

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection has resulted in the current COVID-19 pandemic. Worldwide this disease has infected over 2.5 million individuals with a mortality rate ranging from 5 to 10%. There are several efforts going on in the drug discovery to control the SARS-CoV-2 viral infection. The main protease (MPro) plays a critical role in viral replication and maturation, thus can serve as the primary drug target. To understand the structural evolution of MPro, we have performed phylogenetic and Sequence Similarity Network analysis, that depicted divergence of Coronaviridae MPro in five clusters specific to viral hosts. This clustering was corroborated with the comparison of MPro structures. Furthermore, it has been observed that backbone and binding site conformations are conserved despite variation in some of the residues. These attributes can be exploited to repurpose available viral protease inhibitors against SARS-CoV-2 MPro. In agreement with this, we performed screening of ∼7100 molecules including active ingredients present in the Ayurvedic anti-tussive medicines, anti-viral phytochemicals and synthetic anti-virals against SARS-CoV-2 MPro as the primary target. We identified several natural molecules like δ-viniferin, myricitrin, taiwanhomoflavone A, lactucopicrin 15-oxalate, nympholide A, afzelin, biorobin, hesperidin and phyllaemblicin B that strongly binds to SARS-CoV-2 MPro. Intrestingly, these molecules also showed strong binding with other potential targets of SARS-CoV-2 infection like viral receptor human angiotensin-converting enzyme 2 (hACE-2) and RNA dependent RNA polymerase (RdRp). We anticipate that our approach for identification of multi-target-directed ligand will provide new avenues for drug discovery against SARS-CoV-2 infection.
Communicated by Ramaswamy H. Sarma
Keywords: Coronavirus, COVID-19, hACE-2, MPro, multi-target-directed ligand, protease inhibitor, RdRp, SARS-CoV-2 virus
1. Introduction
Coronaviruses, a large family of viruses, causing upper-respiratory-tract infections in humans and other higher mammals (Bedford et al., 2020). Coronavirus outbreak has been reported three times in the 21st century namely SARS in 2002, MERS in 2012, and COVID-19 in November 2019. The recent outbreak of COVID-19 caused by SARS-CoV-2 has emerged from China. Human-human transmission of this highly infectious zoonotic virus has led to exponential growth in the number of infected cases, which resulted in its pandemic outbreak worldwide (Fahmi, 2020). Currently, more than 225 countries are affected due to COVID-19 with more than 2.5 million people to date which is increasing in thousands per day around the world and approximately 5 to 10% mortality rate (Fahmi, 2020). Additionally, the lives of millions of people have been impacted due to mandatory lockdowns, isolations, and quarantines. Thus the severe effect of the COVID-19 outbreak has imposed major challenges for global health, society and economy (Fry, 2020). Currently, there are no specific antiviral drugs or vaccines available for the treatment and management of COVID-19, which is further making the situation difficult to handle. At present preventive and supportive therapies are being implemented to prevent complications (Salata et al., 2020). Several efforts are going on to target key proteins of SARS-CoV-2 pathogenesis like the main protease (MPro), RNA-dependent RNA polymerase (RdRp), RNA binding N terminal domain (NTD) of Nucleocapsid protein (N protein), viral ion channel (E protein), 2′-O-RiboseMethyltransferase and human Angiotensin-converting-enzyme 2 receptor (hACE-2) (Gao et al., 2020; Gupta et al., 2020; Khan et al., 2020; Sarma et al., 2020; Shang et al., 2020; Zhang et al., 2020). To propose or repurpose drug and/or lead molecules against SARS-CoV-2, it would be effective to target multiple virus pathogenesis specific proteins within a close network of interaction or having dependent functionality (Zhou et al., 2020).
For drug discovery purposes, the MPro of coronaviruses has been studied extensively. They are papain-like proteases involved in the self-maturation and processing of viral replicase enzymes (Zhang et al., 2020). MPro has 11 putative cleavage sites in 790 kD replicase lab indicating its predominance in the proteolytic processing of large polyprotein lab. MPro from SARS-CoV-2 showed high sequence identity and structural similarity to that of the SARS-CoV MPro. It contains two catalytic domains, I (chymotrypsin) and II (picornavirus 3 C protease-like) each containing six-stranded antiparallel β-barrel containing active diad H41 and C145. Since they have a key role in virus replication, these proteases have emerged as important drug targets. Furthermore, with their very low similarity with human proteases, inhibitors of MPro are found to be very less cytotoxic (Anand et al., 2003; Zhang et al., 2020). Preliminary studies have suggested the potential use of protease inhibitor lopinavir/ritonavir, commonly used drugs for human immunodeficiency virus (HIV), for the treatment of COVID-19 patients (Cao et al., 2020; Muralidharan et al., 2020). Unfortunately, in the open-label randomized clinical study, these drugs are not found to be that impressive for COVID-19 treatment. Additionally, several other viral protease inhibitors like HCV Protease Inhibitor Danoprevir, HIV protease inhibitor Darunavir are under in vivo and clinical studies for the treatment of SARS-CoV-2 infection (Dong et al., 2020). To find new or repositioning existing drug molecules, the understanding of sequence and structure of SARS-CoV- MPro in regards to various other coronavirus strains could be important. Their comparative understanding is enigmatic, which could be important to repurpose available antiviral protease inhibitors against SARS-CoV-2 MPro.
Coronavirus uses a multiprotein complex to replicate its RNA based genomes. Cleavage of viral polyproteins (ORF1a and ORF1b) produces a set of non-structural proteins (Nsp). Of these, RdRp or Nsp12 catalyzes the synthesis and is known to play an important role in the replication and transcription cycle of the virus. RdRp is a primary target for nucleotide analog antiviral inhibitors such as Remdesivir, a drug under evaluation for SARS-CoV2 infection in clinics (Gao et al., 2020; Imbert et al., 2006). Remdesivir, a nucleotide analog, has shown broad-spectrum antiviral activities, and preclinical studies have also shown promising human safety data (Amirian & Levy, 2020; Dong et al., 2020). Recently, a report suggested that the significant inhibition of RdRp of SARS-CoV-2 by Remdesivir, and clinical studies for the same are ongoing to evaluate the efficacy of this molecule in the COVID-19 patients (www.clinicaltrials.gov; Study No: NCT04280705).
