

Abstract
The quinoprotein glycine oxidase from the marine bacterium Pseudoalteromonas luteoviolacea (PlGoxA) uses a protein-derived cysteine tryptophylquinone (CTQ) cofactor to catalyze conversion of glycine to glyoxylate and ammonia. This homotetrameric enzyme exhibits strong cooperativity toward glycine binding. It is a good model for studying enzyme kinetics and cooperativity, specifically for being able to separate those aspects of protein function through directed mutagenesis. Variant proteins were generated with mutations in four active-site residues, Phe-316, His-583, Tyr-766, and His-767. Structures for glycine-soaked crystals were obtained for each. Different mutations had differential effects on kcat and K0.5 for catalysis, K0.5 for substrate binding, and the Hill coefficients describing the steady-state kinetics or substrate binding. Phe-316 and Tyr-766 variants retained catalytic activity, albeit with altered kinetics and cooperativity. Substitutions of His-583 revealed that it is essential for glycine binding, and the structure of H583C PlGoxA had no active-site glycine present in glycine-soaked crystals. The structure of H767A PlGoxA revealed a previously undetected reaction intermediate, a carbinolamine product-reduced CTQ adduct, and exhibited only negligible activity. The results of these experiments, as well as those with the native enzyme and previous variants, enabled construction of a detailed mechanism for the reductive half-reaction of glycine oxidation. This proposed mechanism includes three discrete reaction intermediates that are covalently bound to CTQ during the reaction, two of which have now been structurally characterized by X-ray crystallography.
Keywords: enzyme catalysis, enzyme mechanism, enzyme kinetics, cooperativity, structure-function, quinone, oxidase, site-directed mutagenesis, acid-base chemistry, cysteine tryptophylquinone, GoxA, LodA-like protein
Introduction
The phenomenon of cooperativity was initially observed and studied through the allosteric binding of oxygen to hemoglobin over a century ago (1–3). A classical model of positive cooperativity is described as a multimeric protein, in which the affinity for ligand binding to an open site on one subunit is increased by conformational changes induced by binding of a ligand to another subunit. Cooperativity as a mathematical concept can also describe steady-state kinetic behavior when the increase in initial velocity cannot be fit to a hyperbolic curve using the Michaelis-Menten equation. In each case, binding and kinetics, a sigmoidal curve is characteristic of positive cooperativity. If a protein or enzyme has multiple subunits, each of which possess a ligand- or substrate-binding site, the possibility of cooperativity must be considered.
The quinoprotein glycine oxidase from Pseudoalteromonas luteoviolacea (PlGoxA)2 (4) is an ideal enzyme with which to study the interplay between the cooperativity of substrate binding and steady-state kinetic behavior. PlGoxA uses a protein-derived cysteine tryptophylquinone (CTQ) cofactor (Fig. 1A) (5, 6). The complete glycine oxidation reaction is oxygen-dependent, but the several reaction steps in the reductive half-reaction are not (Fig. 1B). Addition of glycine to PlGoxA under anaerobic conditions yields a stable product-reduced CTQ Schiff-base intermediate with a characteristic absorbance spectrum distinct from that of the resting oxidized protein. For WT PlGoxA, the initial binding of glycine is slow relative to the reaction steps that form the product Schiff base. As such, an anaerobic titration of PlGoxA with glycine yielded spectral changes that were used to quantitate the fraction of protein to which glycine has bound. This yielded a Hill coefficient (h value) of 3.7 for this homotetrameric protein, which describes strong cooperative binding (7). Analysis of the steady-state kinetics of glycine oxidation by PlGoxA also exhibited positive cooperativity, but with a smaller h value of 1.8 (4). This result was interpreted to mean that the substrate binding was only partially rate-limiting for the overall reaction, thus obfuscating the binding cooperativity.
Figure 1.

A, protein-derived cysteine tryptophylquinone cofactor. The posttranslational modifications that form CTQ are indicated in red. R signifies the point of attachment to the protein. B, reaction scheme and structures of intermediates for glycine oxidation by PlGoxA. This is based on the results of previous studies (13, 17) and this study.
PlGoxA is a member of the family of proteins, named after a lysine ϵ-oxidase from Marinomonas mediterranea (MmLodA) (8). LodA-like proteins are CTQ-bearing enzymes that, unlike previously-studied tryptophylquinone dehydrogenases, function as oxidases (9). MmLodA has been described as either a homodimer or tetramer (10). Steady-state kinetic studies revealed the enzyme obeyed Michaelis-Menten kinetics with no discernable cooperativity. A second LodA-like protein from the same bacterium is a glycine oxidase, MmGoxA (11), which forms a homodimer. Analysis of the steady-state kinetics of glycine oxidation by MmGoxA did reveal positive cooperativity, with an h value of 1.7 (12). With MmLodA and MmGoxA, it was not possible to monitor substrate binding because no stable product–CTQ adduct with distinct spectroscopic features was detectable.
Recently, it was shown that mutation of Asp-678 to Glu in the active site of PlGoxA eliminated the observed cooperativity of steady-state kinetic behavior, but it did not diminish the observed cooperativity of glycine binding (13). The explanation for this was that the mutation slowed the rate of one or more reaction steps such that binding of glycine was no longer even partially rate-limiting in the overall reaction. As a consequence of this, it was also possible to spectroscopically characterize a substrate-oxidized CTQ Schiff base reaction intermediate that accumulated during the reaction of D678E PlGoxA, which was not observed during the reaction of the WT enzyme (Fig. 1B).
