

Abstract
Kainate and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are two major, closely related receptor subtypes in the glutamate ion channel family. Excessive activities of these receptors have been implicated in a number of central nervous system diseases. Designing potent and selective antagonists of these receptors, especially of kainate receptors, is useful for developing potential treatment strategies for these neurological diseases. Here, we report on two RNA aptamers designed to individually inhibit kainate and AMPA receptors. To improve the biostability of these aptamers, we also chemically modified these aptamers by substituting their 2′-OH group with 2′-fluorine. These 2′-fluoro aptamers, FB9s-b and FB9s-r, were markedly resistant to RNase-catalyzed degradation, with a half-life of ∼5 days in rat cerebrospinal fluid or serum-containing medium. Furthermore, FB9s-r blocked AMPA receptor activity. Aptamer FB9s-b selectively inhibited GluK1 and GluK2 kainate receptor subunits, and also GluK1/GluK5 and GluK2/GluK5 heteromeric kainate receptors with equal potency. This inhibitory profile makes FB9s-b a powerful template for developing tool molecules and drug candidates for treatment of neurological diseases involving excessive activities of the GluK1 and GluK2 subunits.
Keywords: aptamer, RNA, RNA modification, drug development, ionotropic glutamate receptor, inhibitor, neurological disease, 2′-fluoro–modified RNAs, AMPA receptors, kainate receptors, selective antagonist, SELEX
Introduction
Developing selective antagonists against kainate receptors is significant for the following reasons. Kainate receptors are a subtype of the ionotropic glutamate receptor family, which also includes N-methyl-d-aspartate (NMDA)2 receptors and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors (1–4). Ionotropic glutamate receptors mediate the majority of excitatory neurotransmission in the mammalian central nervous system (CNS), and they are essential for brain development and function (1). At the receptor subtype level, AMPA and NMDA receptors have been implicated in a number of CNS diseases and disorders, such as epilepsy, pain, stroke, and neurodegenerative diseases (5–8). However, the role of kainate receptors in the CNS and CNS diseases is the least understood, as compared with either AMPA or NMDA receptors (3). This is mainly attributed to a lack of selective pharmacological ligands and tool molecules targeting kainate receptors (9). Furthermore, kainate and AMPA receptors are more closely related, as compared with NMDA receptors, in both protein sequences and structures (2, 3). Unlike AMPA receptors, however, kainate receptors are expressed not only postsynaptically, where they mediate excitatory neurotransmission, but also presynaptically, where they play a modulatory role in neurotransmission, such as in neurotransmitter release and neuronal excitability (4, 10). Functional kainate receptors are thought to be tetramers and can be assembled from five subunits, GluK1–5. GluK1–3 share 75–80% amino acid sequence homology, whereas their similarity to GluK4–5 is only 40%, and the sequence identity is 68% between the latter two (11–13). Each of the GluK1–3 subunits is capable of forming homomeric channels (11, 14, 15). In contrast, GluK4–5 require heteromeric assembly with GluK1–3 to form functional channels (12). Thus, developing kainate receptor-selective antagonists would provide us with the ability to selectively control and regulate the activity of kainate receptor subtype in vivo.
Most of the compounds and antagonists that have been developed select the GluK1 subunit (9, 16, 17). Some of these GluK1 antagonists, however, inhibit receptors containing the GluK3 subunit (18) and exhibit even appreciable activities toward AMPA receptors (9, 19). To date, virtually no compounds are selective to the GluK2 subunit (9, 17). In fact, compounds that inhibit GluK2 actually show higher potency toward GluK1 (9). Earlier studies have shown that in the developing brain, GluK1 is highly expressed (20–23). However, in the mature brain, the GluK1 subunit is predominantly expressed in hippocampal interneurons and cerebellar Purkinje cells, whereas the GluK2 subunit is highly expressed throughout the brain (24, 25). Furthermore, GluK2/GluK5 heteromeric receptor is the most abundant kainate receptor in the brain (25). Studies have also implicated that GluK2 and GluK2/GluK5 kainate receptors are associated with schizophrenia (25, 26), temporal lobe epilepsy (27–30), and bipolar disorder (31). Despite a high potential of targeting GluK2 and/or GluK2/GluK5 for therapeutic intervention, developing potent antagonists for GluK2 and GluK2/GluK5 has thus far met with significant difficulties. It should be noted that the full-length GluK2 complexed with an agonist has been determined by the use of cryo-EM at 7.6 Å resolution (32), although the GluK2 structure is thought to reflect a desensitized channel (17). However, no crystal structure of GluK2 bound with any antagonist has been reported. Therefore, generating small-molecule compounds as potential drug candidates targeting GluK2 or GluK2-containing channels by the use of structure-based ligand discovery has been just as elusive.
Here we report an RNA inhibitor or RNA aptamer, termed AB9s-b. This aptamer shows a micromolar potency in inhibiting the GluK2 kainate receptor subunit and also GluK2/GluK5 channels. AB9s-b has been derived from aptamer AB9 originally discovered from a large RNA library (∼1014 sequence variations) by the use of systematic evolution of ligands by exponential enrichment (SELEX) (33, 34). RNA aptamers as potential regulatory agents and drug candidates have some unique properties as compared with traditional, small-molecule compounds (35–37). RNA aptamers are water-soluble and in general show higher potency and selectivity. To make these RNA aptamers amenable for in vivo use, we have also developed its chemically modified aptamer, FB9s-b, with satisfactory in vitro stability in the presence of ribonucleases, as in cerebrospinal fluid (CSF).
Results
Experimental procedures
As an alternative to synthetic chemistry, which yields small molecule inhibitors, we have previously used SELEX to “evolve” RNA molecules bound to target from a large library (∼1014 sequence variations). In particular, we have isolated a class of potent aptamers targeting AMPA receptors (38–41), including a GluA2 subunit-selective RNA aptamer (40). In these SELEX experiments, we expressed an AMPA receptor in human embryonic kidney (HEK-293) cells and used the membrane fragments that contain AMPA receptors as the target. A typical SELEX operation in our case involves about a dozen of cycles, and each cycle is composed of RNA binding, elution of the bound RNA molecules, RT-PCR, and regeneration of an enriched RNA library for the next cycle (33, 34). Eventually, molecular cloning and sequencing technique can be used to identify the RNA molecules evolved from these repeated cycles of enrichment. RNAs that exhibit identical or virtually identical sequences are candidates of functional importance. We then carry out a functional assay to screen all of these candidate RNAs to identify those that are capable of inhibiting the target. Despite our success in using SELEX to isolate desired AMPA receptor RNA aptamers, as described above, we have not yet been able to reproduce our success in isolating useful RNA aptamers against GluK2 kainate receptor.3
In the current study, we decided to use a recently isolated RNA aptamer with dual activities on both AMPA and kainate receptors (41) to develop kainate receptor aptamers, instead of continuing with SELEX and a large RNA library to search for a random sequence that may or may not act as a kainate receptor inhibitor. The aptamer with the dual activity, termed AB9s, is a functionally active RNA with 55 nucleotides derived from its parent RNA or AB9 aptamer with 101 nucleotides (41). During the truncation of the original or full sequence to generate the minimal but functional sequence, we noticed two main, secondary sequence segments or domains, which we indicate as red and blue sequences (Fig. 1A). We have also found that some RNA sequences between the two domains can be removed without affecting the function of the shorter RNAs (41). Therefore, we hypothesized that each domain acts independently. As such, two single-domain RNA mutants would function separately in inhibiting AMPA and kainate receptors. In other words, one mutant would only inhibit AMPA, whereas the other mutant would only inhibit kainate receptors.
Figure 1.
