

Abstract
Site-specific arrest of RNA polymerases (RNAPs) is fundamental to several technologies that assess RNA structure and function. Current in vitro transcription “roadblocking” approaches inhibit transcription elongation by blocking RNAP with a protein bound to the DNA template. One limitation of protein-mediated transcription roadblocking is that it requires inclusion of a protein factor extrinsic to the minimal in vitro transcription reaction. In this work, we developed a chemical approach for halting transcription by Escherichia coli RNAP. We first established a sequence-independent method for site-specific incorporation of chemical lesions into dsDNA templates by sequential PCR and translesion synthesis. We then show that interrupting the transcribed DNA strand with an internal desthiobiotin-triethylene glycol modification or 1,N6-etheno-2′-deoxyadenosine base efficiently and stably halts Escherichia coli RNAP transcription. By encoding an intrinsic stall site within the template DNA, our chemical transcription roadblocking approach enables display of nascent RNA molecules from RNAP in a minimal in vitro transcription reaction.
Keywords: transcription, RNA polymerase, RNA, RNA folding, biotin, desthiobiotin, etheno-dA, RNAP roadblocking, site-specific transcription arrest, translesion synthesis
Introduction
In vitro display of nascent RNA molecules from halted transcription elongation complexes (TECs)2 is a powerful tool with proven applications in nascent RNA folding (1–4) and systematic RNA aptamer characterization (5, 6). Historically, transcription arrest at a defined DNA coordinate has been achieved by attaching a protein roadblock to a DNA template to halt the progression of RNA polymerases (RNAPs). Sequence-specific protein roadblocks, such the catalytically inactive EcoRIE111Q mutant (7) and the Escherichia coli DNA replication terminator protein Tus (5), can be directed to a precise DNA location by encoding a binding sequence in the template DNA. Alternatively, the biotin–streptavidin complex is capable of halting some RNAPs when positioned at the downstream DNA template terminus (8) or internally within the transcribed DNA strand (3). Recently, a reversible dCas9 roadblock was developed to enable time-dependent control of transcription arrest sequentially at multiple DNA positions (9). Although these approaches have proven effective in diverse applications, their utility is limited to experimental contexts that tolerate inclusion of extrinsic protein factors in the in vitro transcription reaction.
In contrast to protein roadblocks, chemical DNA lesions have not typically been used for nascent RNA display experiments despite their well-established inhibitory effect on transcription elongation (10). The lack of chemical transcription roadblocking approaches can be attributed to two challenges. First, chemical lesions that efficiently stall RNAPs are also likely to stall DNA polymerases so that preparation of DNA templates for in vitro transcription yields a truncated dsDNA product during PCR amplification. Preparation of internally modified dsDNA has been achieved for short DNA molecules by annealing and ligating modified oligonucleotides (11) and for long DNA molecules by enzymatically generating single-strand DNA gaps that can be filled with modified oligonucleotides (12–14). However, these approaches are sequence-dependent and therefore not suitable for internally modifying the complex DNA sequence libraries that are frequently used in nascent RNA display experiments (4–6). Second, some RNAPs have been shown to bypass nonbulky DNA lesions such as abasic sites (15–19); consequently, RNAP stalling at some chemical lesions is time-dependent. In this work, we address both of the above challenges to develop a simple and versatile chemical approach for halting E. coli RNAP transcription elongation in vitro.
We determined that an internal desthiobiotin-triethylene glycol (desthiobiotin–TEG) modification positioned in the transcribed strand of in vitro transcription DNA templates efficiently and stably halts E. coli RNAP. We first show that internally modified dsDNA can be prepared by sequential PCR amplification and translesion synthesis. We then show that desthiobiotin–TEG alone efficiently halts E. coli RNAP one nucleotide upstream of the modification site, that bypass of the desthiobiotin–TEG modification by RNAP is sufficiently slow to enable manipulation of stalled TECs, and that stalled TECs are stable for at least 2 h at ambient temperature. Last, we demonstrate that our translesion synthesis approach can be used to enzymatically prepare DNA templates containing an internal 1,N6-etheno-2′-deoxyadenosine (etheno-dA) nucleotide, which was recently shown to halt E. coli RNAP transcription (19). Our findings establish a method for chemically encoding a transcription stall site within a dsDNA molecule to enable efficient RNAP roadblocking in a minimal in vitro transcription reaction.
Results
Rationale for DNA modifier selection
We observed that an internal desthiobiotin–TEG modification by itself (Fig. 1A) in the transcribed DNA strand causes E. coli RNAP to stall when attempting to develop a reversible desthiobiotin–streptavidin transcription roadblock. Although the desthiobiotin–streptavidin complex was capable of halting RNAP, RNAP also appeared to stall at the desthiobiotin–TEG modification site following streptavidin elution. Given that E. coli RNAP has been reported previously to bypass abasic sites in the transcribed DNA strand (15), we envisioned that the desthiobiotin–TEG modification might enable protein-free transcription roadblocking at a defined DNA template position. Notably, the desthiobiotin–TEG modification is distinct from abasic lesions because of the presence of the desthiobiotin moiety and because it introduces unnatural spacing (two versus three carbons) in the transcribed DNA strand (Fig. S1). We therefore performed our initial assessment of desthiobiotin–TEG as a transcription roadblock compared with an internal amino linker modification, which preserves the natural three-carbon spacing of the DNA phosphodiester backbone and contains a less perturbative functional moiety (Fig. 1B and Fig. S1). In the sections below, we first describe our characterization of desthiobiotin–TEG as a chemical transcription roadblock. We then describe a similar characterization of the etheno-dA DNA modification in the final section under “Results.”
Figure 1.

Preparation and characterization of chemical roadblocking DNA templates. A and B, chemical structures of internal desthiobiotin–TEG (A) and amino linker (B) modifications. C, layout of internally modified DNA templates. The positions of the forward primer and the internally modified reverse primer are shown. The transcription start site (TSS) and U15 walk site are indicated. D, overview of internally modified (mod.) DNA template preparation. The position of the internally modified reverse primer is shown in purple. E, denaturing PAGE quality analysis of internally modified DNA template preparations (Modification +) alongside an unmodified (Modification −) positive control. The size marker is the Low Range ssRNA Ladder (New England Biolabs). F and G, single-round in vitro transcription of internal desthiobiotin–TEG–modified (F) and amino linker–modified (G) DNA templates. In both cases, RNAP stalls one nucleotide upstream of the modification site. E is representative of several DNA template quality control gels performed throughout this study. The experiments in F and G were performed once to precisely map critical bands for all subsequent experiments.