The structure of PlGoxA reveals other amino acid residues that potentially play roles in catalysis and cooperativity (Fig. 2). Of particular interest are residues Tyr-766 and His-767. The side chains of these residues project from a loop of one subunit into the active site of another subunit of the homotetramer. The crystal structure of the substrate-bound PlGoxA showed that these residues interact with the carboxyl group of the glycine substrate in the covalent CTQ adduct that is formed by reaction of the glycine amino group with CTQ (4). The inter-subunit projection of these residues suggests a role in cooperativity. It was previously shown that Phe-237 in MmGoxA plays a role in its cooperative kinetic behavior (12). The corresponding residue in PlGoxA is Phe-316, which suggests that it may also play a role in cooperativity of PlGoxA. Residue His-583 of PlGoxA is seen in the crystal structure to interact with the glycine–CTQ adduct in the active site. This suggests a possible role in substrate binding or catalysis or both. In this study, these four amino acid residues were altered by site-directed mutagenesis, and the effects of the mutations on glycine binding, glycine oxidation kinetics, and protein structure were determined. The results describe how subtle changes in structure cause dramatic effects in substrate binding, reaction kinetics, and cooperativity. Furthermore, alteration of the binding and kinetic mechanism by a H767A mutation allowed structural characterization of another previously undetectable reaction intermediate.
Figure 2.
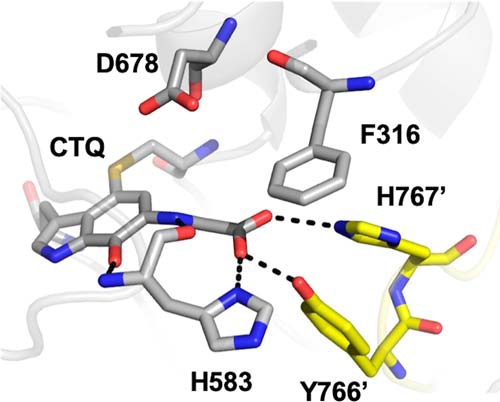
Active site of glycine-soaked crystals of WT PlGoxA (PDB code 6EER) showing the product-reduced CTQ Schiff-base adduct and active-site residues as sticks colored according to element. Carbons from residues of the neighboring protomer are colored yellow.
Results
Expression of PlGoxA variant proteins
The following variant proteins were expressed in and isolated from Escherichia coli: H583A, H583C, F316Y, F316A, Y766F, and H767A. In each case, the amount of protein that was purified was comparable with that of the WT recombinant protein. When subjected to size-exclusion chromatography, each protein eluted with a mass in the range of 350–400 kDa. This size is consistent with each being a homotetramer of four 91-kDa subunits. Each protein exhibited a visible absorbance spectrum that is characteristic of oxidized CTQ and essentially the same as is seen in WT PlGoxA. In contrast to these results, previous mutagenesis studies of MmLodA and MmGoxA, which involved corresponding amino acid residues, compromised CTQ formation and often had lower yields (14, 15). Similarly, mutagenesis of active-site residues in the tryptophan tryptophylquinone (TTQ)-dependent methylamine dehydrogenase compromised TTQ formation (16). Thus, PlGoxA is a particularly robust tryptophylquinone protein, which allows for more detailed structure–function studies of active-site residues than has been possible previously with other CTQ- and TTQ-dependent enzymes. The parameters that were determined in this study that describe glycine binding and steady-state kinetics of glycine oxidation are summarized in Table 1.
Table 1.
Kinetic and binding parameters for PlGoxA and variants
| PlGoxA variant | Steady-state kinetics |
Glycine binding |
|||
|---|---|---|---|---|---|
| kcat | K0.5 | h value | K0.5 | h value | |
| s−1 | μm | μm | |||
| WTa | 6.0 ± 0.2 | 187 ± 13 | 1.8 ± 0.2 | 103 ± 3 | 3.7 ± 0.4 |
| D678Eb | 1.0 ± 0.02 | 151 ± 17 | 1.0 | 26 ± 0.1 | 3.9 ± 0.1 |
| H583A | NRc | NR | |||
| H583C | NR | NR | |||
| F316Y | 8.0 ± 0.2 | 333 ± 17 | 1.5 ± 0.1 | 70 ± 1.2 | 1.7 ± 0.03 |
| F316A | 3.1 ± 0.2 | 783 ± 115 | 1.2 ± 0.1 | 175 ± 9 | 1.4 ± 0.1 |
| Y766F | 8.5 ± 0.2 | 666 ± 33 | 1.2 ± 0.04 | 527 ± 13 | 1.8 ± 0.1 |
| H767A | NDd | ||||
Crystallization and structure determination
Crystals for F316A, H583C, Y766F, and H767A were grown in similar conditions used for the WT (4). The resulting crystals were yellow in color and isomorphous to the WT (Table 2) with very similar overall structures (r.m.s.d. <0.3 Å across backbone atoms). Upon soaking in cryoprotection solution containing 10 mm glycine, F316A and Y766F turned deep blue in color, consistent with the formation of a product-reduced CTQ Schiff base in the crystal (17). H583C exhibited no color change, whereas H767A became nearly transparent, which would be consistent with reduced CTQ (7). Diffraction quality crystals of H583A and F316Y could not be obtained from the WT crystallization conditions.
Table 2.