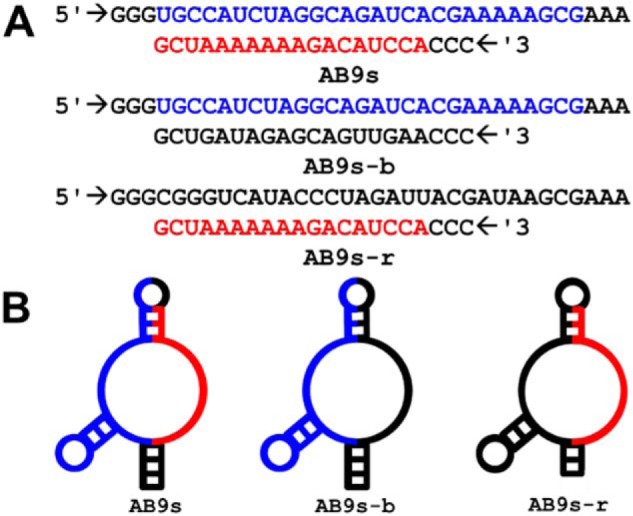
Sequences and secondary structure of AB9s, AB9s-b, and AB9s-r. A, RNA sequences of aptamers AB9s, AB9s-b, and AB9s-r. The 2′-F–modified versions of these aptamers, termed FB9s, FB9s-b, and FB9s-r, share the same sequences. B, secondary structures of these aptamers, predicted by MFold.
To test this hypothesis, we created two mutants, termed AB9s-r and AB9s-b, based on the sequence and the MFold-predicted structure of AB9s (Fig. 1, A and B). In constructing these two mutants, we also used the MFold program (42) to ensure that a mutant was still able to uphold the same secondary structure as the original AB9s (see Fig. 1B and its legend for additional description). We reasoned that both the sequence and the secondary structural motif that surrounds a stretch of the sequence would be essential for the function of the mutant RNA. As shown in Fig. 1, the resulting Mfold structure of AB9s-b is similar to that of AB9s. However, a different set of sequences had to be used to keep the blue region in AB9s-r the same fold as in AB9s. We then characterized the impact of individual mutations, by using whole-cell recording, on a panel of NMDA, AMPA, and kainate receptor subunits.
Enzymatic transcription for preparing AB9s-b and AB9s-r and functional assay
AB9s-b and AB9s-r, along with AB9s, were prepared by enzymatic transcription. Each RNA was purified using a PAGE column (43). The putative activity of an RNA was characterized using whole-cell recording with HEK-293 cells expressing individual subunit of glutamate ion channel receptors. As shown by a pair of representative glutamate-induced whole-cell current traces, the amplitude of whole-cell current response was reduced in the presence of a functional aptamer inhibitor. Based on the ratio of the whole-cell current amplitude in the absence and presence of an aptamer, A/A(I), as a function of aptamer concentration, we further determined the inhibition constant, KI, by using Equations 1 and 2 (“Experimental procedures”). It should be noted that at a lower concentration of glutamate, most of the receptors in a receptor population would be in the closed-channel state, whereas at a saturating concentration of glutamate, virtually all the receptor channels would be in the open-channel state (44). As such, by using different glutamate concentrations, we were able to determine the potency of an aptamer for both the closed- and the open-channel state or form. For example, KI for AB9s-r with the closed-channel state of GluA2Qflip was found to be 4.1 ± 0.5 μm (Fig. 2B, left panel, solid symbols). However, AB9s-r did not inhibit the open-channel state of GluA2Qflip AMPA receptor (Fig. 2B, left panel, open symbols). The KI values for AB9s-b for the open- and closed-channel states of the GluK1 kainate receptor were estimated to be 3.2 ± 0.3 and 3.9 ± 0.3 μm, respectively (Fig. 2B, right panel).
Figure 2.

Representative whole-cell recording assay of AB9s-r and AB9s-b and estimate of inhibition constant on representative receptors. Selected A/A(I) plot versus aptamer concentration for AB9s-r and AB9s-b as well as representative whole-cell traces are shown. A, displayed are a pair of representative whole-cell current responses of GluA2Q to 0.1 mm glutamate in the presence and absence of 2 μm AB9s-r (left panel) and the whole-cell current responses of GluK1 to 0.05 mm glutamate in the absence or presence of 2 μm AB9s-b (right panel). The current amplitude was obtained from whole-cell recording with GluA2Q and GluK1, expressed in HEK-293 cells. B, the inhibition constant, KI, of AB9s-r was determined from the A/A(I) plot versus aptamer concentration, using Equations 1 and 2 (“Experimental procedures”), to be 4.1 ± 0.5 μm for the closed-channel state of GluA2Q (solid symbols, left panel). Likewise, the KI values of AB9s-b were estimated to be 3.2 ± 0.3 and 3.9 ± 0.3 μm for the open-channel (open symbols, right panel) and the closed-channel (solid symbols, right panel) states of GluK1, respectively.
Unique subtype selectivity of AB9s-b and AB9s-r versus dual activity of AB9s
The selectivity of AB9s-b and AB9s-r were characterized against a panel of glutamate ion channel subunits and subtypes (Fig. 3). Shown also on the right of each panel of Fig. 3 is whole-cell current response of a representative receptor to glutamate in the absence and presence of an aptamer. The results, represented by A/A(I) value, showed that AB9s-r preferred AMPA receptors (Fig. 3A), whereas AB9s-b selected kainate receptors (Fig. 3B). As expected, neither AB9s-b nor AB9s-r showed any activity on NMDA receptors (Fig. 3C). Furthermore, AB9s-b inhibited not only the homomeric kainate receptor channels assembled from the GluK1 subunit and separately the GluK2 subunit but also the heteromeric channels assembled from GluK5 with GluK1 and separately GluK2 subunits. It should be noted that GluK5 cannot form a functional, homomeric channel on its own (12, 13). On the other hand, AB9s-r exhibited no appreciable kainate receptor activity, but it inhibited all four AMPA receptor subunits (Fig. 3A). As compared with AB9s, which contains both the blue and the red sequence segments and shows activities on both the AMPA and kainate receptors (41), the two sequence segments, when they are individually presented with a similar secondary structure as the parent aptamer AB9s (Fig. 1, A and B), now exhibit distinct subtype selectivity. These results have shown that the hypothesis we proposed is valid. In other words, the two sequence segments encode separate, distinct subtype inhibitory activity. It should be emphasized that AB9s-b inhibited equally potently both GluK1 and GluK2 homomeric channels and also their respective heteromeric channels with the GluK5, i.e. GluK1/GluK5 and GluK2/GluK5 (Fig. 3B). This type of the inhibitory profile has not been previously reported.
Figure 3.
