

Abstract
Glial cell line–derived neurotrophic factor (GDNF) is a growth factor that regulates the health and function of neurons and other cells. GDNF binds to GDNF family receptor α1 (GFRa1), and the resulting complex activates the RET receptor tyrosine kinase and subsequent downstream signals. This feature restricts GDNF activity to systems in which GFRa1 and RET are both present, a scenario that may constrain GDNF breadth of action. Furthermore, this co-dependence precludes the use of GDNF as a tool to study a putative functional cross-talk between GFRa1 and RET. Here, using biochemical techniques, terminal deoxynucleotidyl transferase dUTP nick end labeling staining, and immunohistochemistry in murine cells, tissues, or retinal organotypic cultures, we report that a naphthoquinone/quinolinedione family of small molecules (Q compounds) acts as RET agonists. We found that, like GDNF, signaling through the parental compound Q121 is GFRa1-dependent. Structural modifications of Q121 generated analogs that activated RET irrespective of GFRa1 expression. We used these analogs to examine RET–GFRa1 interactions and show that GFRa1 can influence RET-mediated signaling and enhance or diminish AKT Ser/Thr kinase or extracellular signal-regulated kinase signaling in a biased manner. In a genetic mutant model of retinitis pigmentosa, a lead compound, Q525, afforded sustained RET activation and prevented photoreceptor neuron loss in the retina. This work uncovers key components of the dynamic relationships between RET and its GFRa co-receptor and provides RET agonist scaffolds for drug development.
Keywords: receptor tyrosine kinase, signal transduction, small molecule, ligand-binding protein, retinal degeneration, structure–function, biased agonist, naphthoquinone, neuroprotection, RET receptor, retinitis pigmentosa
Introduction
Naphthoquinones are structurally and biologically diverse molecules of natural and synthetic origin that have been studied extensively as drug leads (1). The mechanisms of action are quite varied, and the biological outcomes range on a spectrum, with many variables governing the balance between cytotoxicity and cytoprotection (2). Activities include wide and nonselective inhibition of protein-tyrosine phosphatases (PTPs)2 (3), but there are examples of selective PTP inhibition (4). Recent work with the natural quinones shikonin (5) and plumbagin (6) indicated that this class can generate a wide range of cytotoxic or protective effects, from the regulation of inflammatory and stress mediators such as TNFα and pMAPK to the activation of prosurvival signals through growth factors and their receptors.
Hence, analogs of naphthoquinone scaffolds may be used to potentially generate more selective activity. We focused on activation of neurotrophic receptors to regulate neuronal health and function after stress or injury (7). Specifically, we studied mechanisms related to glial cell line–derived neurotrophic factor (GDNF), a growth factor that was tested in trials for Parkinson's (8). GDNF therapy has also been proposed for retinal degenerative diseases and showed early promise in animal models of retinitis pigmentosa (RP) (9–11). RP is a blinding condition resulting from the progressive loss of photoreceptors. Since over 300 different mutations (e.g. in the Rhodopsin gene) can give rise to the RP phenotype, there is a need for a broad-spectrum neuroprotective strategy (12).
So far, GDNF therapy has failed clinically. The reasons for slow therapeutic progress include poor pharmacokinetics, stability, and distribution. This means it must be given continuously to be efficacious, and the potential for side effects is a concern. Another problem is related to the way by which GDNF activates trophic signals. GDNF must bind to a receptor GFRa and only then it can activate a receptor tyrosine kinase RET, which mediates the trophic signals. This requirement by GDNF of a co-receptor narrows the breadth of responding cells to those that can access both subunits. A small molecule selective activator of RET, irrespective of GFRa expression, might circumvent all these therapeutic problems. Moreover, such molecules could be used as chemical-biology tools to evaluate RET–GFRa functional interactions.
In the present work we report the development of novel substituted naphthoquinones along with their corresponding quinoline forms and show that they act as selective agonists for the RET tyrosine kinase receptor, activating the downstream effectors Akt and Erk. Some agents are agonistic regardless of whether or not the GFRa1 receptor is present. For other agents, GFRa1 receptor expression causes biased RET signaling with differential effects upon pAkt and pErk activation, and this can be regulated positively or negatively by the reported GFRa1 modulator XIB4035. A GFRa1-independent RET agonist Q525 was neuroprotective in a mouse model of RP, generating trophic signals in vivo within the Muller glial cell population, thus validating RET as a druggable therapeutic target and suggesting potential utility for therapy of neurodegenerative diseases.
Results
Identification of novel naphthoquinone RET activators/modulators
All compounds and structures are summarized in Table 1 in the order that they are mentioned. Biochemical assays revealed that the chlorinated methoxy 1,4-naphthoquinone Q121 activated pAkt and pErk in MG87 RET/GFRa1 cells. Q121 induced RET phosphorylation in a dose-dependent manner (blots were probed using mAb 4G10 for total pTyr residues) (Fig. 1A). Compound Q128 with an (–OH) substituting the (–OCH3) group of Q121 was inactive (Fig. 1A), indicating the functional relevance of the –OCH3 adduct. These data provide a rationale for further substitutions of the core structure at this position.
Table 1.
Overview and compound structures
Overview and compound structures are shown, listed as they appear to show Structure–activity relationship progression. Letters A–D denote the positions and order in which the modifications took place throughout the paper, and the compounds that resulted. The adjacent text on the core structure describes the parameters that the modification influenced. Starting from Q121 and Q128, side chain substitutions at position A generated compounds that activated RET without GFRa1 expression. Modification at position B through removal of the chlorine atom generated compounds that were much less potent, although toxicity was improved and they potentiated GDNF survival. However, A and B modified compounds exhibited poor selectivity. Modification at position C by Nsubstitution yielded compounds with improved selectivity for RET and were pursued in further studies. Finally, compounds tested with N-substitutions at both positions C and D were found to be generally inactive.

Figure 1.

Screening and structure–activity relationship of naphthoquinone RET activators. A, compound Q121 and Q128 activity in MG87 RET/GFRa1 cells. Following serum starvation, the cells were exposed for 20 min to vehicle, 10 ng/ml GDNF, or compounds at the indicated micromolar range. The lysates were collected and probed for pAkt, pErk, and total phosphotyrosine (4G10). Representative Western blotting data are shown. Actin was probed as internal loading standard. B, representative total RET immunoprecipitation from MG87 RET cells transfected with either GFRa1 or GFP control. Q121 treatment results in RET phosphorylation at concentrations of 50 and 100 μm in the cells expressing GFRa1. Total RET was probed as internal loading standard. For MG76 RET/GFRa1 cells, G represents GDNF as positive control. For MG87 RET/GFP, G+a1 represents GDNF+ GFRa1 co-treatment, which is needed to induce RET phosphorylation in these cells lacking GFRa. C, activities of naphthoquinone derivatives. Side-chain modifications generated compounds active in MG87 RET/GFRa1 as demonstrated by the representative Western blotting. D, the naphthoquinone derivatives are also active in MG87 TrkA cells as demonstrated by the representative Western blotting. E, compound Q151 induces RET phosphorylation in both MG87 RET/GFRa1 and in RET/GFP cells lacking GFRa1. F, MG87 RET/GFRa1 cells in SFM were treated with Q2003 and Q2004 alone or in combination with suboptimal GDNF (GDNF SO, 5 ng/ml). Survival was assessed by MTT after 72 h. Both compounds had low but significantly trophic activity at a concentration of 1 μm. Compounds significantly potentiate suboptimal GDNF. G, MG87 TrkA cells in SFM were treated with Q2003 and Q2004 alone or in combination with suboptimal NGF (NGF SO, 60 pg/ml). Survival was assessed by MTT after 72 h. Both compounds had low but significantly trophic activity at a concentration of 1 μm but did not potentiate suboptimal NGF. The data are expressed as percentages of survival ± S.D. from four experiments, with the respective optimal trophic factor (GF O, 30 ng/ml) standardized to 100% and vehicle to 0%. *, vehicle versus all treatments; #, suboptimal GF versus optimal GF; +, suboptimal GF versus all treatments. One symbol, p < 0.05; two symbols, p < 0.005; three symbols, p < 0.0005 (Bonferroni-corrected t test).