Data collection, processing, and refinement statistics
Numbers in parentheses are for the high-resolution shell.
| H583C GoxA + Gly, pH 7.5 | H767A GoxA + Gly, pH 7.5 | Y766F GoxA + Gly, pH 7.5 | F316A GoxA + Gly, pH 5.5 | |
|---|---|---|---|---|
| Data collection | ||||
| Wavelength (Å) | 1.00000 | 1.00000 | 1.00000 | 1.00000 |
| Space group | P21 | P21 | P21 | P21 |
| Unit cell parameters | ||||
| a, b, c (Å) | 109.8, 93.3, 188.6 | 110.0, 93.1, 187.6 | 107.9, 93.0, 187.1 | 109.6, 91.9, 178.8 |
| α, β, γ (°) | 90.0, 95.1, 90.0 | 90.0, 95.1, 90.0 | 90.0, 95.1, 90.0 | 90.0, 91.4, 90.0 |
| Resolution range (Å) | 49.2–2.14 | 48.2–2.20 | 48.4–2.24 | 47.2–1.99 |
| No. of reflections (measured/unique) | 770,319/207,973 | 665,403/189,420 | 634,586/173,760 | 843,086/240,912 |
| Rmerge | 0.09 (0.61) | 0.11 (0.51) | 0.12 (0.64) | 0.10 (0.56) |
| I/σI | 9.2 (2.1) | 5.9 (2.1) | 5.9 (1.9) | 9.0 (2.2) |
| Completeness (%) | 99.4 (100.0) | 98.9 (99.6) | 98.0 (99.7) | 99.1 (98.5) |
| Redundancy | 3.7 (3.9) | 3.5 (3.3) | 3.7 (3.7) | 3.5 (3.1) |
| Refinement statistics | ||||
| Resolution (Å) | 2.14 | 2.20 | 2.24 | 1.99 |
| Rwork/Rfree | 0.157/0.211 | 0.187/0.239 | 0.202/0.250 | 0.172/0.223 |
| r.m.s.d. | ||||
| Bond lengths (Å) | 0.013 | 0.012 | 0.004 | 0.008 |
| Bond angles (°) | 1.174 | 1.080 | 0.719 | 0.941 |
| Ramachandran statistics | ||||
| Allowed | 99.4% | 99.5% | 99.5% | 99.5% |
| Outliers | 0.6% | 0.5% | 0.5% | 0.5% |
| Average B-factor (Å2) | 47.6 | 51.8 | 51.8 | 32.3 |
His-583 variants
The only PlGoxA variant proteins in this study that did not show any reactivity toward glycine were those in which His-583 was mutated. Mutation of His-583 to either Cys or Ala produced the same result, a CTQ-bearing protein with a characteristic absorbance spectrum that could not be reduced by glycine (Fig. 3A). Consistent with this observation, the crystal structure of H583C PlGoxA, which was soaked with glycine, had no glycine present in the active site (Fig. 4). Rather, a pair of water molecules or perhaps a single water with alternate positions interacts with the quinone oxygen and His-767 and Tyr-766 from a neighboring protomer (Fig. 4B). Ser-681, which interacts with the bound substrate carboxyl group in the WT structure, is oriented away from the active site in H583C, consistent with a lack of bound substrate in this mutant.
Figure 3.

Absorbance spectra of WT PlGoxA (black) and H583C PlGoxA (red). Spectra are shown of the oxidized enzymes before and after addition of 5 mm glycine.
Figure 4.

Active site of glycine-soaked crystals of H583C PlGoxA (PDB code 6VL7). A, omit Fo − Fc electron density contoured at 4.0σ for a model omitting active-site waters. B, 2Fo − Fc electron density contoured at 1.0σ for a model including waters. C, active-site residues shown as sticks colored according to element. Carbons from residues of the neighboring protomer are colored yellow. Waters are shown as red spheres, and putative hydrogen bonds are shown as dotted lines with distances indicated in Å. D, overlay of the H583C (cyan and yellow) and WT (gray) PlGoxA-active sites. The hydrogen bonds indicated are from the WT structure.
It was previously shown that D678A and D678N PlGoxA variant proteins were not reduced by glycine but did have glycine present in the active sites in the glycine-soaked crystals (13). In the structures of the glycine-reduced CTQ adduct of WT PlGoxA and the glycine-soaked crystals of D678A and D678N PlGoxA, a His-583 ring nitrogen interacts with a carboxyl oxygen of glycine. This interaction is not possible in the H583A and H583C variants. In the latter case, Cys-583 is oriented away from the substrate-binding site and stabilizes two water molecules not observed in the WT structure due to the presence of the His-583 side chain (Fig. 4).
Asp-678 is required to deprotonate the glycine NH3+ group for Schiff base formation, either directly or via a water. Thus, it can be concluded that His-583 is critical for initial binding of glycine in the active site and positioning it relative to CTQ and Asp-678 to allow adduct formation. A reason for mutating His-583 to Cys is that Cys is present in this position in MmLodA. However, in the structure of the MmLodA–lysine adduct (10), Cys does not interact with the lysine. In that enzyme the Schiff base with CTQ is formed by the ϵ-amino group of the lysine side chain rather than the backbone amino group of glycine in GoxA. Other residues within the much larger substrate pocket of that enzyme interact with the bound substrate.
Phe-316 variants
F316A and F316Y PlGoxA variants were each reactive toward glycine as judged by glycine-induced changes in the absorbance spectrum, consistent with product Schiff base formation. As such, it was possible to perform a titration with glycine to assess the binding parameters (Fig. 5, A and B). Whereas WT PlGoxA exhibited an h value of 3.7 ± 0.4, the F316Y and F316A variants exhibited h values of 1.7 ± 0.1 and 1.4 ± 0.1, respectively. Each mutation increased the K0.5 for glycine from 187 μm in WT PlGoxA to 333 μm for F316Y PlGoxA and 783 μm for F316A PlGoxA. Thus, removal of the phenyl ring or even a more modest modification of conversion to Tyr each significantly diminishes the cooperativity of glycine binding and its affinity. The variants each exhibited steady-state activity (Fig. 5, C and D). For F316Y PlGoxA, the kcat of 8.0 ± 0.2 s−1 is actually a bit greater than that of WT PlGoxA, whereas the kcat of 3.1 ± 0.2 s−1 for F316A PlGoxA is about half that of WT PlGoxA. For WT PlGoxA, the h value determined from the fit of the kinetic data was 1.8. For the F316Y and F316A variants, it was 1.6 and 1.2, respectively. These values are comparable with the h values determined from the glycine titration. Thus, the F316Y mutation diminished the cooperativity of substrate binding, and it increased K0.5 but did not reduce kcat. The F316A did reduce kcat, as well as affected binding and cooperativity.