Translesion synthesis enables preparation of internally modified dsDNA
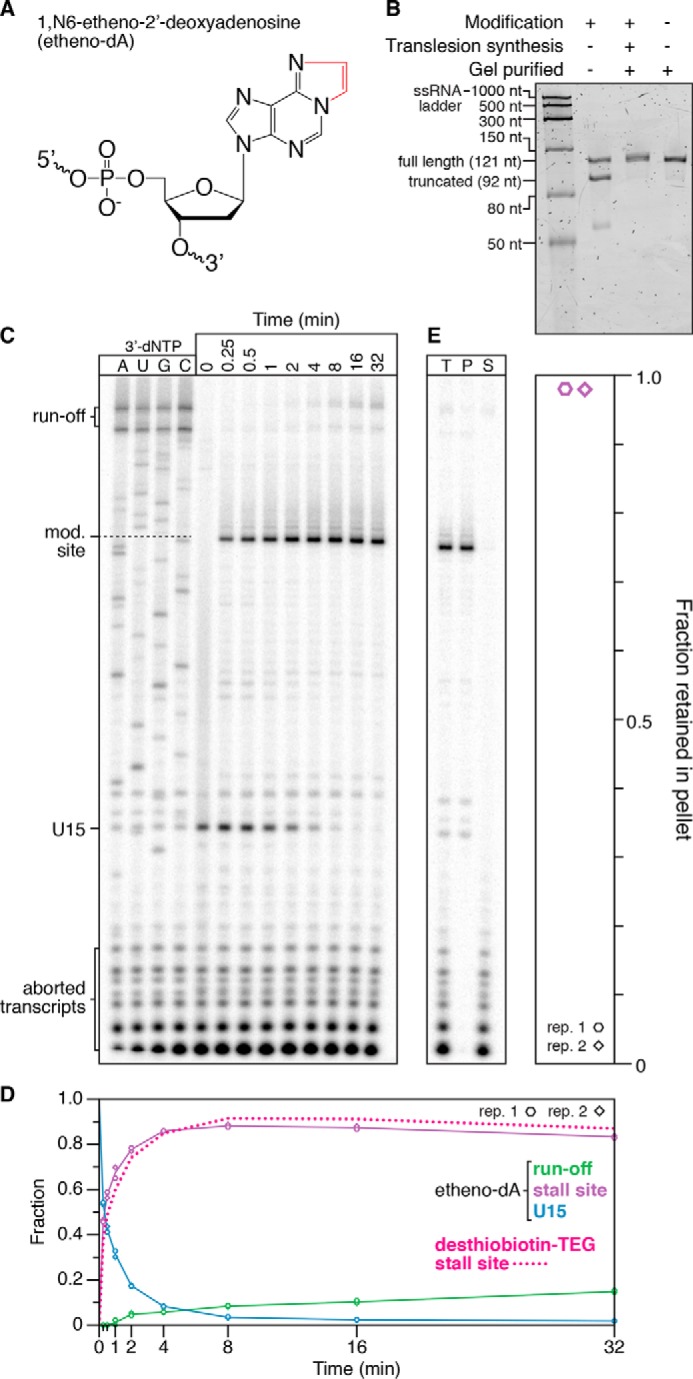
To assess the transcription roadblocking properties of the desthiobiotin–TEG and internal amino linker modifications, we prepared in vitro transcription DNA templates using synthetic oligonucleotide primers that contain an internal modification site 29 bp upstream of the template end (Fig. 1C and Table S1). Preparation of internally modified DNA templates by PCR amplification yielded a dsDNA product with one truncated strand, suggesting that Vent® (exo−) DNA polymerase cannot efficiently bypass the modification site (Fig. 1, D and E). To complete the truncated DNA strand, we performed translesion synthesis using the thermostable Y-family lesion bypass polymerase Sulfolobus islandicus DNA polymerase IV (DNA Pol IV) (Fig. 1D). The related Sulfolobus solfataricus DNA Pol IV has been shown to discriminate between correct and incorrect nucleotides with reported misincorporation frequencies between 8 × 10−3 and 3 × 10−4 (20), and DNA Pol IV from several Sulfolobus species has been used in combination with TaqDNA polymerase to PCR-amplify damaged and ancient DNA (21). In our application, we separate PCR amplification and translesion synthesis into independent steps so that the DNA template promoter and transcribed region are synthesized first using a DNA polymerase with fidelity suitable for downstream applications. Translesion synthesis is then used to bypass the modification site and complete the truncated DNA strand. In our initial protocol, we purified the truncated dsDNA PCR product before performing translesion synthesis. We later determined that, because Vent® (exo−) DNA polymerase and DNA Pol IV are active in the same buffer and under the same nucleotide conditions, efficient translesion synthesis could be achieved by simply adding DNA Pol IV directly to the reaction after PCR cycling without an intermediate cleanup step. Both translesion synthesis protocols yielded internally modified dsDNA templates that were indistinguishable from unmodified templates by denaturing PAGE (Fig. 1E and Fig. S2A). DNA templates containing an internal desthiobiotin–TEG modification were also analyzed by nondenaturing PAGE and showed a slight mobility shift relative to unmodified DNA (Fig. S2B). DNA templates in which translesion synthesis was performed using the thermostability-enhancing dNTPs 2-amino-dATP and 5-propynyl-dCTP showed an identical mobility shift, suggesting that the shift is a consequence of the desthiobiotin–TEG modification and not instability in the DNA downstream of the modification site (Fig. S2C).
Modification bypass exhibits distinct nucleotide incorporation preferences
Translesion DNA synthesis can involve incorporation of a nucleotide opposite the traversed lesion (22). To further characterize our internally modified DNA templates, we determined the nucleotide incorporation preference of DNA Pol IV-mediated lesion bypass for the internal desthiobiotin–TEG, amino linker, and etheno-dA DNA modifications by primer extension (Fig. 2A). On an unmodified DNA template, primer extension with all four dNTPs primarily yielded runoff products with one, and to a lesser extent, two more nucleotides than the expected full-length template-encoded product (Fig. 2, B and C). This is consistent with observations that S. solfataricus DNA Pol IV can extend a blunt DNA terminus by one to two nucleotides with a preference for dATP incorporation (23). The internal desthiobiotin–TEG–modified template yielded a runoff product distribution comparable with that of the unmodified template, suggesting that desthiobiotin–TEG bypass frequently occurs without incorporation of a nucleotide opposite the modification site (Fig. 2, B, C, and F). In contrast, the enrichment of full-length +2 products from amino linker– and etheno-dA–modified templates suggests that bypass of these lesions can involve incorporation of a nucleotide opposite the modification site (Fig. 2, B, C, and F). Notably, the etheno-dA template essentially shifted the product distribution of the unmodified template by one additional nucleotide (Fig. 2C). We confirmed these interpretations by primer extension in the presence of ddNTPs to determine how bypass of each modification distorts the observed sequencing ladder. In agreement with our analysis above, we observed that the desthiobiotin–TEG template yielded a sequencing ladder similar to that of the unmodified control, the amino-linker template yielded the expected +0 sequencing ladder and a ladder with a +1 offset, and the etheno-dA template yielded a mixed +0/+1 sequencing ladder with enrichment for +1 products (Fig. 2D).
Figure 2.

Characterization of Sulfolobus DNA Pol IV-mediated DNA modification bypass. A, overview of the primer extension reaction on an unmodified and internally modified template. The length of DNA nucleotides that are not shown is indicated by the numbers in brackets at the ends of the sequence. B, characterization of full-length (FL) primer extension products for unmodified (−) and desthiobiotin–TEG (dS)–modified, amino linker (aL)–modified, and etheno-dA (ϵA)–modified templates. The primary runoff product is FL+1 because of untemplated incorporation of a 3′dA overhang; some runoff products contain a second overhang nucleotide (23). The amino linker and etheno-dA modifications increase the occurrence of FL+2, suggesting incorporation of a dNTP opposite the modification site upon lesion bypass. C, raw intensity traces of the full-length products from B. D, ddNTP sequencing ladders for unmodified and desthiobiotin–TEG (deSbio)–modified, amino linker (aminoLnk)–modified, and etheno-dA–modified templates. In agreement with B, bypass of the desthiobiotin–TEG modification yields a sequencing ladder that resembles the unmodified control. In contrast, the amino linker and etheno-dA templates yield a mixed sequencing ladder that suggests incorporation of an additional base during modification bypass. For each modified oligonucleotide, ddNTP truncation products within four to five nucleotides of the modification site were obscured by short products that were not fully extended. E, primer extension reactions for unmodified and modified DNA templates performed in the absence of dNTPs (0), with all dNTPs (4), or with only one dNTP (A, T, G, or C). The unmodified control and desthiobiotin–TEG reactions were performed with the sequences shown in A (+1G) and with a template in which the +1 nucleotide is a C (+1C). Quantification of primer extension is shown in the plot below; red points indicate individual dNTP conditions that enable high-efficiency modification bypass. F, schematic of primer extension products resulting from bypass without dA incorporation (top) and bypass with dA incorporation opposite the modification site (bottom). B–D are representative of two independent replicates. The data in B are controls that were performed alongside the reactions in D; the solid line between these two panels indicates a gel splice site used to facilitate clear visualization of the sequencing ladders in D. In E, n = 2.
