

Abstract
Endorepellin, the C-terminal fragment of the heparan sulfate proteoglycan perlecan, influences various signaling pathways in endothelial cells by binding to VEGFR2. In this study, we discovered that soluble endorepellin activates the canonical stress signaling pathway consisting of PERK, eIF2α, ATF4, and GADD45α. Specifically, endorepellin evoked transient activation of VEGFR2, which, in turn, phosphorylated PERK at Thr980. Subsequently, PERK phosphorylated eIF2α at Ser51, upregulating its downstream effector proteins ATF4 and GADD45α. RNAi-mediated knockdown of PERK or eIF2α abrogated the endorepellin-mediated up-regulation of GADD45α, the ultimate effector protein of this stress signaling cascade. To functionally validate these findings, we utilized an ex vivo model of angiogenesis. Exposure of the aortic rings embedded in 3D fibrillar collagen to recombinant endorepellin for 2–4 h activated PERK and induced GADD45α vis à vis vehicle-treated counterparts. Similar effects were obtained with the established cellular stress inducer tunicamycin. Notably, chronic exposure of aortic rings to endorepellin for 7–9 days markedly suppressed vessel sprouting, an angiostatic effect that was rescued by blocking PERK kinase activity. Our findings unravel a mechanism by which an extracellular matrix protein evokes stress signaling in endothelial cells, which leads to angiostasis.
Keywords: angiogenesis, stress response, proteoglycan, endothelial cell, signal transduction, angiogenesis, eIF2α, GADD45, PERK, stress signaling
Introduction
Perlecan, one of the largest heparan sulfate proteoglycans of basement membranes and cell surfaces, plays a major role in vasculogenesis and angiogenesis (1). For instance, morpholino-based knockdown of perlecan in zebrafish causes profound disruption of all vessels formed via angiogenesis from the dorsal aorta and other major blood vessels (2).
In mice, the effect of global perlecan knockdown is even more pronounced, as perlecan-null mice (Hspg2−/−) are embryonic lethal because of intrapericardial hemorrhage and malformation of coronary arteries and cardiac outflow tracts (3). Perlecan expression is finely regulated during cardiovascular development (4, 5) and is one of the few extracellular matrix constituents expressed in vascular and avascular matrices.
Perlecan is a modular proteoglycan composed of five domains that are involved in growth factor regulation, cancer, inflammation, lipid metabolism, basement membrane assembly, cell adhesion, and mechanosensing, among many other reported functions (6–16). The parent protein perlecan is pro-angiogenic because of the presence of three heparan sulfate chains covalently attached at its N terminus, which act as a reservoir of growth factors. In contrast, the C-terminal module of perlecan, called endorepellin (17, 18), is anti-angiogenic and inhibits cancer progression by interfering with the blood supply of the growing tumors (19–21).
Endorepellin interacts with VEGFR2 via its two proximal laminin-like globular (LG1/2)3 domains and to the α2β1 integrin via its terminal LG3 module, evoking dual receptor antagonism (22, 23). Indeed, soluble endorepellin inhibits angiogenesis by interfering with several VEGFR2-evoked signaling pathways, primarily by evoking transcriptional repression of hypoxia-inducible factor 1 (HIF-1α) and vascular endothelial growth factor A (VEGFA) and concurrently inhibiting nuclear factor of activated T cells (NFAT1) (24). We recently discovered a link between endorepellin-induced inhibition of angiogenesis and autophagy in endothelial cells (20). In our quest to find novel autophagic targets for endorepellin through NanoString analysis (25), we discovered that the mRNA of growth arrest and DNA damage–inducible (GADD45α) protein was dynamically regulated by the endorepellin/VEGFR2 axis. This protein has been reported to have a dual role: one in activating autophagy and the other in inhibiting angiogenesis (25). As an angiogenic suppressor, GADD45α interacts with mTOR and prevents Stat3 phosphorylation from driving VEGFA expression, inhibiting angiogenesis (26). As an autophagy inducer, GADD45α inhibits mTOR and induces the levels of LC3-II, an autophagic marker (27).
GADD45α belongs to the canonical PKR-like endoplasmic reticulum kinase (PERK)/eIF2α/activating transcription factor 4 (ATF4)/GADD45α axis, which is usually activated by external stresses such as exposure to UV light, nutrient deprivation, hypoxia, or oxidative stress (28, 29). Notably, ATF4 promotes skeletal muscle atrophy via a unique interaction with the transcriptional regulator C/EBPβ (30). All of these external stimuli are known to trigger the unfolded protein response, which ultimately leads to stress activation in cells (31, 32). The stress pathway was originally considered to help cells cope with stress stimuli by promoting survival. However, under prolonged stress, the survival mechanism can switch to cellular elimination via autophagic activation, which can eventually lead to cell death. For instance, in glioblastoma cells, eIF2α phosphorylation can up-regulate ATF4 levels, which induce autophagy by mTORC1 inhibition, causing cell death (33). Alternatively, ATF4 can lead to accumulation of transcription factor CCAAT-enhancer-binding protein homologous protein and apoptotic induction by GADD34 activation in endothelial cells (34). Protein misfolding was originally understood to be essential for activating intracellular stress; however, more recent discoveries have shown that ligand–receptor interactions (i.e. estrogen receptor 1 with 3,3-bis(4-hydroxyphenyl)-7-methyl-1,3-dihydro-2H-indol-2-one) also activate stress and cellular necrosis (35). Together, these reports suggest that the PERK/eIF2α/ATF4/GADD45α stress axis can be activated in cells without accumulation of misfolded proteins.
Given that endorepellin and GADD45α concurrently evoke autophagy and angiostasis, we hypothesized that the biological repertoire of endorepellin could include activation of a stress signaling pathway in vascular endothelial cells. We discovered that endorepellin evoked profound and sustained stress signaling in in vitro and ex vivo models of angiogenesis primarily by interacting with VEGFR2. Our studies highlight the role of endorepellin in inducing the stress pathway. This, in turn, would significantly curtail the generation of new vessels in the tumor matrix with clear implications for impeding cancer progression.
Results
Endorepellin induces GADD45α by evoking a stress signaling axis
To dissect the mechanism involved in endorepellin-evoked anti-angiogenic activity, we investigated GADD45α, a gene that is highly modulated in a NanoString analysis of endothelial cells exposed to recombinant endorepellin (25). The rationale is based on the fact that GADD45α increases in response to various stressors (28, 36) and suppresses tumor angiogenesis by blocking the mTOR pathway (26) while concurrently inducing LC3 (37). Moreover, the GADD45α–mTOR interaction prevents Stat3 phosphorylation from driving VEGFA expression, inhibiting angiogenesis (26). We performed time course experiments using the same concentrations of endorepellin (200 nm) under nutrient-rich conditions as in the NanoString studies (25). We discovered that endorepellin evoked a rapid and sustained increase in GADD45α (Fig. 1, A and B). As GADD45α is known to be induced by ATF4 (38), we tested ATF4 levels in parallel experiments. We found a significant induction of ATF4 that peaked at 3 h and declined thereafter (Fig. 1, A and B).
Figure 1.

Endorepellin evokes phosphorylation of PERK/eIF2α and up-regulation of ATF-4/GADD45α downstream of VEGFR2. A, representative immunoblots of time course experiments of HUVECs treated with endorepellin (200 nm, ranging from 0–6 h) and probed for ATF4 and GADD45α. Data were obtained from endorepellin-treated HUVECs from three independent experiments (n = 3) and normalized to GAPDH. B, quantification of ATF4 and GADD45α from A. C, representative immunoblots of time course experiments of HUVECs treated with endorepellin (200 nm, 0–6 h) and probed for p-PERKThr980, total PERK, p-eIF2αSer51, and total eIF2α. Data were acquired from three independent experiments (n = 3) and normalized to their respective total protein levels. D, quantification of p-PERKThr980 and p-eIF2αSer51 from C. One-way ANOVA was performed on all data. E, immunoblots of time course experiments of HUVECs pretreated for 30 min with SU5416 (a VEGR2 kinase inhibitor, 30 μm), followed by addition of endorepellin ranging from 0–24 h. The last lane shows the positive control, i.e. endorepellin alone for 2 h. α-Tubulin served as the loading control. F, immunofluorescence images of HUVECs showing cytoplasmic or nuclear distribution of GADD45α (red) seen with respect to DAPI (blue) after treatment with vehicle (PBS), endorepellin (200 nm), or tunicamycin (10 μg/ml) for 4 h. Nuclear localization of GADD45α is seen as a magenta hue under endorepellin or tunicamycin treatment conditions because of the merging of red and blue tones in the nucleus. G, quantification of cells with nuclear localization of GADD45α from F. An average of 100 cells per treatment (vehicle, endorepellin, or tunicamycin) were analyzed from five independent experiments. H, representative immunoblots of cell fractionation experiments of HUVECs treated for 3 h with or without endorepellin (200 nm). The membranes were probed for lamin A/C to label nuclear fractions (Nu), GAPDH to label cytoplasmic fractions (Cy), and GADD45α. Only the nuclear fraction was enriched in the trimeric form of GADD45α, and these levels were increased by exposure to endorepellin. I, immunofluorescence images of HUVECs treated with vehicle, endorepellin, or tunicamycin for 4 h and probed for filamentous actin (red) and ATF4 (green). The nuclei are outlined by dotted lines. J, quantification of cells with nuclear localization of ATF4 from I. An average of 500 cells per treatment (vehicle, endorepellin, or tunicamycin) were analyzed from three independent experiments (n = 3). All statistical analyses were calculated via one-way ANOVA (***, p < 0.001).