Apart from the maturation and replication, the entry of this virus by binding its surface spike protein to the hACE-2 is also a crucial process that can be targeted (Hasan et al., 2020; Kuhn et al., 2004; Shang et al., 2020). Since this host cell receptor is essential for the virus entry; targeting hACE-2 has a promise for preventing SARS-Cov-2 infection. It has been shown that clinically approved serine protease inhibitor (TMPRSS2) can also bind to the SARS-CoV-2 receptor hACE-2 and inhibit viral entry (Hoffmann et al., 2020). To date, the Chinese Clinical Trial Registry has recorded around 550 trials against SARS-CoV-2, mostly evaluating existing drug molecules. These trials include the application of antiviral (favipiravir, adalimumab, dihydroartemisininpiperaquine, leflunomide, lopinavir), antimalarial drugs (chloroquine or hydroxychloroquine), high‑dose vitamin C, etc (www.chictr.org.cn/index.aspx). From the available data of experimental and clinical studies, it has been found that molecules targeting single protein will be ineffective antiviral lead. Thus, to devise an effective strategy, there is a need for molecules that can target multiple key proteins such as RdRp and hACE-2 along with the MPro as a major drug target (Wu et al., 2020).
In silico screening of the drugs is a very vital and useful tool for rapid screening. This enables to shortlist leads and thus meet the urgent demand for repurposing drugs for the treatment of SARS-CoV-2 infection. Therefore, in this study, we have performed a comparative analysis of the major target of SARS-CoV-2, MPro for its structural characteristics to other viruses from the Coronoviridea family. This information was further used for the virtual screening of the custom-made library of phytochemicals, active ingredients present in the commonly used ayurvedic anti-tussive medicines in India, and the synthetic anti-viral drugs against MPro. Top hit molecules from this screen were then docked against SARS-CoV-2 RdRP and hACE2 to find the multi-target-directed molecules. We believe that this study will provide a new approach and a base for the discovery of multi-targeted antiviral molecules against SARS-CoV-2 pathogenesis. Thus these compounds could be potential candidates for in vitro and in vivo anti-viral studies followed by clinical treatment of SARS-CoV-2.
2. Results and discussion
2.1. Main protease shows divergence due to sequence variation
Phylogenetic analysis of proteases (MPro) helped to understand the structural-functional variation of SARS-CoV-2 concerning other viruses from the Coronaviridae family (Figure 1A). This analysis suggested that the MPro’s from the Coronaviridae family are distributed in five clusters. It has been observed that these proteases are separated in different clades depending on their zoonotic sources. In the clade 1 (orange colour), which appears to be the ancestral clade in the phylogenetic tree have sequences from Porcine transmissible gastroenteritis and pandemic diarrhea coronavirus.
Figure 1.

Diversity of protease like proteins in the Coronaviridae family. (A) The phylogenetic tree depicts five clusters. Clade-1 (orange) mainly consists of porcine transmissible coronavirus proteases. Clade-2 (blue) covers avian infectious bronchitis virus proteases. Clade-3 (purple) dominates with bat coronavirus sequences; followed by clade-4 (red) shared by the bat and human proteases. Clade-5 (green) has three distinct sub-clades that attribute to different hosts. The first sub-clade of clade 5 corresponds to bovine coronavirus and the third sub-clade exclusively involves murine coronavirus proteases. (B) The sequence similarity network (SSN) of the Coronaviridae proteases showed five clusters at the E-value of 1e-140. The cluster composition of sequences in the five clusters aligns with that of clades in the phylogeny (highlighted with colour code and symbols).
This clade appears to be evolutionary primitive to avian coronavirus but also showed coevolutionary traits. In the clade 2 (cyan), most of the sequences are from the strains of Avian infectious bronchitis virus. It is then followed by clade 3 (blue), which mainly consists of the sequences from different strains of Bat coronavirus. It is also found to be the parental clade for the clade 4 (red) and the clade 5 (green), which consist of predominantly mammalian host coronaviruses. It is interesting to see that the clade 4, which comprises viruses from bats and the majority of human infectious coronaviruses like SARS, MERS and also recent SARS-CoV-2. Our phylogenetic analysis revalidated the bat origin of the SARS-CoV-2 (Andersen et al., 2020). While the clade 5 has sequences from Bovine coronaviruses, it also has subclades that are indicative of a distinct host range. Part of the Clade 5 is from the bovine coronavirus and the other part is from murine coronavirus. Between these two subclades, there is a set of sequences that forms clad 5.2 comprises of human coronaviruses like strain OC43 (P0C6U7) and HKU1 (P0C6X3) which have bovine or murine as primary or intermediate host (Figure 1A). As evident in the multiple sequence alignment (Supplementary Figure 1), the MPro sequences from various coronaviruses strains showed very high conservation in the active site. Also, sequences of MPro from isolates of SARS-CoV-2 viral strain showed high identity. However, the sequence variation around the active site can lead to the separation of these proteases in distinct clades depending on their hosts.
Furthermore, the sequence similarity network (SSN) analysis was carried out to understand the similarities in MPro sequences from viruses of the Coronaviridae family. It was noted that till e-value 1E-80, all the sequences were clustered together suggesting an extremely high sequence similarity amongst all the Coronaviridae proteases (Supplementary Figure 2). At e-value 1E-140, the sequences were segregated in 5 clusters (Figure 1B). The largest cluster (green) consisted of sequences from bovine coronavirus, murine coronavirus and some human coronavirus such as OC43 and HKU1. The second cluster (orange) exclusively consisted of the porcine coronaviruses and related strains. Bat coronaviruses were grouped in a single cluster (blue), while cyan cluster housed avian coronaviruses. The red cluster consisted of human coronaviruses including SARS, MERS, SARS-CoV-2 along with various other bat coronaviruses. The small cluster of 2 sequences that separated from the red cluster at e-value 1E-125 also belongs to the bat coronaviruses. The clusters of SSN are showing a high correlation with the clades of the phylogenetic tree (Figure 1A) suggesting high evolutionary divergence amongst the coronaviruses depending on their host reservoir. This high specificity of the variations related to the host might have resulted from the natural selection in the animal hosts before human transmission (Andersen et al., 2020).
2.2. Coronavirus main protease structural backbone and active site conformation are conserved despite sequence variations
A comparison of 49 structures of MPro from different coronavirus strains using the DALI server showed five clusters similar to the phylogenetic tree (Figure 2A; Supplementary Data 1). Clusters 1 to 3 contains protease structures from porcine transmissible gastroenteritis and pandemic diarrhea coronavirus. Also, they include the MPro structure from human coronavirus strains like NL63 and HUK. Furthermore, clusters 4 and 5 mostly consist of protease structures from the coronavirus strains causing respiratory infections like SARS, MERS and SARS-CoV-2. The structural similarity matrix with hierarchical clustering also corroborates with the phylogenetic tree and structural cladogram showing the classification of MPro in five major clusters (Figure 2B). These results indicate that sequence variations in the MPro are also reflected in the structural variations resulting in similar clustering at the sequence and structure level.
Figure 2.

