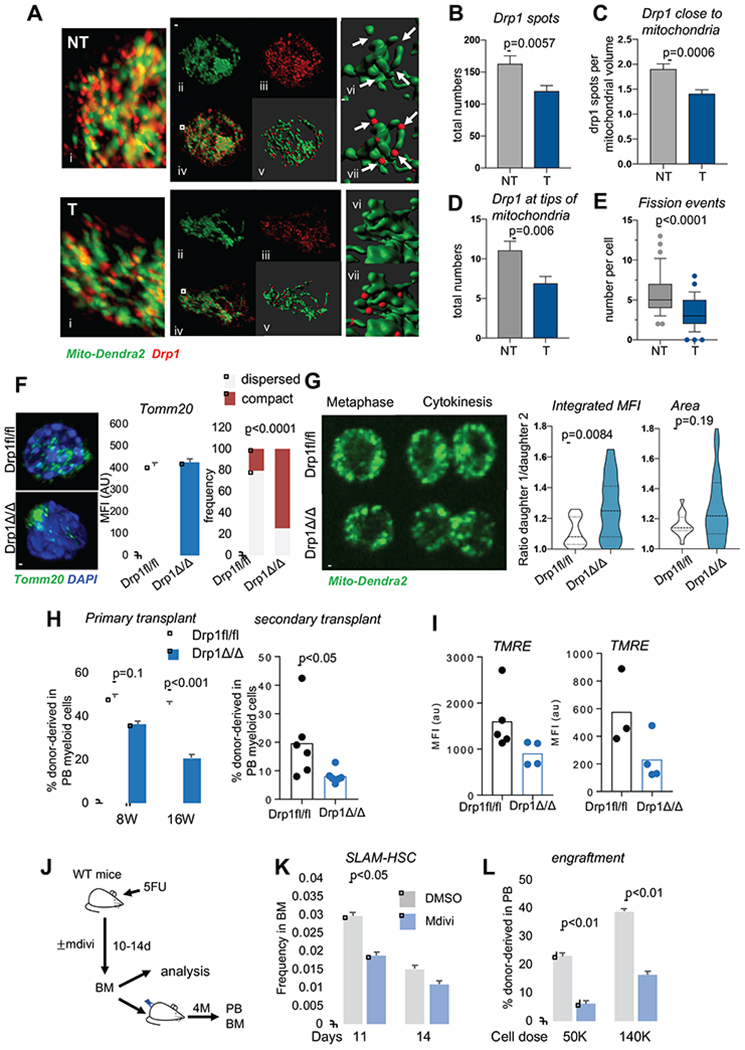
(A-E). Analysis of Drp1 onto mitochondria during NT and T SLAM activation in vitro(24h in culture). Quantification of mito-Dendra2 (green) was done with Imaris surface building. Drp1 signals (in red) were analyzed using Imaris spot detection. (A) Representative IF images Scale bar 5μm. (B) Total number of Drp1 spots per cell. (C) Numbers of Drp1 spots that were within 0.3 microns of a mitochondrial surface. (D) Numbers of Drp1 spots that were at the tip of mitochondrial surfaces. (E) Numbers of fission events identified at mitochondrial segments separated by Drp1 spots per cell. (mean±s.e.m, n=27NT and 28T cells, unpaired T-Test, representative experiment. Analysis was done blind by 2 individuals
(F) Representative IF images, quantification of Tom20 and observed cell frequency with dispersed vs compact mitochondria in Drp1fl/fl and Drp1Δ/Δ SLAMs (n=100-120 cells; exact fisher test). scale bar 5μm
(G) Mito-Dendra2 Drp1fl/fl and Drp1Δ/Δ SLAM divisions were traced in vitro using live IF imaging. Representative images of mitosis, scale bar 5μm. Graphs are ratio of Integrated MFI and area occupied by mitochondria per daughter cell. (n=25 divisions, Mann-Whitney Test)
(H) Serial competitive transplantation of Drp1fl/fl and Drp1Δ/Δ BM cells showing donor chimerism in peripheral blood in myeloid cells 4 months post-primary transplant and 3 months post-secondary transplant. mean±SD, n=12 mice for primary transplant; n=6 for secondary transplant.
(I) TMRE in Drp1fl/fl and Drp1Δ/Δ SLAMs 4 months post-transplantation (n=3-4 mice per experiments, 2 independent experiments are shown).
(J-L) WT mice challenged with 5FU were treated with mdivi or vehicle (DMSO) for 14 days. BM was analyzed and used for subsequent competitive transplant. (J) scheme of experiment. (K) donor-derived SLAM frequency in BM of treated mice. (L) donor chimerism in PB at 16 weeks post-transplantation (mean±SD, n=7-9 mice).
To determine the functional consequences of loss of Drp1 and changes in mitochondrial morphology in HSC, we used conditional genetic deletion of Drp1 (Junko Wakabayashi, 2009) (Figure S2D). Drp1Δ/Δ SLAMs carry severely aggregated mitochondria (Figure 3F). Drp1Δ/Δ SLAMs had low mitochondrial motility during cell activation (Figure S2E) and asymmetrically segregated mitochondria at cytokinesis in vitro, with one daughter cell inheriting more mitochondria than the other. In this case, each Drp1Δ/Δ daughter cell exhibited a more compact mitochondrial network than Drp1fl/fl control, such that the area occupied by mitochondria was not different between the daughter cells, although the area variance was notably higher in Drp1Δ/Δ SLAMs (Figure 3G and S2F). This suggests that mitochondrial inheritance during cytokinesis and their dispersed distribution in daughter cells are Drp1-dependent. Of note, mitochondrial scattering during metaphase was still evident in T-SLAMs (Figure 2G). It also partially occurred in Drp1Δ/Δ SLAMs, and thus compensatory pathways may exist. The hematopoietic parameters in Drp1Δ/Δ mice were normal 4 weeks after Drp1 deletion (not shown). Three-4 months after deletion, Drp1Δ/Δ mice acquired higher frequency and total numbers of SLAMs and MPP than Drp1fl/fl mice (Figure S2G,H). There were no changes in the blood parameters and mature lineage differentiation (Figure S2I–K). Drp1Δ/Δ SLAMs maintained quiescence, MMP and MitoSox similar to littermate controls at steady state (Figure S2L,M). Repopulation was assessed into sublethal irradiated immunodeficient mice due to mixed background of Drp1fl/fl mice. Interestingly, Drp1Δ/Δ cells had lower repopulation activity in serial competitive transplant studies, as seen by significantly lower percent donor-derived hematopoietic cells in the peripheral blood of recipients (Figure 3H and S3A,B). The frequency of regenerated Drp1Δ/Δ SLAMs was unchanged or slightly higher in the BM of transplanted recipients compared with WT cells 4 months post-transplant (Figure S3C–F). MMP was lower in Drp1Δ/Δ SLAMs after transplantation (Figure 3I), and MMP loss in response to rotenone challenge was further decreased compared with Drp1fl/fl SLAMs (Figure S3G). Together, deletion of Drp1 causes mitochondria to aggregate in HSCs, induces an increase in HSC frequency, yet a decrease in HSC regenerative potential, suggesting that loss of Drp1 favors the generation of a low-functioning HSC population, mimicking the function of T-SLAMs and HSCs with history of division.