ATF4 is known to be regulated by phosphorylation of eIF2α, the α subunit of eukaryotic initiation factor 2 (39). Phosphorylation of eIF2α at Ser51 results in global protein synthesis suppression and plays a key role in response to various stresses, such as viral infection, buildup of misfolded proteins, and starvation. Paradoxically, phosphorylation of eIF2α at Ser51 induces transcription and translation of ATF4 (40). There are only four eIF2α mammalian kinases (PERK, heme-regulated inhibitor, PKR, and GCN2) (29, 39, 41), all of which phosphorylate eIF2α at Ser51, leading to similar downstream effects. In initial screenings, we found that human umbilical vein endothelial cells (HUVECs) predominantly express PERK, PKR, and GCN2 (Fig. S1A). Thus, we tested PERK and found time-dependent phosphorylation of PERK at Thr980 within its catalytic site (42), evoked by endorepellin (Fig. 1, C and D). Moreover, we found sustained activation of eIF2α at Ser51 (Fig. 1, C and D). We note that activation of PERK occurred rapidly, peaking at 1 h and preceding that of eIF2α (Fig. 1, C and D). We also saw temporal phosphorylation of PERK by LG1/2 domains of endorepellin, but not LG3, mirroring the effect of endorepellin (Fig. S1, C–F). As an internal positive control, we utilized tunicamycin, an antibiotic that inhibits N-linked glycosylation and induces the unfolded protein response (43) (Fig. S1B).
To address the mechanism of stress signaling induction by exogenous endorepellin, we blocked the VEGFR2 kinase with the small-molecule inhibitor SU5416 (44, 45) and found complete suppression of endorepellin-evoked PERK (Fig. 1E). Thus, it appears that VEGFR2 is directly involved in endorepellin biological activity, in agreement with our previous studies (20, 24, 45).
Next we performed imaging and cell fractionation to ascertain whether GADD45α and its activator ATF4 would translocate into the nuclei of stressed endothelial cells, as shown before in other cells (46, 47). We discovered that endorepellin evoked marked nuclear translocation of GADD45α at 4 h, similar to that induced by tunicamycin (Fig. 1, F and G). Cell fractionation studies followed by endorepellin treatment for 4 h showed that the trimeric form of GADD45α (68–72 kDa doublet, Fig. 1H) that was mainly observed in the nucleus under basal conditions, as seen in other studies (46), was markedly increased by endorepellin (Fig. 1H). In contrast, the monomeric form of GADD45α (∼22 kDa) did not change appreciably (Fig. 1H). In support of these findings, ATF4 levels and nuclear translocation were concurrently increased in HUVECs at 4 h (Fig. 1, I and J). These findings were further validated in two other endothelial cell models, telomerase human aortic endothelial cells (TeloHAECs) and porcine aortic endothelial cells expressing VEGFR2 (PAER2s). Notably, we found even faster induction of PERK phosphorylation, with maximal activation at 30 min in TeloHAECs (Fig. S2, A–C) compared with HUVECs, where p-PERK peaked at 1 h (Fig. 1C). In PAER2s, the kinetics of PERK phosphorylation at Thr980 were similar to that of HUVECs (Fig. S2, E–G). These biochemical data were corroborated by immunofluorescence studies, which showed up-regulation of p-PERK and GADD45α in TeloHAECs and PAER2s together with nuclear translocation of GADD45α (Fig. S2, C and G).
Collectively, our results provide the first evidence that endorepellin activates the PERK/eIF2α/ATF4/GADD45α stress signaling pathway. This could be a dynamic nexus for endorepellin bioactivity connecting stress signaling and angiostasis.
PERK and eIF2α are required for endorepellin-dependent stress axis induction
To confirm the biological relevance of PERK and eIF2α in endorepellin-mediated activation of stress signaling, we transiently depleted these two proteins via RNAi in HUVECs. We observed that PERK depletion (>80% knockdown) led to significant abrogation of eIF2α phosphorylation at Ser51 induced by endorepellin or tunicamycin (Fig. 2, A and B). Notably, even basal eIF2α phosphorylation at Ser51 was suppressed by PERK depletion (Fig. 2, A and B), further indicating a primary role of PERK as the key kinase regulating eIF2α phosphorylation in vascular endothelial cells.
Figure 2.

Endorepellin-dependent PERK and eIF2α activation is essential for downstream GADD45α up-regulation. A, representative immunoblots of HUVECs pretreated with 100 pm scrambled siRNA (siScr) or siRNA targeting PERK (siPERK), followed by treatment with endorepellin (200 nm) or tunicamycin (10 μg/ml). Lysates show RNAi-mediated knockdown of PERK (siPERK) and subsequent suppression of eIF2α phosphorylation. Treatment conditions are indicated in the bottom panel. B, quantification of PERK and p-eIF2α from A, normalized on GAPDH for PERK or total protein level for P-eIF2α. Data are from three independent biological experiments (n = 3). C, representative immunoblots of HUVECs pretreated with 100 pm scrambled siRNA (siScr) or with siRNA targeting eIF2α (sieIF2α), followed by treatment with endorepellin (200 nm) or tunicamycin (10 μg/ml). Lysates show RNAi-mediated knockdown of eIF2α (sieIF2α) and subsequent suppression of GADD45α levels. D, quantification of eIF2α and GADD45α from C, normalized to GAPDH. Data are from three independent experiments (n = 3). Statistical significance was calculated via two-tailed unpaired Student's t test (**, p < 0.01). All statistical analyses were calculated via one-way ANOVA (***, p < 0.001).
Next we found that, when eIF2α was depleted (>90% knockdown), there was marked suppression of GADD45α under basal conditions as well as endorepellin or tunicamycin treatments (Fig. 2, C and D). Collectively, our results underscore the requirement of PERK as the major activator of the endorepellin-evoked stress axis in endothelial cells and provide novel mechanistic insights into sequential activation of the PERK/eIF2α/GADD45α axis by endorepellin.
Endorepellin activates the stress pathway in ex vivo angiogenic assays
To directly test whether the anti-angiogenic activity of endorepellin was associated with induction of stress signaling, we performed ex vivo aortic ring assays in 3D type I fibrillar collagen (48). Fully sprouted explanted aortic rings of C57BL/6J mice were subjected to acute treatment with endorepellin or tunicamycin using the same in vitro concentrations. For p-PERK induction, we used 2-h treatment, the time point at which we observed maximal PERK phosphorylation (cf. Fig. 1C). For confocal imaging, we utilized isolectin IB4, which specifically binds to galactosyl residues on endothelial cells (49, 50), and antibodies against p-PERK at Thr980, considered a marker for its activation status (51–53). We found a marked increase in p-PERK evoked by endorepellin or tunicamycin (Fig. 3A). These effects could be clearly seen using the line-scanning tool, as described under “Experimental procedures” (Fig. 3B). Quantification of four independent experiments using three randomly selected aortic rings per mouse showed significant induction of p-PERK, measured as fluorescence intensity of the sprouted area normalized on the area occupied by the vascular sprouts (p < 0.001, Fig. 3C and Fig. S3B). We confirmed the endothelial nature of the sprouts by staining with a second endothelial cell marker, CD31 (red), and counterstaining with P-PERK (green) (Fig. S3D). We obtained similar results as those shown for IB4 and p-PERK staining (Fig. 3A).
Figure 3.