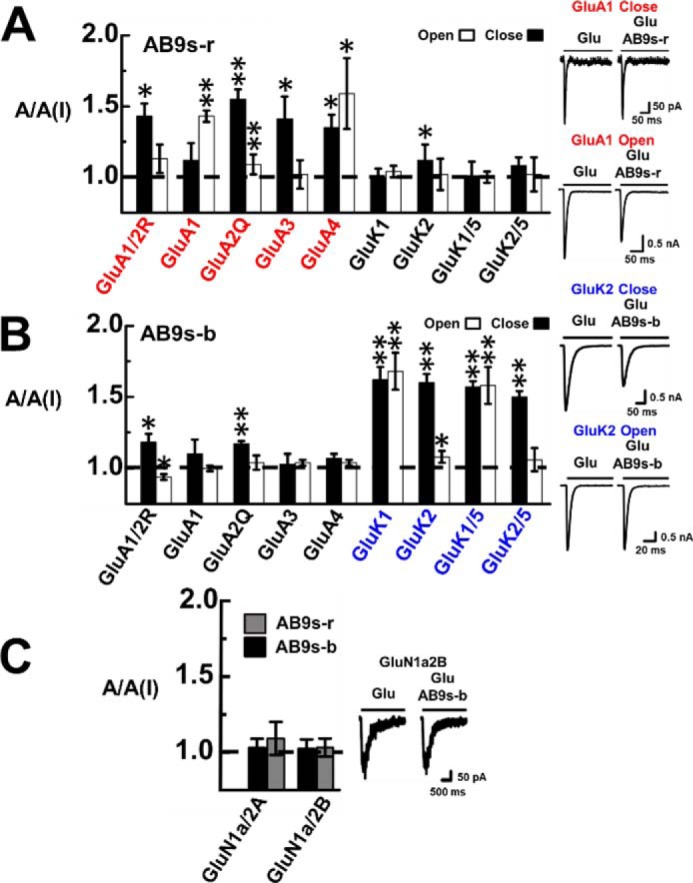
Selective inhibition of glutamate receptor subunits by AB9s-r and AB9s-b. The inhibition is represented by the A/A(I) ratio or the ratio of whole-cell current amplitude in the absence, A, and the presence of 2 μm aptamer, A(I). For each of the receptor subunits and types, the glutamate concentration was chosen to be equivalent to ∼4% and ∼95% fraction of the open channels. Specifically, the glutamate concentration was 0.05 mm for the closed-channel form and 3 mm for the open-channel form for GluA1/2R and GluA1, 0.5 and 10 mm for GluA3, and 0.1 and 3 mm for GluA4. The kainate receptors were tested at 0.05 mm glutamate for closed-channel and 3 mm for the open-channel forms. The difference between open- and closed-channel states was determined with a two-sample, two-tailed Student's t test. Error bars indicate standard deviation from the mean, and all results are based on at least three measurements. A, AB9s-r shows preferential inhibition toward AMPA receptor subunits (red labels) over kainate receptor subunits. Shown on the right are representative whole-cell current responses of GluA1 to glutamate in the absence and presence of aptamer AB9s-r. At the low glutamate concentration, the kdes values of GluA1 in the presence and absence of the aptamer were estimated to be 131 ± 9 and 127 ± 8 s−1, respectively. At high glutamate concentration, the kdes values with and without the aptamer are 166 ± 8 and 160 ± 8 s−1, respectively. One-way ANOVA with post hoc Tukey HSD test analysis comparing closed-channel and the open-channel AMPA and kainate A/A(I) values shows p levels of 0.00042 and 0.06310, respectively. B, AB9s-b shows preferential inhibition toward kainate receptor subunits (blue labels) over AMPA receptors. Also shown on the right is a pair of representative whole-cell current responses of GluK2 to glutamate in the absence and presence of aptamer AB9s-b. At low glutamate concentration, kdes values of 40 ± 2 and 38 ± 4 s−1 were determined for GluK2 with and without the aptamer. At high glutamate concentration, we found kdes to be 226 ± 7 and 214 ± 13 s−1 with and without AB9s-b. One-way ANOVA with post hoc Tukey HSD test analysis comparing closed-channel AMPA and kainate A/A(I) values has p < 0.00001. One-way ANOVA with post hoc Tukey HSD test analysis comparing the open-channel AMPA and kainate A/A(I) values resulted in p = 0.00036. C, the specificity against NMDA channels was tested at a concentration of 5 μm glutamate for GluN1a/2B and 50 μm glutamate for GluN1a/2A. Neither AB9s-r (gray columns) nor AB9s-b (black columns) significantly inhibited the NMDA channels, as seen in the pair of representative whole-cell current response of GluN1a2B in the absence and presence of AB9s-b. From these traces, we estimated the kdes values to be 4 ± 0.3 and 4 ± 0.4 s−1, respectively, with and without AB9s-b.
Enzymatic transcription for preparing 2′-fluoro–modified FB9s-r and FB9s-b
A natural or regular RNA aptamer is degraded quickly through the RNase-catalyzed cleavage of phosphodiester bonds in the RNA backbone. Ribonucleases are very abundant in biological fluids, including CSF. For instance, the half-life (t½) of the degradation of RNA in human blood is typically only a few minutes (45, 46). However, chemical modifications at the 2′ position in the ribose sugar is known to enhance nuclease stability (47–49). In serum, for instance, RNase A targets the 2′-OH group of the ribopyrimidines and cleaves the phosphodiester bond in the sugar-phosphate backbone of RNA strands (50, 51). One of the commonly used chemical modifications is the 2′-fluoro substitution of the 2′-OH group (47–49). To make our RNA aptamers suitable for in vivo use, we chose to replace every 2′-OH group with 2′-fluorine at U, A, and C positions, while leaving the G (guanine) positions unmodified. This was to ensure that the first two nucleotides after the promoter site be canonical GG (or CC in the template strand) so that enzymatic transcription reaction could still proceed with a reasonable yield (52). Experimentally, we made two 2′-fluoro–modified RNA aptamers, i.e. FB9s-r and FB9s-b, by transcribing them in the presence of the 2′-fluoro NTPs (i.e. 2′-F-A, 2′-F-C, and 2′-F-U). FB9s-r and FB9s-b shared the same corresponding sequences with unmodified, regular RNA aptamers, AB9s-r and AB9s-b (Fig. 1). Each of the modified aptamers was purified by PAGE, the same way we purified its regular aptamer counterpart.
Functional characterization of selectivity of FB9s-r and FB9s-b using whole-cell recording
As shown with whole-cell current response of a representative receptor to glutamate in the absence and presence of an aptamer (Fig. 4), FB9s-r only blocked AMPA receptor channels, whereas FB9s-b only inhibited kainate receptors (Fig. 4, A and B). Neither aptamer showed any appreciable activity against NMDA receptors (Fig. 4C). These results are similar to those for the corresponding unmodified aptamers (Fig. 3). We further estimated the KI or the potency for these receptors. The KI for FB9s-r against GluA2 was calculated to be 5.1 ± 0.3 against the closed channel (Fig. 5A). The KI for FB9s-b was found to be 3.0 ± 0.1 μm for GluK2 closed-channel form only, because this aptamer only inhibited the closed-channel form of GluK2 (Fig. 5B). All of these data demonstrated that these two fluorinated aptamers, i.e. FB9s-b and FB9s-r, maintained the desired subtype selectivity, despite the fact that both aptamers contain a significant amount of noncanonical nucleotides. Both FB9s-b and FB9s-r further exhibited similar potency, as compared with their regular RNA aptamer counterparts (Fig. 3).
Figure 4.