To further evaluate RET phosphorylation by Q121, as well as its potential requirement of GFRa1 expression (which GDNF requires), immunoprecipitation experiments were performed. Phospho-RET was significantly increased by treatment of 100 μm. However, in MG87 RET cells lacking GFRa1, there were no increases in RET phosphorylation detected above background vehicle control (Fig. 1C). These data indicate that Q121, like GDNF, requires GFRa1 to signal through RET.
Compounds coded Q101, Q105, Q112, Q141, Q143, Q151, and Q1047 were then synthesized (see “Materials and Methods” and supporting text) and evaluated in biochemical assays. Significant increases in pAkt and pErk were observed with compounds Q105, Q112, Q141, Q151, and Q1047, whereas Q101 and Q143 were inactive (Fig. 1C). However, counterassays using MG87 cells expressing the NGF receptor TrkA tyrosine kinase (instead of RET) showed significant activation of pAkt and pErk, even though RET is absent. This indicates that these quinone derivatives lack selectivity (Fig. 1D).
Despite poor RET selectivity, compounds such as Q151 remained interesting because they induced RET phosphorylation whether or not the GFRa1 co-receptor was present (Fig. 1E). These data suggest that the modifications to the side chain of position A (Table 1) allows GFRa1 independence. One concern with these agents is that significant cytotoxicity was observed in the 1–10 μm range when these compounds were screened via the MTT survival assay; this was therefore addressed in further analogs.
Chlorine substitution affects potency and toxicity and generates GDNF-modulating compounds
To address cytotoxicity, we evaluated substitutions of the chlorine atom at position B (Table 1) because the ring-associated chlorine can enhance chemical reactivity and oxidative toxicity (2). Compounds Q2003 and Q2004, bearing a hydrogen in place of the chlorine, were prepared.
Biochemical assays revealed these compounds to be significantly less potent, with nonsignificant pAkt/pErk activation even when assayed at a high μm concentration range (data not shown). Moreover, in biological assays measuring cell survival, both Q2003 and Q2004 displayed weak trophic activity, which was nonselective and was detectable in MG87 RET/GRFa1 as well as in MG87 TrkA cells. In MTT assays Q2003 and Q2004 supported cell viability to 8–18% of the survival induced by optimal growth factor concentrations (GDNF for RET/GRFa1-expressing cells and NGF for TrkA-expressing cells) (Fig. 1, F and G).
Although Q2003 and Q2004 lacked potency and selectivity, they remained interesting because their activity was additive in MG87 RET/GRFa1 with suboptimal doses of GDNF (Fig. 1F), and this effect was not observed in MG87 TrkA with suboptimal doses of NGF (Fig. 1G). Therefore, although the survival induced by Q2003 and Q2004 can be RET-independent (i.e. the compounds afford survival to MG87 TrkA cells), the RET/GDNF axis is involved in the functional outcome because the agents potentiate GDNF trophic action, but they do not potentiate NGF trophic action.
To assess the basis of toxicity by some compounds, MTT assays were performed with cells growing in serum to assess negative effects on growth/survival over 72 h. At 1 μm, both Q2003 and Q2004 (lacking the chlorine) showed no signs of causing cell death (Fig. 2). In contrast, Q1047, identical to Q2003 but including the chlorine, was significantly toxic. Toxicity was comparable for MG87 RET/GFRa1 and MG87 TrkA cells, indicating that the toxicity is from a RET-independent mechanism.
Figure 2.

Chlorine substitution affects toxicity and directs lead compound development. MG87 RET/GFRa1- and TrkA-expressing cells were exposed to 1 μm compounds in regular growth media and assessed by MTT. Q2003 and Q2004 bearing a hydrogen in place of the chlorine were not toxic, indistinguishable from vehicle control (Veh.). However, Q1047, the chlorine-containing analogue of Q2003, showed significant signs of toxicity under the same conditions. Additionally, the lack of toxicity observed by Q525 demonstrated that the chlorine could be maintained and directed the development of more potent, RET-selective leads. The comparable toxicity between both cell lines indicated that the origin of the toxicity is not due to RET overactivation but rather a generalized mechanism.
Knowing that the presence of the chlorine is tied with potency, we designed structural modifications that preserve that position but reduce toxicity. Synthesis and evaluation of Q525 showed that is significantly less toxic than Q1047. Q525 was selected for further studies.
In summary, simple side-chain substitutions of the naphthoquinone core resulted in RET-activating compounds, and although lacking RET selectivity the compounds did not require GFRa1 co-expression. The chlorine is associated with the potency of the compounds but also with the toxicity. Removal of the chlorine generated less potent compounds, and although RET selectivity was still not achieved, the compounds were considerably less toxic and had the ability to cooperate with GDNF. These observations prompted further chemical design of the naphthoquinone core in efforts to improve RET selectivity.
Quinoline analogs are RET activators with improved selectivity and GFRa1 independence
To improve upon this first generation of molecules, we synthesized quinoline derivatives bearing identical side chains to their naphthoquinone counterparts. This effort generated new chemical entities with a nitrogen at position C (Table 1). These compounds are chemically distinguished from known inhibitors of protein-tyrosine phosphatases (PTPases), to exclude this as a possible factor in the poor selectivity of the first-generation molecules.
Q525 (and Q508) were screened in biochemical assays. Q525 was active in MG87 RET/GFRa1 cells across a broad range of concentrations and generated large and significant increases in pAkt/pErk (Fig. 3A; quantified in Fig. 3B). The compound maintained a high degree of selectivity for RET, because no significant increases in pAkt and pErk were observed in MG87 TrkA cells (Fig. 3C; quantified in Fig. 3D).
Figure 3.

Development of lead quinoline derivatives. A and B, biochemical screening of Q525 in MG87 RET/GFRa1 cells. The compound generated large increases in pAkt/pErk within the 5–20 μm range. Western blotting data were quantified from three experiments and are expressed as means ± S.D. versus vehicle (V or Veh), standardized to 100%. For pAkt; *, p < 0.05; **, p < 0.005; ***, p < 0.0005. For pErk: #, p < 0.05, Dunnett's test. C and D, counter screens with Q525 were performed in MG87 TrkA cells. Only slight increases in pAkt/pErk were observed within the same concentration range. For comparison, the poorly selective Q1047 bearing an identical side chain is also shown. The results were quantified from three experiments and are expressed as means ± S.D. versus vehicle, standardized to 100%. For pAkt: **, p < 0.005. For pErk: #, p < 0.05, Dunnett's test. G, positive control GDNF; N, NGF.