Endorepellin activates PERK in sprouts of ex vivo aortic rings. A, representative confocal images of aortic rings following treatment with endorepellin (200 nm) or tunicamycin (10 μg/ml) for 2 h. Endothelial cells in sprouts were visualized with isolectin IB4 (red), a marker for endothelial cells, and counterstained with an antibody against P-PERKThr980 (green). Insets show magnified sprouts stained for p-PERKThr980. B, line intensity profiles of PERK (green) expression in aortic ring sprouts corresponding to the yellow line traced along the enlarged image of sprouts in the inset in A. C, quantification of the fluorescence intensity of PERK in the sprouted area from A; fluorescence intensity data are from a total of 12 vehicle (Veh)-, nine endorepellin (Endo)-, and 10 tunicamycin (Tunic)-treated aortic rings from four independent experiments (n = 4). D, representative immunoblots of three pooled, sprouted blood vessels probed for p-PERKThr980 and total PERK. E, quantification of p-PERKThr980 levels over total PERK from D. Three aortic rings were pooled per condition (vehicle, endorepellin, or tunicamycin) and repeated four times; therefore, four independent experiments were performed (n = 4). The data are derived only from newly formed sprouts, as the aortic rings were removed before solubilization. All statistical analyses were calculated via one-way ANOVA (***, p < 0.001).
To validate our confocal imaging studies, we next performed biochemical analysis of rings treated with endorepellin or tunicamycin for 2 h. We first pooled two aortic rings per sample and removed the rings after solubilization with radioimmune precipitation assay buffer. Thus, the protein extracts were composed exclusively of sprouted endothelial cells growing in 3D collagen. In agreement with the confocal imaging studies, we found marked induction of p-PERK at Thr980 evoked by either treatment (Fig. 3, D and E).
Next we probed for GADD45α, the ultimate effector protein of the stress axis. Interestingly, we observed that GADD45α was not appreciably altered at 2 h (not shown) but markedly increased at 4 h of treatment with endorepellin or tunicamycin (Fig. 4A). Quantification of four independent experiments using three randomly selected aortic rings per mouse showed significant induction of GADD45α (p < 0.001, Fig. 4B), as determined by the fluorescence intensity of the sprouted area, measured as described under “Experimental procedures” (Fig. S3B). In agreement with the confocal imaging, we found by immunoblotting that endorepellin or tunicamycin markedly induced GADD45α (Fig. 4, C and D). Collectively, these ex vivo functional assays corroborate the in vitro results and show progressive activation of the stress signaling pathway, where PERK is first phosphorylated at Thr980, followed by an increase in GADD45α levels.
Figure 4.

Endorepellin up-regulates the effector protein GADD45α in aortic rings. A, representative confocal images of aortic rings following treatment with endorepellin (200 nm) or tunicamycin (10 μg/ml) for 4 h. Sprouts were labeled with IB4 (red) and GADD45α (green). Insets, magnified sprouts stained for IB4 (red), GADD45α (green), and DAPI (blue). B, quantification of the fluorescence intensity of GADD45α for the sprouted area from A. Fluorescence intensity data are from a total of 10 vehicle (Veh)-, eight endorepellin (Endo)-, and eight tunicamycin (Tunic)-treated aortic rings from four independent experiments (n = 4). C, representative immunoblots of three pooled, sprouted blood vessels probed for GADD45α. GAPDH was used as a loading control. D, quantification of GADD45α levels normalized to GAPDH from C. Data are from four independent biological experiments (n = 4), with each condition comprised of three pooled sprouts. All statistical analyses were calculated via one-way ANOVA (***, p < 0.001).
The endorepellin-activated stress axis inhibits angiogenesis
To further investigate the specificity of endorepellin-evoked PERK activation, we utilized a small-molecule inhibitor, GSK2656157, a cell-permeable, ATP-competitive inhibitor of PERK (PERKi) (54). This small molecule is exquisitely specific for PERK with more than 100-fold selectivity for PERK over other eIF2α kinases (55) and an IC50 of ∼1 nm in various cells, as shown by inhibition of PERK autophosphorylation, eIF2α phosphorylation, and ATF4 suppression (55). Oral administration of this PERKi results in tumor suppression and angiostasis (55), consistent with reduced tumor growth in Perk−/− mice (56). We found that the PERKi efficiently blocked the activity of endorepellin in a dose-dependent manner (Fig. 5A) with an IC50 of ∼10 nm (Fig. 5B). Notably, we observed a parallel reduction in phosphorylation of downstream eIF2α at Ser51 (Fig. 5A) with an IC50 of ∼1 nm (Fig. 5B). We also confirmed that GSK2656157 was 1000-fold more potent in inhibiting PERK phosphorylation in HUVECs with an IC50 of ∼1 nm (Fig. 5A) compared with another PERK inhibitor, AMG44, which was discovered recently and has an IC50 of ∼1000 nm in HUVECs (Fig. S3C).
Figure 5.

PERK inhibition blocks endorepellin-dependent stress signaling and angiostasis in aortic rings. A, representative immunoblots of dose-dependent experiments of HUVECs pretreated with the PERKi for 30 min, followed by endorepellin (200 nm) exposure for 1 h and 30 min. Blots were probed for p-PERK and p-eIF2α. α-Tubulin served as the loading control. B, quantification of p-PERK and p-eIF2α from A, with normalization to their respective total protein. Data are from three independent biological experiments (n = 3), plotted as mean ± S.E. One-way ANOVA was performed on the data. C, representative phase-contrast images of aortic rings treated with endorepellin or endorepellin and the PERKi for 7 days in total after sprouting. D, quantification of the radial distance of endorepellin- or endorepellin and PERKi–treated sprouts from C. Phase-contrast data are from 12 vehicle-, 10 endorepellin-, and 10 endorepellin and PERKi–treated rings from four independent experiments (n = 4). E, representative confocal images of aortic rings treated with endorepellin or endorepellin and PERKi–treated rings. Rings were labeled with IB4 (red) and DAPI (blue). F, quantification of fluorescence intensity of the sprouted area from E. Confocal data are from eight vehicle-, eight endorepellin-, and nine endorepellin and PERKi–treated rings from four independent biological experiments (n = 4). All statistical analyses were calculated via one-way ANOVA (***, p < 0.001).
Next we performed functional aortic ring assays in 3D type I collagen using the PERKi with or without endorepellin. In these experiments, we added the PERKi or endorepellin after 3 days, when we detected the first sprouts. We added fresh PERKi (10 nm) or endorepellin (200 nm) every other day and continued the treatment for 9 days in total. At the end of the chronic treatment, we observed a drastic abrogation of ring sprouting in endorepellin-treated rings (Fig. 5C) vis à vis vehicle (p < 0.001, Fig. 5D). However, the PERKi completely blocked the angiostatic activity of endorepellin (Fig. 5, C and D). We acquired phase-contrast images of the rings and calculated the radial distance of the sprouts by subtracting the background and highlighting sprouts using the threshold function in ImageJ. We then drew circles around the rings, encompassing the edges of the sprouts, and measured the radii (Fig. S3A). Next we labeled the rings with IB4 and DAPI, and, using confocal laser scanning microscopy, we obtained comparable qualitative (Fig. 5E) and quantitative (Fig. 5F) findings. Collectively, our results suggest that endorepellin-dependent inhibition of angiogenesis requires activation of the PERK–stress axis.
Discussion
Classical pathways evoked by protracted activation of the unfolded protein response and ER stress are known to interfere with angiogenesis (57). Specifically, the PERK/eIF2α/ATF4/GADD45α pathway, a major ER stress signaling axis, can generate angiomodulatory and angiostatic cues (57). Although this axis is usually activated under stressful stimuli, such as osmotic/oxidative stress, irradiation, or nutrient deprivation, it may also be activated in the absence of external stresses (58). In this study, we present endorepellin-mediated activation of this stress axis through ligand–receptor interaction in the absence of external stress. We provide a working model summarizing our current findings regarding the mechanism of action for endorepellin-evoked endothelial cell stress (Fig. 6). According to this model, endorepellin engages the ectodomain of VEGFR2 via LG1/2 globular domains, acting as a partial agonist and evoking phosphorylation of the receptor tyrosine kinase Tyr1175, a key residue from which the canonical phospholipase C-γ cascade originates (59). Phosphorylation of VEGFR2 at Tyr1175 is necessary for stress activation because downstream signaling is completely abrogated by inhibiting VEGFR2 kinase activity with SU5416 (44, 45). The signal emanating from VEGFR2 causes activation of the stress-sensing kinase PERK at Thr980. PERK activation subsequently leads to eIF2α phosphorylation at Ser51, enhanced translation of ATF4, and up-regulation of GADD45α (Fig. 6). ATF4 and GADD45α translocate into the nucleus, where, presumably, a number of anti-angiogenic and pro-autophagic genes are induced. Ultimately, endorepellin-mediated activation of the stress axis causes inhibition of angiogenesis.
Figure 6.