Figure 5.

Substrate binding and steady-state kinetic analysis of F316Y PlGoxA (A and C) and F316A PlGoxA (B and D). A and B, changes in absorbance at 600 nm were monitored during titration with glycine. The lines are fits of the data to Equation 1. C and D, steady-state kinetics data were also fit to Equation 5. Each data point is the average of three replicates. When error bars are not evident, it is because they are too small to extend beyond the circle.
The crystal structure of F316A PlGoxA soaked in glycine exhibits electron density consistent with the formation of the product Schiff base (Fig. 6). However, the orientation of the carboxyl group of bound substrate is slightly different from that observed for WT, resulting in altered interactions with active-site residues (Fig. 6C). In the WT protein, Phe-316 is engaged in a face–to–edge π-stacking interaction with His-767 from the neighboring protomer. The absence of this interaction and the steric bulk of the phenyl ring likely account for the subtle perturbations observed in the active site, which result in the decreased cooperativity and kcat.
Figure 6.

Active site of glycine-soaked crystals of F316A PlGoxA (PDB code 6VMW). A, omit Fo − Fc electron density contoured at 5.0σ for the active site modeled as CTQ. B, 2Fo − Fc electron density contoured at 1.0σ. C, overlay of the F316A (cyan and yellow) and WT (gray) PlGoxA-active sites. Active-site residues shown as sticks are colored according to element. Carbons from residues of the neighboring protomer are colored yellow. Putative hydrogen bonds are shown as dotted lines with distances indicated in Å.
Tyr-766 variants
Tyr-766 and His-767 both project from one monomer of the heterotetramer into the active site of another and interact with the glycine carboxyl group in the crystal structure of the product–CTQ Schiff base adduct of PlGoxA. Substitution of Tyr-766 with Phe, which eliminates the interaction with this residue, has a profound effect on substrate binding. Y766F PlGoxA did react with glycine, as judged by changes in the absorbance spectrum. Similar to what was observed with the Phe-316 variants, the h value obtained from the glycine titration of Y766F PlGoxA was decreased to 1.8 ± 0.1 (Fig. 7). However, in this case the K0.5 for binding increased significantly to 527 μm. This decreased affinity was also evident from the steady-state kinetic analysis. The K0.5 for the reaction also significantly increased to 666 ± 33 μm. As was observed for the F316Y PlGoxA, the Y766F mutation did not diminish kcat, rather it increased to 8.5 ± 0.2 s−1. The crystal structure of glycine-soaked Y766F PlGoxA is nearly identical to that of the WT enzyme, except for the absence of the OH group on residue 766 (Fig. 8).
Figure 7.

Substrate binding and reaction kinetics of Y766F PlGoxA. A, changes in absorbance were monitored at 600 nm during titration with glycine. The dashed line is a fit of the data to Equation 1. B, steady-state kinetic analysis of glycine oxidase activity of Y766F PlGoxA. The dotted line is a fit of the data to Equation 5.
Figure 8.

Active site of glycine-soaked crystals of Y766F PlGoxA (PDB code 6VMF). A, omit Fo − Fc electron density contoured at 5.0σ for the active site modeled as CTQ. B, 2Fo − Fc electron density contoured at 1.0σ. C, overlay of the Y766F (cyan and yellow) and WT (gray) PlGoxA-active sites. Active-site residues shown as sticks are colored according to element. Carbons from residues of the neighboring protomer are colored yellow. Putative hydrogen bonds are shown as dotted lines with distances indicated in Å.
His-767 variants
His-767 also interacts with the glycine carboxyl group in the crystal structure of the product–CTQ Schiff base adduct of PlGoxA. On first look, it did not appear that the H767A PlGoxA variant reacted with glycine, as there was no rapid change in the absorbance spectrum. However, after addition of excess glycine a very slow change was observed. In contrast to WT PlGoxA and the other variant proteins in this study, the spectrum did not change to that of the product CTQ Schiff base intermediate. Instead, it appeared to change to that of the reduced CTQ on the minutes time scale (Fig. 9). Given these results, it was not possible to perform glycine titration to assess binding parameters, as was done for the other variant proteins. Steady-state kinetic studies appeared to show no reaction. However, if very high concentrations of glycine were used, a slow reaction above background was observed. It appeared to increase linearly with second-order rate constant of 17 m−1 s−1 with a rate of 0.24 s−1 at 10 mm glycine.
Figure 9.
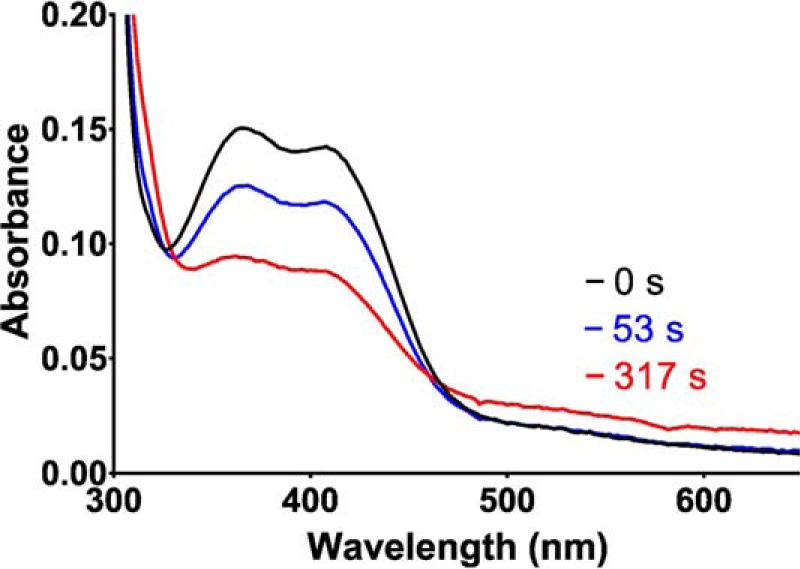
Spectroscopic changes associated with slow reaction of H767A PlGoxA with glycine. Spectra were recorded immediately after addition of 5 mm glycine to 30 μm H767A PlGoxA (black), 53 s later (blue), and 317 s later (red).