Impaired mitochondrial dynamics during HSC division causes HSC functional decline of the progeny.
To confirm that changes in mitochondrial morphology directly cause HSC functional decline, young HSCs were acutely treated with Mdivi, an inhibitor of Drp1 activity that causes mitochondrial stress (Figure S3H) (Quiros et al., 2017; Bordt et al., 2017) but does not alter cell survival (not shown). HSCs were treated for 48h at concentration, then transplanted in vivo in competitive transplantation settings. Remarkably, Mdivi-treatment decreased durable myeloid reconstitution potential of cultured HSCs, compared to vehicle-treated control (Figure S3I), suggesting early decline in long-term repopulation activity (Dykstra et al., 2007). We next tested the ability of Mdivi-treated SLAMs to generate daughter cells that retain multilineage potential at the single cell level using the paired-daughter cell assay in vitro (Ema et al., 2000; Hinge A, 2017). Mdivi-treated SLAMs were no longer able to generate daughter progeny with myeloid multilineage potential (Figure S3J), similarly to T-SLAMs (Hinge A, 2017). During 5FU-induced acute stress in vivo (Figure 3J), mice that were treated with Mdivi had a modest reduction in the frequency of the phenotypically-defined HSPC pool at days 10-14 (Figure 3K). However, the reconstituted HSPC pool had dramatically lower regenerative potential in subsequent transplant studies (Figure 3L). We verified lack to toxicity of Mdivi in vivo treatment under these experimental conditions (not shown). Thus, mitochondrial dynamics is an upstream regulator of HSC regenerative activity. HSC mitochondrial morphology remodels after replicative stress, causing a functional decline of the newly generated HSC pool.
HSC carrying dysfunctional mitochondria fail to synchronize the expression of core component cell cycle and biosynthetic processes genes during division.
To understand why HSC that possess dysfunctional mitochondria are less potent, we analyzed the transcriptome of SLAM and T-SLAM using single-cell RNA sequencing (scRNA-seq), after cell isolation (quiescent; qSLAM and T-qSLAM), 15h after activation in vitro culture (activated: aSLAM and T-aSLAM) under conditions that maintain HSC stemness (Dykstra et al., 2007; Yamamoto et al., 2013), and after a single division in vitro based on dilution of a transgenic-H2B-GFP reporter (divided; divSLAM and T-divSLAM) (Figure 4A). Under these conditions, T-divSLAMs had reduced engraftment ability (Figure S3K). ScRNA-seq analysis was performed using two independent unsupervised algorithms; iterative clustering and guide-gene selection (ICGS) and principal component analysis (PCA)(Olsson A, 2016). Supervised analysis by pairwise comparison was also performed. Unbiased ICGS analysis identified 4 cell populations, which were mainly separated based on their cell-cycle status (Figure 4B). Populations 1 and 2 were predicted to be in G0 and mostly contained qSLAMs and T-qSLAMs. Populations 3 and 4 were in G1/S and S/G2/M, respectively, and composed of aHSCs, divHSCs, T-aSLAMs, T-divSLAMs and few qSLAMs. All cells expressed canonical HSC transcription factors without the coincident expression of genes that define lineage-specific committed progenitors, suggesting that T-SLAMs were not lineage-primed (Figure S3L). Thus, under these experimental conditions, all populations are in a comparable HSPC state even after activation and division in vitro. However, ICGS pairwise comparative and PCA analyses, independently identified distinct regulatory states (Figure 4C,D). Notably, aSLAMs and div-SLAMs increased expression of genes related to ‘cell division’, ‘metabolism’, ‘mRNA processing’, but also genes belonging to ‘mitochondrial organization’, ‘the citrate acid cycle’ and ‘oxidative phosphorylation’ (Figure 4C–E and S3M,N), consistent with mitochondrial activation during HSC cell-cycle entry in vitro (Figure S1). Transcriptional signature of freshly isolated T-qSLAMs showed that these cells have mostly deactivated expression of cell cycle, metabolic and biosynthetic programs after the stress of transplantation, indicating that HSCs had returned to a quiescent and inactive state after replication; albeit some differences exist (Figure S4A–C). In contrast, more profound differences were noted during division. T-aSLAMs and T-divSLAMs failed to upregulate or to maintain the upregulation of genes categorized in ‘biosynthetic process’ ‘mitochondrial part’, ‘the citrate acid cycle’, ‘fatty acid oxidation’ and surprisingly ‘oxidative phosphorylation’ through cell division (compared to aSLAMs and divSLAMs), despite activating expression of cell cycle checkpoint and glycolysis genes (Figure 4D,E and S3M,N). Cells were also separated based on their cell cycle status, ie G1 or SG2M, and independent analyses were run. Interestingly, nuclear-encoded mitochondrial genes were downregulated both in T-SLAMs going through G1 and SG2M cell cycle phases compared with SLAMs (Figure S4D–H). Of note, genes categorized in ‘Electron transport chain’ were downregulated mostly in T-SLAM cells identified to be in G1 (Figure S4G). Consistent with a defect in Drp1 activity during T-SLAM division, Drp1 mRNA expression was significantly lower in T-aSLAMs and T-divSLAMs than controls (Figure S4I). To confirm these findings without in vitro manipulation, we used AltAnalyze to cluster freshly isolated qSLAMs and T-qSLAMs based on their coincident expression of cell cycle genes. Nuclear genes encoding mitochondrial functions were robustly expressed in qSLAMs estimated to be in S/G2/M; however, T-qSLAMs estimated to be in S/G2/M failed to robustly upregulate these same genes (Figure 4F, and S4J). Hence, the in vitro increased expression of mitochondrial genes in aHSCs and their lack of induction in T-aHSCs is physiologic, as it can be reproduced in HSC freshly isolated from the bone marrow. Moreover, PCA analysis comparing only activated SLAM populations confirmed T-divSLAMs had less expression of genes categorized in ‘mitochondrial part and organization’, (eg, oxidative phosphorylation, TCA cycle, fatty acid beta-oxidation), ‘protein translation’ and ‘biosynthetic and catabolic processes’ (Figure 4G). Together the data indicate that T-qSLAMs are transcriptionally similar, albeit not identical, to qSLAMs; however, profound differences are manifested during division. Exit from quiescence in a non-stressed HSC population is coupled with activation of metabolic processes. After replicative stress, HSCs deactivate the expression of metabolic and biosynthetic programs, although they maintain high oxidative stress. They subsequently fail to upregulate pathways for metabolic and proteostatic activation during cell division which contributes to reduced regenerative potential.