We further validated these observations by performing primer extension in the presence of one base at a time to determine the efficiency of lesion bypass using each dNTP. Under the conditions of our assay (incubation at 55 °C for 5 min), starving DNA Pol IV of all but one dNTP appeared to yield homopolymer primer extension products because of misincorporation (Fig. 2E); however, the observation of a readily discernable sequencing ladder when all four dNTPs are present (Fig. 2D) indicates that misincorporation to this degree is specific to nucleotide starvation conditions. Consistent with the observations above, we found that desthiobiotin–TEG bypass is most efficient when dNTP incorporation is templated by the nucleotide immediately downstream of the modification site, amino linker bypass is efficiently mediated by templated dGTP or untemplated dATP incorporation, and etheno-dA bypass is most efficient with dATP (Fig. 2E).
The observation that S. islandicus DNA Pol IV efficiently bypasses the amino linker and etheno-dA modifications in the presence of dATP is consistent with a previous finding that S. solfataricus DNA Pol IV follows the “A rule” (22) and typically incorporates dATP opposite an abasic site (20). One explanation for why desthiobiotin–TEG bypass preferentially occurs without incorporation of a nucleotide opposite the modification site is that its branched triethylene glycol scaffold interrupts the DNA backbone by only two carbons, whereas the amino linker and etheno-dA modifications preserve the natural three-carbon spacing of DNA nucleotides (Fig. S1). Importantly, preparation of DNA templates containing these modifications was efficient despite the difference in DNA Pol IV-mediated lesion bypass, suggesting that our translesion synthesis approach for enzymatic production of internally modified dsDNA will be generalizable to other modifications.
Desthiobiotin–TEG efficiently blocks E. coli RNAP transcription
We evaluated the transcription roadblocking properties of the internal desthiobiotin–TEG and amino linker modifications by single-round in vitro transcription with E. coli RNAP. Transcription was initiated in the absence of CTP to walk RNAP to U15, one nucleotide upstream of the first C in the transcript, before addition of NTPs to 500 μm. Both internal modifications produced a transcription stall site at C42, one nucleotide upstream of the modification position (Fig. 1, F and G). Importantly, an unmodified DNA template control showed no evidence of modification-independent transcription stalling at C42, indicating that the C42 stall site is entirely modification-dependent (Fig. 3A). We next compared the desthiobiotin–TEG and amino-linker roadblocks with the “gold standard” terminal biotin–streptavidin roadblock using a 32-min time course in the presence of 500 μm NTPs. To halt transcription at C42, the terminal biotin–streptavidin roadblock was placed 10 nucleotides downstream of C42 at the transcribed DNA strand 5′ end to account for collision of RNAP with streptavidin (3). After 32 min, there was no indication that RNAP had bypassed the terminal biotin–streptavidin roadblock to produce runoff transcripts (Fig. 3A). In contrast to the terminal biotin–streptavidin complex, retention of TECs at the desthiobiotin–TEG stall site was not absolute; after 32 min, ∼87% of TECs remained at the stall site (Fig. 3, A and B). Nonetheless, desthiobiotin–TEG outperformed the amino linker modification, which retained only ∼40% of TECs at the stall site after 32 min (Fig. 3, A and B). Last, we measured the rate at which RNAP bypasses the desthiobiotin–TEG modification site following promoter escape. Under our standard reaction conditions for internal RNA labeling (200 μm ATP, GTP, and CTP and 50 μm UTP), we observed an initial decay of t½ = 592 min (n = 2, R2 = 0.96, 95% confidence interval [541, 655]) (Fig. 4). In our first replicate, which included time points to 256 min, desthiobiotin–TEG bypass slowed after the 64-min time point. In our second replicate, which included time points to 64 min, modification bypass slowed after the 48-min time point (Fig. 4). Although the origin of this effect is unclear, it suggests that stalled TECs may be heterogeneous. We conclude that virtually all TECs initially stall upon encountering the desthiobiotin–TEG modification and that the vast majority of TECs persist at the stall site well beyond the reaction time of typical nascent RNA display experiments.
Figure 3.

Comparison of biotin–streptavidin and chemical roadblock efficiency. A, single-round in vitro transcription of DNA templates without and with terminal biotin–streptavidin, internal desthiobiotin–TEG, or internal amino linker stall sites in the presence of 500 μm NTPs. Solid lines between gel images denote gel splices. B, quantification of the gels shown in A. All data are from two independent replicates; representative gels are shown. Desthiobiotin–TEG and amino linker experiments were performed together, and unmodified and biotin–streptavidin controls were performed separately.
Figure 4.
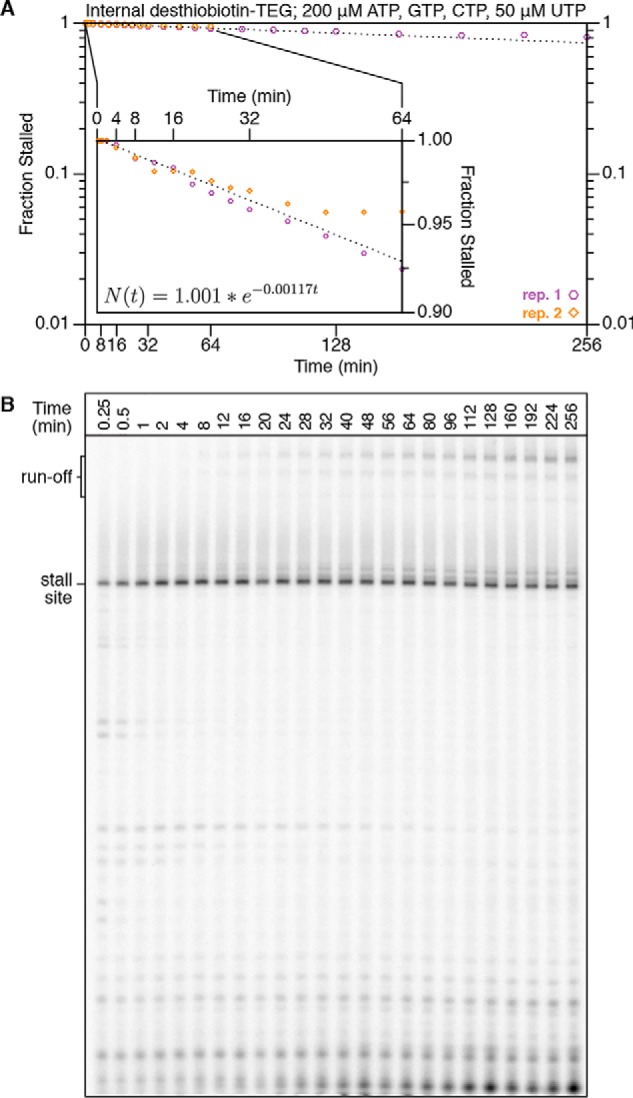
Time-dependent desthiobiotin–TEG bypass by E. coli RNA polymerase. A, plot of the fraction of TECs that reached but did not bypass the desthiobiotin–TEG stall site over 256-min (replicate 1) and 64-min (replicate 2) initiation-synchronized time courses in the presence of 200 μm ATP, GTP, and CTP and 50 μm UTP. The exponential decay curve was fit using time points from 2–48 min. The inset shows time points to 64 min with a truncated y axis to facilitate clear data visualization and shows the decay equation. B, gel showing the 256-min time course from A. Time points from 0.25 min to 64 min are n = 2; time points from 80 min to 256 min are n = 1.