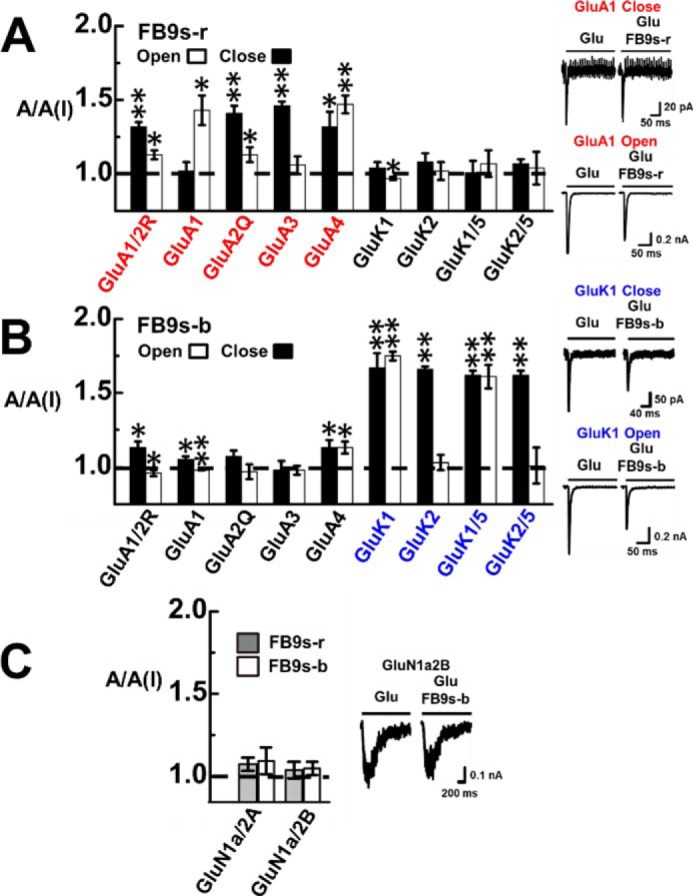
Selective inhibition of glutamate receptor subunits by FB9s-r and FB9s-b. For each 2′-F–modified aptamer, the A/A(I) ratio was collected in the presence of 2 μm aptamer. The glutamate concentrations used for the assay with these receptor subunits were the same as those for unmodified aptamers (see the legend for Fig. 3). Significance between AMPA and kainate receptors was analyzed via a one-way ANOVA. The difference between open- and closed-channel forms was determined with a two-sample, two-tailed Student's t test. Error bars indicate standard deviation from the mean, and all results are based on at least three measurements. A, FB9s-r preferentially inhibited AMPA over the kainate receptor subtype. A one-way ANOVA with post hoc Tukey HSD test analysis comparing the mean A/A(I) values for AMPA and kainate receptor subunits in the open-channel and the closed-channel conformation of FB9s-r yielded p = 0.00422 and p = 0.00527, respectively. Furthermore, from whole-cell current responses of GluA1 as a representative receptor, shown on the right, we estimated the kdes values of GluA1 to be 121 ± 17 and 125 ± 6 s−1 with and without the aptamer at low glutamate concentration. At high glutamate concentration, the kdes values of GluA1 were estimated to be 221 ± 8 and 240 ± 11 s−1 with and without aptamer FB9s-r. B, FB9s-b selectively inhibited kainate over AMPA receptors. Two pairs of representative traces are shown on the right. A one-way ANOVA with post hoc Tukey HSD test analysis comparing the mean A/A(I) values for AMPA and kainate receptor subunits in the open-channel and the closed-channel conformation of FB9s-b yielded p = 0.00136 and p < 0.00001, respectively. Furthermore, at low glutamate concentration, the kdes values of GluK1 were found to be 156 ± 16 and 146 ± 14 s−1, respectively, with and without the aptamer. At high glutamate concentration, kdes values of 202 ± 5 and 221 ± 9 s−1 were estimated, respectively, in the absence and presence of the aptamer. C, neither aptamer showed significant inhibition of NMDA channels, as shown on the right by a pair of representative whole-cell current response of GluN1a2B with and without FB9s-b; the kdes values were found to be 4 ± 0.1 and 4 ± 0.2 s−1, respectively.
Figure 5.

Determination of inhibition constant, KI, for FB9s-b on GluK2 kainate receptor (A) and FB9s-r on GluA2 AMPA receptor (B). Neither aptamer inhibited the open channel state of its respective receptor. The KI was determined from the A/A(I) plot as a function of aptamer concentration using Equations 1 and 2. Specifically, from A (left panel), the KI of FB9s-r was estimated to be 5.1 ± 0.3 μm for Glu2Q AMPA receptor (precisely, the closed-channel state). Also shown on the right is a pair of representative whole-cell current responses of GluA2Q to 0.1 mm glutamate in the presence and absence of 2 μm FB9s-r. From B (left panel), the KI of FB9s-b was determined to be 3.0 ± 0.1 μm for GluK2 kainate receptor (precisely, the closed-channel state). Shown in the right panel is a pair of representative whole-cell current responses of GluK2 to 0.05 mm glutamate in the presence and absence of 2 μm FB9s-b.
In vitro stability of FB9s-b and FB9s-r in cerebrospinal fluid and serum medium
The putative in vitro stability of FB9s-b and FB9s-r was assessed in two types of RNase-containing media, i.e. rat CSF and a serum medium, i.e. Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, which we used for cell culture. We reasoned that if these aptamers are used in vivo as potential CNS drug candidates, they would have to be exposed at least to CSF. Both CSF and serum contain ribonucleases (45). Thus, characterizing the half-life of FB9s-b and FB9s-r in both fluids would reveal their putative stability and the suitability of using these aptamers in vivo.
Using PAGE to follow an intact aptamer, we found that FB9s-r and FB9s-b both showed a t½ of ∼5 days in CSF (Fig. 6, A and B), when the temperature of the digestion reaction was held at 37 °C. In the serum-containing medium, FB9s-r and FB9s-b exhibited a similar stability, i.e. t½ ≈ 4 days (Fig. 6, C and D). As a control, unmodified RNA aptamers AB9s-b and AB9s-r, along with AB9s, were degraded virtually completely, even after just a few minutes at 37 °C in either CSF or serum-containing medium (Fig. S1). Furthermore, FB9s, the chemically modified, predecessor RNA aptamer for FB9s-r and FB9s-b, also showed a prolonged stability, as expected, with a similar t½ to FB9s-r and FB9s-b in either CSF or serum-containing cell culture medium (Fig. S2). Together, these results have shown that by replacing the 2′-OH group with 2′-F on all A, C, and U positions, while leaving all G positions unmodified, we have already significantly improved the stability of the resulting RNA aptamers or their resistance to RNase-catalyzed degradation.
Figure 6.
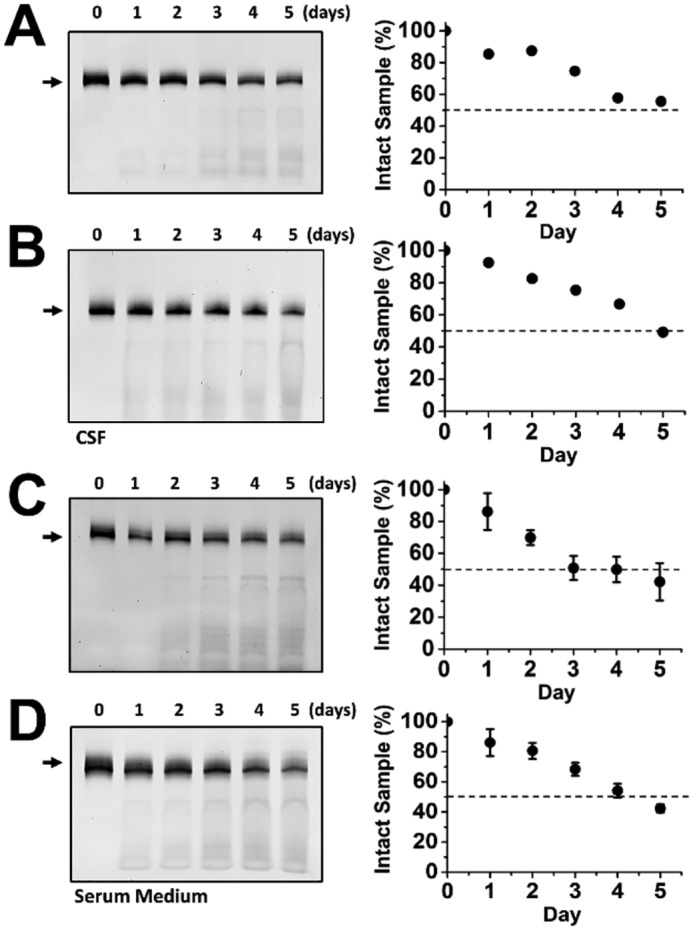
Stability of FB9s-r (A) and FB9s-b in rat CSF (B) and the stability of FB9s-r (C) and FB9s-b (D) in serum medium. The degradation reaction for an aptamer in triplicate was kept at 37 °C in rat CSF (A and B) or fetal bovine serum-containing DMEM (C and D). PAGE was used to visualize the time course of the RNase-catalyzed degradation of an aptamer sample; the arrow on the left of a representative PAGE image represents the full-length aptamer. The digitized band intensity, which was the average of the three experiments, is plotted on the right, where the time at which 50% of the sample was degraded (t½) was estimated to be ∼5 days for both FB9s-b and FB9s-r in rat CSF. In serum medium, the t½ for both FB9s-b and FB9s-r was estimated to be ∼4 days.