(A) Dendrogram of the 49 structures of SARS-CoV-2 MPro like proteins from the Coronoviridea family (B) Structural similarity matrix of Coronaviridea proteases. Cluster 1,2,3 predominantly have porcine protease structures and cluster 4,5 have structures corresponding to human infectious coronavirus. The dendrogram and matrix both display five clusters (labeled as similar to Figure 1A and B), similar to that observed in phylogenetic analysis, indicating that the sequence variations also depict in the structural analysis of proteases. (C) Structural superimposition of selected proteases from Coronaviridae family, showed RMSD <1Å and the high conservation of the backbone structure (D) Superimposition of the active site and binding site residues displayed conservation of active site pocket shape despite few differences in the residues. This indicated that the substrate recognition and binding efficiency or function of the MPro are conserved across the virus family.
Structural superimposition of representative proteases from each cluster of the structure cladogram and the similarity matrix depicted that the backbone of these proteases is conserved with RMSD less than 1 Å in the majority of cases (Figure 2C). It suggests that the sequence variation leads to minor structural changes without major changes in the conformation. Furthermore, a comparison of the substrate-binding pocket of these selected proteases shows that active site residue conformation is highly conserved (Figure 2D). There are some variations in the binding pocket residues as shown in the multiple sequence alignment, but these variations are subtle, and overall binding pocket conformation is conserved. Structural conservation of backbone and binding pocket of MPro from various coronavirus strains suggest that substrate recognition and inhibitor binding would more or less likely to be similar in them. Hegyi and Ziebuhr (2002) have shown the conservation in the substrate specificities and proteolytic processing of replicase polyproteins in the main proteases across coronaviruses (Hegyi & Ziebuhr, 2002).
This similarity highlights the development of universal MPro assays and the design of broad-spectrum inhibitors of MPro from different coronavirus strains. This also suggests that the MPro inhibitor-based drug repositioning would be the rapid and near to accurate approach to screen the drugs against SARS-CoV-2 infection.
2.3. Inter-residue contacts and the structural flexibility is located dominantly in the N-terminal catalytic site harboring beta-barrels
Besides structural conservation, it is important to understand inter-secondary elements and inter-residue contacts within the protein to unravel the mechanism of these proteases. Secondary structure topology indicates that proteins are asymmetrically populated by the beta-sheets at the N- terminus and helix at C- terminus (Figure 3A). The chord plot between pairs of secondary structural elements (arc of a circle) displays the number of interactions (chord thickness) between the corresponding elements. In MPro, the contacts between the secondary structural elements indicated that the majority of contacts are present in the N-terminus beta-sheets, which makes the juxtaposed beta barrels connected with the loop that harbors substrate binding site.
Figure 3.

(A) Secondary structure topology indicated that most of the beta-sheets towards the N-terminal end and helices to the C-terminal (B) The chord plot of secondary structure contacts in COVID-19 MPro wherein S indicate beta sheets and H indicate helices. We observed that beta-sheets exhibit more contacts than helices (C) Inter-residue contact map within the MPro. It shows the presence of more interactions in the first 200 residues i.e. towards N-terminus that mainly forms active sites (D) Superimposed topology with colours corresponding to secondary structure elements (E) Root Means Square Fluctuations of the residues from its mean position. High fluctuation (1 Å<RMSF) is seen in the N-terminus as compared to C-terminal. Active site residues (H41, C145) are highlighted and they showed less fluctuation.
Whereas, C-terminus globular cluster of helix showed comparatively less number of contacts indicating the rigid nature of these helix cluster involved in the protease dimerization (Figure 3B). Inter-residue contact map showed that the population of inter-residue contact is far denser in the first 200 amino acids, which consist of beta barrels having the catalytic role. While C-terminus residues show less number of contacts suggesting the involvement of this part of the protein (globular cluster of helices) in a structural role (Figure 3C). To validate the flexibility of the beta-barrels, the normal mode simulation was performed to map the Root Means Square Fluctuations of the residues from its mean position. Structural trajectory and the fluctuation map indicate that most of the residues present in the beta barrels have fluctuation above 1 Å, while the C-terminus is mostly rigid with very few residues having fluctuation above 1 Å (Figure 3D and 3E). Though the N-terminus show high fluctuations, the active sites (H41 and C145) showed very less variation, which is important to maintain the required conformation of these residues in the substrate-binding pocket.
The high frequency of inter secondary elements and residue contacts in the N terminus might provide structural flexibility for the proteolytic processing of large replicase polypeptide and they are potential sites to be targeted while designing or screening the inhibitors (C. Li et al., 2016).
2.4. Anti-tussive molecules like δ-viniferin and myricitrin showed higher affinity for the active site of SARS-CoV-2 MPro and other targets
Screening of the custom made the library of ∼7100 molecules comprising of different types such as flavonoids, glucosinolates, anti-tussive, anti-influenza, anti-viral, terpenes, terpenoids, alkaloids and other know and/or predicated anti-SARS-CoV-2 molecules was done against SARS-CoV-2 MPro as the main target (Supplementary Data 2). Sequence similarity search of MPro against human protein sequences was performed, which suggests that the viral proteases do not have any homolog in the human, thus targeting MPro will be specific to viral proteases only. Docking analysis results demonstrated that several phytochemicals and anti-viral have strong binding in the active site of SARS-CoV-2 MPro. The selected molecules also displayed a high binding affinity to SARS-CoV-2 RdRp and hACE-2 (Supplementary Data 3). Likewise MPro, other viral targets considered in this study, RdRp, also showed that there is no human homolog present, thus could be a potential viral target with MPro. Top molecules with the highest binding energy and with acceptable absorption, distribution, metabolism, and excretion (ADME) criteria were further selected for interaction analysis (Figure 4). To validate our docking strategy and obtained outcomes we have used a set of known or predicted anti-SARS-CoV-2 molecules reported from the literature (Wu et al., 2020). For further interaction analysis, we have selected the molecules with a high binding score against all the targets. Also, we have taken into account of earlier reports that support the antiviral properties of these molecules against different viruses.
Figure 4.

Heatmap of binding energies of top hits for different types of molecules used for screening, namely flavonoids, glucosinolates, anti-tussive, anti-influenza, synthetic anti-viral, terpenes, terpenoids and alkaloids. In general, we noticed strong binding of ligands towards MPro and RdRp as compared to the hACE 2 receptor. Flavonoids showed promising results with better binding affinities than existing synthetic anti-viral drugs. For details of the docking score and names of the ligands check Supplementary Data 3.