The E. coli RNAP footprint blocks desthiobiotin–TEG from binding streptavidin
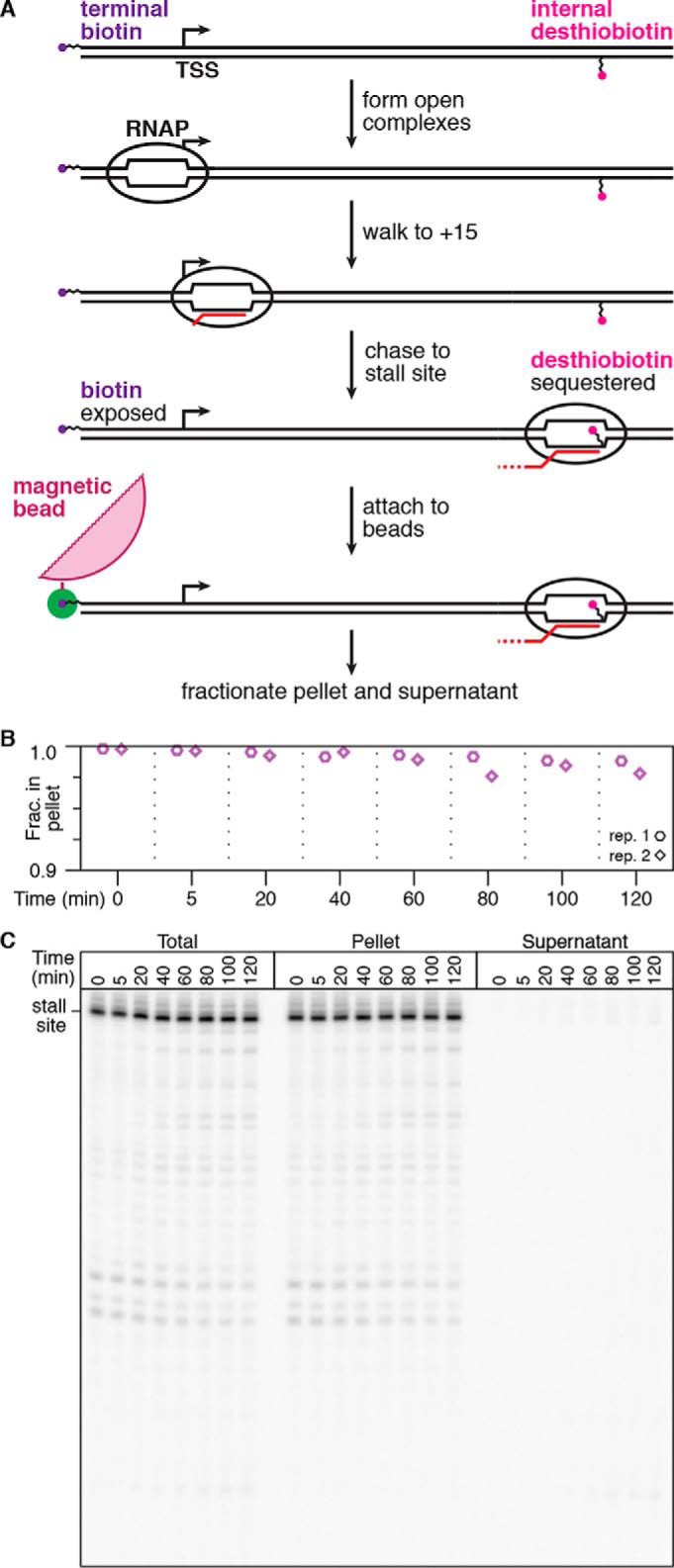
The primary caveat for using the desthiobiotin–TEG modification as a transcription stall site is that it is not functionally inert. Many applications of chemical transcription roadblocking also depend on DNA template immobilization, which is typically achieved by attachment of a 5′-biotin–modified DNA template to streptavidin-coated magnetic beads. It is therefore desirable for the DNA template to contain only one attachment point. One solution for this limitation is to sequester the desthiobiotin–TEG modification within the RNAP footprint before DNA immobilization. To test this approach, we first positioned RNAP at the U15 walk site so that the desthiobiotin–TEG modification is exposed (Fig. 5A). After incubation with streptavidin-coated magnetic beads, ∼89% of U15 complexes partitioned into the bead pellet (Fig. 5B). In contrast, when RNAP was positioned at the desthiobiotin–TEG stall site, only ∼4% of stall site complexes were recovered in the bead pellet fraction (Fig. 5, A and B). Because the ability of desthiobiotin–TEG to bind streptavidin can be controlled by RNAP position, stall site–proximal elongation complexes can be enriched by exclusion from streptavidin-coated magnetic beads. To determine how the E. coli RNAP footprint interferes with desthiobiotin–streptavidin binding at nucleotide resolution, we distributed TECs using 3′-dNTP chain terminators before incubating the arrested complexes with streptavidin-coated magnetic beads and separating pellet and supernatant fractions. In agreement with the established downstream border of E. coli RNAP on dsDNA (24), elongation complexes 15 nucleotides or less upstream of the desthiobiotin–TEG modification were predominantly excluded from the bead pellet, whereas complexes upstream of this boundary were retained (Fig. 5C). Thus, use of desthiobiotin–TEG as a chemical transcription roadblock additionally provides a straightforward means of enrichment for stall site–proximal TECs by exclusion from streptavidin-coated magnetic beads.
Figure 5.

Characterization of desthiobiotin sequestration by E. coli RNAP. A, overview of the desthiobiotin sequestration experiment in B. If RNAP is positioned at U15, then the desthiobiotin–TEG modification is predicted to be exposed and capable of binding streptavidin-coated magnetic beads. If RNAP is positioned at the stall site, then the desthiobiotin–TEG modification is predicted to be sequestered and incapable of binding streptavidin-coated magnetic beads. B, fractionation of internal desthiobiotin–TEG–modified DNA templates with streptavidin-coated magnetic beads in the presence of the TECs described in A. Total reaction (T), pellet (P), and supernatant (S) fractions are shown. The plot shows the fraction of TECs retained in the bead pellet. C, fractionation of internal desthiobiotin–TEG–modified DNA templates with streptavidin-coated magnetic beads in the presence of intermediate TECs. Intermediate TECs were generated by transcription in the presence of chain-terminating 3′-dNTPs. Comparison of P and S fractions indicates that the E. coli RNAP footprint blocks desthiobiotin from binding streptavidin when the nascent RNA 3′ end is 15 nucleotides or less upstream of the modification site. Background-subtracted intensity traces are shown for the pellet and supernatant fractions alongside the gel. All data are from two independent replicates.
Desthiobiotin-halted elongation complexes are stably bound to DNA
The observation that the RNAP footprint sequesters desthiobiotin function enabled us to assess the stability of desthiobiotin–TEG–stalled TECs over long incubation times. We prepared a DNA template containing an internal desthiobiotin–TEG modification to stall RNAP and a 5′-biotin–TEG modification upstream of the promoter for DNA immobilization by attachment to streptavidin-coated magnetic beads (Fig. 6A). After TECs were positioned at the desthiobiotin–TEG stall site, the transcription reaction was incubated with streptavidin-coated magnetic beads for 10 min, washed twice with transcription buffer supplemented with 1 mm MgCl2 to remove free NTPs, and resuspended in this same buffer. Separation of bead pellet and supernatant fractions indicated that ∼98% of stalled TECs remained attached to the beads after 2 h of incubation at room temperature (Fig. 6, B and C). Thus, elongation complexes that are stalled at a desthiobiotin–TEG modification site retain the exceptional stability that is expected for ternary elongation complexes.
Figure 6.
Quantification of elongation complex stability at a desthiobiotin–TEG stall site. A, overview of the experiment shown in B and C. Using a DNA template with an internal desthiobiotin–TEG stall site and a 5′-terminal biotin, RNAP is positioned at the stall site to sequester desthiobiotin, but the 5′-biotin remains exposed. These complexes are immobilized on streptavidin-coated magnetic beads, washed to remove free NTPs, and incubated at room temperature before separation into bead pellet and supernatant fractions. B, quantification of the fraction of desthiobiotin–TEG–stalled TECs retained in the bead pellet over time. The y axis range is 0.9–1.0 to facilitate clear data visualization. C, gel showing replicate 1 from B. Pellet and supernatant data points in B and C are from two independent replicates; the total reaction control was performed once.