Soaking of H767A PlGoxA crystals in 10 mm glycine also seemed to show a bleaching of color. Electron density at the active site showed that a glycine–CTQ adduct was indeed present (Fig. 10). However, the electron density was best fit with the carbinolamine intermediate (see Fig. 1B) with an oxygen bound to the α-carbon of glycine. Attempts to model this structure as the product Schiff-base intermediate resulted in positive difference density at the position of the hydroxyl oxygen and the carboxyl group (Fig. 10C), supporting the assignment of the carbinolamine intermediate. This species has not been observed with WT PlGoxA or any other variant proteins, but it is consistent with the spectral changes observed after glycine addition. Because the α-carbon has been hydroxylated, there is no longer a carbon–nitrogen double bond that is present in the product-reduced CTQ adduct. Without this extended conjugation from CTQ through the bound glycine, the spectrum of this species should be similar to that of reduced CTQ, as was observed in Fig. 9. Comparison with the WT active site shows very little structural perturbation except the closer approach of Asp-678 to form a hydrogen bond with the hydroxyl group (Fig. 10D).
Figure 10.

Active site of glycine-soaked crystals of H767A PlGoxA (PDB code 6VMV). A, omit Fo − Fc electron density contoured at 4.0σ for the active site modeled as CTQ. B, 2Fo − Fc electron density contoured at 1.0σ and Fo − Fc density contoured at 4.0σ are shown for the active-site adduct modeled as a carbinolamine, or C, as the product Schiff-base. D, overlay of the H767A PlGoxA (cyan and yellow) and WT PlGoxA (gray) PlGoxA-active sites. Putative hydrogen bonds are shown as dotted lines with distances indicated in Å. The hydrogen bonds indicated are from the H767A structure. Residues Tyr-766′ and His-767′ are of the neighboring protomer.
Discussion
Previous studies of tryptophylquinone-bearing enzymes have shown that mutation of active-site residues is not well-tolerated. Mutation of the active-site Asp residue that corresponds to Asp-678 in the TTQ-dependent methylamine dehydrogenase (16) and the CTQ-dependent MmLodA (14) and MmGoxA (12) prevented the formation of the quinone cofactor. Mutation of some other active-site residues in these enzymes also prevented or reduced the levels of cofactor biosynthesis. In contrast, the mutagenesis of active-site residues in PlGoxA did not compromise the completion of CTQ biosynthesis. As such, the results of this and previous studies of PlGoxA now allow a detailed description of the kinetic reaction mechanism, identification of reaction intermediates, and the roles of active-site residues in the reaction mechanism. It also describes the basis for the observed differences in the extent of cooperativity of substrate binding and of steady-state kinetics.
An unusual feature of the reaction of WT PlGoxA is that addition of glycine to the enzyme under anaerobic conditions yields a stable Schiff base adduct with the product bound to the reduced CTQ (17). Spectral changes associated with formation of this adduct upon anaerobic titration of PlGoxA with glycine yielded an h value of 3.7, indicative of strong cooperative binding (7). Steady-state kinetic analysis yielded a smaller h value of 1.8 (4), indicating that a reaction step other than binding was partially rate-limiting. Mutagenesis of Asp-678 revealed that it is involved in both the deprotonation of glycine to allow formation of the substrate-oxidized CTQ adduct and the subsequent deprotonation of that adduct during conversion to the product-reduced CTQ adduct. Studies of a D678E variant revealed that steady-state kinetics no longer exhibited cooperativity, even though the cooperativity of binding was retained with an h value of 3.9 (13). Furthermore, it was possible to observe transient formation of the initial substrate-oxidized CTQ Schiff base adduct. This was because the mutation slowed the rate of conversion to the product-reduced CTQ Schiff base to allow its observation, and it was sufficiently slow to be rate-limiting for the overall reaction to eliminate the cooperativity of the steady-state kinetics.
The crystal structure of the glycine-soaked H767A PlGoxA in this study revealed the structure of another reaction intermediate, a carbinolamine species that is still bound to CTQ. This results from addition of water to the product Schiff base. Whereas carbinolamines are typically unstable in solution, the intermediate observed in H767A PlGoxA is quite stable in the active site in the crystal structure, indicating that the hydroxyl moiety is not deprotonated to initiate release of the glyoxylate product (Fig. 1B). This suggests that loss of the interaction of His-767 with the CTQ-bound glycine and reaction intermediates has perturbed the position of the carbinolamine species relative to that in the WT enzyme, such that it is not in a position for facile deprotonation by Asp-678 or an activated water. Thus, as a consequence of this mutation, addition of water to the otherwise stable product-reduced CTQ Schiff base is enhanced, but the deprotonation of the resulting intermediate is deterred. A similar carbinolamine intermediate was identified during the reaction of the TTQ-dependent aromatic amine dehydrogenase. In that study, crystals of the enzyme were soaked with substrate and flash-cooled, and it was observed in one of the resultant crystal forms (18). Hydrolysis of the glyoxylate product occurs during oxidation of CTQ yielding an iminoquinone, which is subsequently hydrolyzed to yield the quinone and ammonia. A similar iminoquinone has been shown to be the initial product of the oxidative half-reaction in the TTQ-dependent methylamine dehydrogenase (19) and aromatic amine dehydrogenase (18), as well tyrosylquinone-dependent copper amine oxidases (20).