As one of the symptoms of COVID-19 is cough, a library of ayurvedic anti-tussive molecules in Indian medicine (82 molecules) are screened against SARS-CoV-2 MPro. The resulting top 20 molecules were further screened against other targets- RdRp and hACE 2 receptors. Amongst these molecules, δ-viniferin, chrysanthemin, myritilin and myricitrin showed strong binding with SARS-CoV-2 MPro. The δ-viniferin and myricitrin showed high solubility and bioavailability in computational ADME analysis; thus, analyzed for intermolecular interaction. With SARS-CoV-2 MPro, δ-viniferin exhibited binding energy of -8.4 Kcal/mol with several non-covalent interactions in the active site and substrate-binding site. THR24 and HIS163 form polar contact with hydroxyl groups of δ-viniferin, while active site residue HIS41 shows Pi-Pi interaction with the central aromatic scaffold of a ligand. Apart from this, MET165 is involved in Pi-sulfur interaction with ligand and other residues from the substrate-binding pocket, which are involved in either attractive van der Waal interaction or carbon-hydrogen bond (Figure 5A). In addition to MPro, δ-viniferin appeared to have a strong binding with the SARS-CoV-2 RdRp and hACE2 receptor with a score of -8.3 and -8.4 Kcal/mol, respectively. In the case of RdRp, binding pocket residues ALA762, TRP800, SER814 showed hydrogen bonds with the hydroxyl group of the ligand. While, active site and other key residues namely ARG553, ASP618, ASP623 and ASP761 display Pi interactions with the aromatic scaffolds of δ-viniferin.
Figure 5.

δ-Viniferin with active site residues of (A) MPro (B) RdRp and (C) hACE-2. It is followed by interaction map of myricitrin with (D) MPro (E) RdRp and (F) hACE-2. The interaction type is distinguished by coloured circles (residues). Dashed lines direct to the specific moiety in the ligand. Green residues symbolize van der Waals forces. Pink residues indicate those are involved in Pi-Pi stacking. Light pink indicates alkyl group interactions. Light orange colour indicates Pi-sulphur interactions. Dark orange show pi-anion interactions. Aromatic rings are involved in Pi-Pi, Pi-anion and Pi-sulphur interactions. Intermolecular interaction between Taiwanhomoflavone A with active site residues of (G) MPro, (H) RdRp and (I) hACE-2. Next is the interaction map of Lactucopicrin 15-oxalate with (J) MPro, (K) RdRp and (L) hACE-2.
The high density of these intermolecular non-covalent interactions is an indicator of strong and specific binding of δ-viniferin with RdRp active pocket. Furthermore, hydroxyl and aromatic groups of δ-viniferin also interacts through polar contacts and Pi-Alkyl interaction with the GLU35, LEU39, LYS68 and ALA71 residues of hACE-2 (Figure 5B and 6C). The interaction of δ-viniferin with MPro, RdRp and hACE-2 suggests its high potential as a multi-target directed ligand against SARS-CoV-2. δ-Viniferin is a resveratrol dimer, a major stilbene produced by stressed grapevine leaves (Aresta et al., 2003). It is also one of the major stilbenes present along with resveratrol in the red wine (Vitrac et al., 2005). Previous studies have also suggested that δ-viniferin shows potent antiviral activity against a variety of viruses viz. rotavirus (B. Yu et al., 2018), Human Immunodeficiency Virus (HIV) (Pflieger et al., 2013); hepatitis C virus (HCV) (Lee et al., 2019).
Figure 6.

Schematic representing multi-target-directed drug ligands against SARS-CoV-2 infection.
Another anti-tussive molecule, myricitrin, found to have a strong binding with the active site residues of the MPro with a binding score of -8.9 Kcal/mol. Active site residues HIS41 and CYS145 form Pi-Alkyl and Pi-Sulfur interactions with the aromatic scaffold of ligand, respectively. TYR54, PHE140, GLY143, HIS163 and GLU166 are involved in the formation of 6 hydrogen bonds with hydroxyl and carboxyl oxygen of ligand. Apart from this, there are other additional interactions like van der Waals forces that stabilize the binding of the ligand to the enzyme (Figure 5D). Myricitrin exhibited equally strong interaction with RdRp and hACE-2 with a binding score of -7.9 and -7.5 Kcal/mol, respectively. For RdRp, the carboxyl group of myricitrin forms two hydrogen bonds with ARG553, ARG555 and THR556. While the active site residue ASP623 is having Pi-Anion interaction with the aromatic rings of the ligand. Similarly, this ligand binds to hACE-2 by forming polar interaction with ALA348, PHE390, ARG393 and ASN394. This interaction is then further stabilized by Pi-stacking of PHE40 and Pi-anion interactions of ASP350, ASP382 with aromatic rings of the ligand (Figure 5E and F).
Myricitrin is a glycosylated analog of myricetin, present in the Myrica esculenta. Although in our docking study, myricetin has shown relatively lesser binding affinity when compared to myricitrin, previous studies have shown that it possesses potent antiviral activity. Myricetin inhibited VP35 protein with double-stranded RNA interaction in Ebola virus (Daino et al., 2018). The glycosylated myricetin, i.e. myricitrin has been found to inhibit HIV (Ortega et al., 2017), herpes simplex virus (Li et al., 2020), SARS coronavirus (Keum & Jeong, 2012; Yu et al., 2012).
Together, our results suggest that δ-viniferin and myricitrin could be potent molecules for the mitigation of SARS-CoV-2. Interestingly, top anti-tussive molecules in the present study mainly δ-viniferin, chrysanthemin, myritilin and myricitrin are present in the extract of black grapes, which is one of the key ingredients of ayurvedic antitussive medicines, and energy and immune booster, Chyawanprash (Georgiev et al., 2014). Thus, these medicines could be helpful in the management and mitigation of COVID-19. Particularly, the herbal formulation and beverages containing Vitis vinifera might be helpful in the management of COVID-19.
2.5. Potential of bioactive molecules from the medicinal plant as SARS-Cov-2 MPro inhibitor and multi-targeted drug ligand
The tapping of natural molecule libraries to predict inhibitors of MPro showed that several active biomolecules from different medicinal plants have a strong affinity against SARS-CoV-2 MPro. The flavonoid group of compounds showed a high binding score with all three targets suggesting this group of phytochemicals most bioactive against SARS-CoV-2 infection. One of the molecules from this type, Taiwanhomoflavone A, isolated from Cephalotaxus wilsoniana showed a strong binding affinity with MPro (-9.6 Kcal/mol). In silico ADME study indicated that this molecule possesses high bioavailability and likeliness to be the lead like compound.