Figure 4. Asynchrony in expression of nuclear-encoded mitochondrial genes in HSC after division.
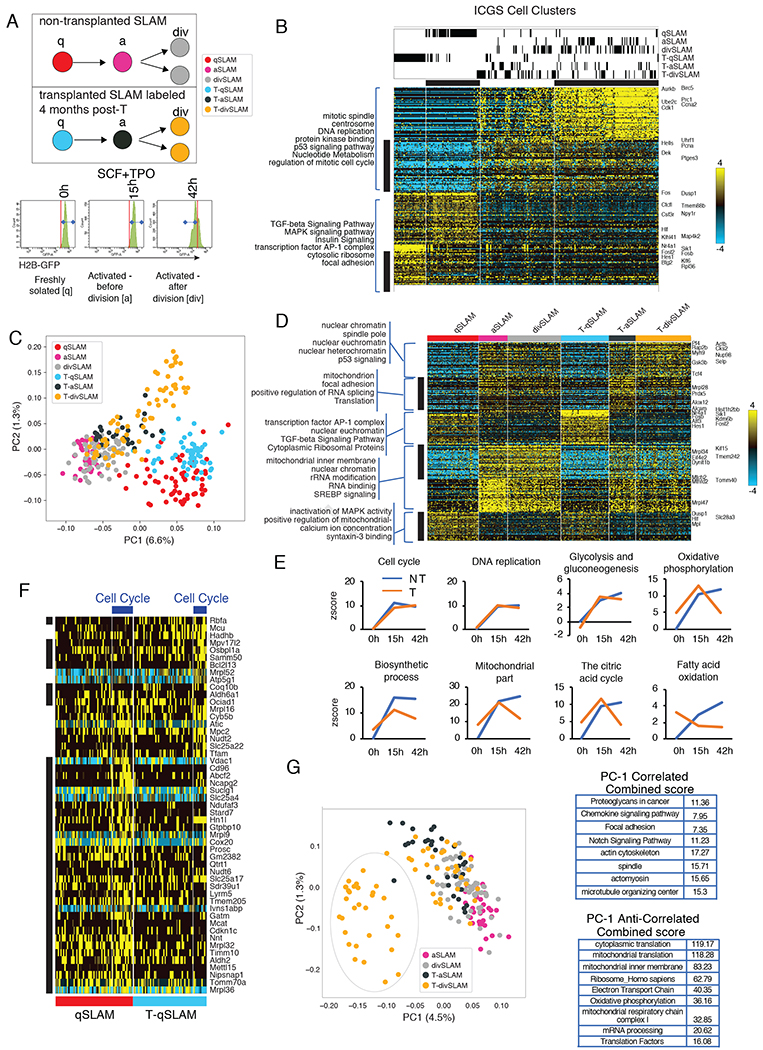
(A) Schema of gene expression analysis by sc-RNA-seq using H2BeGFP labeled SLAM from naïve animals or after transplantation on freshly isolated cells, activated cells (15 hours) and on cells that have divided once (42 hours, see Figure S4 and Table S1 for datasets).
(B) Heatmap of genes delineated by ICGS in scRNA-Seq data (n=301 cells). Columns represent cells. Rows represent genes. ICGS cell clusters are indicated and cell populations comprised in each cluster is indicated at the top. ICGS guide genes are displayed (right). (see Table S1)
(C) PCA visualization of indicated cells.
(D) Hierarchical clustering of differentially expressed genes in indicated population using pairwise comparative analysis. (see Table S1)
(E) Differential gene expression of each group of cells was compared to qSLAM. Graphs are z-scores of indicated GO category.
(F) Hierarchical clustering of differentially expressed mitochondrial genes in freshly isolated SLAM from naive non-transplanted [qSLAM] and transplanted animals [T-qSLAM]. (see Table S1)
(G) PCA visualization of indicated cells. Bar graph is top gene ontology category of differentially expressed genes represented in PC1.
Discussion
Permanent mitochondrial remodeling with division can explain why HSCs that have divided are less potent than HSCs that have never divided in similar microenvironment (Qiu et al., 2014; Wilson A, 2008) or why the newly regenerated HSC pool after bone marrow transplantation is much less potent than the initially engrafted HSC pool (Hinge A, 2017; Yamamoto et al., 2013). We propose that HSCs use mitochondria as a natural checkpoint to remember their divisional history and limit their self-renewal ability.
Our data underscore the importance of examining HSC functionality not only when the cells are quiescent but also during active cycling when more profound differences are manifested. Enhancing, but not lowering biosynthetic process or oxidative phosphorylation may be needed during stress to prolong HSC functions. Interestingly, although MFN2 is important to maintain HSCs with lymphoid potential (Luchsinger LL, 2016), T-HSCs or Drp1Δ/Δ HSCs do not have lineage-bias. These complementary findings underscore the complexity of mitochondrial regulation in HSCs that will require further investigation.
The role of mitochondria in asymmetric cell division is cell-type dependent, and differs in yeast and other mammalian cell types (BenediktWestermann, 2014) (Pekkai Katajisto, 2015). In HSC, mitochondrial fatty acid oxidation controls asymmetric division (Ito et al., 2012) and the lysosomal compartment can be asymmetrically segregated (Loeffler et al., 2019). Intriguingly, our data strongly suggest that HSC keep dysfunctional mitochondria, since HSC carry dysfunctional mitochondria whereas LSK-CD48+ have an unperturbed mitochondrial functions. Drp1 loss does not deplete but expand the HSC pool, indicating that HSC tolerate and keep dysfunctional mitochondria.
Why do HSCs keep dysfunctional mitochondria? Since there is a tight correlation between HSC functional decline with divisional history (Qiu et al., 2014), and mitochondrial remodeling (Figure 1), keeping dysfunctional mitochondria may be one aspect of HSC divisional memory division, although other cellular mechanisms likely contribute. The high mitochondrial mass of HSC (de Almeida MJ, 2017) may thus constitute a ‘reserve’ of mitochondria endowed to young HSCs. The rate of accumulation of dysfunctional mitochondria in HSCs at each round of division may determine the ‘lifespan’ of the stem cell pool. Keeping dysfunctional mitochondria through asymmetric division may be one – of many – physiological mechanism of cellular memory ensuring that HSCs have only limited self-renewal capacity to prevent unlimited numbers of self-renewing division (Bernitz JM, 2016; Qiu et al., 2014).
STAR Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marie-Dominique Filippi (Marie-Dominique.Filippi@cchmc.org). This study did not generate new unique reagents.