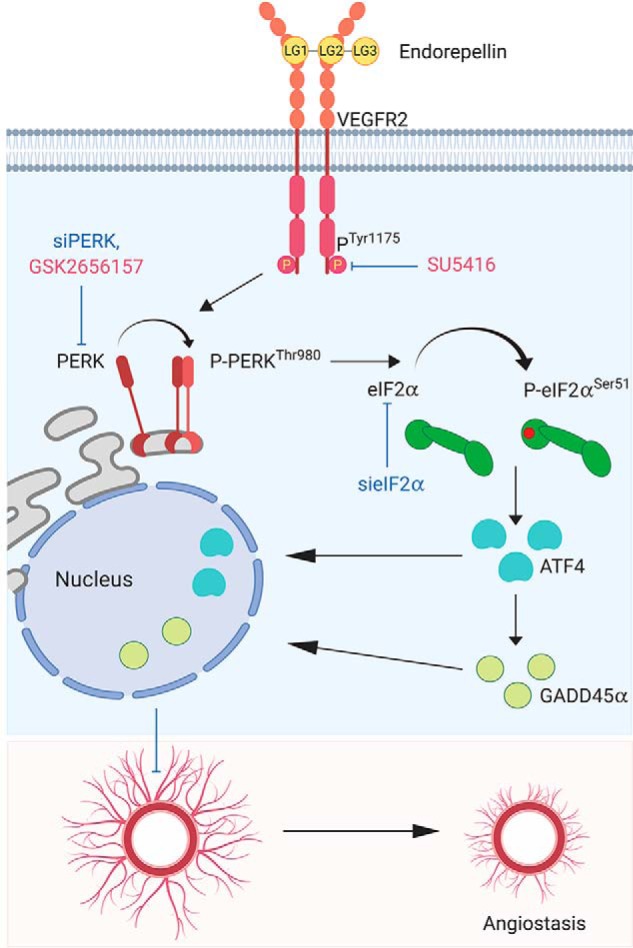
Working model elucidating the mechanism of endorepellin-based stress activation via VEGFR2 and subsequent inhibition of angiogenesis. See text for details.
The path to this study was paved by identification of GADD45α as one the targets of endorepellin in endothelial cells, as discovered by NanoString analysis (25). As GADD45α is a part of the stress axis and is modulated by endorepellin in endothelial cells, we hypothesized that GADD45α may affect angiogenesis. We found that GADD45α up-regulation inhibits angiogenesis. These data are in congruence with a previous study that showed that GADD45α inhibits tumor angiogenesis in mice injected with transformed mouse embryonic fibroblasts (26). Interestingly, we saw up-regulation of GADD45α and upstream stress axis proteins upon endorepellin treatment but a down-regulation of GADD45α mRNA evoked by endorepellin. A plausible cause of this observation may be mRNA degradation by microRNA, which may act on surplus GADD45α being formed. Notably, a GADD45α-repressing microRNA, miR-362-5p, promotes malignancy of chronic myelocytic leukemia cells (60). Another possibility is that there is simultaneous stabilization of GADD45α, which usually requires an accessory protein, such as S7 ribosomal protein, to prevent its ubiquitination and degradation (61). In future studies, it would be interesting to unravel how the endorepellin–VEGFR2 interaction leads to stabilization of GADD45α in endothelial cells; this could help us to better understand vascular stress.
A critical observation in this study was sustained nuclear translocation of GADD45α evoked by soluble endorepellin. Previous studies have shown that nuclear GADD45α interacts with proliferating cell nuclear antigen, protein 21 (p21Waf1), and cell division control 2 (Cdc2), playing a role in DNA repair as a checkpoint protein to prevent a severely damaged cell from proliferating (28). More recently, a novel role of GADD45α has been reported in causing DNA demethylation (62). Specifically, GADD45α recruits the demethylase TET1 at the promoter of the powerful transcription factor TCF21 (62). This bioactivity provides an explanation for the mechanism of epigenetic regulation by GADD45α. We hypothesize that GADD45α may play a direct role in demethylating anti-angiogenic and pro-autophagic gene promoters. Especially important are genes that are known to be controlled by differential methylation, including paternally expressed gene 3 (Peg3) which is differentially methylated in paternal and maternal alleles (63). Notably, we discovered that Peg3 is a master regulator of autophagy (64) and is markedly induced at the mRNA and protein levels in endothelial cells exposed to endorepellin (45). Moreover, we found that overexpression of Peg3 concurrently evokes transcriptional up-regulation of pro-autophagic Beclin 1 and anti-angiogenic thrombospondin 1 (65).
To unravel the mechanism of action of endorepellin-induced activation of the canonical PERK/eIF2α/ATF4/GADD45α stress axis, we performed inhibitory studies of PERK, the most upstream molecule of the stress axis. We used two independent approaches to inhibit PERK action: we knocked down total PERK using RNAi, and we blocked PERK phosphorylation with a specific-kinase small-molecule inhibitor. Notably, genetic and pharmacological inhibitors of PERK markedly suppressed the downstream stress signaling pathway. These results were further validated by knockdown of eIF2α, which also efficiently blocked the downstream effectors of this stress axis. Use of PERK inhibitors required careful selection of the most efficient inhibitory molecule from the cohort of available drugs. Of the two most recently reported PERK inhibitors, GSK2656157 and AMG44, we selected GSK2656157 (55, 66). Although GSK2656157 is the most extensively studied PERK inhibitor and tumor repressor (55), with an IC50 of ∼10 nm (55) and kinase inhibition, AMG44 has a higher specificity for PERK. Unfortunately, the IC50 for AMG44 is ∼84 nm, which is eight times that of GSK2656157, making GSK2656157 the more effective inhibitor (66). To confirm these findings, using both inhibitors in dose–response studies in HUVECs revealed a greater difference in their efficiencies, with GSK2656157 having a 1000-fold higher capability of inhibiting PERK phosphorylation (IC50, ∼1 nm) compared with AMG44 (IC50, ∼1000 nm). Because the advantages of GSK2656157 outweighed that of AMG44, we used GSK2656157 to block PERK phosphorylation and study its impact on angiogenesis. Overall, our results support a high degree of specificity for GSK2656157 because we had the same effects by suppressing PERK with a pool of three to five PERK-specific siRNAs.
The mode of activation of PERK determines its ultimate mechanism of action. For instance, VEGFA-based PERK activation leads to induction of angiogenesis (58), whereas endorepellin-based PERK induction causes angiostasis. This opposing effect could in part be attributed to allosteric inhibition of VEGFA by endorepellin (20). This response could also be due to differential binding of VEGFA and endorepellin to VEGFR2. Indeed, VEGFA binds VEGFR2 at position 2–3 of the immunoglobulin-like sub-domains' on the extracellular domain, whereas endorepellin binds at position 3–4 of the immunonoglobulin-like sub-domains (20, 22). This raises the strong possibility that they may evoke distinct VEGFR2 phosphorylation signatures, leading to activation of diverse downstream signaling pathways. Stress axes are mainly comprised of three pathways: PERK, IRE1, and ATF6. Notably, VEGF simultaneously activates two of these stress pathways, PERK (eIF2α) and ATF6 (58), in contrast to endorepellin, which activates the former. However, we cannot exclude the possibility that endorepellin may also be involved in the ATF6 pathway.
A novel and unique aspect of our study is the combination of biochemical and confocal imaging assays performed on sprouted capillary-like vessels emanating from aortic rings grown in a 3D collagen network. This ex vivo assay is a useful model to study angiogenesis insofar as the sprouts emerging from the aorta are purely endothelial in nature, with only a minute layer of pericytes supporting the sprouting vessels (67). This allowed us to efficiently dissect the angiogenic vessels and the effects of soluble endorepellin, tunicamycin, and PERK inhibitor. We successfully demonstrated endorepellin-based activation of the stress axis in the rings and concurrent abrogation of sprouting by performing imaging and biochemical analysis, providing robust evidence of protracted stress inhibiting angiogenesis in the mouse system.
One of the limitations of the aortic ring assay is the lack of blood flow through the newly formed vessels, which is essential for hemodynamic and mechanochemical functions (67). However, as this study focuses only on endorepellin's effect on formation of neovessels and activation of stress proteins, the absence of blood flow does not have a negative impact on our results. Another challenge concerning the aortic ring assay is reliable quantification of neovessel formation. Different techniques have been described previously (68–70). These methods mainly include counting the sprouts or evaluating the branching patterns. However, counting sprouts can be challenging, especially when the sprouts are numerous and densely packed. A hurdle of quantifying branching pattern is inconsistency in network formation, which may lead to erroneous calculations. To circumvent these issues, we modified the quantification process by using fluorescently labeled rings (marked with IB4 or CD31) to trace the area of vascularization or sprouting and compared the angiogenic footprint between different conditions. As our approach excludes the ring area and exclusively focuses on the newly formed vessel sprouts and branches, it is very sensitive in obtaining detailed information. The margin of error is also primarily reduced because of digital processing and quantification of fluorescence and not manual evaluation of phase-contrast images.