Interaction analysis depicted that reactive groups of Taiwanhomoflavone A form of hydrogen bonds with THR199 and stabilizing forces are provided by electrostatic interaction with LEU189 residue of MPro. The bulkiness of this ligand leads to a large area of interaction that may lead to a higher contribution of attractive van der Waal’s force between ligand and binding site residues (Figure 5G). Furthermore, taiwanhomoflavone A also showed extremely strong binding with the RdRp and hACE2 receptor, with a binding score of – 9.8 Kcal/mol and -7.8 Kcal/mol, respectively. The RdRp binding pocket residues LYS621 and TRP800 showed hydrogen bonds with the carboxyl group of the ligand. While, active site residues namely ASP760 and ASP761 form Pi interactions with the aromatic groups of Taiwanhomoflavone A. In the case of hACE-2, taiwanhomoflavone A makes polar contact with GLU75 and Pi interaction with GLU35 (Figure 5H and I). Taiwanhomoflavone A has been explored for its anticarcinogenic, anti-inflammatory and anti-neoplastic properties(Kuo et al., 2002). It would be interesting to consider this molecule further for anti-viral evaluation in vitro and in vivo.
In the terpenoids group, lactucopicrin 15-oxolate, a derivative of lactucin and oxalic acid showed higher interaction. The lactucarium compounds are found in Lactuca virosa (wild lettuce) and also in the latex of Lactuca species. The Lactucopirin 15-oxolate has a binding energy of -8.2 Kcal/mol with MPro by the virtue of three hydrogen bonds to SER144, GLY143, and CYS145 of the active site. It also showed a high binding affinity with RdRp and hACE2 receptor with the binding score of -7.6 Kcal/mol and -8.3 kcal/mol, respectively (Figure 5J). With RdRp lactucopirin 15-oxolate makes hydrogen bonds at ARG553, ARG555, and THR556 residues, and it is stabilized by hydrophobic interactions of ASP623 and THR687 (Figure 5K). In the case of the hACE2 receptor, it forms three hydrogen bonds with residues ALA99, TYR385, ASN394 and Pi-stacking interaction with PHE40 (Figure 5L). The lactucopicrin and lactucin are known for their anti-malarial activity (Bischoff et al., 2004). Based on the in silico ADME studies, the compound is found to be highly soluble, bioavailable, and high GI absorption criteria which can be utilized for further drug development against novel-SARS-CoV-2. Apart from this, biorobin, pedunculagin, afzelin, hysperidin, Baicalin and other several molecules from alkaloid, flavonoids, and terpenoid that showed strong interaction with SARS-CoV-2 targets (Table 1; Supplementary Data 3). These findings highlight that the traditional medicines and/or nutraceuticals along with synthetic drug molecules could be effective ways to tackle COVID-19.
Table 1.
Potential multi-target-directed ligands against SARS-CoV-2 MPro, RdRp and hACE-2.
| Molecule name | Structure | Pharmacological functions | Source |
|---|---|---|---|
| δ-viniferin |  |
Anti-tussive, Anti-oxidant, Anti-viral |
Vitis vinifera |
| Myricitrin |  |
Anti-tussive, Anti-oxidant Anti-inflammatory Anti-Viral |
Myristica fragrans |
| Taiwanhomoflavone A |  |
Anti-carcinogenic, Anti-inflammatory and Anti-neoplastic |
Cephalotaxus wilsoniana |
| Lactucopicrin 15-oxalate |  |
Anti-oxidant, Anti-malaria |
Lactuca virosa |
| Nympholide-A | 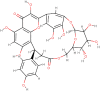 |
Anti-influenza, Analgesic, Sedative |
Nymphaea lotus |
| Saquinavir | 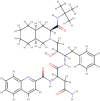 |
Anti-retroviral | Synthetic |
| Biorobin | 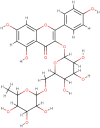 |
Anti-virus | Ficus benjamina |
| Phyllaemblicin B | 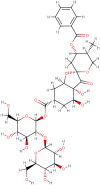 |
Anti-virus |
Phyllanthus emblica |
| -(-)asperlicin |  |
Antagonist: cholecystokinin receptor CCKA | Aspergillus alliaceus |
| Cassameridin |  |
Antifungal activity | Litsea kawakamii |
| Chrysanthemin |  |
Food additives | Roselle plant |
| Scalarane |  |
Anti-viral Anti-bacterial Anti-inflammatory |
Hyrtios erectus |
| Baicalin |  |
Anti-cancerous Anti-inflammatory Anti bacterial |
Scutellaria baicalensis |
| Hesperidin |  |
Anti-oxidant Anti-bacterial Anti-viral |
Citrus fruits |
| Afzelin |  |
Anti-viral | Nymphaea odorata |
2.6. Binding study repurposes application of anti-influenza and anti-HIV molecule against SARS-CoV-2 infection
Drug re-purposing is a strategy to investigate existing drugs for new uses. The advantage of repurposing is that the drugs are preclinically approved, and it helps in the formulation and development of the drug in ease. In this case, we have used the existing anti-influenza and anti-viral drugs to study the treatment strategy for the SARS-CoV-2. We screened 17 compounds and found a macrocyclic derivative Nympholide A, a phytochemical present in the aquatic plant of Nymphaeaceae family (Elegami et al., 2003). It showed the highest binding energy with Mpro and RdRp with a score of -7.8 and -9.3 Kcal/mol, respectively. Mpro residues Met165, GLU166, LEU 167 have hydrogen bond interaction with the ligand. In the case of RdRp, residues LYS551, ARG553, LYS798, TYR 619 side chains have strong hydrogen bonding with the ligand and GLU811 makes the Pi-anion interaction which improves the binding in RdRp compared to protease. The hACE2 receptor has fewer interacting residues, as a result, shows a weaker affinity to Nympholide A (Supplementary Figure 3 A-C).
In the case of antiviral compounds, Saquinavir has the highest binding energy with Mpro (-9 Kcal/mol). RdRp and hACE2 targets have a binding energy of -8.7 and -7.5 Kcal/mol, respectively. Saquinavir aromatic scaffold makes Pi-sulfur and Pi-Pi stacking interaction with the Met49 and His41 residues of Mpro. These interactions help in strong binding to protease active site and further the interaction is stabilized by hydrogen bonding of GLY143, SER144, CYS145, GLU166 of Mpro residues (Supplementary Figure 3 D). Similar to protease, the viral RdRp also have aromatic Pi-alkyl and Pi-anion interaction with ARG624 and ARG553 residues. The hydrogen bonding of ASP618, LYS551, LYS 621 residues of RdRp with the hydroxyl group of ligand give similar stability to protease. The hACE2 showed Pi-Pi stacking interaction of PHE40, PHE390 and one hydrogen bond of ASN394 with the ligand (Supplementary Figure 3E and F). Saquinavir has been known to inhibit the HIV-1 protease. In our study, the Saquinavir has higher interaction with protease compared to RdRp and hACE-2 (Stefanidou et al., 2012). As there are other viral protease inhibitors like Danoprevir, Ritonavir and Lopinavir are being tested in clinical trials for potential treatment against SARS-CoV-2, it would be interesting to consider Saquinavir for further studies (Cao et al., 2020).