The reason that different mutations allow the observation of different intermediates is because each affects the rate of a different reaction step, thus altering the kinetic mechanism. In WT PlGoxA, glycine binding is relatively slow, as the steady-state kinetics show cooperativity meaning that this step is partially rate-limiting to the overall reaction. Formation of the substrate-oxidized CTQ Schiff base is slow relative to conversion of the product-reduced CTQ Schiff base, as only the latter is observed. In D678E PlGoxA, the formation of the substrate Schiff base and its conversion to the product Schiff base are both observed, indicating that the mutation slowed the rate of the conversion. In H767A PlGoxA, the observation of the carbinolamine intermediate may be explained by the following. The initial deprotonation of the glycine and formation of the substrate-oxidized CTQ Schiff base are extremely slow. The subsequent reactions, conversion to the product-reduced CTQ Schiff base and then formation of the carbinolamine are relatively fast. Then the deprotonation of the carbinolamine and its release from the reduced CTQ adduct is extremely slow. This is consistent with the spectral changes that look like a slow conversion from oxidized to reduced CTQ. This is also consistent with the negligible steady-state glycine oxidation activity.
The residues being studied extend aromatic side chains into the active site, partially surrounding the CTQ. His-583 is to one side of the CTQ and the other residues are “in front” of the quinone oxygen to which glycine binds. Phe-316 is angled downward and the loop residues Tyr-766 and His-767 are angled upward in this orientation. The side chain of Asp-678, above the CTQ and about 5 Å away, meets with the glycine adduct through rotation; the planar aromatic residues appear to stay in place and form a stabilizing “pocket” for the glycine to enter and be oriented. Compromising this stabilizing pocket by altering or removing aromatic rings results in a range of effects, from preventing entrance of glycine to relatively minor changes in binding and reaction kinetics. Results of Y766F PlGoxA also suggest that the presence of the aromatic side chain in this position is more important than the OH group for binding and cooperativity than catalysis, as kcat is not diminished. Mutating the Phe-316 residue affects the reaction in a way similar to Y766F in that it is the binding kinetics rather than the kcat that are primarily affected.
Clearly, interactions between His-767 and Tyr-766 from one subunit with the active site of a neighboring subunit are critical for cooperative substrate binding and catalysis. Among these are direct hydrogen bond interactions between these residues and bound substrate as well as π-stacking interactions between Tyr-766 and Phe-316. We had previously identified a hydrated channel connecting the two active sites, into which the loops bearing His-767 and Tyr-766 project (13). This suggested the binding of substrate to one active site may control access to the other. The crystal structures do not show obvious changes that may impact substrate access resulting from the mutations. However, this is likely to be a dynamic process, which may be difficult to capture by X-ray crystallography.
PlGoxA provides a good model for studying enzyme kinetics and cooperativity, specifically for being able to at least partially tease apart those aspects of the protein through directed mutations. It shares active-site residues with many other quinoproteins, some that are dimers and some tetramers, with and without positive cooperativity. This situation provides for the ability to compare similar residues, in some cases nearly identically overlaying arrays of active sites, between related proteins that nonetheless differ in reaction specifics. PlGoxA has uniquely robust protein expression among LodA-like proteins, forming CTQ in variants with mutations that prevented expression in other systems, thus allowing the functions of those residues to be studied beyond their involvement in CTQ formation. In addition, PlGoxA has, along with the surface–to–surface interactions shared by all multimeric proteins, unique inter-subunit projections directly into a neighboring unit's active site. The position of these loop residues, Tyr-766 and His-767, suggests roles in both cooperativity, through communicating ligand binding in one subunit to another, and regulating the reaction within the active site into which they project.
Experimental procedures
Design, expression, and purification of PlGoxA variant proteins
Primers used to generate site-directed mutations are shown in Table 3. Mutagenesis was performed with the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Amplified mutant gene products were transformed into XL-10 ultra-competent E. coli cells, and mutations were verified by Sanger sequencing of harvested plasmids by Genewiz (South Plainfield, NJ). Plasmids harboring the mutated genes were then transformed into E. coli Rosetta cells, from which the protein variants were expressed and purified as described previously (4). In each case, cells were transformed with a pET15 vector containing the gene to be expressed with a hexa-histidine tag at the N terminus. Cells were grown overnight to an OD600 of 0.8, at which point they were induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h. Cells were harvested by centrifugation and then lysed via sonication in 50 mm potassium phosphate, pH 7.5. The cell lysate was centrifuged, and the supernatant was applied to a nickel-nitrilotriacetic acid affinity column. The column was washed with an imidazole gradient in the same buffer to isolate the tagged proteins.
Table 3.