Etheno-dA is a functionally inert chemical transcription roadblock
For some applications, it may be desirable to block transcription using a smaller and less disruptive DNA modification, such as etheno-dA, which has been shown recently to halt E. coli RNAP efficiently in the presence of 100 μm NTPs (19) (Fig. 7A). To confirm the suitability of etheno-dA as a chemical transcription roadblock, we first asked whether dsDNA transcription templates could be prepared by PCR with an internally etheno-dA–modified primer followed by translesion synthesis using Sulfolobus DNA Pol IV. Etheno-dA bypass by DNA Pol IV is efficient (Figs. 7B and Fig. S3) and appears to be predominantly mediated by incorporation of a dA nucleotide opposite the modification site (Fig. 2). Transcription roadblocking by etheno-dA was virtually identical to desthiobiotin–TEG; ∼84% of TECs were retained at the etheno-dA stall site after 32 min in the presence of 500 μm NTPs (Fig. 7, C and D). Last, nearly all TECs that were arrested at the etheno-dA stall site remained stably associated with the DNA template (Fig. 7E). We therefore conclude that etheno-dA is an ideal functionally inert chemical transcription roadblock because of its compatibility with sequence-independent modified DNA template preparation and its ability to efficiently stall E. coli RNAP.
Figure 7.
Transcription roadblocking by etheno-dA. A, chemical structure of etheno-dA. The etheno bridge that occludes the adenine Watson–Crick face is highlighted in red. B, denaturing PAGE quality analysis of an internal etheno-dA–modified DNA template preparation (Modification +) alongside an unmodified (Modification −) positive control. The size marker is the Low Range ssRNA Ladder (New England Biolabs). C, single-round in vitro transcription of an internal etheno-dA–modified DNA template in the presence of 500 μm NTPs. RNAP stalls one nucleotide upstream of the modification (mod.) site. D, quantification of the gel shown in C. The decay of TECs stalled at a desthiobiotin–TEG modification is shown for comparison and is from Fig. 3. E, fractionation of TECs stalled at an etheno-dA modification site using streptavidin-coated magnetic beads. DNA templates were attached to streptavidin-coated magnetic beads by a 5′-biotin–TEG modification upstream of the promoter. T, P, and S fractions are shown. The plot shows the fraction of TECs retained in the bead pellet. All data are from two independent replicates. A comparison of DNA template preparation replicates is shown in Fig. S3.
Discussion
Nascent RNA display is a powerful tool with proven applications in mapping RNA structure and folding (1–4) and systematic characterization of RNA function (5, 6). Established approaches for halting RNAP have typically relied on protein roadblocks that are not easily navigable by transcribing polymerases. Although these approaches have proven effective for placing RNAP at a defined DNA template position (7–9) and for systematically distributing RNAP across every DNA template position (3), each depends on including an extrinsic protein component in the transcription reaction. Although this requirement is compatible with many applications, the chemical transcription roadblocking approach described here provides a straightforward alternative that expands the repertoire of nascent RNA display tools.
The primary advantage of chemical transcription roadblocking is that no extrinsic factors are needed to efficiently halt RNAP. The cost of this advantage is that, in contrast to the gold standard terminal biotin–streptavidin roadblock, transcription arrest at chemical lesions is time-dependent; in the presence of NTPs, some TECs can eventually bypass the modification site (Figs. 3, 4, and 7). Nonetheless, the slow rate at which RNAP bypasses the desthiobiotin–TEG and etheno-dA modifications allows ample time for making biochemical measurements or depleting NTPs, after which the halted ternary elongation complex persists for hours (Fig. 6). Furthermore, the persistence of most stalled TECs for at least 32 min in the presence of 500 μm NTPs suggests that our approach is generalizable to many experimental contexts. Nonetheless, use of a chemical transcription roadblock should begin with kinetic characterization of lesion bypass under the conditions of the intended application to confirm satisfactory performance.
Our chemical approach for transcription roadblocking is enabled by the facile preparation of internally modified DNA templates of arbitrary length using translesion synthesis (Figs. 1, 2, and 7). Because this approach is sequence-independent, it is suitable for preparation of dsDNA templates with a defined sequence and complex sequence libraries. Importantly, the lesion bypass reaction is simple and efficient. Under our reaction conditions, translesion synthesis by DNA Pol IV resulted in DNA template preparations that were indistinguishable from an unmodified template by denaturing gel electrophoresis and remained fully intact after several freeze–thaw cycles throughout data collection (Figs. 1E and 7B and S2, A, D, and E, and S3). Furthermore, our approach is likely to be generalizable to many DNA modifications. In this work, we successfully applied a general translesion synthesis protocol to three chemically distinct DNA modifications. Critically, the preparation and detailed characterization of the resulting DNA templates uses common biochemical techniques and reagents and will therefore be readily accessible for many laboratories.
Our study has established the desthiobiotin–TEG and etheno-dA DNA modifications as potent transcription blockades that are compatible with enzymatic dsDNA template preparation. These DNA modifications have complementary properties that are likely to be advantageous for specific applications. In experiments where enrichment of TECs that have transcribed to the stall site is crucial, the ability of RNAP to sequester desthiobiotin from binding streptavidin provides a means to purify stall site–proximal TECs by exclusion from streptavidin-coated magnetic beads (Fig. 5). In contrast, etheno-dA has no inherent conflicts with immobilization of 5′-biotinylated DNA templates, and its small size may prove useful in applications that benefit from a minimally disruptive transcription stall site. It is likely that other readily available oligonucleotide modifications are compatible with chemical transcription roadblocking and may have functional properties that complement those of desthiobiotin–TEG and etheno-dA. We suggest that a DNA modification is suitable for chemical transcription roadblocking when it satisfies four criteria: the modification should efficiently halt the target RNAP, should be efficiently bypassed by a translesion DNA polymerase to facilitate DNA template preparation; should be chemically stable, and should not interfere with downstream applications. We envision that the chemical transcription roadblocking approach presented in this work will facilitate increasingly demanding nascent RNA display applications by enabling RNA synthesis in a minimal transcription reaction.
Experimental procedures
Oligonucleotides
All oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA) and are described in Table S1. All modified oligonucleotides were HPLC-purified to ensure efficient modification. The oligonucleotide used as a template for PCR amplification was PAGE-purified to enrich for complete product.
Unmodified DNA template preparation
Linear DNA templates without internal modifications were prepared by PCR amplification essentially as described previously (3). Briefly, five 100-μl reactions containing 81.5 μl of nuclease-free water, 10 μl Thermo Pol buffer (New England Biolabs, Ipswich, MA), 2 μl of 10 mm dNTPs (New England Biolabs), 2.5 μl of 10 μm oligonucleotide A (unmodified forward primer (Table S1)), 2.5 μl of 10 μm oligonucleotide C (unmodified reverse primer (Table S1)) or oligonucleotide G (5′-biotinylated reverse primer (Table S1)), 1 μl of Vent® (exo−) DNA polymerase (New England Biolabs), and 0.5 μl of 0.1 nm oligonucleotide H (template oligonucleotide (Table S1)) were amplified for 30 PCR cycles. Following amplification, 100-μl reactions were combined into two 250-μl pools, precipitated by adding 25 μl of 3 m sodium acetate (pH 5.5) and 750 μl of cold 100% EtOH, chilled at −80 °C for 15 min, and centrifuged at 20,000 × g for 15 min. DNA pellets were washed with 70% EtOH (v/v), dried using a SpeedVac, dissolved in 30 μl of nuclease-free water, run on a 1% (w/v) agarose gel, and extracted using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. DNA concentration was determined by a Qubit 1.0 Fluorometer (Life Technologies). The fully assembled DNA template sequence is shown in Table S1.