Discussion
Here we report two new RNA aptamers: one targets kainate receptor selectively, and the other selectively inhibits AMPA receptors. We further report that their chemically modified counterparts with a mixture of 2′-fluro pyrimidines and purines show the same potency (i.e. KI ≈ 2–3 μm) and maintains the same desired selectivity as the unmodified aptamers but with much extended stability (i.e. t½ changes from a few min to 4–5 days). Therefore, the two, 2′-F–modified aptamers are suitable for in vivo use.
In terms of improved stability due to 2′-F modification, both FB9s-b and FB9s-r have a longer t½ than Macugen (pegaptanib), a Food and Drug Administration–approved RNA aptamer drug for treatment of macular degeneration (53, 54). In serum, Macugen has a t½ of 9.3 h (55). The higher stability of our aptamers is especially noteworthy, given the fact that FB9s-r and FB9s-b still have 16 and 12 Gs or 29% and 22% unmodified positions, respectively, among the 55 nucleotides in their sequences (Fig. 1). Macugen, however, has been heavily chemically modified. Among the 27 nucleotides in Macugem, only two are unmodified or 93% of its nucleotide positions are modified, including every U and C nucleotide position, which is substituted with the corresponding 2′-F–modified analog. Furthermore, our stability results suggest that both FB9s-b and FB9s-r should be suitable for animal testing, for instance, without further modification. If necessary, the stability of the two aptamers could be further improved by, for instance, solid-state synthesis. Such a strategy can replace some or even all of the regular G positions.
A significant finding from this study is that FB9s-b not only is a potent and selective inhibitor of kainate receptors but also inhibits GluK1 and GluK2 and their respective GluK5 complex channels equally potently. Of note, developing kainate receptor-selective antagonists has been challenging, especially those antagonists that are selective to the GluK2 subunit. Over the last few decades, kainate receptor selective antagonists and agonists have been synthesized, but these agents are generally only selective to the GluK1 subunit and GluK1-containing kainate receptors (9). The utility of subunit-selective, kainate receptor antagonists has been clearly demonstrated in the case of GluK1 for its involvement in a number of neurological diseases (56–60). Therefore, FB9s-b may now be used as a potential drug candidate for these neurological diseases involving GluK2 and GluK2/GluK5.
In vivo co-application of the two aptamers, FB9s-b and FB9s-r, with varying proportions, may be also therapeutically useful. It is known that epilepsy, stroke, and amyotrophic lateral sclerosis have all been linked to excessive activity/elevated surface expression of both AMPA and kainate receptors (29, 61–64). The roles of AMPA and kainate receptors have also been identified to locate outside of the CNS, such as their expression in human arthritis tissue (65). In rat models of antigen-induced arthritis, the use of the AMPA/kainate receptor antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline (NBQX) has been shown to reduce pain, inflammation, and joint degeneration (65). Currently, most of these small molecule compounds, such as NBQX, suffer from several serious limitations, such as poor water solubility and lack of subtype and/subunit selectivity (66–68). RNA aptamers are water-soluble by nature and may provide a powerful alternative to small molecule compounds.
From RNA structure–function point of view, we can draw two conclusions based on our results from this study. First, the parent RNA molecule, namely AB9s, can be functionally decoupled to generate the AB9s-b/AB9s-r pair. That AB9s-b, which contains only a single stretch of sequence or blue sequence, possesses a similar potency on both GluK1 and GluK2, as compared with AB9s, which contains additionally the red sequence for AMPA receptors, suggests that both the blue and red sequences can function independently. Second, separate chemical modifications of the blue and red sequences have yielded FB9s-b and FB9s-r, strictly based on the corresponding sequences of AB9s-b and AB9s-r. More importantly, these two chemically modified RNA aptamers have inherited or retained the individual subtype selectivity with essentially the same potency, as compared with the corresponding, unmodified counterparts. This result is, again, consistent with the notion that the blue and the red sequences function entirely independently. In fact, FB9s, which is the chemically modified parent molecule AB9s, shows a broad inhibition of both the AMPA and the kainate receptors (Fig. S3), similar to AB9s (41). This is entirely predictable because FB9s, like AB9s, contains both the blue and red sequence motifs. Furthermore, it is assumed that the secondary structures of the blue and the red sequence motifs in the overall structural framework of AB9s are also important in retaining the functionality of the resulting chemically modified aptamers.
Based on the fact that we are apparently able to decouple the subtype selectivity of AB9s and FB9s as well without reducing their respective potencies against the intended subtypes, we predict that FB9s-b can now be used as a unique structural template in that mutations can be made within the blue sequence region alone for developing newer aptamers with potentially single subunit selectivity. In particular, we are hopeful that newer aptamers could be generated to uniquely select the GluK2 subunit without any appreciable effect on GluK1. Such an inhibitor does not exist today. Therefore, designing mutant aptamers based on the FB9s-b sequence motif could be a viable approach toward meeting that goal.
Experimental procedures
Transcription and purification of 2′-fluoro aptamers
Regular RNA aptamers and 2′-fluoro–modified aptamers were transcribed overnight at 37 °C using WT T7 RNA polymerase and a T7 RNA polymerase mutant, Y639F/H784A, respectively. 2′-ATP, 2′-CTP, and 2′-UTP used for making chemically modified aptamers were purchased from TriLink BioTechnologies. An aptamer sample was purified, using a cylindrical PAGE apparatus (43) under a denatured condition. The RNA elution was monitored at 254 nm, and the pooled fractions were concentrated using an Amicon filtration centrifuge tube (3-kDa molecular mass cutoff). The Tris–borate–EDTA acid buffer in the sample was exchanged with extracellular buffer for cell assay (see below), and the concentration of the RNA sample was determined using a Nanodrop 1000 spectrophotometer.
Cell culture, transient receptor expression, and whole-cell recording
Each of the glutamate receptors was transiently expressed in HEK-293S cells (41). The cDNAs encoding the rat GluA1–4 AMPA receptors, GluK1 and GluK2 kainate receptors, and NMDA receptors GluN1a/2A and GluN1a/2B were prepared as described (38–40). The cell line was maintained in DMEM supplemented with 10% fetal bovine serum in the presence of 1% penicillin in a 37 °C, 5% CO2, humidified incubator. The receptors were co-transfected with large T-antigen, whereas those used for recording were also co-transfected with the plasmid encoding green fluorescent protein. After 48 h, the transfected cells were used for electrophysiology.
Whole-cell current recording was used to assay the activity of an aptamer (41). The intracellular electrode solution contains 110 mm CsF, 30 mm CsCl, 4 mm NaCl, 0.5 mm CaCl2, 5 mm EGTA, and 10 mm HEPES (pH 7.4, adjusted using CsOH). The extracellular buffer contained 150 mm NaCl, 3 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 10 mm HEPES (pH 7.4). For recording of the NMDA channels, the intracellular solution contained 140 mm CsCl, 1 mm MgCl2, 0.1 mm EDTA, and 10 mm HEPES (pH 7.2 adjusted by Mg(OH)2), and the extracellular solution contained 2 μm of glycine, 135 mm NaCl, 5.4 mm KCl, 1.8 mm CaCl2, 10 mm glucose, and 5 mm HEPES (pH 7.2 adjusted by NaOH). The glutamate-induced whole-cell current was recorded using an Axopatch-200B amplifier at a cutoff frequency of 2–20 kHz, by a built-in four-pole Bessel filter. The measured current was digitized at a sampling frequency of 5–50 kHz using a Digidata 1322A instrument from Axon Instruments. All whole-cell recordings were at −60 mV and 22 °C.