Primers used to generate site-directed mutations
Changed bases are underlined.
| Mutation | Primers |
|---|---|
| H583C | Forward: 5′-GGT GGT TTT TGC CCT GGC GTT-3′ |
| Reverse: 5′-AAC GCC AGG GCA AAA ACC ACC-3′ | |
| H583A | Forward: 5′-GGT GGT TTT GCC CCT GGC GTT-3′ |
| Reverse: 5′-AAC GCC AGG GGC AAA ACC ACC-3′ | |
| F316Y | Forward: 5′-AAT ACC GAC TAT GCA GAT AAC TCA AAC TGG-3′ |
| Reverse: 5′-CCA GTT TGA GTT ATC TGC ATA GTC GGT AAT-3′ | |
| F316A | Forward: 5′-TTA AAT ACC GAC GCT GCA GAT AAC TCA-3′ |
| Reverse: 5′-TGA GTT ATC TGC AGC GTC GGT ATT TAA-3′ | |
| Y766F | Forward: 5′-CTT GGG TTT GTT GGC TTT CAT GCC GAA GGG-3′ |
| Reverse: 5′-CCC TTC GGC ATG AAA GCC AAC AAA CCC AAG-3′ | |
| H767A | Forward: 5′-CTT GGG TTT GTT GGC TAT GCT GCC GAA GGG-3′ |
| Reverse: 5′-CCC TTC GGC AGC ATA GCC AAC AAA CCC AAG-3′ |
Analytical techniques
Protein purity and native molecular weight were determined by 7.5% SDS-PAGE and size-exclusion chromatography using a HiPrep 16/60 column packed with Sephacryl S-300 HR collected in an ÄKTA Pure FPLC system (GE Healthcare). Absorption spectra were recorded using an HP 8452 diode array spectrophotometer controlled with Olis Globalworks software (Olis, Bogart, GA).
Binding and activity assays
Glycine-binding titrations were monitored via spectrophotometry from the changes in absorbance from 300 to 650 nm at 20 °C in 50 mm potassium phosphate buffer, pH 7.5, under anaerobic conditions as described previously (7). Data from the spectroscopic changes that occurred during the titration were fit to Equation 1, which describes allosteric binding. The fraction of PlGoxA with glycine bound was determined from the ΔA/ΔAmax at appropriate wavelengths during the titration.
| (Eq. 1) |
Glycine oxidase activity was determined using a previously described (15) coupled-enzyme assay in which the formation of NH3 released from glycine (Equation 2) is monitored through coupling to the oxidation of NADH by glutamate dehydrogenase (Equation 3).
| (Eq. 2) |
| (Eq. 3) |
The standard assay mixture contained 0.5 μm PlGoxA, 5 mm 2-oxoglutarate, 0.25 mm NADH, and 20 units/ml glutamate dehydrogenase. Reactions were performed in 50 mm potassium phosphate, pH 7.5, at 30 °C. Initial velocity was determined by monitoring the rate of disappearance of NADH at 340 nm using the ϵ340 of NADH of 6220 m−1 cm−1. Data were analyzed by the Michaelis-Menten equation (Equation 4) and the Hill equation (Equation 5), in which h is the Hill coefficient.
| (Eq. 4) |
| (Eq. 5) |
Crystallization and structure determination
Crystals of the PlGoxA variant proteins were grown in batch mode under paraffin oil. Protein at 10 mg/ml was combined with mother liquor containing either 20–25% PEG 3350, 0.1 m ammonium sulfate, and 0.1 m HEPES, pH 7.5, or 20–25% PEG 3350, 0.1 m citric acid, pH 5.5. Crystals were transferred to mother liquor containing 10% v/v PEG 400 as a cryoprotectant for ∼1 min prior to cryocooling in liquid nitrogen. For glycine soak experiments, 10 mm glycine was included in the cryoprotectant solution.
Diffraction data were collected at 100 K on beamline 5.0.2 at the Advanced Light Source at Berkeley National Laboratory, indexed, and integrated with XDS (21, 22) and scaled using Aimless (23). The structure was solved by molecular replacement using Phaser-MR (24) or by Fourier synthesis, using the native PlGoxA structure (PDB code 6BYW). Manual model building was done in Coot (25) with further rounds of refinement performed using the Phenix suite (26). Figures were prepared using PyMOL (RRID:SCR_ 000305), which was also used for pairwise structural alignments.
Data availability
The data related to x-ray crystallography are deposited in the Protein Data Bank and assigned the following accession codes: H583C GoxA + glycine, pH 7.5, is PDB code 6VL7; F316A GoxA + glycine, pH 5.5, is PDB code 6VMW; Y766F GoxA + glycine, pH 7.5, is PDB code 6VMF; and H767A GoxA + glycine, pH 7.5, is PDB code 6VMV. All other data are contained in the text.
Author contributions
K. J. M. and V. L. D. conceptualization; K. J. M., E. T. Y., and V. L. D. formal analysis; K. J. M., E. T. Y., and V. L. D. validation; K. J. M., E. T. Y., and V. L. D. investigation; K. J. M., E. T. Y., and V. L. D. methodology; K. J. M., E. T. Y., and V. L. D. writing-original draft; K. J. M., E. T. Y., and V. L. D. writing-review and editing; E. T. Y. and V. L. D. resources; E. T. Y. and V. L. D. data curation; V. L. D. funding acquisition; V. L. D. project administration.
Acknowledgment
We thank Yu Tang for technical assistance.
This work was supported by National Institutes of Health NIGMS Award R35GM130173 (to V. L. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PlGoxA
- glycine oxidase from P. luteoviolacea
- CTQ
- cysteine tryptophylquinone
- MmLodA
- l-lysine-ϵ-oxidase from M. mediterranea
- MmGoxA
- glycine oxidase from M. mediterranea
- TTQ
- tryptophan tryptophylquinone
- r.m.s.d.
- root mean square deviation.