Experimental Model and Subject Details
Mice:
Young (6-12 weeks) and middle-aged (15 months) C57BI/6 mice of both gender were bred and aged in house. Transgenic R26-M2rtTA;Col1a1-tetO-H2B-GFP (H2B-EGFP) and B6;129S-Gt(ROSA)26Sortm1.1(CAG-COX8A/Dendra2)Dcc (mito-Dendra2) (Pham et al., 2012) were purchased from Jackson laboratory. In, mito-Dendra2 mice, the mitochondrial targeting element of cox8, a mitochondrial outer membrane protein, is fused with eGFP-dendra2 to specifically label mitochondria. Conditional Drp1flox/flox mice (SV129 background) were obtained from Dr Hiromi Sesaki (Junko Wakabayashi, 2009) and crossed with MxCre1 mice to generate Drp1flox/flox:MxCre mice. Cre recombination was induced with polyI-C (GE Healthcare) injections (10μg/g body weight, three intraperitoneal injections every alternate day for one week). PolyIC-injected Drp1flox/flox mice were used as controls. Drp1flox/flox:MxCre and Drp1flox/flox mice mice were used 4 weeks after poyIC injection. All animals were bred in house in pathogen-free environment. B6.SJL- Ptprca (BoyJ) mice were used as recipients when donor cells were isolated from C57BI/6 (Ptprcb) mouse background. NOD/SCID gammac−/− (NSG) mice were used as recipients when donor cells were obtained from mixed mouse background. In the study, both male and female mice were used. All studies were conducted with a protocol approved by the Animal Care Committee of Cincinnati Children’s Hospital Medical Center.
Method Details
Single cell RNA sequencing:
Naïve H2B-eGFP mice (6-10 weeks old) were treated with doxycycline for one week to label the cells. Lin−Sca-1+c-Kit+CD48−CD150+ H2B-eGFP+ cells (LSK-SLAM eGFP+) were isolated (BD FACS Aria II, BD Biosciences). Freshly isolated cells were used immediately to perform single cell RNA sequencing (0 hour time point). LSK-SLAM H2B-eGFP + cells were also incubated in stemspan medium (Stem cell technology) supplemented with murine stem cell factor (SCF; 100ng/ml; Peprotech) and murine thrombopoietin (TPO; 100ng/ml; Peprotech) at 37°C for up to 42 hours in 5% CO2 incubator. Cells were harvested after 15 hours in culture for single cell RNA sequencing to examine transcriptome in activated HSCs. After 38-40 hours in culture, cells were re-isolated based on H2B-eGFP dilution for single cell RNA sequencing to analyze transcriptome of cells that have divided once. The cells were loaded on 5-10micron chip. Single cells were confirmed visually under the microscope; cDNA and libraries were prepared by the CCHMC Gene Expression Core. Deep-sequencing was done by the CCHMC DNA core. Similar experiment was performed after transplanting H2B-eGFP bone marrow (BM) cells. In this case, non-pre labeled unfractionated BM cells from H2B-eGFP mice (one million) were transplanted with BoyJ competitor cells (one million) into in lethally irradiated BoyJ recipient mice. Four months after transplantation, mice were treated with doxycycline for one week and H2B-eGFP LSK-SLAM were isolated. scRNA-seq was performed at 0 hour, at 14 hours of culture and after cell division (42 hours in culture). Gene-expression clusters were generated in AltAnalyze using the HOPACH algorithm (including cell cycle). Bioinformatic analyses – unsupervised ICGS, pairwise comparative analyses and PCA were performed to compare transcriptome between the groups using AltAnalyze. (Olsson A, 2016). Two tailed empirical Bates moderated t-test from AltAnalyze was used with p<0.05 and fold>2 and minimum number of changed genes was 3. Gene ontology analyses of PCA genes was performed using ToppGene Suite and EnrichR softwares (Chen et al., 2013; Chen et al., 2007; Kuleshov et al., 2016). For analysis of nuclear-encoded genes in SLAM going through cell cycle in vivo, qSLAM and T-qSLAM cells were segregated based on their cell cycle status, which was identified in AltAnalyze using G1-S and G2M cell cycle phase gene expression. Coincident expression of nuclear-encoded genes important for mitochondria are shown as ‘Mitocarta’.
Flow cytometry staining:
Hematopoietic stem cells were analyzed and isolated as described previously. (Hinge A 2017) BM cells were obtained by flushing the bones in Hanks’ Buffered Saline Solution (HBSS) containing 10% heat inactivated fetal bovine serum (Atlanta). BM cells were either used unfractionated or separated by density gradient Ficoll histopaque separation (Sigma 1083). For LSK-SLAM isolation, low density bone marrow cells were enriched for c-Kit+ cells using c-Kit magnetic microbeads. C-Kit+ cells were separated using Automacs pro cell separator machine (Miltenyi biotec). C –Kit enriched cells were immuno-stained for specific surface markers for LSK-SLAM. All antibodies were from BD bioscience unless specified. Briefly, cells were incubated with biotin labeled anti-mouse lineage antibodies (Ter119 (Cat 51-09082J, lot number- 6169946), B220 (Catalogue number – 51-01122J, lot number – 6169942), Gr-1 (catalogue number- 51-01212J, lot number- 6169943), CD11b (catalogue number- 51-01712J, lot number- 6169944), CD3e (catalogue number- 51-01082J, lot number – 6169941) followed by staining for streptavidin, Sca-1, c-Kit, CD48, CD150 (eBioscience) and CD45.2. Different combinations of flurochromes were used, such as lineage V500/ APCCy7 (catalogue number- 561419, lot number – 80118820 and for APCCy7 catalogue number- 554063, lot number – 6287843), Sca-1 PECy7 (clone- D7, catalogue number- 558162, lot number- 7153905), c-Kit – APCCy7/PE (clone-2B8, catalogue number-47-1171-82, lot number – 4337566, Invitrogen and for PE catalogue number – 553355, lot number – 15226, BD biosciences), CD48 AF700/FITC/BV605 (clone- HM48-1, catalogue number- 103426, lot number- B236445, Biolegend, for FITC catalogue number – 557484, lot number – 6168976, BD biosciences and for BV605 catalogue number- 740353, lot number – 8073937, BD bioscience), CD150 APC/FITC/PE (Clone – 9D1, catalogue number -17-1501-81, lot number- 1950705, Invitorgen, for FITC catalogue number – 11-1501-82, lot number – 4278033, eboscience and for PE catalogue number – 12-1501-82, lot number – E01517-1632, ebioscience) and CD45.2 Percpcy5.5/AF700 (Clone –104, catalogue number- 560693, lot number – 7034806 and for percpcy5.5 catalogue number – 552950, lot number – 6021537, BD bioscience) These cells were further sorted using BD FACSAria II, Cell sorter (BD Bioscience).