The discovery of activation of the stress pathway by endorepellin is important because the effector molecule GADD45α is the convergence point for various pathways (28). For instance, GADD45α is known to inhibit mTOR and may have a role in evoking autophagy in endothelial cells, leading to angiostasis (26). GADD45α could be the connector protein between the two processes, induction of autophagy and suppression of angiogenesis, which we have proposed in our previous studies (20, 45). It would be interesting to evaluate the role of GADD45α in enabling cross-talk between these two pathways. This study reinforces previous findings that endorepellin induces broad activation of many anti-angiogenic pathways. We showed previously that endorepellin inhibits the PI3K/PDK1/Akt and PKC/c-Jun N-terminal kinase/AP1 axes, which subsequently inhibit HIF1α- and AP1-based VEGFA promoter activation, inhibiting angiogenesis (24). Additionally, we recently discovered that endorepellin induces autophagic degradation of hyaluronan synthase 2 (HAS2), the major producer of pro-angiogenic and pro-inflammatory hyaluronan. This, in turn, evokes dynamic interaction of HAS2 and ATG9A and downstream angiogenic inhibition in vascular endothelia (71). Thus, endorepellin could evoke multiple pathways in endothelial cells emanating from its interaction with VEGFR2, which would synergistically block angiogenesis.
Endorepellin-based shotgun activation of these pathways ensures failsafe inhibition of angiogenesis, making it a viable candidate for combinatorial treatment. Overactivation of stress in the vasculature using endorepellin could be used to enhance susceptibility of endothelial cells to antiangiogenic drugs. Notably, hyperactivation of the stress axis in myeloma cells evoked by proteasome inhibitors enhances susceptibility to chemotherapy (72). Even though endorepellin does not target cancer cells directly, the concept of stress hyperactivation making cells susceptible to annihilation by drugs could be applied in the antiangiogenic paradigm.
Collectively, our findings posit endorepellin as an activator of cellular stress that simultaneously inhibits angiogenesis and evokes autophagy. Given this information, endorepellin may prove to be a protein-based therapeutic agent for overcoming drug resistance against angiogenic inhibitors and, thus, improve cancer therapy. Furthermore, we believe that subsequent investigation will add more extracellular matrix proteins to the list of stress activators and bring to the surface many more key players associated with the stress axis. Thus, we attribute an additional meaningful function to endorepellin's already extensive repertoire and further underscore the importance of the extracellular matrix in improving cancer therapy.
Experimental procedures
Cells and materials
HUVECs were procured from Lifeline Cell Technology (Frederick, MD) and maintained in basal medium supplemented with the VascuLife EnGS LifeFactors Kit (Lifeline Cell Technology). HUVECs were cultured in cell culture dishes treated with 0.2% gelatin (Thermo Fisher Scientific, Waltham, MA) and used within the first four passages. In all experiments, excluding the knockdown experiments, HUVECs were cultured to full confluence for 2 days to make them quiescent. TeloHAECs were procured from the ATCC (Manassas, VA) and grown in Endothelial Cell Growth Medium 2 (EGM-2 SingleQuot Kit Supplements and Growth Factors, Lonza, Walkersville, MD). Porcine aortic endothelial cells expressing VEGFR2 (PAER2) have been described before (73, 74). They were cultured in DMEM (Gibco) with 10% FBS, 1% PenStrep containing G418 (250 μg/ml) (Thermo Fisher Scientific) as the antibiotic selection agent. HeLa and Epstein–Barr nuclear antigen cells were grown in DMEM with 10% FBS and 1% PenStrep. Primary rabbit monoclonal antibodies against PERK (D11A8), eIF2α (D7D3), ATF4 (D4B8), GADD45α (D17E8), and GAPDH (14C10) were obtained from Cell Signaling Technology (Danvers, MA), and rabbit monoclonal for p- eIF2α (ab32157) were from Abcam (Cambridge, MA). Primary rabbit mAb against p-PERK at Thr980 (3179) was obtained from Cell Signaling Technology. Primary mouse mAb against α-tubulin (TU-02), heme-regulated inhibitor (sc-365239), PKR (sc-6282), and GCN2 (sc-374609) were purchased from Santa Cruz Biotechnology. Mouse mAb against CD31 was obtained from BD Pharmingen (553370). For immunoblots, all primary antibodies were used at 1:1000 dilution, except GAPDH, which was used at 1:10,000. Secondary antibodies were used at 1:4000 dilution. For immunofluorescence studies, primary antibodies were used at a concentration of 1:200 and secondary antibodies at a dilution of 1:400. HRP-conjugated goat anti-rabbit secondary antibody (AP307P) was obtained from Millipore (Billerica, MA), and HRP-conjugated goat anti-mouse secondary antibody (W4021) was obtained from Promega (Madison, WI). Alexa Fluor 488 goat anti-mouse (A11001), Alexa Fluor 568 goat anti-mouse (A11004), Alexa Fluor 488 donkey anti-rabbit (A21206), and Alexa Fluor 488 goat anti-rabbit (A11008) secondary antibodies were obtained from Thermo Fisher. Isolectin GS-IB4 Alexa Fluor 594 (I21413) was obtained from Thermo Fisher and used at a dilution of 1:400. Recombinant human endorepellin, LG1/2, and LG3 were produced and purified in the laboratory as described previously (17, 45). Tunicamycin was obtained from Sigma (T7765), PERK inhibitor II (GSK2656157, 504651) from Millipore, AMG44 from Tocris (5517), and SU5416 from Calbiochem (676487).
Nuclear fractionation and immunoblotting
HUVECs (∼106 cells) treated with vehicle or endorepellin for 4 h were harvested and spun down at 500 × g for 5 min. Pelleted cells were washed in PBS, and fractionation was performed using NE-PER nuclear and cytoplasmic extraction reagent (78833, Thermo Scientific). The nuclear pellets were washed twice with PBS to remove any cytoplasmic contaminants before extraction. To perform Western blotting, following treatment, endothelial cells were lysed using radioimmune precipitation assay buffer (50 mm Tris-HCl, 50 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 0.5% sodium deoxycholate, 1 mm sodium orthovanadate, 0.5% SDS, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mm PMSF, 100 μm l-1-tosylamido-2-phenylethyl chloromethyl ketone, and 1 EDTA-free protease inhibitor tablet) for 20 min while rocking on ice. Proteins were separated on an SDS-PAGE gel, transferred to a nitrocellulose membrane (Bio-Rad), incubated with the appropriate antibodies, and visualized using SuperSignal West Pico chemiluminescence substrate (Thermo Scientific) and an Image Quant LAS-4000 (GE Healthcare).
Immunofluorescence and confocal laser microscopy
HUVECs (∼5 × 104 cells) were grown on gelatin-coated four-chambered slides (Thermo Fisher Scientific) and given the stipulated treatments, and then the cells were briefly washed with PBS and fixed with 4% (w/v) paraformaldehyde for 20 min on ice (75). Cells were blocked in 1% BSA for 1 h, washed three times in PBS, incubated with primary antibodies for 1 h, washed three times in PBS (76), and then incubated with secondary antibodies for 1 h, as described before (77, 78). Stained cells were mounted using a hard-set mounting medium containing DAPI (Vector Laboratories, Burlingame, CA) and sealed with a coverslip. Immunofluorescence images were captured using ×63 objective on a Leica DM5500B microscope installed with Leica Application Suite v1.8 software (Leica Microsystems, Frankfurt, Germany). For higher-resolution images, confocal microscopy was used, and images were captured using a ×63, 1.3 oil immersion objective of a Zeiss LSM-780 confocal laser-scanning microscope equipped with the Zen Imaging software. Images were acquired as single optical sections of 1 μm and collected with the pinhole set to 1 Airy unit for all channels. Post-acquisition, images were analyzed/processed using ImageJ and Photoshop CS6 (Adobe Systems).
siRNA-mediated knockdown
Individual transient knockdown of PERK and eIF2α was achieved in HUVECs using siRNA specific for PERK (sc-36213) or eIF2α (sc-35272). Scrambled siRNA (sc-37007) was used as the negative control. HUVECs (∼2 × 105 cells/well) were seeded in 6-well plates to achieve ∼80% confluence after attachment and transfected with 100 pm siScr, siPERK, or sieIF2α and 10 μl of Lipofectamine RNAiMax (Life Technologies) diluted in 150 μl of serum-free Opti-MEM medium (Gibco) overnight. The following day, medium containing siRNA was aspirated, and cells were incubated in 250 μl/well Lifeline Cell Technology medium containing 4% FBS and 1% PenStrep for 1 day. On day 3, 4% medium was replaced with 2% FBS-containing medium. On day 4, we started respective treatments with vehicle (PBS), endorepellin (200 nm), or tunicamycin (10 μg/ml). siRNA-mediated knockdown of PERK or eIF2α was verified by immunoblotting. Cell lysates were further analyzed by immunoblotting for proteins downstream of PERK and eIF2α.