Data analysis
The ratio of whole-cell current amplitude in the absence and presence of an aptamer, A/A(I), was used to plot and compare the potency and selectivity. In some instances, an apparent inhibition constant (KI,app, or simply KI) was determined as a function of aptamer concentration using (Equations 1 and 2) (41). Equations 1 and 2 were derived based on the assumption by which an inhibitor binds to one site on the receptor (41). AL2 represents the open-channel conformation. I is the concentration of the inhibitor, whereas L is the ligand concentration. Φ represents the channel opening equilibrium constant.
| (Eq. 1) |
| (Eq. 2) |
Furthermore, we measured the KI value of an aptamer for the closed- and open-channel states of an receptor by using two ligand concentrations, which corresponded ∼4% and ∼96% fractions of the open-channel state, respectively (41, 44). This is because when glutamate concentration was low (L ≪ K1), most of the receptors in the population were in the closed-channel state. In contrast, when glutamate concentration was high or saturating (L ≫ K1), the majority of the receptors were in the open-channel conformation. Thus, KI value for a respective state could be determined by varying the concentration of glutamate.
Each data point used for an A/A(I) plot was an average of at least three measurements, each of which was from an individual cell. Uncertainties refer to the standard deviation from the mean. The significance of inhibition was evaluated by a one-sample two-tailed Student's t test with the assumption that H0:μ = μ0 = 1; 1 is the theoretical value of no inhibition. An asterisk indicates p ≤ 0.05, whereas two asterisks indicate p ≤ 0.01. All analysis, including ANOVA (analysis of variance) and post hoc Tukey HSD (honestly significant difference) test were performed using software package R-Studio (version 1.0.136).
RNA digestion assay
An RNA sample (3 μg) prepared in 15 μl of 1× extracellular buffer was mixed with either 135 μl of HEK-293 cell culture medium containing 10% FBS or 135 μl of rat CSF (rat CSF was provided by Dr. Jacqueline Sagen at the University of Miami). The mixture was kept at 37 °C for the duration of the experiment. Sample aliquots were drawn at specific time points and mixed with equal volume of the loading dye composed of 95% formamide, 18 mm EDTA, 0.025% SDS, and bromphenol blue. The stability of the RNA was examined on a denaturing 10% urea-PAGE and stained with ethidium bromide. The gel was digitized, and the relative intensities of the bands were estimated.
Author contributions
W. J., Z. H., N. K., and L. N. conceptualization; W. J., Z. H., N. K., V. D. P., J. L., and L. N. data curation; W. J., Z. H., and L. N. formal analysis; W. J. and L. N. supervision; L. N. funding acquisition; W. J., Z. H., N. K., and L. N. investigation; W. J., Z. H., and L. N. methodology; W. J. and Z. H. writing-original draft; L. N. project administration; W. J. and L. N. writing and editing.
Supplementary Material
Acknowledgment
We thank Wei Wen for collecting some data.
This work was supported by National Institutes of Health/NINDS Grant R01 NS060812 and R21 NS106392 (to L. N.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
J. S. Park, Z. Huang, and L. Niu, unpublished data.
- NMDA
- N-methyl-d-aspartate
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CNS
- central nervous system
- CSF
- cerebrospinal fluid
- SELEX
- systematic evolution of ligands by exponential enrichment
- DMEM
- Dulbecco's modified Eagle's medium
- ANOVA
- analysis of variance
- HSD
- honestly significant difference.
References
- 1. Traynelis S. F., Wollmuth L. P., McBain C. J., Menniti F. S., Vance K. M., Ogden K. K., Hansen K. B., Yuan H., Myers S. J., and Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496 10.1124/pr.109.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dingledine R., Borges K., Bowie D., and Traynelis S. F. (1999) The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–61 [PubMed] [Google Scholar]
- 3. Evans A. J., Gurung S., Henley J. M., Nakamura Y., and Wilkinson K. A. (2019) Exciting times: new advances towards understanding the regulation and roles of kainate receptors. Neurochem. Res. 44, 572–584 10.1007/s11064-017-2450-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lerma J., and Marques J. M. (2013) Kainate receptors in health and disease. Neuron 80, 292–311 10.1016/j.neuron.2013.09.045 [DOI] [PubMed] [Google Scholar]
- 5. Lemoine D., Jiang R., Taly A., Chataigneau T., Specht A., and Grutter T. (2012) Ligand-gated ion channels: new insights into neurological disorders and ligand recognition. Chem. Rev. 112, 6285–6318 10.1021/cr3000829 [DOI] [PubMed] [Google Scholar]
- 6. Zhuo M. (2017) Ionotropic glutamate receptors contribute to pain transmission and chronic pain. Neuropharmacology 112, 228–234 10.1016/j.neuropharm.2016.08.014 [DOI] [PubMed] [Google Scholar]
- 7. Benussi A., Alberici A., Buratti E., Ghidoni R., Gardoni F., Di Luca M., Padovani A., and Borroni B. (2019) Toward a glutamate hypothesis of frontotemporal dementia. Front. Neurosci. 13, 304 10.3389/fnins.2019.00304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Low S. J., and Roland C. L. (2004) Review of NMDA antagonist-induced neurotoxicity and implications for clinical development. Int. J. Clin. Pharmacol. Ther. 42, 1–14 10.5414/CPP42001 [DOI] [PubMed] [Google Scholar]
- 9. Jane D. E., Lodge D., and Collingridge G. L. (2009) Kainate receptors: pharmacology, function and therapeutic potential. Neuropharmacology 56, 90–113 10.1016/j.neuropharm.2008.08.023 [DOI] [PubMed] [Google Scholar]
- 10. Contractor A., Mulle C., and Swanson G. T. (2011) Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci. 34, 154–163 10.1016/j.tins.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Egebjerg J., Bettler B., Hermans-Borgmeyer I., and Heinemann S. (1991) Cloning of a cDNA for a glutamate receptor subunit activated by kainate but not AMPA. Nature 351, 745–748 10.1038/351745a0 [DOI] [PubMed] [Google Scholar]
- 12. Herb A., Burnashev N., Werner P., Sakmann B., Wisden W., and Seeburg P. H. (1992) The KA-2 subunit of excitatory amino acid receptors shows widespread expression in brain and forms ion channels with distantly related subunits. Neuron 8, 775–785 10.1016/0896-6273(92)90098-X [DOI] [PubMed] [Google Scholar]
- 13. Werner P., Voigt M., Keinänen K., Wisden W., and Seeburg P. H. (1991) Cloning of a putative high-affinity kainate receptor expressed predominantly in hippocampal CA3 cells. Nature 351, 742–744 10.1038/351742a0 [DOI] [PubMed] [Google Scholar]
- 14. Paternain A. V., Herrera M. T., Nieto M. A., and Lerma J. (2000) GluR5 and GluR6 kainate receptor subunits coexist in hippocampal neurons and coassemble to form functional receptors. J. Neurosci. 20, 196–205 10.1523/JNEUROSCI.20-01-00196.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schiffer H. H., Swanson G. T., and Heinemann S. F. (1997) Rat GluR7 and a carboxy-terminal splice variant, GluR7b, are functional kainate receptor subunits with a low sensitivity to glutamate. Neuron 19, 1141–1146 10.1016/S0896-6273(00)80404-3 [DOI] [PubMed] [Google Scholar]