3. Conclusion
This study provides a comprehensive overview of the structural characteristics of the Coronaviridea proteases and their potential inhibitors. Phylogenetic and SSN network analysis has illustrated the divergence in the proteases depending on their host reservoir suggesting the role of natural selection of host in conditioning the virus. Structural comparison depicted that the high structural similarities in the backbone and substrate binding pocket indicating conservation in the substrate specificities over different strains of Coronavirus. Asymmetrical structural flexibility in the MPro might be crucial for its activity, thus it can be targeted for inhibition. The structural similarity in the binding pocket assisted the prediction of repositioning of known protease inhibitors as SARS-CoV-2 MPro. Several molecules were identified as of the potent SARS-CoV-2 MPro inhibitors like delta δ-Viniferin, myricitrin, chrysanthemin, myritilin, taiwanhomoflavone A, Lactucopicrin 15-oxalate, nympholide A, afzelin, biorobin, herperidin and phyllaemblicin B. These molecules also equally showed strong binding with other targets of SARS-CoV-2, like RdRp and hACE-2 (Figure 6). Interestingly, most of the potential lead natural molecules of our study are anti-tussive and anti-virals, thus we speculate that the herbal formulation containing these bioactive phytochemicals might be applicable in the management of COVID-19. These molecules could be potential lead molecules for multi-targeted drug development against SARS-CoV-2.
4. Materials and methods
4.1. Sequences retrieval and phylogenetic analysis
The amino acid sequences of main proteases from the Coronaviridae family were retrieved from the UniProtKB database (www.uniprot.org) depending on their similarity with COVID-19 MPro. The criteria for the selection of the sequences were more than 90% coverage and similarity more than 50%. A total of 57 sequences (Supplementary Data 4) were selected for the phylogenetic analysis by maximum likelihood (ML) method using MEGA6 software (Tamura et al., 2013). Jones-Taylor-Thornton (JTT) model was used for amino acid substitution with a bootstrap value of 1000.
The ML tree was developed with a heuristic method with Nearest-Neighbor-Interchange (NNI) as a model. Tree representation was done using FigTree v1.4.4 software (http://tree.bio.ed.ac.uk/software/figtree/). SSN was carried out using SSNpipe (https://github.com/ahvdk/SSNpipe). The protocol was followed as per the online manual (https://github.com/ahvdk/SSNpipe/wiki/SSNpipe-Usage-Examples) (Halary et al., 2010). The networks generated were visualized using Cytoscape 3.7.2(Manual, 2018).
4.2. Structural alignment and superimposition of MPro
The crystal structure of SARS-Cov-2 MPro (PDB ID: 6Y2E) is compared with the 49 crystal structure of the proteases from various coronaviruses (Supplementary data 1) using the DALI server with all against all option (Holm & Rosenström, 2010). The dendrogram was built based on similarity Z-score and the similarity matrix was built based on structural conservation. Representation of the similarity matrix was obtained using Multiple Experimental Viewer (Mev) software. Furthermore, the hierarchical clustering of the similarity matrix was performed and compared with the dendrogram. Representative structure from each cluster was then superimposed and compared with COVID MPro using MatchMaker in Chimera 1.10.2 software. The structural backbone and active site conservation representation was obtained by Chimera 1.10.2 software (Pettersen et al., 2004).
4.3. Secondary structure element and inter-residue contacts analysis
Secondary structure topology and the non-covalent contact in COVID MPro was analyzed by the Protein Contact Atlas tool (https://www.mrc-lmb.cam.ac.uk/rajini/index.html) (Kayikci et al., 2018). Topology and the chord plot of MPro are obtained as server output. Inter-residue contacts were analyzed by the Fast Coarse-Grained protein dynamics simulation using the CABS-flex server (http://biocomp.chem.uw.edu.pl/CABSflex2/) (Jamroz et al., 2013). The output trajectory of the CABS-flex server was used to plot the fluctuation map of MPro.
4.4. Library preparation
Phytochemicals belonging to alkaloids, flavonoids, glucosinolates, phenolics, terpenes and terpenoids were prepared using available structures from PubChem (Kim et al., 2018). Similarly, a library of active ingredients from the plants used in ayurvedic antitussive medicine was prepared. This library was further enriched with antiviral molecules available in PubChem, other databases and literature. We have also included several molecules that are predicted as anti-SARS-CoV-2 molecules in recent reports. All the molecules were checked for stereochemical properties and then converted to *.pdbqt format using Autodock Tools (Morris et al., 2009). This library was used for docking studies.
4.5. Preparation of the target molecules
Crystal structure of liganded MPro (PDB ID: 6Y2F) and hACE-2 complexed with viral spike protein (PDB ID: 2AJF) was downloaded from the RCSB Protein DataBank(Shang et al., 2020; Zhang et al., 2020). Water and other heteroatoms were deleted from these structures. Grid for MPro was set around active site residues H41 and C145 with dimension on 36 × 56 x 40 Å using the AutoGrid program of AutoDock Tools (Morris et al., 2009). The protein is converted to *.pdbqt for further docking studies. A similar process was performed on SARS-CoV-2 RdRp crystal structure (PDB: 7BTF) with a grid around the RNA binding pocket with a dimension of 34 × 34 x 36 Å (Gao et al., 2020). Furthermore, hACE2 was prepared for docking, with a grid dimension of 20 × 38 x 24 Å around the viral spike protein recognition residues (K31, E35, D38, M82, K353)(Shang et al., 2020). These target molecules were then further used for virtual screening.
4.6. Virtual screening using autodock vina and ADME studies
Prepared receptor molecules from the custom-made libraries were set for the virtual screening by AutoDock Vina based Lamarckian Genetic Algorithm (LGA) parameter for ligand tethering of the proteins using 10 runs criteria (Trott & Olson, 2010). Top hits of ligands were selected based on their docking score against all targets namely, MPro, RdRp and hACE2. A comparison of the docking score for these targets is represented as a heatmap of the binding score using MeV software (http://www.tm4.org/). Top ligands were then submitted to the SwissADME server for in silico prediction of absorption, distribution, metabolism and excretion parameters of these ligands (http://www.swissadme.ch/)(Daina et al., 2017). Ligands with high solubility and bioavailability were further taken for the interaction analysis. Ligand binding position and interaction analysis were performed using PyMOL visualization software (The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC) and two-dimensional ligand interaction images are made using Biovia Discovery Studio 4.5 (Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2017, San Diego: Dassault Systèmes, 2016).