Aortic ring assay
For all animal-based experiments, instructions were followed according to the Guide for Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee of Thomas Jefferson University. Thoracic aortae from 6- to 7-week-old WT C57/BL6 mice (The Jackson Laboratory) were surgically isolated, cleaned off the fibro-adipose tissue, and sectioned serially into 0.5- to 1-mm rings. Rings were sandwiched between two 3D collagen type I gel layers (1 mg/ml) in 48-well plates and incubated in EGM-2 medium (Lonza) at 37 °C as described elsewhere (48, 79). Following initial sprouting, i.e. 3 days after embedding, the rings were given acute or chronic treatment. For immediate measurement of stress induction, rings were treated acutely (2–4 h) with vehicle (PBS), endorepellin (200 nm), or tunicamycin (10 μg/ml). They were subsequently processed for immunofluorescence or Western blotting. For observing long-term effects of the reagents on sprouting, the rings were grown over 10 days with alternative-day treatment of vehicle (PBS), endorepellin (200 nm), tunicamycin (10 μg/ml), or PERKi. The rings were then processed for immunofluorescence or Western blotting.
Processing the aortic ring data
Sprouting of the rings was measured as radial distance using low-magnification (×5) phase-contrast images (79). We subtracted the background and highlighted sprouts using the threshold function. Then we manually encircled the edges of the sprouts and measured the radii (Fig. S3A).
During the immunofluorescence analyses, the rings were labeled with IB4, CD31, p-PERK, and GADD45α. Z-stack images of the rings were captured with a Zeiss LSM780 NLO confocal/multiphoton microscope, and the images were analyzed using ImageJ software. Two methods of analyses were used: fluorescence of the traced-out sprouts (79) and intensity profiles of the sprouts, both using ImageJ. Briefly, fluorescence intensity was uniformly normalized across all conditions using the threshold function, and regions of interest outlining the sprouts were selected using the polygon drawing tool. We then measured the integrated intensity of the traced areas (Fig. S3B) and normalized on the frame areas. The normalized fluorescence intensity of each protein was plotted as the mean fluorescence intensity. IB4 or CD31 expression was also used to quantify the extent of sprouting in the rings. We also performed line-scanning analyses of protein levels in the sprouts using the profile plot tool in ImageJ. A line was drawn along the sprout, and fluorescence intensity was measured along the length.
Quantification and statistical analysis
The immunoblots were analyzed by scanning densitometry using ImageJ. Statistical significance was calculated by two-tailed, unpaired Student's t test. One-way ANOVA was performed on data with multiple groups. All experiments were conducted at least three times, and the mean differences were considered significant at p < 0.05.
Data availability
All data are contained in this manuscript.
Author contributions
A. K., C. G. C., and R. V. I. conceptualization; A. K., C. G. C., and R. V. I. data curation; A. K. and C. G. C. software; A. K., C. G. C., and R. V. I. formal analysis; A. K., C. G. C., and R. V. I. validation; A. K., C. G. C., and R. V. I. investigation; A. K., C. G. C., and R. V. I. visualization; A. K. and C. G. C. methodology; A. K. and R. V. I. writing-original draft; C. G. C. and R. V. I. writing-review and editing; R. V. I. supervision; R. V. I. funding acquisition; R. V. I. project administration.
Supplementary Material
Acknowledgments
We thank L. Claesson-Welsh for providing the PAE-VEGFR2 cells, M. Mongiat for providing LG1/2 in the initial stages of this work, S. Buraschi for help with immunostaining, and T. Neill for critical reading of the manuscript.
This work was supported in part by National Institutes of Health Grants RO1 CA039481 and RO1 CA047282 (to R. V. I.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- LG
- laminin G-like domain
- VEGF
- vascular endothelial growth factor
- PERK
- PKR-like endoplasmic reticulum kinase
- PKR
- protein kinase R
- HUVEC
- human umbilical vein endothelial cell
- TeloHAEC
- telomerase immortalized human aortic endothelial cell
- PAER2
- porcine aortic endothelial cell expressing VEGFR2
- PERKi
- PERK inhibitor
- ER
- endoplasmic reticulum
- PenStrep
- penicillin/streptomycin
- ANOVA
- analysis of variance.
References
- 1. Sparro G., Galdenzi G., Eleuteri A. M., Angeletti M., Schroeder W., and Fioretti E. (1997) Isolation and N-terminal sequence of multiple forms of granulins in human urine. Protein Expr. Purif. 10, 169–174 10.1006/prep.1997.0726 [DOI] [PubMed] [Google Scholar]
- 2. Zoeller J. J., McQuillan A., Whitelock J., Ho S.-Y., and Iozzo R. V. (2008) A central function for perlecan in skeletal muscle and cardiovascular development. J. Cell Biol. 181, 381–394 10.1083/jcb.200708022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Costell M., Gustafsson E., Aszódi A., Mörgelin M., Bloch W., Hunziker E., Addicks K., Timpl R., and Fässler R. (1999) Perlecan maintains the integrity of cartilage and some basement membranes. J. Cell Biol. 147, 1109–1122 10.1083/jcb.147.5.1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cohen I. R., Grässel S., Murdoch A. D., and Iozzo R. V. (1993) Structural characterization of the complete human perlecan gene and its promoter. Proc. Natl. Acad. Sci. U.S.A. 90, 10404–10408 10.1073/pnas.90.21.10404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Handler M., Yurchenco P. D., and Iozzo R. V. (1997) Developmental expression of perlecan during murine embryogenesis. Dev. Dyn. 210, 130–145 [DOI] [PubMed] [Google Scholar]
- 6. Cohen I. R., Murdoch A. D., Naso M. F., Marchetti D., Berd D., and Iozzo R. V. (1994) Abnormal expression of perlecan proteoglycan in metastatic melanomas. Cancer Res. 54, 5771–5774 [PubMed] [Google Scholar]
- 7. Iozzo R. V., and Cohen I. (1993) Altered proteoglycan gene expression and the tumor stroma. Experientia 49, 447–455 10.1007/BF01923588 [DOI] [PubMed] [Google Scholar]
- 8. Fuki I. V., Iozzo R. V., and Williams K. J. (2000) Perlecan heparan sulfate proteoglycan: a novel receptor that mediates a distinct pathway for ligand catabolism. J. Biol. Chem. 275, 25742–25750 10.1074/jbc.M909173199 [DOI] [PubMed] [Google Scholar]
- 9. Iozzo R. V. (2005) Basement membrane proteoglycans: from cellar to ceiling. Nat. Rev. Mol. Cell Biol. 6, 646–656 10.1038/nrm1702 [DOI] [PubMed] [Google Scholar]
- 10. Gubbiotti M. A., Neill T., and Iozzo R. V. (2017) A current view of perlecan in physiology and pathology: a mosaic of functions. Matrix Biol. 57–58, 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farach-Carson M. C., and Carson D. D. (2007) Perlecan: a multifunctional extracellular proteoglycan scaffold. Glycobiology 17, 897–905 10.1093/glycob/cwm043 [DOI] [PubMed] [Google Scholar]
- 12. Farach-Carson M. C., Warren C. R., Harrington D. A., and Carson D. D. (2014) Border patrol: insights into the unique role of perlecan/heparan sulfate proteoglycan 2 at cell and tissue borders. Matrix Biol. 34, 64–79 10.1016/j.matbio.2013.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schaefer L., Tredup C., Gubbiotti M. A., and Iozzo R. V. (2017) Proteoglycan neofunctions: regulation of inflammation and autophagy in cancer biology. FEBS J. 284, 10–26 10.1111/febs.13963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karamanos N. K., Piperigkou Z., Theocharis A. D., Watanabe H., Franchi M., Baud S., Brézillon S., Götte M., Passi A., Vigetti D., Ricard-Blum S., Sanderson R. D., Neill T., and Iozzo R. V. (2018) Proteoglycan chemical diversity drives multifunctional cell regulation and therapeutics. Chem. Rev. 118, 9152–9232 10.1021/acs.chemrev.8b00354 [DOI] [PubMed] [Google Scholar]
