

Abstract
Background
Lysosomes digest extracellular material from the endocytic pathway and intracellular material from the autophagic pathway. This process is performed by the resident hydrolytic enzymes activated by the highly acidic pH within the lysosomal lumen. Lysosome pH gradients are mainly maintained by the vacuolar (H+) ATPase (or V-ATPase), which pumps protons into lysosomal lumen by consuming ATP. Dysfunction of V-ATPase affects lysosomal acidification, which disrupts the clearance of substrates and leads to many disorders, including neurodegenerative diseases.
Main body
As a large multi-subunit complex, the V-ATPase is composed of an integral membrane V0 domain involved in proton translocation and a peripheral V1 domain catalyzing ATP hydrolysis. The canonical functions of V-ATPase rely on its H+-pumping ability in multiple vesicle organelles to regulate endocytic traffic, protein processing and degradation, synaptic vesicle loading, and coupled transport. The other non-canonical effects of the V-ATPase that are not readily attributable to its proton-pumping activity include membrane fusion, pH sensing, amino-acid-induced activation of mTORC1, and scaffolding for protein-protein interaction. In response to various stimuli, V-ATPase complex can reversibly dissociate into V1 and V0 domains and thus close ATP-dependent proton transport. Dysregulation of pH and lysosomal dysfunction have been linked to many human diseases, including neurodegenerative disorders such as Alzheimer disease, Parkinson’s disease, amyotrophic lateral sclerosis as well as neurodegenerative lysosomal storage disorders.
Conclusion
V-ATPase complex is a universal proton pump and plays an important role in lysosome acidification in all types of cells. Since V-ATPase dysfunction contributes to the pathogenesis of multiple neurodegenerative diseases, further understanding the mechanisms that regulate the canonical and non-canonical functions of V-ATPase will reveal molecular details of disease process and help assess V-ATPase or molecules related to its regulation as therapeutic targets.
Keywords: Vacuolar-type ATPase, Lysosomal acidification, Neurodegeneration, Autophagy
Background
Lysosomes are the primary degradative compartment of the cell that executes the final destruction and recycling of organelles and molecules. Extracellular materials are delivered to the lysosomes via the endocytic pathway while intracellular materials reach the organelle by the autophagic process [1]. Since the discovery of lysosome by Belgian biochemist Christian de Duve [2], significant progress has been made about the function of the organelle. We now know that lysosomes have diverse functions including turnover of cellular molecules, nutrient sensing, regulation of receptor distribution, antigen presentation, and response to cellular stress etc. [3–5].
The degradative function of the lysosomes is carried out by the resident hydrolytic enzymes within their lumen. These enzymes are activated by highly acidic pH (less than pH 5.0) in the luminal compartment. The acidic pH is primarily mainly maintained by a proton pump, the V-type proton ATPase (V-ATPase), which pumps protons into the lysosomal lumen, a process that requires the consumption of ATP [6]. Dysregulated acidification and increased intraluminal pH suppress the activities of various enzymes in lysosome and disrupt the clearance of various substrates including protein aggregates. Thus, it is not surprising that conditions impairing lysosomal acidification may contribute to a range of diseases, many of which are severe or life threatening. The most well-known diseases related to lysosomes are lysosomal storage disorders (LSDs) [7], a family of disorders caused by inherited gene mutations of lysosomal and non-lysosomal proteins, including the resident hydrolytic enzymes inside the lysosome [8]. The deficiencies of those lysosomal enzymes lead to defective processing of substrates critical for cell survival and the abnormal storage of macromolecular substrates that can cause further lysosomal damage [8]. The resulting abnormal accumulation of protein aggregates are the hallmarks of multiple major neurodegenerative diseases including Alzheimer’s Disease (AD) and Parkinson’s Disease (PD) etc. [9–12]. In this review, we will first introduce the general features of lysosome in neurons, describe the structure and functions of V-ATPase, and highlight the evidence linking deficiencies of V-ATPase components and neurodegenerative diseases.