Internally modified DNA template preparation
Internally modified linear DNA templates were PCR-amplified as above, except that oligonucleotide A or B (unmodified and 5′-biotinylated forward primer, respectively (Table S1)) was used as a forward primer, and oligonucleotide D, E, or F (internal desthiobiotin–TEG, internal amino linker, and internal etheno-dA reverse primer, respectively (Table S1)) was used as a reverse primer. Two protocols were used for translesion synthesis. In our initial protocol (used for experiments shown in Figs. 1 and 3–6), 10 100-μl PCRs were individually purified using the QIAquick PCR Purification Kit (Qiagen) according to the manufacturer's protocol, eluted in 30 μl of nuclease-free water, and combined with 100 μl of Thermo Pol buffer, 20 μl of 10 mm dNTPs, 10 μl of Sulfolobus DNA polymerase IV (New England Biolabs), and nuclease-free water to 1 ml. The 1-ml master mixture was split into 100-μl aliquots and incubated at 55 °C for 1 h. We later determined that, because Vent® (exo−) DNA polymerase and Sulfolobus DNA polymerase IV use the same buffer and dNTP conditions, DNA purification after PCR was unnecessary, and efficient translesion synthesis could be achieved by pooling the initial 100-μl PCRs, adding 1 μl of Sulfolobus DNA polymerase IV per 100-μl reaction volume, splitting the reaction master mixture into 100-μl aliquots, and incubating at 55 °C for 1 h; this protocol was used to prepare DNA templates for Fig. 7 and Fig. S2A. For both protocols, the 1-ml translesion synthesis master mixture contained ∼4.5 μg of the truncated PCR product. Translesion synthesis with thermostability-enhancing dNTPs was performed using our original protocol but with a dNTP mixture in which dATP and dCTP were completely substituted with 2-amino-dATP and 5-propynyl-dCTP (TriLink Biotechnologies, San Diego, CA). Following translesion synthesis, template DNA was precipitated, gel-extracted, and quantified as described for unmodified DNA templates.
DNA template quality control
To verify success of the translesion synthesis reaction, all internally modified DNA templates were subjected to quality control by denaturing urea-PAGE using the UreaGel System (National Diagnostics, Atlanta, GA) (Figs. 1E and 7B and Figs. S2, A, D, and E, and S3). Each quality control gel included a reaction aliquot that had not undergone translesion synthesis as a negative control and an unmodified positive control. In some quality control gels, we observed a smear beneath the full-length unmodified positive control and the modified template that had undergone translesion synthesis. We believe this smear was due to renaturation of a small fraction of the 121-bp dsDNA, which results in faster migration than single-stranded DNA because of the increased charge of the dsDNA duplex. This effect was typically resolved in technical replicates that were run at higher voltage but remained somewhat variable. For this reason, it is critical to always include an unmodified positive control when performing translesion synthesis DNA template quality control and to ensure a sufficiently high gel running temperature. Quality control was performed as follows. A 10% urea-PAGE gel was prerun at 300 V (Figs. 1E and 7B and Fig. S2, A and E) or 250 V (Fig. S2D) for 30 min on a Mini-Protean Tetra Cell (Bio-Rad) that was assembled so that 1× Tris borate-EDTA buffer in the outer chamber covered only the bottom ∼1 cm of the gel plates to help maintain a hotter running temperature. Immediately prior to loading, 1 μl of 50 nm DNA template (50 fmol) was mixed with 15 μl of formamide loading dye (1× transcription buffer, described under “Single-round in vitro transcription,” 80% (v/v) formamide, 0.025% (w/v) bromphenol blue, and xylene cyanol) and boiled for 5 min before placing on ice for 2 min. All nondenaturing PAGE quality control gels were 8% polyacrylamide in 1× Tris borate-EDTA buffer. Gels were stained with SYBR Gold (Life Technologies) and imaged using an Amersham Biosciences Typhoon 9400 Variable Mode Imager. Following collection of all data, a second denaturing PAGE quality control gel was performed to confirm that the internal desthiobiotin-modified DNA template remained fully intact (Fig. S2D).
End labeling and primer extension reactions
The 32P end–labeled primer was prepared by incubating 10 pmol of oligonucleotide I (Table S1) in a 50-μl reaction containing 1× polynucleotide kinase (PNK) buffer (New England Biolabs), 20 units of PNK (New England Biolabs), and 100 μCi of [γ-32P]ATP (PerkinElmer Life Sciences) at 37 °C for 1 h, followed by 20-min incubation at 65 °C to heat-inactivate PNK. Free [γ-32P]ATP was removed by applying the end-labeling reaction to a Tris buffer Bio-Spin P-30 Gel Column (Bio-Rad) according to the manufacturer's protocol; the final sample volume was raised to 100 μl for an approximate final primer concentration of 100 nm. The end-labeled oligonucleotide I (Table S1) primer was annealed to oligonucleotide C, D, E, F, J, or K (Table S1) at a ratio 1:1.5 (0.05 pmol primer, 0.075 pmol template) in 1× Thermo Pol buffer by heating at 95 °C for 3 min, cooling to 55 °C at a rate of 0.1 °C/s, incubating at 55 °C for 5 min, and cooling to 4 °C at a rate of 0.1 °C/s. All 10-μl primer extension reactions contained 5 nm end-labeled primer, 7.5 nm template oligonucleotide (preannealed), 1× Thermo Pol buffer, and 0.02 units/μl Sulfolobus DNA polymerase IV. For dNTP starvation experiments, the specified dNTP mixture (Invitrogen) was included at a final concentration of 100 μm, and reactions were performed by placing all samples on a thermal cycler block precooled to 4 °C, raising the temperature to 55 °C for 5 min, and cooling the block to 4 °C. For ddNTP sequencing ladder experiments, dNTPs were included at a final concentration of 100 μm. The specified ddNTP was included at 500 μm or 1 mm, and reactions were performed by placing all samples on a thermal cycler block precooled to 4 °C, raising the temperature to 55 °C for 30 min, and cooling the block to 4 °C. After primer extension, 90 μl of stop solution (0.6 m Tris-HCl (pH 8.0), 12 mm EDTA) was added to each reaction. Reactions were then extracted by adding 100 μl of phenol/chloroform/isoamyl alcohol (25:24:1), vortexing, centrifugation, and collection of the aqueous phase and then ethanol-precipitated by adding 300 μl of 100% ethanol, 10 μl 3 m sodium acetate (pH 5.5), 1.2 μl of GlycoBlue coprecipitant (Thermo Fisher Scientific, Waltham, MA) and stored at −20 °C overnight. After centrifugation and removal of bulk and residual ethanol, precipitated RNA was resuspended in formamide loading dye and fractionated by urea-PAGE using a 15% (dNTP starvation experiments) or 10% (sequencing ladder experiments) polyacrylamide sequencing gel prepared with the UreaGel system.
Quantification of radiolabeled primer extension reactions
Reactive nucleotides were detected by an Amersham Biosciences Typhoon 9400 Variable Mode Imager and quantified using ImageQuant (GE Life Sciences). In Fig. 2E, “fraction extended” was calculated by dividing the band intensity of primers that were extended by at least one nucleotide by the total of band intensity of extended and unextended primer.
Single-round in vitro transcription
Single-round in vitro transcription reactions were performed essentially as described previously (4, 25). All single-round transcription reactions contained 5 nm DNA template and 0.016 units/μl E. coli RNA polymerase holoenzyme (New England Biolabs) in transcription buffer (20 mm Tris-HCl (pH 8.0), 0.1 mm EDTA, 1 mm DTT, and 50 mm KCl) and 0.2 mg/ml BSA in a total volume of 25 μl. All NTP mixtures were prepared using high-purity ATP, GTP, CTP, and UTP (GE Life Sciences, Chicago, IL).
Two protocol variations were performed. For experiments in which transcription was initiated by walking RNAP to +15 relative to the transcription start site (Figs. 1, F and G, 3, and 7C), TECs were stalled at U15 by incubating reactions containing 2.5 μm ATP and GTP, 1.5 μm UTP, 0.2 μCi/μl [α-32P]UTP (PerkinElmer Life Sciences), and 10 mm MgCl2 at 37 °C for 10 min before adding NTPs to 500 μm and rifampicin (Gold Biotechnology, St. Louis, MO) to 10 μg/ml; 25-μl aliquots were removed at the specified time points and added to 125 μl of stop solution. RNA sequencing ladders were generated by walking RNAP to +15 before adding NTPs supplemented with a chain terminating 3′-dNTP (TriLink Biotechnologies) to 100 μm; reactions proceeded at 37 °C for 5 min before addition of 125 μl of stop solution. For Fig. 3, the biotin–streptavidin roadblock was prepared by adding streptavidin monomer (Promega, Madison, WI) to 100 nm during reaction master mixture preparation.