The improved selectivity of the quinoline series (e.g. Q525) is exemplified by a lack of selectivity in Q1047 (the naphthoquinone form of Q525), which activates signals in MG87 TrkA cells (Fig. 2C). Additionally, analogs Q1041 and Q1048 bearing a quinoxaline motif with N-substitutions at both positions C and D (Table 1) were synthesized and found to be inactive at all concentrations tested. Together, these data suggest that the N-substitution in the ring system has a significant impact on compound selectivity, generating agents that maintain RET activity and GFRa1 independence.
Differential signaling profiles through pAkt and pErk in combination with the GFRa1 modulator XIB4035
We used the GFRa1-independent RET agonists as chemical-biology tools to study the influence of the GFRa1 co-receptor upon RET signaling. We evaluated the quinone/quinoline pairs Q1047/Q525 and Q112/Q508 in combination with XIB4035, which is a reported GFRa1 modulator with no agonistic activity on its own (19).
Suboptimal doses (10 μm) of Q1047 (nonselective agonist) or Q525 (selective RET agonist) generated low but significant increases in both pAkt and pErk, compared with control vehicle or to 4 μm XIB4035, which did not activate signals (Fig. 4A). Compared with single treatments, a combination of 4 μm XIB4035 + 10 μm of Q1047 or Q525 resulted in significant increases of pErk signals (50 and 100%, respectively) (Fig. 4B), whereas pAkt levels were unaffected (Fig. 4C). These data suggest a RET-mediated pErk bias when GFRa1 is modulated by XIB4035.
Figure 4.
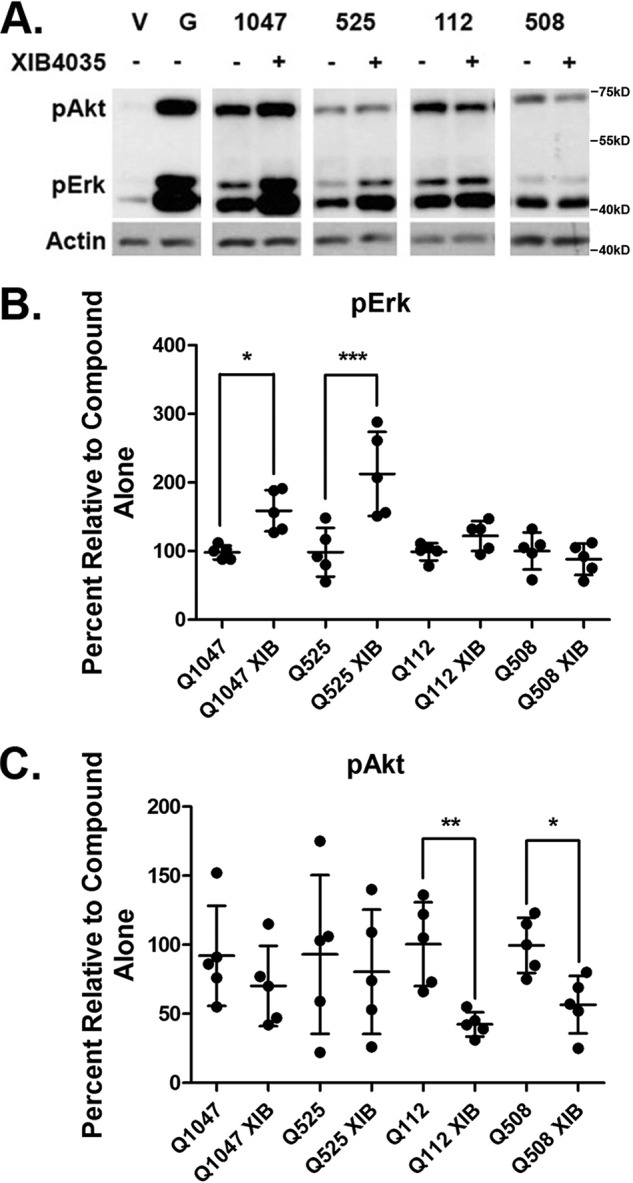
Differential signaling through pAkt/pErk is mediated by XIB4035-induced modulation of GFRa1. A, representative blots taken from separate experiments showing pAkt and pErk signaling of compounds Q1047, Q525, Q112, and Q508 with or without XIB4035 pretreatment. V, vehicle. G, positive control GDNF. B, quantification of pErk signals of compounds Q1047, Q525, Q112, and Q508 in combination with the GFRa1 modulator XIB4035. XIB4035 pretreatment did not affect pErk signals induced by Q112 and Q508. However, Q1047 and Q525 signals were significantly increased. The data are expressed as means ± S.D. from five experiments standardized to compound alone, which was set at 100%. *, p < 0.05; ***, p < 0.0005, Bonferroni-corrected t test. C, quantification of pAkt signals of compounds Q1047, Q525, Q112, and Q508 in combination with the GFRa1 modulator XIB4035. XIB4035 pretreatment did not affect pAkt signals induced by Q1047 and Q525. However, Q112 and Q508 signals were significantly decreased. The data are expressed as means ± S.D. from five experiments standardized to compound alone, which was set at 100%. *, p < 0.05; **, p < 0.005, Bonferroni-corrected t test.
Suboptimal doses (10 μm) of Q112 (nonselective agonist) or Q508 (selective RET agonist) generated low but significant increases in both pAkt and pErk, compared with control vehicle or to 4 μm XIB4035. A combination with 4 μm XIB4035 caused a reduction by 50% of the pAkt that was induced by Q112 or Q508 alone (Fig. 4C) while leaving pErk unaffected (Fig. 4B).
Hence, a GFRa1 modulator XIB4035 alters the signals induced by RET agonists, even when these agonists do not require GFRa1 to signal on their own. In one case (Q1047 or Q525) it enhances pErk signals but does not affect pAkt, and in another case (Q112 or Q508) it decreases pAkt signals but does not affect pErk. The end result for both families of compounds is a pErk signaling bias; however, this is likely achieved through different mechanisms that involve a ligand-mediated regulation of RET by the GFRa1 receptor. Also, it is of interest that N-substitution in the ring of structurally related quinone/quinoline pairs does not affect regulation of signaling by XIB4035.
RET activators are not inhibitors of protein-tyrosine phosphatases
Several naphthoquinones have been reported as inhibitors of PTPases, some nonselective (3) and some with a degree of selectivity (4). RET activation could potentially stem from inhibition of PTPases, and we evaluated this possibility.
Select compounds were tested in assays of PTPase activity to evaluate their impact upon the enzymatic activity of five PTPase enzymes (LAR, PTP-sigma, PTP-1B, MKPX, and SHP-1) as described (4). The compounds did not affect the activity of purified PTPases compared with DMSO vehicle, whereas the positive control sodium orthovanadate showed significant inhibition of >80% (Table 2). The 40 μm compound concentrations evaluated in these PTPase assays are at least 3-fold higher than the compound concentrations that induce biological signals in cultured cells and likely much higher than compound concentrations inside the cell, where PTPases are present.
Table 2.