- 15. Theocharis A. D., Skandalis S. S., Neill T., Multhaupt H. A., Hubo M., Frey H., Gopal S., Gomes A., Afratis N., Lim H. C., Couchman J. R., Filmus J., Sanderson R. D., Schaefer L., Iozzo R. V., and Karamanos N. K. (2015) Insights into the key roles of proteoglycans in breast cancer biology and translational medicine. Biochim. Biophys. Acta 1855, 276–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Neill T., Schaefer L., and Iozzo R. V. (2015) Decoding the matrix: instructive roles of proteoglycan receptors. Biochemistry 54, 4583–4598 10.1021/acs.biochem.5b00653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mongiat M., Sweeney S. M., San Antonio J. D., Fu J., and Iozzo R. V. (2003) Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J. Biol. Chem. 278, 4238–4249 10.1074/jbc.M210445200 [DOI] [PubMed] [Google Scholar]
- 18. Bix G., Fu J., Gonzalez E. M., Macro L., Barker A., Campbell S., Zutter M. M., Santoro S. A., Kim J. K., Höök M., Reed C. C., and Iozzo R. V. (2004) Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through the α2β1 integrin. J. Cell Biol. 166, 97–109 10.1083/jcb.200401150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bix G., Castello R., Burrows M., Zoeller J. J., Weech M., Iozzo R. A., Cardi C., Thakur M. L., Barker C. A., Camphausen K., and Iozzo R. V. (2006) Endorepellin in vivo: targeting the tumor vasculature and retarding cancer growth and metabolism. J. Natl. Cancer Inst. 98, 1634–1646 10.1093/jnci/djj441 [DOI] [PubMed] [Google Scholar]
- 20. Goyal A., Gubbiotti M. A., Chery D. R., Han L., and Iozzo R. V. (2016) Endorepellin-evoked autophagy contributes to angiostasis. J. Biol. Chem. 291, 19245–19256 10.1074/jbc.M116.740266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poluzzi C., Iozzo R. V., and Schaefer L. (2016) Endostatin and endorepellin: a common route of action for similar angiostatic cancer avengers. Adv. Drug Deliv. Rev. 97, 156–173 10.1016/j.addr.2015.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goyal A., Pal N., Concannon M., Paul M., Doran M., Poluzzi C., Sekiguchi K., Whitelock J. M., Neill T., and Iozzo R. V. (2011) Endorepellin, the angiostatic module of perlecan, interacts with both the α2β1 integrin and vascular endothelial growth factor receptor 2 (VEGFR2). J. Biol. Chem. 286, 25947–25962 10.1074/jbc.M111.243626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Willis C. D., Poluzzi C., Mongiat M., and Iozzo R. V. (2013) Endorepellin laminin-like globular repeat 1/2 domains bind Ig3–5 of vascular endothelial growth factor(VEGF) receptor 2 and block pro-angiogenic signaling by VEGFA in endothelial cells. FEBS J. 280, 2271–2284 10.1111/febs.12164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goyal A., Poluzzi C., Willis C. D., Smythies J., Shellard A., Neill T., and Iozzo R. V. (2012) Endorepellin affects angiogenesis by antagonizing diverse VEGFR2-evoked signaling pathways: transcriptional repression of HIF-1α and VEGFA and concurrent inhibition of NFAT1 activation. J. Biol. Chem. 287, 43543–43556 10.1074/jbc.M112.401786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neill T., Andreuzzi E., Wang Z.-X., Peiper S. C., Mongiat M., and Iozzo R. V. (2018) Endorepellin remodels the endothelial transcriptome toward a pro-autophagic and pro-mitophagic gene signature. J. Biol. Chem. 293, 12137–12148 10.1074/jbc.RA118.002934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang F., Zhang W., Li D., and Zhan Q. (2013) Gadd45a suppresses tumor angiogenesis via inhibition of the mTOR/STAT3 protein pathway. J. Biol. Chem. 288, 6552–6560 10.1074/jbc.M112.418335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holczer M., Bánhegyi G., and Kapuy O. (2016) GADD34 keeps the mTOR pathway inactivated in endoplasmic reticulum stress related autophagy. PLoS ONE. 11, e0168359 10.1371/journal.pone.0168359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liebermann D. A., and Hoffman B. (2008) Gadd45 in stress signaling. J. Mol. Signal. 3, 15 10.1186/1750-2187-3-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wek R. C., Jiang H. Y., and Anthony T. G. (2006) Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34, 7–11 10.1042/BST0340007 [DOI] [PubMed] [Google Scholar]
- 30. Ebert S. M., Bullard S. A., Basisty N., Marcotte G. R., Skopec Z. P., Dierdorff J. M., Al-Zougbi A., Tomcheck K. C., DeLau A. D., Rathmacher J. A., Bodine S. C., Schilling B., and Adams C. M. (2020) Activating transcription factor 4 (ATF4) promotes skeletal muscle atrophy by forming a heterodimer with the transcriptional regulator C/EBPβ. J. Biol. Chem. 295, 2787–2803 10.1074/jbc.RA119.012095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Walter P., and Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- 32. Wek R. C., and Cavener D. R. (2007) Translational control and the unfolded protein response. Antioxid. Redox Signal. 9, 2357–2371 10.1089/ars.2007.1764 [DOI] [PubMed] [Google Scholar]
- 33. Salazar M., Carracedo A., Salanueva I. J., Hernández-Tiedra S., Lorente M., Egia A., Vázquez P., Blázquez C., Torres S., García S., Nowak J., Fimia G. M., Piacentini M., Cecconi F., Pandolfi P. P., et al. (2009) Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Invest. 119, 1359–1372 10.1172/JCI37948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marciniak S. J., Yun C. Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H. P., and Ron D. (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077 10.1101/gad.1250704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Livezey M., Huang R., Hergenrother P. J., and Shapiro D. J. (2018) Strong and sustained activation of the anticipatory unfolded protein response induces necrotic cell death. Cell Death. Differ. 25, 1796–1807 10.1038/s41418-018-0143-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liebermann D. A., Tront J. S., Sha X., Mukherjee K., Mohamed-Hadley A., and Hoffman B. (2011) Gadd45 stress sensors in malignancy and leukemia. Crit. Rev. Oncog. 16, 129–140 10.1615/CritRevOncog.v16.i1-2.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ebert S. M., Dyle M. C., Kunkel S. D., Bullard S. A., Bongers K. S., Fox D. K., Dierdorff J. M., Foster E. D., and Adams C. M. (2012) Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J. Biol. Chem. 287, 27290–27301 10.1074/jbc.M112.374777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adams C. M., Ebert S. M., and Dyle M. C. (2017) Role of ATF4 in skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 20, 164–168 10.1097/MCO.0000000000000362 [DOI] [PubMed] [Google Scholar]
- 39. Donnelly N., Gorman A. M., Gupta S., and Samali A. (2013) The eIF2α kinases: their structures and functions. Cell Mol. Life Sci. 70, 3493–3511 10.1007/s00018-012-1252-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vattem K. M., and Wek R. C. (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 101, 11269–11274 10.1073/pnas.0400541101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Proud C. G. (2005) eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 16, 3–12 10.1016/j.semcdb.2004.11.004 [DOI] [PubMed] [Google Scholar]
- 42. Su Q., Wang S., Gao H. Q., Kazemi S., Harding H. P., Ron D., and Koromilas A. E. (2008) Modulation of the eukaryotic initiation factor 2 α-subunit kinase PERK by tyrosine phosphorylation. J. Biol. Chem. 283, 469–475 10.1074/jbc.M704612200 [DOI] [PubMed] [Google Scholar]
- 43. Xu C., Bailly-Maitre B., and Reed J. C. (2005) Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 115, 2656–2664 10.1172/JCI26373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fong T. A., Shawver L. K., Sun L., Tang C., App H., Powell T. J., Kim Y. H., Schreck R., Wang X., Risau W., Ullrich A., Hirth K. P., and McMahon G. (1999) SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 59, 99–106 [PubMed] [Google Scholar]
- 45. Poluzzi C., Casulli J., Goyal A., Mercer T. J., Neill T., and Iozzo R. V. (2014) Endorepellin evokes autophagy in endothelial cells. J. Biol. Chem. 289, 16114–16128 10.1074/jbc.M114.556530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kovalsky O., Lung F. D., Roller P. P., and Fornace A. J. Jr. (2001) Oligomerization of human Gadd45α protein. J. Biol. Chem. 276, 39330–39339 10.1074/jbc.M105115200 [DOI] [PubMed] [Google Scholar]
- 47. Weigel P. H. (2015) Hyaluronan synthase: the mechanism of initiation at the reducing end and a pendulum model for polysaccharide translocation to the cell exterior. Int. J. Cell Biol. 2015, 367579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baker M., Robinson S. D., Lechertier T., Barber P. R., Tavora B., D'Amico G., Jones D. T., Vojnovic B., and Hodivala-Dilke K. (2011) Use of the mouse aortic ring assay to study angiogenesis. Nat. Protoc. 7, 89–104 10.1038/nprot.2011.435 [DOI] [PubMed] [Google Scholar]