For cell analyses, unfractionated cells were used for mitochondrial staining. Cells were first stained for LSK-SLAM, as above. Then cells were washed and incubated at 37°C and 5% CO2 for 30 minutes with mitotracker dyes such as mito sox (1μM to measure mitochondrial superoxide anions, Invitrogen) or TMRE (0.1μM to measure mitochondrial membrane potential, Sigma) or mitotracker green (25nM, Invitrogen). Cells were washed and were acquired on BD FACS Canto flow cytometer (340558) within 60 minutes of mitochondrial staining. Mitochondrial parameters were analyzed in different HSC population using FACS diva software (BD Bioscience). For Drp1 quantification by flowcytometry, cells were first stained for LSK-SLAM cell surface markers and then cells were fixed and permeabilized with BD fixation/permeabilization solution kit (Catalogue number- 554714). Cells were then incubated in rabbit recombinant monoclonal Drp1 antibody (abcam-184247) (1:100) overnight. Cells were stained with anti-rabbit Alexa 546 (Invitrogen-A11010) secondary for 1 hour at 4 degrees. For combined HSC and MPP analysis, unfractionated BM cells stained with various combination of flurochromes such as lineage V500/ APCCy7, Sca-1 PECy7, c-Kit – APCCy7/PE, CD48 AF700/FITC/BV605, CD150 APC/FITC/PE and CD45.2 Percpcy5.5/AF700, CD34 efluor 450 (clone- RAM34, catalogue number – 48-0341-82, lot number – E10919-1637, e bioscience) and CD16/32 AF700 (clone-93, catalogue number- 12-0161-82, lot number- 4274520).
For mature cell analysis in peripheral blood, peripheral blood was collected after 8 and 16 weeks of transplant by retro-orbital puncture. Red blood cells were lysed using lysis buffer (BD bioscience). When BoyJ mice were used as recipients, cells were immuno-stained for CD45.1 PE/BV605 (clone-A20, catalogue number- 553776, lot number- 5230561 and for BV605 catalogue number- 563010, lot number- 7075978, both BD Bioscience) , CD45.2 Percpcy5.5/AF700/FITC (Clone –104, catalogue number – 552950, lot number – 6021537, for AF700 catalogue number- 560693, lot number – 7034806 and for FITC catalogue number- 553772, lot number – 5356548 both BD bioscience) or when NSG mice were used as recipients, H2Kd FITC/PE (clone- SF1-1.1, catalogue number- 553565, lot number – 0000069718 and for PE catalogue number- 553566, lot number – 6053774 both BD bioscience) pan CD45 APCCy7 (clone – 30-F11, catalogue number- 557659, lot number – 4341648, BD bioscience) to identify recipient versus donor population. Cells were immune-stained for blood mature lineages Gr-1 AF700/ FITC (clone- RB6-8C5, catalogue number- 557979, lot number- 7125812, B D Bioscience and for FITC, catalogue number- 108419, lot number –B163253, Biolegend), Mac1 AF700/FITC (clone- M1-70, catalogue number – 557960, lot number – 7046745, and for FITC catalogue number – 553310, lot number – 3010776, BD bioscience), B220 PECy7 (clone -CD45R/B220, catalogue number- 552772, lot number 6105665, BD bioscience) , CD3e APC/PE (clone – 145-2C11, catalogue number- 561826, lot number – 8005903 and for PE catalogue number- 553048, lot number – 6007736, both BD bioscience) and CD8 APC/PE (clone – 53-6.7, B catalogue number - 553035, lot number – 5341621 and for PE catalogue number – 553032, lot number 6110823 both BD bioscience). Similar protocol was used for mature cell analysis in BM.
In vivo treatment:
5 fluorouracil was injected intravenously, 5-FU (150mg/kg). Transgenic H2B-eGFP mice were treated with doxycycline (1g/500ml, Sigma) and sucrose (5g/500ml, Sigma) in drinking water for 1 or 2 weeks as per experimental requirements. Mice were injected with mdivi-1 (2.5mg/kg of mouse, Sigma) or DMSO intraperitoneally every day for two weeks following 5-FU challenge. All mice were sacrificed at required time interval and iliac crest, tibia and femur bones were harvested from the hind limbs of the mice.
Transplantation:
106 unfractionated bone marrow cells from C57BI/6 or mito-dendra2 mice were mixed with 106 unfractionated BM cells from BoyJ mice and were transplanted into lethally irradiated BoyJ recipient mice (1175 rads irradiation dose, 700+475 split dose 3 hours apart). Transplanted mice were used for experiments 4 months after transplantation. In another experiment, unfractionated BM cells from Drp-1f/f and Drp-1Δ/Δ mice (100x103 or 250x103 cells) were transplanted in sub-lethally irradiated (250 rads) NSG recipient mice due to mixed background of donor cells. Peripheral blood was collected by retro-orbital bleeding at 8 and 16 weeks post transplantation to analyze percent donor chimerism in the myeloid population by flow cytometry (percent-derived in the lymphoid population is 100 under these conditions). For secondary transplant, CD45+H2Kd-lin− population was sorted and 15x103 cells per mouse were transplanted.
Ex vivo culture:
To test response to mitochondrial stress, low density bone marrow cells isolated from naïve and transplanted mice. Cells were incubated with rotenone (1μM) or oligomycin (0.1μM) in Stemspan containing SCF+TPO (100 ng/ml each) at 37°C in 5% CO2 incubator, for 15 hours or 30 minutes, respectively. Cells were then used to examine TMRE in LSK-SLAM populations, as described previously. To test effect of Mdivi-1 on HSC repopulation potential, LSK-SLAM (500 from naïve mice) were isolated as above and were treated with mdivi-1 (1μM) or DMSO control for 48 hours in Stemspan containing SCF+TPO (100 ng/ml each) at 37°C in 5% CO2 incubator. Cells were then mixed with 250,000 unfractionated BoyJ BM cells and transplanted into lethally irradiated BoyJ recipients. We calculated the ratio of donor contribution to myeloid, B and T cells in each recipient mouse, and classified the reconstitution as myeloid-dominant, balanced or lymphoid-dominant according to the criteria previously assigned to similar patterns. (Dykstra et al., 2007)
In vitro paired daughter cell assay:
It was performed as described before. (Ema et al., 2000; Hinge A, 2017) Briefly, single LSK-SLAM cells were sorted into 96-well plate containing mdivi-1 (1μM) or vehicle control DMSO. Single cells were visually confirmed under light microscope and cultured in serum free Stemspan medium (Stem Cell Technology) supplemented with murine SCF and murine TPO (100 ng ml−1, each, Peprotech) during the first cell division only at 37°C in 5% CO2 incubator. After the first division, daughter pairs were separated and individually cultured in Iscove’s Modified Dulbeco’s Medium (IMDM) containing 10% fetal bovine serum (Omega) and a cocktail of cytokines allowing for myeloid differentiation (murine SCF, murine TPO, human G-CSF (each 20 ng ml−1), murine IL-3 (50 ng ml−1) and EPO (4 U ml−1; Espogen)) for 14 days at 37°C in 5% CO2 incubator. Paired-clones were harvested and used for cytospin preparation. Cells of various lineages were identified based on their morphology after Diff-quick staining (Siemens). Clones were examined for the presence of neutrophils (n), erythroid cells (e), macrophages (m) and megakaryocytes (M; nemM) and scored as symmetric if each daughter clones contains nemM cells, asymmetric is only one daughter clone is nemM and committed if none of the daughter clones are nemM.