Ability of select compounds to inhibit phosphatases
Select compounds were screened for their ability to inhibit the phosphatases LAR, Sigma, PTP1B, MKPX, and SHP-1. The data are shown as the percentages of inhibition relative to DMSO control. Sodium orthovanadate (Na3VO4) was used as positive control as a broad-spectrum PTP inhibitor.
| Q number | LARD1D2 | SigmaD1D2 | PTP1B | MKPX | SHP-1 |
|---|---|---|---|---|---|
| 105 | 97 | 98 | 77 | 99.5 | 87 |
| 112 | 91.5 | 96.5 | 121.5 | 97 | 80 |
| 121 | 96.5 | 97.5 | 93 | 98 | 90 |
| 143 | 96 | 99 | 101 | 96.5 | 103.5 |
| 508 | 72 | 71 | 92.5 | 96.5 | 91 |
| 525 | 97.5 | 99.5 | 96 | 99 | 94 |
| Na3VO4 | 16.5 | 24 | 4 | 5 | |
| DMSO | 99 | 97.5 | 99.5 | 99 | 93 |
In particular we note lack of inhibition by the quinone/quinoline pairs Q1047/Q525 and Q112/Q508, neither of which affected any of the five PTPases tested. Moreover, the fact that XIB4035 (a GFRa1 modulator with no reported link to PTPases) regulates pErk signals induced by Q1047/Q525 and Q112/Q508 (Fig. 4) is consistent with the notion that these agents are not PTPase inhibitors.
Q525 is neuroprotective by activation of Müller glial cells
We selected Q525 for pharmacological evaluation in vivo in the transgenic mutant RHOP347S mice (expressing a mutant rhodopsin gene that causes retinitis pigmentosa). These mice undergo rapid photoreceptor apoptosis that peaks in vivo between postnatal days 18 and 22 (20). The rate of photoreceptor apoptosis is fully replicated ex vivo in cultured whole retinal explants from these mice, allowing rapid testing of many compounds and doses.
Retinal explants were prepared from 18-day-old mice and incubated for 24 h with either compounds or vehicle, followed by quantification of TUNEL staining. Representative confocal images of the central retinal region are shown (Fig. 5A). Q525 at a 20 μm dose affords robust preservation of the photoreceptors, as indicated by a 40% reduction in TUNEL+ cells in this layer (Fig. 5B). In controls, the inactive compound Q143, assayed previously (Fig. 1C), was indistinguishable from vehicle-treated retinas. The data support the notion that RET activation by Q525 may be a disease-modifying strategy for degenerative conditions like RP.
Figure 5.

Q525 is neuroprotective ex vivo in a mouse model of retinal degeneration and activates trophic signals in Muller glial cells. A, representative confocal images of retinal flat mounts following treatment with Q525 and Q143. Freshly dissected retinas from 18-day-old RHOP347S mice were incubated in culture medium for 24 h: one retina with the indicated compounds and the contralateral retina of the same mouse with vehicle control (e.g. each individual had its own internal control). The tissues were processed for TUNEL staining and flat-mounted. 12 images were taken per retina and quantified semiautomatically. B, quantification of TUNEL-positive (pos.) cells after treatment. Cell counts from n = 3 mice/group were standardized versus vehicle control (Veh). Q525 treatment affords a significant reduction in TUNEL staining, whereas Q143 treatment was comparable to vehicle. *, p < 0.05, Student's t test. C and D, images of retinal sections stained for pErk and pAkt following intravitreal injection of compound Q525 in vivo. Eyecups were collected 1 h after injection. Significant increases in staining for pErk and pAkt co-localized with the Muller cell marker CRALBP, suggesting activation within the glial population. RGC, retinal ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bar, 25 μm. E, pErk quantification of Q525 and Q143 retinal sections (n = 3/group) expressed as the fold change in pixel area over vehicle ± S.D. ***, p < 0.0005, Student's t test. F, pAkt quantification of Q525 and Q143 retinal sections (n = 3/group) expressed as the fold change in pixel area over vehicle ± S.D., *, p < 0.05, Student's t test. DAPI, 4′,6-diamidino-2-phenylindole.
To understand the neuroprotective mechanism of action, we studied activation of biochemical pathways in the retina. Mice (n = 3) received intravitreally administered test compound in one eye and control vehicle in the contralateral eye. Eyecups were collected 1 h later and examined by immunohistochemistry to quantify pErk and pAkt signals. The results show that the Q525-treated eyes had elevated levels of pErk and pAkt by 1.8- and 2.0-fold, respectively, whereas no significant elevations in pErk or pAkt were observed in animals injected with the inactive Q143 (Fig. 5, E and F). This is consistent with biochemical assays using cell lines (Figs. 1 and 3). The pAkt and pErk signals co-localized with the Müller cell marker CRALBP. This indicates that the Müller glial cells are among the primary cellular targets for the RET agonists, and these cells are known to express RET and to support the health of photoreceptors.
Discussion
We discovered a series of new chemical entities, naphthoquinone/quinoline derivatives with agonistic activity at the RET receptor tyrosine kinase. Employing them as biological tools, we explored functional regulation between RET and its co-receptor GFRa1. A summary of screening and biochemical data is provided in Table 3.
Table 3.
Summary of pharmacological profiles
Comparison of signaling (maximal efficacy of activation of Akt/Erk in RET-expressing cells) by the compounds relative to each other and to GDNF. Potency is not indicated (e.g. GDNF is nm, and compounds are μm). RET selectivity was gauged by quantification of signaling in cells not expressing RET but expressing other RTKs (negative Akt/Erk activation is wanted). Culture of various cell types with the compounds in serum-containing media gauged general toxicity, and culture in serum-free conditions gauged accelerated or delayed cell death. The use of various cell types in such assays evaluated RET dependence. Q525 is highlighted as a potential lead with the best balance overall. +, low/poor; ++, moderate; +++, high; —, not tested/not applicable. The summary illustrates the interplay that exists between the different parameters and profiles and the challenges of choosing a lead agent.
| Test | Potency | RET selectivity | GFRa1-dependent | Toxicity |
|---|---|---|---|---|
| GDNF | ++++ | +++ | Yes | — |
| 121 | ++ | + | Yes | ++ |
| 128 | + | + | — | ++ |
| 101 | + | + | — | +++ |
| 105 | ++ | + | No | +++ |
| 112 | +++ | + | No | +++ |
| 141 | ++ | + | No | +++ |
| 143 | + | — | — | +++ |
| 151 | ++ | + | No | +++ |
| 1047 | +++ | + | No | +++ |
| 2003 | + | + | — | + |
| 2004 | + | + | — | + |
| 508 | ++ | +++ | No | + |
| 525 | +++ | +++ | No | + |
| 1041 | + | — | — | — |
| 1048 | + | — | — | — |
We demonstrated that although these agents do not require the presence of GFRa1, their signaling profiles can be nuanced by the presence and bound state of GFRa1. The regulatory functions that the GFRa subunits exert over RET provide additional points to consider when using RET agonists for therapy. We also showed the neuroprotective efficacy of a lead agent, Q525, in a model of RP neurodegeneration; further validating RET as a potential therapeutic target.
Screening and optimization of novel derivatives
The finding that the relatively simple naphthoquinone Q121 was a weak RET agonist but required GFRa1 expression was surprising, given its extremely small size compared with GDNF and the large surface of interactions of the receptor complex. Large growth factors like GDNF are typically envisioned as mediating a “bridge” between receptor components, bringing them in close proximity through multiple contact points and stabilizing an active conformation. It is unlikely that small molecules are capable of fully replicating this. The literature suggests that RET and GFRa1 associate in a ligand-independent manner (21), and it is therefore possible that our agents cause RET conformational changes through a preformed receptor complex.