- 16. Alt A., Weiss B., Ogden A. M., Knauss J. L., Oler J., Ho K., Large T. H., and Bleakman D. (2004) Pharmacological characterization of glutamatergic agonists and antagonists at recombinant human homomeric and heteromeric kainate receptors in vitro. Neuropharmacology 46, 793–806 10.1016/j.neuropharm.2003.11.026 [DOI] [PubMed] [Google Scholar]
- 17. Møllerud S., Frydenvang K., Pickering D. S., and Kastrup J. S. (2017) Lessons from crystal structures of kainate receptors. Neuropharmacology 112, 16–28 10.1016/j.neuropharm.2016.05.014 [DOI] [PubMed] [Google Scholar]
- 18. Perrais D., Pinheiro P. S., Jane D. E., and Mulle C. (2009) Antagonism of recombinant and native GluK3-containing kainate receptors. Neuropharmacology 56, 131–140 10.1016/j.neuropharm.2008.08.002 [DOI] [PubMed] [Google Scholar]
- 19. Pallesen J., Møllerud S., Frydenvang K., Pickering D. S., Bornholdt J., Nielsen B., Pasini D., Han L., Marconi L., Kastrup J. S., and Johansen T. N. (2019) N1-substituted quinoxaline-2,3-diones as kainate receptor antagonists: X-ray crystallography, structure–affinity relationships, and in vitro pharmacology. ACS Chem. Neurosci. 10, 1841–1853 10.1021/acschemneuro.8b00726 [DOI] [PubMed] [Google Scholar]
- 20. Lauri S. E., Segerstråle M., Vesikansa A., Maingret F., Mulle C., Collingridge G. L., Isaac J. T., and Taira T. (2005) Endogenous activation of kainate receptors regulates glutamate release and network activity in the developing hippocampus. J. Neurosci. 25, 4473–4484 10.1523/JNEUROSCI.4050-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lauri S. E., Vesikansa A., Segerstråle M., Collingridge G. L., Isaac J. T., and Taira T. (2006) Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate receptor-mediated inhibition of glutamate release. Neuron 50, 415–429 10.1016/j.neuron.2006.03.020 [DOI] [PubMed] [Google Scholar]
- 22. Vesikansa A., Sakha P., Kuja-Panula J., Molchanova S., Rivera C., Huttunen H. J., Rauvala H., Taira T., and Lauri S. E. (2012) Expression of GluK1c underlies the developmental switch in presynaptic kainate receptor function. Sci. Rep. 2, 310 10.1038/srep00310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carta M., Fièvre S., Gorlewicz A., and Mulle C. (2014) Kainate receptors in the hippocampus. Eur. J. Neurosci. 39, 1835–1844 10.1111/ejn.12590 [DOI] [PubMed] [Google Scholar]
- 24. Bahn S., Volk B., and Wisden W. (1994) Kainate receptor gene expression in the developing rat brain. J. Neurosci. 14, 5525–5547 10.1523/JNEUROSCI.14-09-05525.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Porter R. H., Eastwood S. L., and Harrison P. J. (1997) Distribution of kainate receptor subunit mRNAs in human hippocampus, neocortex and cerebellum, and bilateral reduction of hippocampal GluR6 and KA2 transcripts in schizophrenia. Brain Res. 751, 217–231 10.1016/S0006-8993(96)01404-7 [DOI] [PubMed] [Google Scholar]
- 26. Greenwood T. A., Lazzeroni L. C., Calkins M. E., Freedman R., Green M. F., Gur R. E., Gur R. C., Light G. A., Nuechterlein K. H., Olincy A., Radant A. D., Seidman L. J., Siever L. J., Silverman J. M., Stone W. S., et al. (2016) Genetic assessment of additional endophenotypes from the Consortium on the Genetics of Schizophrenia Family Study. Schizophr. Res. 170, 30–40 10.1016/j.schres.2015.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Das A., Wallace G. C. 4th, Holmes C., McDowell M. L., Smith J. A., Marshall J. D., Bonilha L., Edwards J. C., Glazier S. S., Ray S. K., and Banik N. L. (2012) Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 220, 237–246 10.1016/j.neuroscience.2012.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Melyan Z., Lancaster B., and Wheal H. V. (2004) Metabotropic regulation of intrinsic excitability by synaptic activation of kainate receptors. J. Neurosci. 24, 4530–4534 10.1523/JNEUROSCI.5356-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mulle C., Sailer A., Pérez-Otaño I., Dickinson-Anson H., Castillo P. E., Bureau I., Maron C., Gage F. H., Mann J. R., Bettler B., and Heinemann S. F. (1998) Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature 392, 601–605 10.1038/33408 [DOI] [PubMed] [Google Scholar]
- 30. Ben-Ari Y. (2012) Kainate and Temporal Lobe Epilepsies: 3 decades of progress. In Jasper's Basic Mechanisms of the Epilepsies (Noebels J. L., Avoli M., Rogawski M. A., Olsen R. W., and Delgado-Escueta A. V., eds) 4th Ed., National Center for Biotechnology Information, Bethesda, MD: [PubMed] [Google Scholar]
- 31. Gratacos M., Costas J., de Cid R., Bayes M., Gonzalez J. R., Baca-Garcia E., de Diego Y., Fernandez-Aranda F., Fernandez-Piqueras J., Guitart M., Martin-Santos R., Martorell L., Menchon J. M., Roca M., Saiz-Ruiz J., et al. (2009) Identification of new putative susceptibility genes for several psychiatric disorders by association analysis of regulatory and non-synonymous SNPs of 306 genes involved in neurotransmission and neurodevelopment. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 808–816 10.1002/ajmg.b.30902 [DOI] [PubMed] [Google Scholar]
- 32. Mayer M. L., Ghosal A., Dolman N. P., and Jane D. E. (2006) Crystal structures of the kainate receptor GluR5 ligand binding core dimer with novel GluR5-selective antagonists. J. Neurosci. 26, 2852–2861 10.1523/JNEUROSCI.0123-06.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ellington A. D., and Szostak J. W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822 10.1038/346818a0 [DOI] [PubMed] [Google Scholar]
- 34. Tuerk C., and Gold L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510 10.1126/science.2200121 [DOI] [PubMed] [Google Scholar]
- 35. Huang Z., and Niu L. (2019) Developing RNA aptamers for potential treatment of neurological diseases. Future Med. Chem. 11, 551–565 10.4155/fmc-2018-0364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kaur H., Bruno J. G., Kumar A., and Sharma T. K. (2018) Aptamers in the therapeutics and diagnostics pipelines. Theranostics 8, 4016–4032 10.7150/thno.25958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Y., Lai B. S., and Juhas M. (2019) Recent advances in aptamer discovery and applications. Molecules 24, E941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang Z., Han Y., Wang C., and Niu L. (2010) Potent and selective inhibition of the open-channel conformation of AMPA receptors by an RNA aptamer. Biochemistry 49, 5790–5798 10.1021/bi100690k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang Z., Pei W., Jayaseelan S., Shi H., and Niu L. (2007) RNA aptamers selected against the GluR2 glutamate receptor channel. Biochemistry 46, 12648–12655 10.1021/bi701036p [DOI] [PubMed] [Google Scholar]