Confocal microscopy:
Analysis on fixed cells: LSK-SLAM cells were sorted and incubated at 37°C on retronectin coated slides for one hour or 24 hours as required for different experimental conditions (details mentioned in the respective figure legends and in results). After incubation, cells were fixed with 4% paraformaldehyde at room temperature for 20 minutes, permeabilized with 0.1% TritonX 100 and blocked with 2% bovine serum albumin in PBS. For tubulin staining, cells were fixed with 4% paraformaldehyde at 37°C in 5% CO2 incubator for 30 minutes. Cells were immuno-stained with rabbit anti Tomm20 antibody (1:100 dilution, clone – EPR5581-39 catalogue number- ab186734, lot number – GR3195073-4, abcam), mouse anti Drp-1 antibody (1:100 dilution, clone-8-DLP-1, catalogue number- 611112, lot number – 6214737, abcam BD transduction laboratories), rabbit anti alpha tubulin (1:100 dilution, polyclonal, catalogue number- ab18251, lot number-GR324760-1, abcam) at room temperature for 60 minutes. Secondary antibodies in anti-rabbit AF488 (catalogue number–A21206, lot number–1874771, Invitrogen) or anti-rabbit AF546 (1:100 dilution, catalogue number – A11010, lot number – 1733163, life technologies) and anti-mouse AF555 (1:100 dilution, atalogue number – A21422, lot number – 1685557, life technologies) were incubated at room temperature for 60 minutes. The cells were mounted using slowfade gold antifade reagent with DAPI (Invitrogen). Images were captured using 60X lens objective with Z stacks (Nikon confocal microscope, model 552541 T1-HUBC/A).
For high resolution imaging, single cell was imaged using Nyquist limit setting (0.1um XY pixel size) at 60X magnification using 1.2 AU pinhole to give an optical section thickness of 0.39 um. Z-stacks were acquired with a step size of 0.1 um. Typically 100-120 Z sections were taken. Deconvolution analysis of the images was done using NIS elements Image analysis was performed using Imaris or NIS elements softwares.
Live Imaging:
To understand mitochondrial inheritance to daughter cells, SLAM-HSC were isolated and incubated on slide in StemSpan plus SCF and TPO. Live Imaging was performed using confocal microscope (Nikon A1R GaAsP inverted confocal microscope) equipped with chamber with 37°C, 5% CO2 between time 30h to 45 hours to capture cell divisions. Images were captured using 20X lens objective with Z stacks using multipoint settings. Each point was captured every 10 minutes for 12 – 16 hours. Live imaging videos were edited using Nikon NIS elements software.
Transmission electron microscopy was performed as recently published using 10,000 SLAMs or CP isolated cells from NT and T mice. (Kumar and Filippi, 2016)
Quantification and Statistical Analysis
Analysis of images:
To calculate mean fluorescence intensity Nikon elements software was used. To analyze organization of mitochondrial network, visual analysis of Z stack images was done. To analyze number of surfaces, surface building using enable touching object feature of Imaris software was used. Total volume was extracted from detailed statistical analysis of the software. Total volume was divided with number of surfaces of each cell to obtain volume per surface. Polarization analysis: A reference point was positioned to the center of the nucleus (DAPI). This allowed the cell to be defined as 8 sections in accordance to the X, Y, and Z axes. Using IMARIS spot analysis mitochondria were positioned along the X axis and spots were created for the brightest fluorescence intensity along the Y and Z axes. Spots were then quantified per section. A cell was deemed polarized if one or more sections had a statistically higher number of spots. For analysis of Drp1 association with mitochondria, mitochondrial segments were identified using surface building algorithm. Drp1 oligomers were detected using spot detection algorithm and total numbers of spots and numbers of Drp1 spots close to mitochondrial surface (<0.3um) were measured using Matlab analysis in Imaris. Images were visualized and number of Drp1 spots at the tip of mitochondrial segments was numerated per cell. In addition, numbers of Drp1 spots separating 2 mitochondrial segments were numerated and presented as ‘fission event’ per cell. All quantifications were performed by 2 individuals and from independent experiments. Fission events were numerated in a blinded fashion by 2 individuals. For mitochondrial motility analysis from live imaging, 5-6 frames during interphase were extracted using Nikon elements software. Number of surfaces per frame were measured using surface building algorithm. For mitosis analysis from live imaging, 2-3 frames before and after cell division were extracted using Nikon elements software. Number of surfaces and volume of each daughter cell was analyzed as described above. Number of surfaces and volume of each daughter cell were compared with subsequent daughter cell to analyze differences between the cells. Volume based statistical coded analysis was performed using Imaris software between daughter cells. Maximum intensity 3D projection using NIS elements was also used for analysis of daughter cells. In addition, post-division images were analyzed using ImageJ threshold analysis and ratio of integrated MFI and area occupied by mitochondria were measured.
Data exclusion:
For quantification mitochondrial volume and number of surface in Imaris software, cells with fluorescence that is too low or too high (over-saturated) does not allow for detailed view of the mitochondrial network and for accurate quantification. These cells were thus excluded from the analysis.
Figures 1K, 1R, S2A, S3A, S3F, S3G, S2F–G, S1D, S1M, S2A, S2B, S2E, S2F, S3M are composite images.
Statistical analyses:
The results are presented as mean ± SD or as mean ± sem. Unless specified in legend all data are from 2-3 independent experiments. Unless specifically indicated, data were analyzed using unpaired student T- test; p values are indicated for each analysis. Analysis of response to mitochondrial stress was analyzed using paired student T-Test of rotenone or oligomycin relative to vehicle control. Mann-Whitney test was used when variances were significantly different between groups. Relative percent of cells with dispersed or aggregated mitochondrial network was analyzed using 2x contingency table Fisher exact test. Results of transplantation of cultured SLAM treated with mdivi-1 were analyzed using chi-square 3x contingency table comparing frequency of mice presenting as myeloid-dominant, balanced or lymphoid-dominant reconstitution. Data presentation: Data with more than 5 values are presented as bar graph and SD or s.e.m. Data with 4 values or less are presented as bar graphs with overlay corresponding dot plots.