The substitution of the methoxy group for a hydroxyl abrogated RET activity and clearly implicated this position for synthesis of analogs. However, further screening of these naphthoquinone derivatives, although biochemically interesting, did not possess the RET selectivity worthy of pursuit. It is unclear how the compounds activate pAkt and pErk in non–RET-expressing cells, but the potential molecular targets of the quinone family are broad. Their GFRa1 independence was unexpected but highlights one of the many strengths in small molecule development in that minor structural modifications can have major biochemical impacts. This is again reflected by the impact of the chlorine to hydrogen substitution, yielding mildly prosurvival, GDNF-modulating effects in vitro.
To differentiate the core structure from those reported previously as being phosphatase inhibitors (4), we made additional modifications to the ring system, generating novel quinolinediones which demonstrated much improved characteristics. The quinoline scaffold has a rich pharmacological background, particularly in regard to anti-cancer therapeutics (22), and in fact a family of RET kinase inhibitors with this motif exists (23). Others also reported a series of quinolines with neurotrophic activity (24), but their molecular target and signals remain unknown.
Significance of GFRa1-dependent signaling bias
Biased signaling is a relatively new concept in the field of pharmacology, and even more recent is the notion that this property can be exploited to improve therapeutic efficacy (25). Some of the first biased ligands were synthetically derived, and it was largely unknown whether this type of signaling occurred endogenously as an added regulatory mechanism. The chemokines and their receptors are one such example, revealing the presence and utility of signaling bias at the biological level as a means to fine-tune responses to a wide array of stimuli (26).
Our findings with the compounds in combination with XIB4035 demonstrate a unique aspect of ligand bias working through a co-receptor, and there are few examples of this in the literature (27, 28). The compounds reported display a balanced activation of both pAkt and pErk pathways through RET by themselves but become biased in the presence of a GFRa1 modulator. In contrast GDNF activation of the RET tyrosine kinase functions solely in conjunction with a membrane-bound or soluble co-receptor GFRa, a feature of this receptor that is conserved among vertebrates (29).
It is tempting to speculate that GFRas may be a gatekeeper of signaling bias. Through shifting their expression levels and patterns, GFRas may allow a single ligand to generate a series of orchestrated responses within the same tissue. Ligand–GFRa complexes may also control RET distribution into and out of lipid rafts, as is the case when comparing persephin versus GDNF (30), and this could dictate which adaptor proteins are most accessible to the activated kinase. It is also known that receptor–receptor interactions on the membrane can create allosteric regulatory sites at which ligands may bind to alter the functional outcome (31). Studies with constitutively active p75 mutants reveal that very slight changes in subunit positioning affect the resulting signaling profile and therefore suggest that different ligands could stabilize unique conformations of the receptor complex to drive signaling bias (32).
Although we did not address this within the present work, soluble GFRas can also be liberated from the membrane surface, and the biological roles of this process are understudied but are likely to be diverse (33–35). This could provide another way for bias control via the RET–GFRa complex.
Additionally, there may be other endogenous ligands such as GDF15 (36) or ligands of GFRa that can modulate biased signals through RET to mediate important processes. In that regard, it would be interesting to use GFRa modulators in combination with RET agonists, but unfortunately XIB4035 cannot be used in vivo in the retina because of poor solubility and high toxicity (19). There are GFRa modulators under development (e.g. XIB4035 analogs) that eventually can be tested to answer this question.
Our findings have therapeutic relevance, especially in disease where RET expression remains stable, but GFRa levels vary considerably (37, 38). A RET agonist may provide benefits in some diseases where dynamic changes occurring at GFRa in vivo are difficult to assess or control. As more work is done to validate RET and its co-receptors as targets, the ability to take advantage of signaling bias to yield highly specific outcomes with lower side effects may be a promising strategy.
Materials and methods
Commercially available compounds
Compounds Q121 (CAS registry no. 6956-96-3), Q128 (CAS registry no. 1526-73-4), and Q151 (CAS registry no. 84348-90-3) are known and described.
Chemistry and synthesis of novel naphthoquinone derivatives
All chemicals were purchased from Aldrich, J&K, or Otava. The reactions were monitored by TLC using aluminum-backed silica gel plates (Aldrich, silica gel matrix with fluorescent indicator), products visualized under UV light (254 and 365 nm). Flash chromatography purifications were performed in columns using Silica Gel 60A, 230–400 mesh (ACP). NMR spectra were recorded on Bruker 400 MHz spectrometer. Mass spectra were obtained with a Bruker mass spectrometer AmaZon SL direct infusion and Bruker UltraFlextreme MALDI-TOF. Chemical synthesis, MS, and NMR spectra and other characterization of the compounds are presented in the supporting information.
Cell lines
MG87 RET are murine MG87 fibroblasts stably transfected with RET proto-oncogene cDNA (13) and were cultured in DMEM containing 10% FBS, 2 mm l-glutamine, 10 mm HEPES, 100 units/ml penicillin/streptomycin, and 2 μg/ml puromycin. MG87 RET cells were transfected with a GFRa1 cDNA construct containing Blasticidin resistance to generate the MG87 RET/GFRa1 cell line and were cultured in the same medium with the addition of 5 μg/ml Blasticidin S. MG87 TrkA cells were transfected with human TrkA cDNA and cultured in DMEM with 10% FBS supplemented with 250 μg/ml G418. MG87RET/GFRa1 cell lines were stably transfected with the PathDetect Elk-1 system (Stratagene) harboring Luciferase reporter under the control of Erk activity. The cells were cultured in DMEM, 10% FBS, 15 mm HEPES, pH 7.2, 100 μg/ml normocin (Invivogen), 2 μg/ml puromycin (14). Growth factors GDNF and NGF were purchased from Peprotech (catalog nos. 450-10 and 450-01).
RET phosphorylation assays
Phosphorylation of RET was assessed as previously described (15). MG87 RET cells were plated on 35-mm tissue culture dishes, left to attach to the surface overnight, and then transfected with 4 μg/dish of GFRa1-expressing plasmid using Lipofectamine 2000 (Invitrogen) for DNA delivery as described by manufacturer. The next day the cells were starved for 4 h in serum-free DMEM containing 15 mm HEPES, pH 7.2, and treated with 1% DMSO vehicle, compounds, or GDNF (200 ng/ml) for 15 min. The cells were lysed using radioimmune precipitation assay–modified buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 1% Triton X-100, 10% glycerol, EDTA-free EASYpack, protease inhibitor mixture (Roche), 1 mm Na3VO4, 2.5 mg/ml of sodium deoxycholate). Cleared lysates were immunoprecipitated with 2 μg/ml of anti-RET C-20 (Santa Cruz Biotechnology, Inc., sc-1290), and the beads were conjugated with protein G (Thermo Fisher Scientific catalog no. 10004D). Eluted samples were resolved on 7.5% SDS-PAGE, and total phosphotyrosine residues were then probed in Western blots using the 4G10 antibody (Millipore). To confirm equal loading, the membranes were reprobed with anti-RET C-20 antibodies (1:500, Santa Cruz Biotechnology, Inc.).
pAkt, pErk1,2 biochemical studies
The cells were seeded onto 6-well plates (0.4 × 106 cells/well) and cultured overnight. The cells were serum-starved for 2 h and treated with vehicle, compounds, or growth factor (GF) as indicated in the text (generally for 20 min). Cell lysates were prepared (20 mm Tris-HCl, pH 7.5, 137 mm NaCl, 2 mm EDTA, 1% Nonidet P-40 containing a protease inhibitor mixture). Protein was quantified in cleared lysates using the Bradford assay (Bio-Rad). After SDS-PAGE, Western blotting was evaluated with the primary antibodies pAkt, pErk1,2 (Cell Signaling, catalog nos. 4060 and 4370), or Actin control for loading (Sigma–Aldrich, catalog A2066). Signals were developed using Western Lightning Plus ECL (PerkinElmer), and films were scanned and quantified using ImageJ software.