Supplementary Material
Funding Statement
The project work is supported by the research grant from the Council of Scientific and Industrial Research (CSIR), New Delhi, India and CSIR-National Chemical Laboratory, Pune, India.
Acknowledgments
Authors would like to thank Council of Scientific and Industrial Research (CSIR), New Delhi, India for infrastructure and manpower support. RSJ would like to acknowledge CSIR-National Chemical Laboratory, Pune, India for research start-up grant. SSJ and SBS would like to thankDBT for their research fellowships. SSS acknowledge CSIR for research fellowship and VKP thanks SERB-NPDF (PDF/2017/001544) for fellowship.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Amirian E. S., & Levy J. K. (2020, March). Current knowledge about the antivirals remdesivir (GS-5734) and GS-441524 as therapeutic options for coronaviruses. One Health, 9, 100128 10.1016/j.onehlt.2020.100128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J. R., & Hilgenfeld R. (2003). Coronavirus main proteinase (3CLpro) Structure: Basis for design of anti-SARS drugs. Science, 300(5626), 1763–1767. 10.1126/science.1085658 [DOI] [PubMed] [Google Scholar]
- Andersen K. G., Rambaut A., Lipkin W. I., Holmes E. C., & Garry R. F. (2020). The proximal origin of SARS-CoV-2. Nature Medicine, 26(4), 448–450. 10.1038/s41591-020-0820-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aresta A., Cioffi N., Palmisano F., & Zambonin C. G. (2003). Simultaneous determination of ochratoxin A and cyclopiazonic, mycophenolic, and tenuazonic acids in cornflakes by solid-phase microextraction coupled to high-performance liquid chromatography. Journal of Agricultural and Food Chemistry, 51(18), 5232–5237. 10.1021/jf034385r [DOI] [PubMed] [Google Scholar]
- Bedford J., Enria D., Giesecke J., Heymann D. L., Ihekweazu C., Kobinger G., Lane H. C., Memish Z., Oh M-d., Sall A. A., Schuchat A., Ungchusak K., & Wieler L. H. (2020). COVID-19: Towards controlling of a pandemic. The Lancet, 395(10229), 1015–1018. 10.1016/S0140-6736(20)30673-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff T. A., Kelley C. J., Karchesy Y., Laurantos M., Nguyen-Dinh P., & Arefi A. G. (2004). Antimalarial activity of lactucin and lactucopicrin: Sesquiterpene lactones isolated from Cichorium intybus L. Journal of Ethnopharmacology, 95(2-3), 455–457. 10.1016/j.jep.2004.06.031 [DOI] [PubMed] [Google Scholar]
- Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan, L., Song, B., Cai, Y., Wei, M., Li, X., Xia, J., Chen, N., Xiang, J., Yu, T., Bai, T., Xie, X., Zhang, L., Li, C., et al (2020). A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. The New England Journal of Medicine, 1–13. 10.1056/NEJMoa2001282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Michielin O., & Zoete V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7(1), 42717 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daino G. L., Frau A., Sanna C., Rigano D., Distinto S., Madau V., Esposito F., Fanunza E., Bianco G., Taglialatela-Scafati O., Zinzula L., Maccioni E., Corona A., & Tramontano E. (2018). Identification of myricetin as an ebola virus VP35–double-stranded RNA interaction inhibitor through a novel fluorescence-based assay. Biochemistry, 57(44), 6367–6378. 10.1021/acs.biochem.8b00892 [DOI] [PubMed] [Google Scholar]
- Dong L., Hu S., & Gao J. (2020). Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discoveries & Therapeutics, 14(1), 58–60. 10.5582/ddt.2020.01012 [DOI] [PubMed] [Google Scholar]
- Elegami A. A., Bates C., Gray A. I., Mackay S. P., Skellern G. G., & Waigh R. D. (2003). Two very unusual macrocyclic flavonoids from the water lily Nymphaea lotus. Phytochemistry, 63(6), 727–731. 10.1016/S0031-9422(03)00238-3 [DOI] [PubMed] [Google Scholar]
- Fahmi I. (2020, March). #Covid19 coronavirus disease 2019. DroneEmprit, 2019, 1–19. https://pers.droneemprit.id/covid19/ [Google Scholar]
- Fry H. (2020, March 26). The coronavirus, by the numbers. Nature, 579, 482–483.32203366 [Google Scholar]
- Gao Y., Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao, L., Tao, W., Sun, Q., Ming, Z., Zhang, L., Ge, J., Zheng, L., Zhang, Y., Wang, H., Zhu, Y., Zhu, C., Hu, T., Hua, T., et al. (2020, April). Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science, 7498, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev V., Ananga A., & Tsolova V. (2014). Recent advances and uses of grape flavonoids as nutraceuticals. Nutrients, 6(1), 391–415. 10.3390/nu6010391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M. K., Vemula S., Donde R., Gouda G., Behera L., & Vadde R. (2020). In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. Journal of Biomolecular Structure and Dynamics, 1–17. 10.1080/07391102.2020.1751300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halary S., Leigh J. W., Cheaib B., Lopez P., & Bapteste E. (2010). Network analyses structure genetic diversity in independent genetic worlds. Proceedings of the National Academy of Sciences of Sciences, 107(1), 127–132. 10.1073/pnas.0908978107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan A., Paray B. A., Hussain A., Qadir F. A., Attar F., Aziz F. M., Sharifi, M., Derakhshankhah, H., Rasti, B., Mehrabi, M., Shahpasand, K., Saboury, A. A., Falahati M. (2020). A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1754293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi A., & Ziebuhr J. (2002). Conservation of substrate specificities among coronavirus main proteases. Journal of General Virology, 83(3), 595–599. 10.1099/0022-1317-83-3-595 [DOI] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N.-H., Nitsche A., Müller M. A., Drosten C., & Pöhlmann S. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell, 181(2), 210–271. 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L., & Rosenström P¿i. (2010). Dali server: Conservation mapping in 3D. Nucleic Acids Research, 38(suppl_2), W545–W549. 10.1093/nar/gkq366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbert I., Guillemot J.-C., Bourhis J.-M., Bussetta C., Coutard B., Egloff M.-P., Ferron F., Gorbalenya A. E., & Canard B. (2006). A second, non-canonical RNA-dependent RNA polymerase in SARS Coronavirus. The EMBO Journal, 25(20), 4933–4942. 10.1038/sj.emboj.7601368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamroz M., Kolinski A., & Kmiecik S. (2013). CABS-flex: Server for fast simulation of protein structure fluctuations. Nucleic Acids Research, 41(W1), W427–W431. 