Supplementary Material
Table S1. Expression files of scRNA-seq datasets related to Figure 4
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies: Flow cytometry | ||
| Ter119 – biotin labeled | B D Bioscience | 51-09082J |
| B220 - biotin labeled | B D Bioscience | 51-09082J |
| B220 – PECy7 | B D Bioscience | 552772 |
| Gr-1 - biotin labeled | B D Bioscience | 51-09082J |
| Gr-1 – AF700 | B D Bioscience | 557979 |
| Gr-1 - FITC | Biolegend | 108419 |
| Mac1 - biotin labeled | B D Bioscience | 51-09082J |
| Mac1 – AF700 | B D Bioscience | 557960 |
| Mac1 - FITC | B D Bioscience | 553310 |
| CD3e - biotin labeled | B D Bioscience | 51-09082J |
| CD3e - APC | B D Bioscience | 561826 |
| CD3e - PE | B D Bioscience | 553048 |
| CD8 - APC | B D Bioscience | 553035 |
| CD8 - PE | B D Bioscience | 553032 |
| Streptavidin – V500 | B D Bioscience | 561419 |
| Streptavidin – APCCy7 | B D Bioscience | 554063 |
| Sca-1 - PECy7 | B D Bioscience | 558162 |
| c-Kit – APCef780 | Invitrogen | 47-1171-82 |
| c-Kit -PE | B D Bioscience | 553355 |
| CD48 – AF700 | Biolegend | 103426 |
| CD48 - FITC | B D Bioscience | 557484 |
| CD48 – BV605 | B D Bioscience | 740353 |
| CD150 - FITC | eBioscinece | 11-1501-82 |
| CD150 - PE | eBioscinece | 12-1501-82 |
| CD150 - APC | Invitrogen | 17-1501-81 |
| CD45.2 – percpcy5.5 | B D Bioscience | 552950 |
| CD45.2 – AF700 | B D Bioscience | 560693 |
| CD45.2 - FITC | B D Bioscience | 553772 |
| CD34 – ef450 | eBioscinece | 48-0341-82 |
| CD16/32 | Invitrogen | 12-0161-82 |
| CD45.1 - PE | B D Bioscience | 553776 |
| CD45.1 – BV605 | B D Bioscience | 563010 |
| H2Kd - FITC | B D Bioscience | 553565 |
| H2Kd - PE | B D Bioscience | 553566 |
| CD45 – APCCy7 | B D Bioscience | 557659 |
| Anti-Drp1 | abcam | Ab56788 |
| Mitotracker dyes: Flow cytometry | ||
| TMRE | Sigma- Aldrich | 87917 |
| MitoSox™ red | Invitrogen | M36008 |
| Mitotracker™ green FM | Invitrogen | M7514 |
| Antibodies: Microscopy / Imaging | ||
| tomm20 | Abcam | ab186734 |
| drp-1 | BD transduction laboratories | 611112 |
| alpha tubulin | Abcam | ab18251 |
| anti-rabbit – AF488 | Invitrogen | A21206 |
| anti-rabbit AF546 | Life technologies | A11010 |
| anti-mouse AF555 | Life technologies | A21422 |
| Chemicals | ||
| Polyinosinic:polycytidylic acid | GE health care | Tlrl-pic |
| Doxycycline | Sigma- Aldrich | D9891-25G |
| Sucrose | Sigma- Aldrich | 501212905 |
| Mdivi-1 | Sigma- Aldrich | M0199 |
| DMSO | Fisher Scientific | BP231-100 |
| Rotenone | Sigma- Aldrich | R8875 |
| Oligomycin | Sigma- Aldrich | 75351 |
| Ficol histopcque | Sigma- Aldrich | 10831 |
| Paraformaldehyde | Electron Microscopy Sciences | 15713 |
| Triton X-100 | Sigma-Aldrich | X10-100ml |
| Bovine serum albumin | Roche | 3117332001 |
| SlowFade gold antifade mountant with DAPI | Invitrogen | S36939 |
| Fetal bovine serum | Atlanta | S11550 |
| Fetal bovine serum | Omega | FB-01 |
| Diffquick staining | Siemens | B4132-A |
| Media: | ||
| Stemspan medium | Stem cell technology | 9650 |
| Hank’s buffered saline solution | 21-020-CV | |
| Iscove’s Modified Dulbeco’s medium | 10-016-CV | |
| Cytokines: | ||
| Murine stem cell factor | Peprotech | 250-03 |
| Murine thrombopoietin | Peprotech | 315-14 |
| Human granulocyte colony stimulating factor | GF05-20ug | |
| Murine interleukin-3 | Peprotech | 213-13 |
| Erythropoietin | Espogen | NDC55513-126-10 |
| Experimental Models: Strains | ||
| C57BI/6 | In house, CCHMC | n/a |
| B6.SJL-Ptprca (BoyJ) | In house, CCHMC | n/a |
| Transgenic R26-M2rtTA;Col1a1-tetO-H2B-GFP | Jackson laboratories | 016836 |
| B6;129S-403 Gt(ROSA)26Sortm1.1(CAG-COX8A/Dendra2)Dcc | Jackson laboratories | 018397 |
| Drp1flox/flox mice | Dr. Hiromi Sesaki laboratory | n/a |
| MxCre1 mice | In house, CCHMC | n/a |
| NOD/SCID gammac−/− | In house, CCHMC | n/a |
| Deposited Data. | ||
| RNA-seq data | This paper | GSE141472 |
| Software and Algorithms | ||
| FlowJo | FlowJo™ Software | https://www.flowjo.com/ |
| GraphPad Prism 5.0 | GraphPad Software Inc. | http://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Imaris | Oxford Instruments | https://imaris.oxinst.com |
| AltAnalyze | Emig, et al | http://www.altanalyze.org |
| Toppgene | Chen et al | https://toppgene.cchmc.org |
| EnrichR | Chen et al; Kuleshov et al | https://amp.pharm.mssm.edu/Enrichr/ |
Highlights.
HSC permanently remodel the mitochondrial network after replicative stress
HSC keep dysfunctional mitochondria due to Drp1 loss causing functional decline
HSC accumulate dysfunctional mitochondria through asymmetric division
HSC attrition is due to asynchrony in cell cycle and biosynthetic gene expression
ACKNOWLEDGEMENTS
We thank Drs Jose Cancelas, Damien Reynaud and Daniel Lucas (Division of Experimental Hematology and Cancer Biology – CCHMC) for important comments on the manuscript. We thank Dr David Muench for technical assistance. We thank mouse the core staff, Jeff Bailey and Victoria Summey, for bone marrow transplants and the flow cytometry core for assistance in cell sorting at Cincinnati Children’s Hospital Medical Center. We thank Olena Kolesnichenko for careful reading of the manuscript. The work was supported by NIH (DK102890 [MDF]; HL090676 [MDF]; a Pilot Innovative Project (CCHMC [MDF]; American Society of Hematology bridge award [MDF]; R01 HL122661 [HLG] and R21 AI35595 [HLG}, and the Evans MDS Foundation [HLG]).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no conflict of interest.
Data and Code Availability.
RNA-seq raw data supporting this work have been deposited in GEO, under accession number GSE141472. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141472 All original data are available upon reasonable request to Lead Contact.