Phosphatase inhibition screens
PTP activity/inhibition assays are as we reported previously (4). The tagged catalytic domain or the full length of the following phosphatases were used for the screen: GST–PTP1B aa 1–321, GST–LAR–D1D2, GST–Sigma–D1D2, GST–MKPx aa 1–184, and His-SHP-1 aa 1–595. Enzyme reactions were performed in assay buffer 50 mm HEPES, pH 7.0, for PTP1B, LAR, Sigma, and MKPx and buffer 50 mm Bis-Tris, pH 6.3, 2 mm EDTA for SHP-1. DiFMUP (Invitrogen) was used as substrate for all assays, in black 96-well plates (Corning) in a final volume of 100 μl at 25 °C. The reaction was monitored by measuring the fluorescence (excitation wavelength, 358 nm; emission, 455 nm) with the Varioskan plate reader (Thermo Electron). For the kinetic assays, fluorescence was monitored over 10 min in 30-s intervals, and rates were calculated in fluorescent units/min. Km was determined from rates at various substrate concentrations using Michaelis–Menten equation from GraphPad Prism software. A substrate concentration equivalent to the Km value for each PTP was used for the screening of the compound. Inhibitors were diluted in DMSO, and then a dilution to 10 μm or 40 μm was made in assay buffer. Controls contain 1–2% DMSO final. Phosphatases and compounds were preincubated 2–5 min prior to addition of DiFMUP for enzyme inhibition quantification.
Cell survival assays
Cell survival was measured by the MTT assay (Sigma–Aldrich) using optical density readings as the end point. 2000–5000 cells were plated in 96-well format in SFM containing 0.03% FBS (HCell-100, Wisent). The indicated test agents or DMSO vehicle were added to the wells. The respective GFs (30 ng/ml GDNF for MG87 RET/GFRa1 and 30 ng/ml NGF for MG87 TrkA) at optimal concentrations were added as control. Assay length was typically 72 h because this time point yields an optimal signal-to-noise ratio between vehicle and growth factor controls. The assays were repeated at least three times (each assay n = 4–6 wells/condition). MTT optical density data were standardized to growth factor = 100%, and SFM = 0%, using the following formula: (ODtest − ODSFM) × 100/(ODGF − ODSFM).
Animal models
All animal procedures respected the institutional animal care and use committee guidelines for use of animals in research and protocols approved by McGill University Animal Welfare Committees. All animals were housed with a 12-h dark–light cycle with food and water ad libitum. We used the RHOP347S transgenic mouse (expressing the human Rhodopsin mutated at amino acid position 347) in a C57BL/6J (B6) background (kindly donated by Dr. T. Li). This model of RP faithfully replicates the features of disease progression in humans.
Retinal organotypic cultures
Whole eyes were enucleated, and whole retinas dissected from WT and RHOP347S mice at postnatal day 18 were used for organotypic culture experiments. Following enucleation, the eyes were placed in a Petri dish with PBS. The cornea was perforated and cut away along the ora serrata, leaving room to remove the lens. Whole intact retinas were then freely dissected away from the sclera and immediately transferred into 24-well plates containing 500 μl of culture medium (DMEM/Ham's F-12 supplemented with 10 mm NaHCO3, 100 μg/ml transferrin, 100 μm putrescine, 20 nm progesterone, 30 nm Na2SeO3, 0.05 mg/ml gentamicin, 2 mm l-glutamine, and 1 mm sodium pyruvate). Under sterile conditions, the medium was gently removed, replaced with fresh medium containing the treatments or controls, and incubated at 37 °C and 5% CO2 for 24 h. The compounds were tested at a concentration of 20 μm. Cell grade DMSO was used for vehicle treatments and was 0.5% by volume. The retinas were then used for TUNEL staining.
TUNEL staining
Staining was performed using the DeadEnd Fluorometric TUNEL system (Promega) as per manufacturer's instructions and as described by us (16, 17). RHOP347S retinas in culture were first fixed in 4% paraformaldehyde in PBS and kept at 4 °C overnight, followed by permeabilization using 2% Triton-X in PBS and refixed in paraformaldehyde for 30 min. Retinas were incubated with 50 μl of equilibration buffer for 20 min and then 25 μl of terminal deoxynucleotidyl transferase reaction mixture for 2.5 h at 37 °C, and the reaction was then terminated. The retinas were washed and mounted using Vectashield with 4′,6-diamidino-2-phenylindole. For image acquisition (17, 18), the retinas were divided into four quadrants, and three pictures with a 20× objective were taken in each area (central, mid, peripheral) for a total of 12 images of the outer nuclear layer per retina. Total TUNEL-positive cells were counted in each image semiautomatically (ImageJ). Counts were verified by at least one other person blinded to the experimental conditions. Retinal flat mounts from WT mice (where there is no mutation-driven photoreceptor death) were used as negative controls.
Immunohistochemistry
Staining was performed as described by us (17). After enucleation, the cryoprotected eyes were mounted in Tissue-TEK (Sakura), and cryostat sections were cut and mounted onto gelatin-coated glass slides. Sections (14 μm thick) were incubated in PBS containing 3% normal goat serum, 0.2% Triton X-100, and 3% BSA for 2 h. The retinal sections were incubated overnight at 4 °C with primary antibody (1:250 p-MAPK, Cell Signaling no. 4370; 1:250 p-Akt, Cell Signaling no. 4060; 1:400 CRALBP, Abcam ab183728). The sections were rinsed and incubated with secondary antibody for 1 h at room temperature. Then the sections were washed and mounted (Vectashield mounting medium with 4′,6-diamidino-2-phenylindole).
Image acquisition (fluorescence microscopy) and data analysis
Pictures were taken as Z-stacks of confocal optical sections using a Leica confocal microscope at a 20× objective. The images were equally adjusted using Adobe Photoshop CS 8.0 to remove background signals. For each experimental condition, a minimum of six images were acquired from three sections cut from different areas of the retina (n = 3 retinas/group). The area of the profiles of the cells expressing pErk and pAkt was measured using ImageJ software.
Intravitreal injections
Briefly, the mice were anesthetized with isoflurane delivered through a gas anesthetic mask. The treatments were delivered using a Hamilton syringe. Injections were done using a surgical microscope to visualize the Hamilton entry into the vitreous chamber and confirm delivery of the injected solution (3 μl of a 2 mm stock solution). After the injection, the syringe was left in place for 30 s and slowly withdrawn from the eye to prevent reflux. In each animal, the right eye was injected with the test agents (experimental eye), and the left eye was untreated (internal control).