- 40. Park J. S., Wang C., Han Y., Huang Z., and Niu L. (2011) Potent and selective inhibition of a single alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subunit by an RNA aptamer. J. Biol. Chem. 286, 15608–15617 10.1074/jbc.M111.229559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jaremko W. J., Huang Z., Wen W., Wu A., Karl N., and Niu L. (2017) Identification and characterization of RNA aptamers: a long aptamer blocks the AMPA receptor and a short aptamer blocks both AMPA and kainate receptors. J. Biol. Chem. 292, 7338–7347 10.1074/jbc.M116.774752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang Z., Jayaseelan S., Hebert J., Seo H., and Niu L. (2013) Single-nucleotide resolution of RNAs up to 59 nucleotides by high-performance liquid chromatography. Anal. Biochem. 435, 35–43 10.1016/j.ab.2012.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li G., and Niu L. (2004) How fast does the GluR1Qflip channel open? J. Biol. Chem. 279, 3990–3997 10.1074/jbc.M310410200 [DOI] [PubMed] [Google Scholar]
- 45. Schieven G. L., Blank A., and Dekker C. A. (1982) Ribonucleases of human cerebrospinal fluid: detection of altered glycosylation relative to their serum counterparts. Biochemistry 21, 5148–5155 10.1021/bi00264a007 [DOI] [PubMed] [Google Scholar]
- 46. Zhou J., and Rossi J. (2017) Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov. 16, 181–202 10.1038/nrd.2016.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pieken W. A., Olsen D. B., Benseler F., Aurup H., and Eckstein F. (1991) Kinetic characterization of ribonuclease-resistant 2′-modified hammerhead ribozymes. Science 253, 314–317 10.1126/science.1857967 [DOI] [PubMed] [Google Scholar]
- 48. Cummins L. L., Owens S. R., Risen L. M., Lesnik E. A., Freier S. M., McGee D., Guinosso C. J., and Cook P. D. (1995) Characterization of fully 2′-modified oligoribonucleotide hetero- and homoduplex hybridization and nuclease sensitivity. Nucleic Acids Res. 23, 2019–2024 10.1093/nar/23.11.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aurup H., Williams D. M., and Eckstein F. (1992) 2′-Fluoro- and 2′-amino-2′-deoxynucleoside 5′-triphosphates as substrates for T7 RNA polymerase. Biochemistry 31, 9636–9641 10.1021/bi00155a016 [DOI] [PubMed] [Google Scholar]
- 50. Breslow R., and Chapman W. H. Jr. (1996) On the mechanism of action of ribonuclease A: relevance of enzymatic studies with a p-nitrophenylphosphate ester and a thiophosphate ester. Proc. Natl. Acad. Sci. U.S.A. 93, 10018–10021 10.1073/pnas.93.19.10018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fattal E., and Bochot A. (2006) Antisense oligonucleotides, aptamers and siRNA: promises for the treatment of ocular diseases. Arch. Soc. Esp. Oftalmol. 81, 3–6 [PubMed] [Google Scholar]
- 52. Cazenave C., and Uhlenbeck O. C. (1994) RNA template-directed RNA synthesis by T7 RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 91, 6972–6976 10.1073/pnas.91.15.6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ruckman J., Green L. S., Beeson J., Waugh S., Gillette W. L., Henninger D. D., Claesson-Welsh L., and Janjic N. (1998) 2′-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 273, 20556–20567 10.1074/jbc.273.32.20556 [DOI] [PubMed] [Google Scholar]
- 54. Bell C., Lynam E., Landfair D. J., Janjic N., and Wiles M. E. (1999) Oligonucleotide NX1838 inhibits VEGF165-mediated cellular responses in vitro. In Vitro Cell Dev. Biol. Anim. 35, 533–542 10.1007/s11626-999-0064-y [DOI] [PubMed] [Google Scholar]
- 55. Tucker C. E., Chen L. S., Judkins M. B., Farmer J. A., Gill S. C., and Drolet D. W. (1999) Detection and plasma pharmacokinetics of an anti-vascular endothelial growth factor oligonucleotide-aptamer (NX1838) in rhesus monkeys. J. Chromatogr. B Biomed. Sci. Appl. 732, 203–212 10.1016/S0378-4347(99)00285-6 [DOI] [PubMed] [Google Scholar]
- 56. Gill R. (1994) The pharmacology of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA)/kainate antagonists and their role in cerebral ischaemia. Cerebrovasc. Brain Metab. Rev. 6, 225–256 [PubMed] [Google Scholar]
- 57. O'Neill M. J., Bogaert L., Hicks C. A., Bond A., Ward M. A., Ebinger G., Ornstein P. L., Michotte Y., and Lodge D. (2000) LY377770, a novel iGlu5 kainate receptor antagonist with neuroprotective effects in global and focal cerebral ischaemia. Neuropharmacology 39, 1575–1588 10.1016/S0028-3908(99)00250-6 [DOI] [PubMed] [Google Scholar]
- 58. Smolders I., Bortolotto Z. A., Clarke V. R., Warre R., Khan G. M., O'Neill M. J., Ornstein P. L., Bleakman D., Ogden A., Weiss B., Stables J. P., Ho K. H., Ebinger G., Collingridge G. L., Lodge D., et al. (2002) Antagonists of GLU(K5)-containing kainate receptors prevent pilocarpine-induced limbic seizures. Nat. Neurosci. 5, 796–804 10.1038/nn880 [DOI] [PubMed] [Google Scholar]
- 59. Braga M. F., Aroniadou-Anderjaska V., Li H., and Rogawski M. A. (2009) Topiramate reduces excitability in the basolateral amygdala by selectively inhibiting GluK1 (GluR5) kainate receptors on interneurons and positively modulating GABAA receptors on principal neurons. J. Pharmacol. Exp. Ther. 330, 558–566 10.1124/jpet.109.153908 [DOI] [PubMed] [Google Scholar]
- 60. Bhangoo S. K., and Swanson G. T. (2013) Kainate receptor signaling in pain pathways. Mol. Pharmacol. 83, 307–315 10.1124/mol.112.081398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Borbély S., Czégé D., Molnár E., Dobó E., Mihály A., and Világi I. (2015) Repeated application of 4-aminopyridine provoke an increase in entorhinal cortex excitability and rearrange AMPA and kainate receptors. Neurotox Res 27, 441–452 10.1007/s12640-014-9515-7 [DOI] [PubMed] [Google Scholar]
- 62. Nadler J. V. (2003) The recurrent mossy fiber pathway of the epileptic brain. Neurochem. Res. 28, 1649–1658 10.1023/A:1026004904199 [DOI] [PubMed] [Google Scholar]
- 63. Represa A., Tremblay E., and Ben-Ari Y. (1987) Kainate binding sites in the hippocampal mossy fibers: localization and plasticity. Neuroscience 20, 739–748 10.1016/0306-4522(87)90237-5 [DOI] [PubMed] [Google Scholar]
- 64. Sutula T. P., and Dudek F. E. (2007) Unmasking recurrent excitation generated by mossy fiber sprouting in the epileptic dentate gyrus: an emergent property of a complex system. Prog. Brain Res. 163, 541–563 10.1016/S0079-6123(07)63029-5 [DOI] [PubMed] [Google Scholar]
- 65. Bonnet C. S., Williams A. S., Gilbert S. J., Harvey A. K., Evans B. A., and Mason D. J. (2015) AMPA/kainate glutamate receptors contribute to inflammation, degeneration and pain related behaviour in inflammatory stages of arthritis. Ann. Rheum. Dis. 74, 242–251 10.1136/annrheumdis-2013-203670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nussinov R., and Tsai C. J. (2012) The different ways through which specificity works in orthosteric and allosteric drugs. Curr. Pharm. Des. 18, 1311–1316 10.2174/138161212799436377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Waterhouse R. N. (2003) Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 5, 376–389 10.1016/j.mibio.2003.09.014 [DOI] [PubMed] [Google Scholar]
- 68. Weiser T. (2005) AMPA receptor antagonists for the treatment of stroke. Curr. Drug Targets CNS Neurol. Disord. 4, 153–159 10.2174/1568007053544129 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.