Figure 1–4 have associated raw data. Raw Imaging are available upon requests.
References
- Adachi Y, Itoh K, Yamada T, Cerveny KL, Suzuki TL, Macdonald P, Frohman MA, Ramachandran R, Iijima M, and Sesaki H. 2016. Coincident Phosphatidic Acid Interaction Restrains Drp1 in Mitochondrial Division. Molecular cell 63:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anesti V, and Scorrano L. 2006. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim Biophys Acta 1757:692–699. [DOI] [PubMed] [Google Scholar]
- Benard G, and Karbowski M. 2009. Mitochondrial fusion and division: Regulation and role in cell viability. Seminars in cell & developmental biology 20:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BenediktWestermann. 2014. Mitochondrial inheritance in yeast. BBA Bioenergetics 1837:1039–1046. [DOI] [PubMed] [Google Scholar]
- Bernitz JM, K. H, MacArthur B, Sieburg H, Moore K 2016. Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell 167:1296–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, Francis TC, Kennedy NW, Picton LK, Kumar T, Uppuluri S, Miller AM, Itoh K, Karbowski M, Sesaki H, Hill RB, and Polster BM. 2017. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev Cell 40:583–594 e586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, and Zheng P. 2008. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med 205:2397–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A. 2013. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Xu H, Aronow BJ, and Jegga AG. 2007. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics 8:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida MJ LL, Corrigan DJ, Williams LJ, Snoeck HW 2017. Dye-Independent Methods Reveal Elevated Mitochondrial Mass in Hematopoietic Stem Cells. Cell stem cell 6:725–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykstra B, Kent D, Bowie M, McCaffrey L, Hamilton M, Lyons K, Lee SJ, Brinkman R, and Eaves C. 2007. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell stem cell 1:218–229. [DOI] [PubMed] [Google Scholar]
- Ema H, Takano H, Sudo K, and Nakauchi H. 2000. In vitro self-renewal division of hematopoietic stem cells. J Exp Med 192:1281–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch AL, Gurel PS, and Higgs HN. 2014. Novel roles for actin in mitochondrial fission. Journal of cell science 127:4549–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinge A XJ, Javier J, Mose E, Kumar S, Kapur R, Srour EF, Malik P, Aronow BJ, Filippi MD. 2017. p190-B RhoGAP and intracellular cytokine signals balance hematopoietic stem and progenitor cell self-renewal and differentiation. Nat Commun 8:8:14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, and Passegue E. 2017. Autophagy maintains the metabolism and function of young and old stem cells. Nature 543:205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P, and Qu CK. 2013. Metabolic plasticity and hematopoietic stem cell biology. Current opinion in hematology 20:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT, Evans RM, Suda T, Lee CH, and Pandolfi PP. 2012. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med 18:1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, and Suda T. 2006. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 12:446–451. [DOI] [PubMed] [Google Scholar]
- Ito K, and Suda T. 2014. Metabolic requirements for the maintenance of self-renewing stem cells. Nature reviews. Molecular cell biology 15:243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, T R, Cui J, Zimmerman SE, Pinho S, Mizoguchi T, Arai F, Runnels JM, Alt C, Teruya-Feldstein J, Mar JC, Singh R, Suda T, Lin CP, Frenette PS, Ito K. 2016. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 354:1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junko Wakabayashi, Z Z, Nobunao Wakabayashi, Yasushi Tamura, Masahiro Fukaya, Thomas W. Kensler, Miho Iijima, Hiromi Sesaki. 2009. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. JCB 186:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, and Ma’ayan A. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44:W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, and Filippi MD. 2016. An Alternative Approach for Sample Preparation with Low Cell Number for TEM Analysis. J Vis Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler D, Wehling A, Schneiter F, Zhang Y, Muller-Botticher N, Hoppe PS, Hilsenbeck O, Kokkaliaris KD, Endele M, and Schroeder T. 2019. Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 573:426–429. [DOI] [PubMed] [Google Scholar]
- Luchsinger LL, d.A M, Corrigan DJ, Mumau M, Snoeck HW. 2016. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 529:528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R, Dufour E, Spelbrink JN, Weinberg SE, Zhao Y, DeBerardinis RJ, and Chandel NS. 2016. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Molecular cell 61:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryanovich M, Z Y, Ruggiero A, Goldman A, Shachnai L, Zaidman SL, Porat Z, Golan K, Lapidot T, Gross A. 2015. An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat Commun 6:7901: [DOI] [PubMed] [Google Scholar]
- Norddahl GL, Pronk CJ, Wahlestedt M, Sten G, Nygren JM, Ugale A, Sigvardsson M, and Bryder D. 2011. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell stem cell 8:499–510. [DOI] [PubMed] [Google Scholar]
- Olsson A, V M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, Grimes HL. 2016. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 537:698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekkai Katajisto, J D, Christine Chaffer, Nalle Pentinmikko, Nemanja Marjanovic, Sharif Iqbal, Roberto Zoncu, Walter Chen, Robert A. Weinberg, and David M. Sabatin. 2015. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348:340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham AH, McCaffery JM, and Chan DC. 2012. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis 50:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Papatsenko D, Niu X, Schaniel C, and Moore K. 2014. Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Reports 2:473–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Prado MA, Zamboni N, DAmico D, Williams RW, Finley D, Gygi SP, and Auwerx J. 2017. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216:2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santel A, and Frank S. 2008. Shaping mitochondria: The complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB life 60:448–455. [DOI] [PubMed] [Google Scholar]
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC, and Sadek HA. 2010. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell stem cell 7:380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson RS, Soga T, Hirao A, Suematsu M, and Suda T. 2013. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell stem cell 12:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takubo K, and Suda T. 2012. Roles of the hypoxia response system in hematopoietic and leukemic stem cells. International journal of hematology 95:478–483. [DOI] [PubMed] [Google Scholar]
- Umemoto T, Hashimoto M, Matsumura T, Nakamura-Ishizu A, and Suda T. 2018. Ca(2+)-mitochondria axis drives cell division in hematopoietic stem cells. J Exp Med 215:2097–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini N, G M, Naveiras O, Nikitin G, Campos V, Giger S, Roch A, Auwerx J, Lutolf MP. 2016. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat Commun 7:13125: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, L E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, Lio P, Macdonald HR, Trumpp A. 2008. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135:1118–1129. [DOI] [PubMed] [Google Scholar]
- Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, Ema H, and Nakauchi H. 2013. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154:1112–1126. [DOI] [PubMed] [Google Scholar]
- Yu WM, L X, Shen J, Jovanovic O, Pohl EE, Gerson SL, Finkel T, Broxmeyer HE, Qu CK. 2013. Metabolic Regulation by the Mitochondrial Phosphatase PTPMT1 Is Required for Hematopoietic Stem Cell Differentiation. Cell stem cell 12:62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Expression files of scRNA-seq datasets related to Figure 4