Statistical analyses
The quantitative data were subjected to statistical analyses using GraphPad Prism 5 software and are presented as means ± S.D. for all studies. The differences between groups were determined by analysis of variance (multiple groups) followed by Dunnett's or Bonferroni corrections as indicated in the figure legends. Student's t tests were performed to compare two groups. p values below 0.05 were considered to indicate statistically significant differences between groups.
Author contributions
S. J. and H. U. S. conceptualization; S. J., Y. S., and H. N. data curation; S. J., Y. S., and H. N. formal analysis; S. J., Y. S., and H. N. investigation; S. J. writing-original draft; S. J., Y. S., M. S., and H. U. S. writing-review and editing; Y. S. and H. N. validation; H. N. methodology; M. S. and H. U. S. supervision; M. S. and H. U. S. funding acquisition; H. U. S. project administration.
Supplementary Material
Acknowledgments
We are thankful to Dr. Michel Tremblay and Isabelle Aubry (McGill University) for the protein-tyrosine phosphatase assays.
This work was supported by grants from the Canada Institutes of Health Research and the Canadian Consortium on Neurodegeneration and Aging and by Optic Nerve Trauma Grant W81XWH1910853 from the U.S. Department of Defense (to H. U. S.). Patents have been filed by McGill University on behalf of the authors, covering agents reported in this work.
This article contains supporting text.
- PTP
- protein-tyrosine phosphatase
- CRALBP
- cellular retinaldehyde-binding protein
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- GDNF
- glial cell line–derived neurotrophic factor
- GFRa1
- GDNF family receptor α1
- NGF
- nerve growth factor
- RP
- retinitis pigmentosa
- SFM
- serum-free medium
- TrkA
- tropomyosin receptor kinase A
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling
- Erk
- extracellular signal-regulated kinase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- GF
- growth factor
- aa
- amino acids.
References
- 1. Widhalm J. R., and Rhodes D. (2016) Biosynthesis and molecular actions of specialized 1,4-naphthoquinone natural products produced by horticultural plants. Hortic. Res. 3, 16046 10.1038/hortres.2016.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bolton J. L., and Dunlap T. (2017) Formation and biological targets of quinones: cytotoxic versus cytoprotective effects. Chem. Res. Toxicol. 30, 13–37 10.1021/acs.chemrestox.6b00256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Klotz L. O., Hou X., and Jacob C. (2014) 1,4-naphthoquinones: from oxidative damage to cellular and inter-cellular signaling. Molecules 19, 14902–14918 10.3390/molecules190914902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perron M. D., Chowdhury S., Aubry I., Purisima E., Tremblay M. L., and Saragovi H. U. (2014) Allosteric noncompetitive small molecule selective inhibitors of CD45 tyrosine phosphatase suppress T-cell receptor signals and inflammation in vivo. Mol. Pharmacol. 85, 553–563 10.1124/mol.113.089847 [DOI] [PubMed] [Google Scholar]
- 5. Wang L., Li Z., Zhang X., Wang S., Zhu C., Miao J., Chen L., Cui L., and Qiao H. (2014) Protective effect of shikonin in experimental ischemic stroke: attenuated TLR4, p-p38MAPK, NF-κB, TNF-α and MMP-9 expression, up-regulated claudin-5 expression, ameliorated BBB permeability. Neurochem. Res. 39, 97–106 10.1007/s11064-013-1194-x [DOI] [PubMed] [Google Scholar]
- 6. Yuan J. H., Pan F., Chen J., Chen C. E., Xie D. P., Jiang X. Z., Guo S. J., and Zhou J. (2017) Neuroprotection by plumbagin involves BDNF-TrkB-PI3K/Akt and ERK1/2/JNK pathways in isoflurane-induced neonatal rats. J. Pharm. Pharmacol. 69, 896–906 10.1111/jphp.12681 [DOI] [PubMed] [Google Scholar]
- 7. Josephy-Hernandez S., Jmaeff S., Pirvulescu I., Aboulkassim T., and Saragovi H. U. (2017) Neurotrophin receptor agonists and antagonists as therapeutic agents: an evolving paradigm. Neurobiol. Dis. 97, 139–155 10.1016/j.nbd.2016.08.004 [DOI] [PubMed] [Google Scholar]
- 8. Nutt J. G., Burchiel K. J., Comella C. L., Jankovic J., Lang A. E., Laws E. R. Jr., Lozano A. M., Penn R. D., Simpson R. K. Jr., Stacy M., Wooten G. F., and ICV GDNF Study Group (2003) Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 60, 69–73 10.1212/WNL.60.1.69 [DOI] [PubMed] [Google Scholar]
- 9. Frasson M., Picaud S., Léveillard T., Simonutti M., Mohand-Said S., Dreyfus H., Hicks D., and Sabel J. (1999) Glial cell line-derived neurotrophic factor induces histologic and functional protection of rod photoreceptors in the rd/rd mouse. Invest. Ophthalmol. Vis. Sci. 40, 2724–2734 [PubMed] [Google Scholar]
- 10. Touchard E., Heiduschka P., Berdugo M., Kowalczuk L., Bigey P., Chahory S., Gandolphe C., Jeanny J. C., and Behar-Cohen F. (2012) Non-viral gene therapy for GDNF production in RCS rat: the crucial role of the plasmid dose. Gene Therapy 19, 886–898 10.1038/gt.2011.154 [DOI] [PubMed] [Google Scholar]
- 11. García-Caballero C., Lieppman B., Arranz-Romera A., Molina-Martínez I. T., Bravo-Osuna I., Young M., Baranov P., and Herrero-Vanrell R. (2018) Photoreceptor preservation induced by intravitreal controlled delivery of GDNF and GDNF/melatonin in rhodopsin knockout mice. Mol. Vis. 24, 733–745 [PMC free article] [PubMed] [Google Scholar]
- 12. Baranov P., Lin H., McCabe K., Gale D., Cai S., Lieppman B., Morrow D., Lei P., Liao J., and Young M. (2017) A novel neuroprotective small molecule for glial cell derived neurotrophic factor induction and photoreceptor rescue. J. Ocul. Pharmacol. Ther. 33, 412–422 10.1089/jop.2016.0121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eketjäll S., Fainzilber M., Murray-Rust J., and Ibáñez C. F. (1999) Distinct structural elements in GDNF mediate binding to GFRα1 and activation of the GFRα1–c-Ret receptor complex. EMBO J. 18, 5901–5910 10.1093/emboj/18.21.5901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sidorova Y. A., Mätlik K., Paveliev M., Lindahl M., Piranen E., Milbrandt J., Arumäe U., Saarma M., and Bespalov M. M. (2010) Persephin signaling through GFRα1: the potential for the treatment of Parkinson's disease. Mol. Cell. Neurosci. 44, 223–232 10.1016/j.mcn.2010.03.009 [DOI] [PubMed] [Google Scholar]