- 49. Ernst C., and Christie B. R. (2006) Isolectin-IB 4 as a vascular stain for the study of adult neurogenesis. J. Neurosci. Methods 150, 138–142 10.1016/j.jneumeth.2005.06.018 [DOI] [PubMed] [Google Scholar]
- 50. Benton R. L., Maddie M. A., Minnillo D. R., Hagg T., and Whittemore S. R. (2008) Griffonia simplicifolia isolectin B4 identifies a specific subpopulation of angiogenic blood vessels following contusive spinal cord injury in the adult mouse. J. Comp Neurol. 507, 1031–1052 10.1002/cne.21570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harding H. P., Zhang Y., and Ron D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- 52. Shi Y., Vattem K. M., Sood R., An J., Liang J., Stramm L., and Wek R. C. (1998) Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 18, 7499–7509 10.1128/MCB.18.12.7499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Harding H. P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., and Ron D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 10.1016/S1097-2765(00)00108-8 [DOI] [PubMed] [Google Scholar]
- 54. Atkins C., Liu Q., Minthorn E., Zhang S. Y., Figueroa D. J., Moss K., Stanley T. B., Sanders B., Goetz A., Gaul N., Choudhry A. E., Alsaid H., Jucker B. M., Axten J. M., and Kumar R. (2013) Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 73, 1993–2002 10.1158/0008-5472.CAN-12-3109 [DOI] [PubMed] [Google Scholar]
- 55. Axten J. M., Romeril S. P., Shu A., Ralph J., Medina J. R., Feng Y., Li W. H., Grant S. W., Heerding D. A., Minthorn E., Mencken T., Gaul N., Goetz A., Stanley T., Hassell A. M., et al. (2013) Discovery of GSK2656157: an optimized PERK inhibitor selected for preclinical development. ACS Med. Chem. Lett. 4, 964–968 10.1021/ml400228e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gupta S., McGrath B., and Cavener D. R. (2009) PERK regulates the proliferation and development of insulin-secreting β-cell tumors in the endocrine pancreas of mice. PLoS ONE 4, e8008 10.1371/journal.pone.0008008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Binet F., and Sapieha P. (2015) ER stress and angiogenesis. Cell Metab. 22, 560–575 10.1016/j.cmet.2015.07.010 [DOI] [PubMed] [Google Scholar]
- 58. Karali E., Bellou S., Stellas D., Klinakis A., Murphy C., and Fotsis T. (2014) VEGF signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol. Cell 54, 559–572 10.1016/j.molcel.2014.03.022 [DOI] [PubMed] [Google Scholar]
- 59. Olsson A.-K., Dimberg A., Kreuger J., and Claesson-Welsh L. (2006) VEGF receptor signalling in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371 10.1038/nrm1911 [DOI] [PubMed] [Google Scholar]
- 60. Yang P., Ni F., Deng R. Q., Qiang G., Zhao H., Yang M. Z., Wang X. Y., Xu Y. Z., Chen L., Chen D. L., Chen Z. J., Kan L. X., and Wang S. Y. (2015) MiR-362-5p promotes the malignancy of chronic myelocytic leukaemia via down-regulation of GADD45α. Mol. Cancer 14, 190 10.1186/s12943-015-0465-3 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61. Gao M., Li X., Dong W., Jin R., Ma H., Yang P., Hu M., Li Y., Hao Y., Yuan S., Huang J., and Song L. (2013) Ribosomal protein S7 regulates arsenite-induced GADD45α expression by attenuating MDM2-mediated GADD45α ubiquitination and degradation. Nucleic Acids Res. 41, 5210–5222 10.1093/nar/gkt223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Arab K., Karaulanov E., Musheev M., Trnka P., Schäfer A., Grummt I., and Niehrs C. (2019) GADD45A binds R-loops and recruits TET1 to CpG island promoters. Nat. Genet. 51, 217–223 10.1038/s41588-018-0306-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bretz C. L., Frey W. D., Teruyama R., and Kim J. (2018) Allele and dosage specificity of the Peg3 imprinted domain. PLoS ONE 13, e0197069 10.1371/journal.pone.0197069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Buraschi S., Neill T., Goyal A., Poluzzi C., Smythies J., Owens R. T., Schaefer L., Torres A., and Iozzo R. V. (2013) Decorin causes autophagy in endothelial cells via Peg3. Proc. Natl. Acad. Sci. U.S.A. 110, E2582–E2591 10.1073/pnas.1305732110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Torres A., Gubbiotti M. A., and Iozzo R. V. (2017) Decorin-inducible Peg3 evokes Beclin 1-mediated autophagy and thrombospondin 1-mediated angiostasis. J. Biol. Chem. 292, 5055–5069 10.1074/jbc.M116.753632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Smith A. L., Andrews K. L., Beckmann H., Bellon S. F., Beltran P. J., Booker S., Chen H., Chung Y. A., D'Angelo N. D., Dao J., Dellamaggiore K. R., Jaeckel P., Kendall R., Labitzke K., Long A. M., et al. (2015) Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 58, 1426–1441 10.1021/jm5017494 [DOI] [PubMed] [Google Scholar]
- 67. Nicosia R. F. (2009) The aortic ring model of angiogenesis: a quarter century of search and discovery. J. Cell Mol. Med. 13, 4113–4136 10.1111/j.1582-4934.2009.00891.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Blacher S., Devy L., Burbridge M. F., Roland G., Tucker G., Noël A., and Foidart J. M. (2001) Improved quantification of angiogenesis in the rat aortic ring assay. Angiogenesis 4, 133–142 10.1023/A:1012251229631 [DOI] [PubMed] [Google Scholar]
- 69. Iqbal F., Szaraz P., Librach M., Gauthier-Fisher A., and Librach C. L. (2017) Angiogenic potency evaluation of cell therapy candidates by a novel application of the in vitro aortic ring assay. Stem Cell Res. Ther. 8, 184 10.1186/s13287-017-0631-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Van Valckenborgh E., De Raeve H., Devy L., Blacher S., Munaut C., Noël A., Van Marck E., Van Riet I., Van Camp B., and Vanderkerken K. (2002) Murine 5T multiple myeloma cells induce angiogenesis in vitro and in vivo. Br. J. Cancer 86, 796–802 10.1038/sj.bjc.6600137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen C. G., Gubbiotti M. A., Kapoor A., Han X., Yu Y., Linhardt R. J., and Iozzo R. V. (2020) Autophagic degradation of HAS2 in endothelial cells: a novel mechanism to regulate angiogenesis. Matrix Biol. pii: S0945-053X(20)30011-1 10.1016/j.matbio.2020.02.001 [DOI] [PubMed] [Google Scholar]
- 72. Tameire F., Verginadis I. I., and Koumenis C. (2015) Cell intrinsic and extrinsic activators of the unfolded protein response in cancer: mechanisms and targets for therapy. Semin. Cancer Biol. 33, 3–15 10.1016/j.semcancer.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Waltenberger J., Claesson-Welsh L., Siegbahn A., Shibuya M., and Heldin C.-H. (1994) Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 269, 26988–26995 [PubMed] [Google Scholar]
- 74. Ito N., and Claesson-Welsh L. (1999) Dual effects of heparin on VEGF binding to VEGF receptor-1 and transduction of biological responses. Angiogenesis 3, 159–166 10.1023/A:1009008926710 [DOI] [PubMed] [Google Scholar]
- 75. Chen C., Kapoor A., and Iozzo R. V. (2019) Methods for monitoring matrix-induced autophagy. Methods Mol. Biol. 1952, 157–191 10.1007/978-1-4939-9133-4_14 [DOI] [PubMed] [Google Scholar]
- 76. Goldoni S., Owens R. T., McQuillan D. J., Shriver Z., Sasisekharan R., Birk D. E., Campbell S., and Iozzo R. V. (2004) Biologically active decorin is a monomer in solution. J. Biol. Chem. 279, 6606–6612 10.1074/jbc.M310342200 [DOI] [PubMed] [Google Scholar]
- 77. Rudnicka L., Varga J., Christiano A. M., Iozzo R. V., Jimenez S. A., and Uitto J. (1994) Elevated expression of type VII collagen in the skin of patients with systemic sclerosis. J. Clin. Invest. 93, 1709–1715 10.1172/JCI117154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Alvarez R. J., Sun M. J., Haverty T. P., Iozzo R. V., Myers J. C., and Neilson E. G. (1992) Biosynthetic and proliferative characteristics of tubulointerstitial fibroblasts probed with paracrine cytokines. Kidney Int. 41, 14–23 10.1038/ki.1992.3 [DOI] [PubMed] [Google Scholar]
- 79. Kapoor A., Chen C. G., and Iozzo R. V. (2020) A simplified aortic ring assay: a useful ex vivo method to assess biochemical and functional parameters of angiogenesis. Matrix Biol. Plus 10.1016/j.mbplus.2020.100025 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained in this manuscript.