10.1093/nar/gkt332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayikci M., Venkatakrishnan A. J., Scott-Brown J., Ravarani C. N. J., Flock T., & Babu M. M. (2018). Visualization and analysis of non-covalent contacts using the Protein Contacts Atlas. Nature Structural & Molecular Biology, 25(2), 185–194. https://doi.org/ 10.1038/s41594-017-0019-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keum Y. S., & Jeong Y. J. (2012). Development of chemical inhibitors of the SARS coronavirus: Viral helicase as a potential target. Biochemical Pharmacology, 84(10), 1351–1358. 10.1016/j.bcp.2012.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan R. J., Jha R. K., Amera G., Jain M., Singh E., Pathak A., Singh, R. P., Muthukumaran, J., Singh A. K. (2020). Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2’-O-Ribosemethyltransferase. Journal of Biomolecular Structure and Dynamics, 1–40. 10.1080/07391102.2020.1753577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B. A., Thiessen P. A., Yu B., Zaslavsky L., Zhang J., & Bolton E. E. (2018). PubChem 2019 update: Improved access to chemical data. Nucleic Acids Research, 47(D1), D1102–D1109. 10.1093/nar/gky1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn J. H., Li W., Choe H., & Farzan M. (2004). Angiotensin-converting enzyme 2: A functional receptor for SARS coronavirus. Cellular and Molecular Life Sciences Cmls, 61(21), 2738–2743. 10.1007/s00018-004-4242-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y.-H., Hwang S.-Y., Yang Kuo L.-M., Lee Y.-L., Li S.-Y., & Shen Y.-C. (2002). A Novel Cytotoxic C-methylated biflavone, taiwanhomoflavone-B from the twigs of Cephalotaxus wilsoniana. Chemical & Pharmaceutical Bulletin, 50(12), 1607–1608. 10.1248/cpb.50.1607 [DOI] [PubMed] [Google Scholar]
- Lee S., Mailar K., Kim M. I., Park M., Kim J., Min D.-H., Heo T.-H., Bae S. K., Choi W., & Lee C. (2019). Plant-derived purification, chemical synthesis, and in vitro/in vivo evaluation of a resveratrol dimer, viniferin, as an HCV Replication inhibitor. Viruses, 11(10), 890–818. 10.3390/v11100890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Teng X., Qi Y., Tang B., Shi H., Ma X., & Lai L. (2016). Conformational flexibility of a short loop near the active site of the SARS-3CLpro is essential to maintain catalytic activity. Scientific Reports, 6(1), 1–9. 10.1038/srep20918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Xu C., Hao C., Zhang Y., Wang Z., Wang S., & Wang W. (2020, January). Inhibition of herpes simplex virus by myricetin through targeting viral gD protein and cellular EGFR/PI3K/Akt pathway. Antiviral Research, 177, 104714 10.1016/j.antiviral.2020.104714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manual, C. U. (2018). Cytoscape User Manual.
- Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., & Olson A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. 10.1002/jcc.21256.AutoDock4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan N., Sakthivel R., Velmurugan D., & Gromiha M. M. (2020). Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 Protease against COVID-19. Journal of Biomolecular Structure and Dynamics, 1–7. 10.1080/07391102.2020.1752802 [DOI] [PubMed] [Google Scholar]
- Ortega J. T., Suárez A. I., Serrano M. L., Baptista J., Pujol F. H., & Rangel H. R. (2017). The role of the glycosyl moiety of myricetin derivatives in anti-HIV-1 activity in vitro. AIDS Research and Therapy, 14(1), 1–6. 10.1186/s12981-017-0183-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., & Ferrin T. E. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Pflieger A., Waffo Teguo P., Papastamoulis Y., Chaignepain S., Subra F., Munir S., Delelis O., Lesbats P., Calmels C., Andreola M.-L., Merillon J.-M., Auge-Gouillou C., & Parissi V. (2013). Natural stilbenoids isolated from grapevine exhibiting inhibitory effects against HIV-1 integrase and eukaryote MOS1 transposase in vitro activities. PLoS One., 8(11), e81184 10.1371/journal.pone.0081184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salata C., Calistri A., Parolin C., & Palù G. (2020). Coronaviruses: A paradigm of new emerging zoonotic diseases. Pathogens and Disease, 77(9), 1–5. 10.1093/femspd/ftaa006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma P., Sekhar N., Prajapat M., Avti P., Kaur H., Kumar S., Singh, S., Kumar, H., Prakash, A., Dhibar, D.P., Medhi B. (2020). In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure & Dynamics, 1–11. 10.1080/07391102.2020.1753580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Gemg, Q., Auerbach, A., Li F. (2020). Structural basis of receptor recognition by SARS-CoV-2. Nature, 1–8. 10.1038/s41586-020-2179-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanidou M., Herrera C., Armanasco N., & Shattock R. J. (2012). Saquinavir inhibits early events associated with establishment of HIV-1 infection: Potential role for protease inhibitors in prevention. Antimicrobial Agents and Chemotherapy, 56(8), 4381–4390. 10.1128/AAC.00399-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., & Kumar S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Molecular Biology and Evolution, 30(12), 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O., & Olson A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), NA–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitrac X., Bornet A., Vanderlinde R., Valls J., Richard T., Delaunay J.-C., Mérillon J.-M., & Teissédre P.-L. (2005). Determination of Stilbenes (δ-viniferin, trans-astringin, trans-piceid, cis- and trans-resveratrol, ε-viniferin) in Brazilian Wines. Journal of Agricultural and Food Chemistry, 53(14), 5664–5669. 10.1021/jf050122g [DOI] [PubMed] [Google Scholar]
- Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., & Li H. (2020). Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B. 10.1016/j.apsb.2020.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B., Jiang Y., Zhang B., Yang H., & Ma T. (2018). Resveratrol dimer trans-ε-viniferin prevents rotaviral diarrhea in mice by inhibition of the intestinal calcium-activated chloride channel. Pharmacological Research, 129, 453–461. 10.1016/j.phrs.2017.11.016 [DOI] [PubMed] [Google Scholar]
- Yu M.-S., Lee J., Lee J. M., Kim Y., Chin Y.-W., Jee J.-G., Keum Y.-S., & Jeong Y.-J. (2012). Identification of myricetin and scutellarein as novel chemical inhibitors of the SARS coronavirus helicase, nsP13. Bioorganic & Medicinal Chemistry Letters, 22(12), 4049–4054. 10.1016/j.bmcl.2012.04.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., … Hilgenfeld R. (2020, March). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science (New York, N.Y.), 3405, 1–9. 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Hou Y., Shen J., Huang Y., Martin W., & Cheng F. (2020). Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discovery, 6(1), 1-18. 10.1038/s41421-020-0153-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.