- 15. Leppänen V. M., Bespalov M. M., Runeberg-Roos P., Puurand U., Merits A., Saarma M., and Goldman A. (2004) The structure of GFRα1 domain 3 reveals new insights into GDNF binding and RET activation. EMBO J. 23, 1452–1462 10.1038/sj.emboj.7600174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Platón-Corchado M., Barcelona P. F., Jmaeff S., Marchena M., Hernández-Pinto A. M., Hernández-Sánchez C., Saragovi H. U., and de la Rosa E. J. (2017) p75(NTR) antagonists attenuate photoreceptor cell loss in murine models of retinitis pigmentosa. Cell Death Dis. 8, e2922 10.1038/cddis.2017.306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Galán A., Jmaeff S., Barcelona P. F., Brahimi F., Sarunic M. V., and Saragovi H. U. (2017) In retinitis pigmentosa TrkC.T1-dependent vectorial Erk activity upregulates glial TNF-α, causing selective neuronal death. Cell Death Dis. 8, 3222 10.1038/s41419-017-0074-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barcelona P. F., Sitaras N., Galan A., Esquiva G., Jmaeff S., Jian Y., Sarunic M. V., Cuenca N., Sapieha P., and Saragovi H. U. (2016) p75NTR and its ligand ProNGF activate paracrine mechanisms etiological to the vascular, Inflammatory, and neurodegenerative pathologies of diabetic retinopathy. J. Neurosci. 36, 8826–8841 10.1523/JNEUROSCI.4278-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hedstrom K. L., Murtie J. C., Albers K., Calcutt N. A., and Corfas G. (2014) Treating small fiber neuropathy by topical application of a small molecule modulator of ligand-induced GFRα/RET receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 2325–2330 10.1073/pnas.1308889111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li T., Snyder W. K., Olsson J. E., and Dryja T. P. (1996) Transgenic mice carrying the dominant rhodopsin mutation P347S: evidence for defective vectorial transport of rhodopsin to the outer segments. Proc. Natl. Acad. Sci. U.S.A. 93, 14176–14181 10.1073/pnas.93.24.14176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanicola M., Hession C., Worley D., Carmillo P., Ehrenfels C., Walus L., Robinson S., Jaworski G., Wei H., Tizard R., Whitty A., Pepinsky R. B., and Cate R. L. (1997) Glial cell line-derived neurotrophic factor-dependent RET activation can be mediated by two different cell-surface accessory proteins. Proc. Natl. Acad. Sci. U.S.A. 94, 6238–6243 10.1073/pnas.94.12.6238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Afzal O., Kumar S., Haider M. R., Ali M. R., Kumar R., Jaggi M., and Bawa S. (2015) A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 97, 871–910 10.1016/j.ejmech.2014.07.044 [DOI] [PubMed] [Google Scholar]
- 23. Graham Robinett R., Freemerman A. J., Skinner M. A., Shewchuk L., and Lackey K. (2007) The discovery of substituted 4-(3-hydroxyanilino)-quinolines as potent RET kinase inhibitors. Bioorg. Med. Chem. Lett. 17, 5886–5893 10.1016/j.bmcl.2007.07.104 [DOI] [PubMed] [Google Scholar]
- 24. Schmidt F., Champy P., Séon-Méniel B., Franck X., Raisman-Vozari R., and Figadère B. (2009) Chemicals possessing a neurotrophin-like activity on dopaminergic neurons in primary culture. PLoS One 4, e6215 10.1371/journal.pone.0006215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Whalen E. J., Rajagopal S., and Lefkowitz R. J. (2011) Therapeutic potential of β-arrestin– and G protein–biased agonists. Trends Mol. Med. 17, 126–139 10.1016/j.molmed.2010.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rajagopal S., Bassoni D. L., Campbell J. J., Gerard N. P., Gerard C., and Wehrman T. S. (2013) Biased agonism as a mechanism for differential signaling by chemokine receptors. J. Biol. Chem. 288, 35039–35048 10.1074/jbc.M113.479113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao P., Metcalf M., and Bunnett N. W. (2014) Biased signaling of protease-activated receptors. Front. Endocrinol. (Lausanne) 5, 67 10.3389/fendo.2014.00067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ivanisevic L., Banerjee K., and Saragovi H. U. (2003) Differential cross-regulation of TrkA and TrkC tyrosine kinase receptors with p75. Oncogene 22, 5677–5685 10.1038/sj.onc.1206864 [DOI] [PubMed] [Google Scholar]
- 29. Ibáñez C. F. (2013) Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 5, a009134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang J., Lindahl M., Lindholm P., Virtanen H., Coffey E., Runeberg-Roos P., and Saarma M. (2004) PSPN/GFRα4 has a significantly weaker capacity than GDNF/GFRα1 to recruit RET to rafts, but promotes neuronal survival and neurite outgrowth. FEBS Lett. 569, 267–271 10.1016/j.febslet.2004.06.007 [DOI] [PubMed] [Google Scholar]
- 31. Zaccaro M. C., Ivanisevic L., Perez P., Meakin S. O., and Saragovi H. U. (2001) p75 Co-receptors regulate ligand-dependent and ligand-independent Trk receptor activation, in part by altering Trk docking subdomains. J. Biol. Chem. 276, 31023–31029 10.1074/jbc.M104630200 [DOI] [PubMed] [Google Scholar]
- 32. Vilar M., Charalampopoulos I., Kenchappa R. S., Reversi A., Klos-Applequist J. M., Karaca E., Simi A., Spuch C., Choi S., Friedman W. J., Ericson J., Schiavo G., Carter B. D., and Ibáñez C. F. (2009) Ligand-independent signaling by disulfide-crosslinked dimers of the p75 neurotrophin receptor. J. Cell Sci. 122, 3351–3357 10.1242/jcs.055061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paratcha G., Ledda F., Baars L., Coulpier M., Besset V., Anders J., Scott R., and Ibáñez C. F. (2001) Released GFRα1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron 29, 171–184 10.1016/S0896-6273(01)00188-X [DOI] [PubMed] [Google Scholar]
- 34. Mikaels-Edman A., Baudet C., and Ernfors P. (2003) Soluble and bound forms of GFRα1 elicit different GDNF-independent neurite growth responses in primary sensory neurons. Dev. Dyn. 227, 27–34 10.1002/dvdy.10280 [DOI] [PubMed] [Google Scholar]
- 35. Fleming M. S., Vysochan A., Paixão S., Niu J., Klein R., Savitt J. M., and Luo W. (2015) Cis and trans RET signaling control the survival and central projection growth of rapidly adapting mechanoreceptors. Elife 4, e06828 10.7554/eLife.06828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mullican S. E., Lin-Schmidt X., Chin C. N., Chavez J. A., Furman J. L., Armstrong A. A., Beck S. C., South V. J., Dinh T. Q., Cash-Mason T. D., Cavanaugh C. R., Nelson S., Huang C., Hunter M. J., and Rangwala S. M. (2017) GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 23, 1150–1157 10.1038/nm.4392 [DOI] [PubMed] [Google Scholar]
- 37. Zhang J., and Huang E. J. (2006) Dynamic expression of neurotrophic factor receptors in postnatal spinal motoneurons and in mouse model of ALS. J. Neurobiol. 66, 882–895 10.1002/neu.20269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jomary C., Darrow R. M., Wong P., Organisciak D. T., and Jones S. E. (2004) Expression of neurturin, glial cell line-derived neurotrophic factor, and their receptor components in light-induced retinal degeneration. Invest. Ophthalmol. Vis. Sci. 45, 1240–1246 10.1167/iovs.03-1122 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.