For Fig. 4, open promoter complexes were formed by incubating reactions containing 200 μm ATP, GTP, and CTP; 50 μm UTP; and 0.2 μCi/μl [α-32P]UTP (PerkinElmer Life Sciences) at 37 °C for 10 min and initiated by adding magnesium chloride (MgCl2) to 10 mm and rifampicin to 10 μg/ml; 25-μl aliquots were removed at the specified time points and added to 125 μl of stop solution. A mineral oil overlay was applied to the reaction master mix after taking the 4-min time point to prevent evaporation over the 256-min and 64-min time courses.
All reactions were extracted by adding 150 μl of phenol:chloroform:isoamyl alcohol (25:24:1), vortexing, centrifugation, and collection of the aqueous phase and then ethanol-precipitated by adding 450 μl of 100% ethanol and 1.2 μl of GlycoBlue coprecipitant (Thermo Fisher Scientific), and stored at −20 °C overnight. After centrifugation and removal of bulk and residual ethanol, precipitated RNA was resuspended in formamide loading dye and fractionated by urea-PAGE using a 12% polyacrylamide sequencing gel prepared with the UreaGel system.
Equilibration of streptavidin-coated magnetic beads
All transcription reactions with magnetic separation were performed using 10 μl of Dynabeads MyOne Streptavidin C1 beads (Invitrogen) per 25-μl transcription volume. For each experiment, magnetic beads were prepared in bulk by removing storage buffer, incubating with 500 μl of hydrolysis buffer (100 mm sodium hydroxide and 50 mm NaCl) for 10 min at room temperature with rotation, washing once with 1 ml of high-salt wash buffer (50 mm Tris-HCl (pH 7.5), 2 m NaCl, and 0.5% Triton X-100), washing once with 1 ml of binding buffer (10 mm Tris-HCl (pH 7.5), 300 mm NaCl, and 0.1% Triton X-100), and washing twice with 1 ml of 1× transcription buffer supplemented with Triton X-100 to 0.1% and resuspended in 1× transcription buffer with 0.1% Triton X-100 to a volume equivalent to that of the total transcription master mixture in the respective experiment for storage until use.
Desthiobiotin protection assay
For the desthiobiotin protection experiment in Fig. 5B, transcription was initiated by walking RNAP to U15 as described above. To assess desthiobiotin–streptavidin binding when RNAP was at U15, one half of the reaction was mixed with rifampicin to 10 μg/ml and immediately mixed with pre-equilibrated streptavidin-coated magnetic beads. To assess desthiobiotin–streptavidin binding when RNAP was at the desthiobiotin–TEG stall site, the second half of the reaction was mixed with NTPs to 500 μm and rifampicin to 10 μg/ml and incubated at 37 °C for 5 min before mixing with pre-equilibrated streptavidin-coated magnetic beads. After 10 min of incubation with streptavidin-coated magnetic beads at room temperature, 25 μl of the reaction was added to 125 μl stop solution as a “total” sample control, and an additional 25 μl was added to a new tube and placed on a magnetic stand for 1 min to separate the bead pellet and supernatant. The supernatant was added to 125 μl of stop solution, and the pellet was resuspended in 25 μl of 1× transcription buffer supplemented with 10 mm MgCl2 before 125 μl of stop solution was added. Samples were phenol/chloroform-extracted, precipitated, and fractionated by urea-PAGE as described above.
For the experiment in Fig. 5C, transcription was initiated by walking RNAP to U15 as described above before adding NTPs to 100 μm, 3′-dNTPs to 25 μm, and rifampicin to 10 μg/ml and incubating at 37 °C for 5 min to distribute TECs across the DNA template; the total reaction sample was added to 125 μl of stop solution immediately after this incubation. Distributed TECs were then mixed with pre-equilibrated streptavidin-coated magnetic beads. After 10 min of incubation at room temperature, 25 μl of the reaction was added to a new tube and placed on a magnetic stand for 1 min to separate the bead pellet and supernatant, which were then processed as described above.
Elongation complex stability assays
The elongation complex stability assay in Fig. 6 was performed using a DNA template containing an internal desthiobiotin–TEG modification and a 5′-biotin–TEG modification upstream of the promoter. Stalled TECs were prepared by first walking RNAP to U15 as described above and then chasing RNAP to the desthiobiotin–TEG stall site by adding NTPs to 500 μm and rifampicin to 10 μg/ml and incubating at 37 °C for 5 min. The transcription reaction was then mixed with pre-equilibrated streptavidin-coated magnetic beads and incubated at room temperature for 10 min. Following DNA template immobilization, beads were washed twice with 1 ml of 1× transcription buffer supplemented with 1 mm MgCl2 to remove free NTPs. After removing a zero time point for fractionation, the transcription reaction was incubated at room temperature for 2 h with rotation, and time points were taken as indicated. At each time point, 25 μl of the reaction was added to 125 μl stop solution as a total sample control, and a second 25 μl was added to a new tube and placed on a magnetic stand for 1 min to separate the bead pellet and supernatant. The supernatant was added to 125 μl of stop solution, and the pellet was resuspended in 25 μl of 1× transcription buffer supplemented with 1 mm MgCl2 before 125 μl of stop solution was added. Samples were phenol/chloroform-extracted, precipitated, and fractionated by urea-PAGE as described above.
For Fig. 7E, a DNA template containing an internal etheno-dA modification and a 5′-biotin–TEG modification upstream of the promoter was used. For each reaction volume, 0.125 pmol of DNA template was incubated with pre-equilibrated streptavidin-coated magnetic beads in 1× transcription buffer at room temperature for 10 min. After immobilizing the DNA template, the beads were washed once with 1× transcription buffer supplemented with 0.1% Triton X-100 and resuspended in the transcription master mixture described above for walking RNAP to U15. After walking RNAP to U15, TECs were chased to the desthiobiotin–TEG stall site by adding NTPs to 500 μm and rifampicin to 10 μg/ml and incubating at 37 °C for 5 min. Reactions were then fractionated into bead pellet and supernatant and processed as described above for the desthiobiotin protection assay.
Quantification of radiolabeled in vitro transcription reactions
Reactive nucleotides were detected by an Amersham Biosciences Typhoon 9400 Variable Mode Imager and quantified using ImageQuant (GE Life Sciences). For experiments in which RNAP was walked to U15, transcripts were considered end-labeled because of the high probability of radiolabel incorporation during the initial walk (∼4.25% per U nucleotide) and the low probability of radiolabel incorporation during the chase (∼0.013% per U nucleotide), and no normalization was applied. For the experiments in Fig. 4, band intensity was divided by transcript U content to normalize for the incorporation of [α-32P]UTP. In Figs. 3 and 7, the fraction of runoff, stall site, and U15 transcripts was determined by dividing the band intensity of each transcript class by the total band intensity of all three classes. In Fig. 4, the fraction stalled was determined by dividing the normalized band intensity of desthiobiotin–TEG–stalled transcripts by the sum of the normalized band intensity of stalled and runoff transcripts; t½ was determined by applying a one-phase exponential decay fit to time points from 2 min (after new arrival of TECs at the stall site is negligible) to 48 min (before the semilogarithmic plot deviates from a straight line) (26) using GraphPad Prism 8. In Figs. 5–7, the fraction of transcripts in the bead pellet was determined by dividing the band intensity of the indicated complex in the pellet fraction by the sum of the band intensities for the indicated complex in the pellet and supernatant fractions.
Data Availability
All data are contained in the manuscript as plotted values or representative gels. Source files in .gel format are available from the corresponding author (E. J. S.) upon request.