References
- 1. Edsall J. T. (1980) Hemoglobin and the origins of the concept of allosterism. Fed. Proc. 39, 226–235 [PubMed] [Google Scholar]
- 2. Koshland D. E. Jr., Némethy G., and Filmer D. (1966) Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5, 365–385 10.1021/bi00865a047 [DOI] [PubMed] [Google Scholar]
- 3. Monod J., Wyman J., and Changeux J. P. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118 10.1016/S0022-2836(65)80285-6 [DOI] [PubMed] [Google Scholar]
- 4. Andreo-Vidal A., Mamounis K. J., Sehanobish E., Avalos D., Campillo-Brocal J. C., Sanchez-Amat A., Yukl E. T., and Davidson V. L. (2018) Structure and enzymatic properties of an unusual cysteine tryptophylquinone-dependent glycine oxidase from Pseudoalteromonas luteoviolacea. Biochemistry 57, 1155–1165 10.1021/acs.biochem.8b00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davidson V. L. (2018) Protein-derived cofactors revisited: empowering amino acid residues with new functions. Biochemistry 57, 3115–3125 10.1021/acs.biochem.8b00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Klinman J. P., and Bonnot F. (2014) Intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem. Rev. 114, 4343–4365 10.1021/cr400475g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma Z., and Davidson V. L. (2019) The redox properties of a cysteine tryptophylquinone-dependent glycine oxidase are distinct from those of tryptophylquinone-dependent dehydrogenases. Biochemistry 58, 2243–2249 10.1021/acs.biochem.9b00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lucas-Elio P., Hernandez P., Sanchez-Amat A., and Solano F. (2005) Purification and partial characterization of marinocine, a new broad-spectrum antibacterial protein produced by Marinomonas mediterranea. Biochim. Biophys. Acta 1721, 193–203 10.1016/j.bbagen.2004.11.002 [DOI] [PubMed] [Google Scholar]
- 9. Campillo-Brocal J. C., Chacón-Verdú M. D., Lucas-Elío P., and Sánchez-Amat A. (2015) Distribution in microbial genomes of genes similar to lodA and goxA which encode a novel family of quinoproteins with amino acid oxidase activity. BMC Genomics 16, 231 10.1186/s12864-015-1455-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Okazaki S., Nakano S., Matsui D., Akaji S., Inagaki K., and Asano Y. (2013) X-ray crystallographic evidence for the presence of the cysteine tryptophylquinone cofactor in l-lysine ϵ-oxidase from Marinomonas mediterranea. J. Biochem. 154, 233–236 10.1093/jb/mvt070 [DOI] [PubMed] [Google Scholar]
- 11. Campillo-Brocal J. C., Lucas-Elio P., and Sanchez-Amat A. (2013) Identification in Marinomonas mediterranea of a novel quinoprotein with glycine oxidase activity. Microbiologyopen 2, 684–694 10.1002/mbo3.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sehanobish E., Williamson H. R., and Davidson V. L. (2016) Roles of conserved residues of the glycine oxidase GoxA in controlling activity, cooperativity, subunit composition, and cysteine tryptophylquinone biosynthesis. J. Biol. Chem. 291, 23199–23207 10.1074/jbc.M116.741835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mamounis K. J., Avalos D., Yukl E. T., and Davidson V. L. (2019) Kinetic and structural evidence that Asp-678 plays multiple roles in catalysis by the quinoprotein glycine oxidase. J. Biol. Chem. 294, 17463–17470 10.1074/jbc.RA119.011255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sehanobish E., Chacón-Verdú M. D., Sanchez-Amat A., and Davidson V. L. (2015) Roles of active-site residues in LodA, a cysteine tryptophylquinone dependent ϵ-lysine oxidase. Arch. Biochem. Biophys. 579, 26–32 10.1016/j.abb.2015.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sehanobish E., Campillo-Brocal J. C., Williamson H. R., Sanchez-Amat A., and Davidson V. L. (2016) Interaction of GoxA with its modifying enzyme and its subunit assembly are dependent on the extent of cysteine tryptophylquinone biosynthesis. Biochemistry 55, 2305–2308 10.1021/acs.biochem.6b00274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones L. H., Pearson A. R., Tang Y., Wilmot C. M., and Davidson V. L. (2005) Active site aspartate residues are critical for tryptophan tryptophylquinone biogenesis in methylamine dehydrogenase. J. Biol. Chem. 280, 17392–17396 10.1074/jbc.M500943200 [DOI] [PubMed] [Google Scholar]
- 17. Avalos D., Sabuncu S., Mamounis K. J., Davidson V. L., Moënne-Loccoz P., and Yukl E. T. (2019) Structural and spectroscopic characterization of a product Schiff-base intermediate in the reaction of the quinoprotein glycine oxidase, GoxA. Biochemistry 58, 706–713 10.1021/acs.biochem.8b01145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Masgrau L., Roujeinikova A., Johannissen L. O., Hothi P., Basran J., Ranaghan K. E., Mulholland A. J., Sutcliffe M. J., Scrutton N. S., and Leys D. (2006) Atomic description of an enzyme reaction dominated by proton tunneling. Science 312, 237–241 10.1126/science.1126002 [DOI] [PubMed] [Google Scholar]
- 19. Davidson V. L., and Sun D. (2003) Evidence for substrate activation of electron transfer from methylamine dehydrogenase to amicyanin. J. Am. Chem. Soc. 125, 3224–3225 10.1021/ja0297133 [DOI] [PubMed] [Google Scholar]
- 20. Brazeau B. J., Johnson B. J., and Wilmot C. M. (2004) Copper-containing amine oxidases. Biogenesis and catalysis; a structural perspective. Arch. Biochem. Biophys. 428, 22–31 10.1016/j.abb.2004.03.034 [DOI] [PubMed] [Google Scholar]
- 21. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 10.1107/S0907444909047374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 26. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data related to x-ray crystallography are deposited in the Protein Data Bank and assigned the following accession codes: H583C GoxA + glycine, pH 7.5, is PDB code 6VL7; F316A GoxA + glycine, pH 5.5, is PDB code 6VMW; Y766F GoxA + glycine, pH 7.5, is PDB code 6VMF; and H767A GoxA + glycine, pH 7.5, is PDB code 6VMV. All other data are contained in the text.