Author contributions
E. J. S. conceptualization; E. J. S. formal analysis; E. J. S., J. T. L., and J. B. L. funding acquisition; E. J. S. validation; E. J. S. investigation; E. J. S. methodology; E. J. S. writing-original draft; E. J. S., J. T. L., and J. B. L. writing-review and editing.
Supplementary Material
Acknowledgment
We thank Jeffrey W. Roberts for thoughtful discussions and critical reading of the manuscript.
This work was supported by an Arnold O. Beckman postdoctoral fellowship (to E. J. S.), NIGMS, National Institutes of Health Grant GM025232 (to J. T. L.)., and Searle Funds at The Chicago Community Trust (to J. B. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3 and Table S1.
- TEC
- transcription elongation complex
- RNAP
- RNA polymerase
- TEG
- triethylene glycol
- etheno-dA
- 1,N6-etheno-2′-deoxyadenosine
- DNA Pol IV
- DNA Polymerase IV
- PNK
- polynucleotide kinase
- FL
- full-length
- T
- total
- P
- pellet
- S
- supernatant.
References
- 1. Watters K. E., Strobel E. J., Yu A. M., Lis J. T., and Lucks J. B. (2016) Cotranscriptional folding of a riboswitch at nucleotide resolution. Nat. Struct. Mol. Biol. 23, 1124–1131 10.1038/nsmb.3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chauvier A., Picard-Jean F., Berger-Dancause J. C., Bastet L., Naghdi M. R., Dubé A., Turcotte P., Perreault J., and Lafontaine D. A. (2017) Transcriptional pausing at the translation start site operates as a critical checkpoint for riboswitch regulation. Nat. Commun. 8, 13892 10.1038/ncomms13892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Strobel E. J., Watters K. E., Nedialkov Y., Artsimovitch I., and Lucks J. B. (2017) Distributed biotin-streptavidin transcription roadblocks for mapping cotranscriptional RNA folding. Nucleic Acids Res. 45, e109 10.1093/nar/gkx233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Strobel E. J., Cheng L., Berman K. E., Carlson P. D., and Lucks J. B. (2019) A ligand-gated strand displacement mechanism for ZTP riboswitch transcription control. Nat. Chem. Biol. 15, 1067–1076 10.1038/s41589-019-0382-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tome J. M., Ozer A., Pagano J. M., Gheba D., Schroth G. P., and Lis J. T. (2014) Comprehensive analysis of RNA-protein interactions by high-throughput sequencing-RNA affinity profiling. Nat. Methods 11, 683–688 10.1038/nmeth.2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buenrostro J. D., Araya C. L., Chircus L. M., Layton C. J., Chang H. Y., Snyder M. P., and Greenleaf W. J. (2014) Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nat. Biotechnol. 32, 562–568 10.1038/nbt.2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pavco P. A., and Steege D. A. (1990) Elongation by Escherichia coli RNA polymerase is blocked in vitro by a site-specific DNA binding protein. J. Biol. Chem. 265, 9960–9969 [PubMed] [Google Scholar]
- 8. Frieda K. L., and Block S. M. (2012) Direct observation of cotranscriptional folding in an adenine riboswitch. Science 338, 397–400 10.1126/science.1225722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Widom J. R., Rai V., Rohlman C. E., and Walter N. G. (2019) Versatile transcription control based on reversible dCas9 binding. RNA 25, 1457–1469 10.1261/rna.071613.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sancar A. (1996) DNA excision repair. Annu. Rev. Biochem. 65, 43–81 10.1146/annurev.bi.65.070196.000355 [DOI] [PubMed] [Google Scholar]
- 11. Van Houten B., Gamper H., Sancar A., and Hearst J. E. (1987) DNase I footprint of ABC excinuclease. J. Biol. Chem. 262, 13180–13187 [PubMed] [Google Scholar]
- 12. Wang H., and Hays J. B. (2001) Simple and rapid preparation of gapped plasmid DNA for incorporation of oligomers containing specific DNA lesions. Mol. Biotechnol. 19, 133–140 10.1385/MB:19:2:133 [DOI] [PubMed] [Google Scholar]
- 13. Luzzietti N., Brutzer H., Klaue D., Schwarz F. W., Staroske W., Clausing S., and Seidel R. (2011) Efficient preparation of internally modified single-molecule constructs using nicking enzymes. Nucleic Acids Res. 39, e15 10.1093/nar/gkq1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luzzietti N., Knappe S., Richter I., and Seidel R. (2012) Nicking enzyme-based internal labeling of DNA at multiple loci. Nat. Protoc. 7, 643–653 10.1038/nprot.2012.008 [DOI] [PubMed] [Google Scholar]
- 15. Zhou W., and Doetsch P. W. (1993) Effects of abasic sites and DNA single-strand breaks on prokaryotic RNA polymerases. Proc. Natl. Acad. Sci. U.S.A. 90, 6601–6605 10.1073/pnas.90.14.6601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Viswanathan A., and Doetsch P. W. (1998) Effects of nonbulky DNA base damages on Escherichia coli RNA polymerase-mediated elongation and promoter clearance. J. Biol. Chem. 273, 21276–21281 10.1074/jbc.273.33.21276 [DOI] [PubMed] [Google Scholar]
- 17. Viswanathan A., Liu J., and Doetsch P. W. (1999) E. coli RNA polymerase bypass of DNA base damage: mutagenesis at the level of transcription. Ann. N.Y. Acad. Sci. 870, 386–388 10.1111/j.1749-6632.1999.tb08909.x [DOI] [PubMed] [Google Scholar]
- 18. Clauson C. L., Oestreich K. J., Austin J. W., and Doetsch P. W. (2010) Abasic sites and strand breaks in DNA cause transcriptional mutagenesis in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 107, 3657–3662 10.1073/pnas.0913191107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pupov D., Ignatov A., Agapov A., and Kulbachinskiy A. (2019) Distinct effects of DNA lesions on RNA synthesis by Escherichia coli RNA polymerase. Biochem. Biophys. Res. Commun. 510, 122–127 10.1016/j.bbrc.2019.01.062 [DOI] [PubMed] [Google Scholar]
- 20. Boudsocq F., Iwai S., Hanaoka F., and Woodgate R. (2001) Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4): an archaeal DinB-like DNA polymerase with lesion-bypass properties akin to eukaryotic poleta. Nucleic Acids Res. 29, 4607–4616 10.1093/nar/29.22.4607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McDonald J. P., Hall A., Gasparutto D., Cadet J., Ballantyne J., and Woodgate R. (2006) Novel thermostable Y-family polymerases: applications for the PCR amplification of damaged or ancient DNAs. Nucleic Acids Res. 34, 1102–1111 10.1093/nar/gkj512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Strauss B. S. (1991) The “A rule” of mutagen specificity: a consequence of DNA polymerase bypass of non-instructional lesions? Bioessays 13, 79–84 10.1002/bies.950130206 [DOI] [PubMed] [Google Scholar]
- 23. Fiala K. A., Brown J. A., Ling H., Kshetry A. K., Zhang J., Taylor J. S., Yang W., and Suo Z. (2007) Mechanism of template-independent nucleotide incorporation catalyzed by a template-dependent DNA polymerase. J. Mol. Biol. 365, 590–602 10.1016/j.jmb.2006.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Metzger W., Schickor P., and Heumann H. (1989) A cinematographic view of Escherichia coli RNA polymerase translocation. EMBO J. 8, 2745–2754 10.1002/j.1460-2075.1989.tb08416.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Strobel E. J., and Roberts J. W. (2015) Two transcription pause elements underlie a sigma70-dependent pause cycle. Proc. Natl. Acad. Sci. U.S.A. 112, E4374–4380 10.1073/pnas.1512986112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Landick R., Wang D., and Chan C. L. (1996) Quantitative analysis of transcriptional pausing by Escherichia coli RNA polymerase: his leader pause site as paradigm. Methods Enzymol. 274, 334–353 10.1016/S0076-6879(96)74029-6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained in the manuscript as plotted values or representative gels. Source files in .gel format are available from the corresponding author (E. J. S.) upon request.