Main text
Lysosome function
Lysosomes degrade a broad range of substrates including DNA, RNA, proteins, and lipid. The size of those substrates varies from single molecule to a whole organelle. Lysosomes, as the final site of destruction, accept and degrade substrate proteins from several distinct processes, including the secretory, endocytic and autophagic pathways. For example, excess secretory granules containing insulin could fuse with lysosomes through a process called crinophagy in which the secretory proteins located in the secretory vesicles are degraded in β-cell [13, 14]. During developmentally programmed crinophagy in Drosophila, the excessive secretory glue granules fuse with lysosomes, and the degradation of secretory material depends on the activity of the Uvrag-containing Vps34 lipid kinase complex and the V-ATPase proton (H+) pump [15]. In eukaryotic cells, extracellular macromolecules can be taken up by cells through clathrin-mediated endocytosis. The extracellular macromolecules or ligands bind to cell surface receptors that are concentrated in clathrin-coated pits. The pits then bud from the plasma membrane to form intracellular clathrin-coated vesicles, which are sorted by the early endosome system. Substrates are finally delivered to the lysosomes for degradation [16]. This process regulates the interaction of cells with their environment and provides cells with energy sources such as amino acids, carbohydrates, and lipids as well as substrates utilized for biosynthesis [17]. Endocytic and lysosomal pathway is fundamental to neurotransmission, signal transduction and the regulation of many plasma membrane activities [16, 18]. In contrast, autophagy targets intracellular cytosolic components such as long-lived proteins, protein aggregates, stress RNA granules, and organelles including mitochondria for degradation by lysosomes, and is essential for maintaining cellular homeostasis. There are three defined forms of autophagy, macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [13, 19, 20]. Macroautophagy involves wrapping the substrates in the cytosolic compartment into double-membrane vesicles named autophagosomes, transporting autophagosomes and fusing them with lysosomes for degradation [21]. Microautophagy is less well characterized but involves the internalization of cytosolic substrate proteins through invagination of the lysosomal membrane [13]. Microautophagy-like process that delivers soluble cytosolic proteins to the late endosomes/multivesicular bodies (MVBs) has been reported [22]. CMA is a unique form of autophagy that selectively degrades individual proteins. This involves the binding of chaperone Hsc70 to the KFERQ-like pentapeptide motif in substrate proteins [23], interaction of Hsc70/substrate complex with the lysosome-associated membrane protein type 2A (LAMP-2A) on the lysosome, and the subsequent translocation of the substrate proteins into the lysosomal lumen for degradation [24, 25].
Lysosomes need to maintain a highly acidic pH in its lumen in order to successfully perform its digestive function and to drive efflux of digested materials [6]. The acidic environment of the lysosome is critical not only for the function of lysosomes themselves but also for many cellular processes related to lysosomes. One of the most important roles involving the acidic pH is to guarantee the delivery of various enzymes to lysosomes and their proper maturation inside the organelle. The newly synthesized enzymes such as pro-form of cathepsin D are first transported to the late endosomes via a series of steps and the acidic environment of the endosomes is needed for its dissociation from mannose-6-phosphate receptor involved in its proper transportation [26]. Maturation of lysosomal enzymes, which involves sequential cleavage of immature pro-forms to mature forms of hydrolases, depends on the acidification of lysosome [27]. Similarly, normal dissociation of cargoes from their carrier proteins, which is essential for many cellular processes, also requires the acidic environment of lysosome. For example, the dissociation of cholesterol and other lipids from their receptors occurs inside the acidic environment of lysosome [28]. Ferritin is a cytosolic protein that stores excess iron and protects cells from iron toxicity [29]. Lysosomal acidification is crucial for the extraction of iron from ferritin for its degradation in lysosomes [30]. Inhibition of lysosomal acidification triggers cellular iron deficiency, which can cause neuronal stress by impairing mitochondrial function and triggering inflammation in cultured primary neurons [31]. The lysosomes constantly fuse with other organelles including endosomes and autophagsomes. The endosomal and lysosomal lumen pH varies between a range of 4.5 to 6.5 due to the activity of the ATP-dependent proton pumps present on their membrane. This unique acidic environment is for the sequential activation of hydrolases, uncoupling ligands from receptors, inactivation of microbicidal factors, cargo transportation, membrane trafficking, and efficient degradation of various substrates [9, 32]. Dysregulation of the endosomal/autophagosome-lysosomal system has been implicated in several human diseases including neurodegenerative diseases (discuss later) as well as atherosclerosis [33].
Lysosomes in neurons
The brain has abundant amount of lysosomes and this lysosomal network offers neurons a constitutively and highly efficient degradative system [34]. Lysosome-mediated continuous clearance of cellular proteins through autophagy is important for maintaining neuronal homeostasis. This basal process is essential since deficiency of autophagic genes causes the accumulation of intracellular proteins and inclusion bodies, and ultimately neurodegeneration [35, 36]. Disruption of cathepsin- mediated proteolysis leads to marked accumulation of autophagosome in the brain, which resembles the vesicles in the brain of AD patients and model mice [34]. Since neurons are postmitotic and polarized structurally, they are particularly sensitive to the impairment of lysosomal function [37]. The lysosomal system in neurons is easily affected when lysosomal hydrolysis is impaired, leading to rapid accumulation of the materials bound for degradation [38–40]. Thus, abnormal or defective lysosomal function affects many aspects of neuronal processes and has been identified as a critical factor contributing to or even driving the pathogenic process of many neurodegenerative diseases, including AD, PD, and Huntington’s disease (HD) etc. [41–45]. It’s worth noting that multiple lysosomal functions including its proteolytic activity decline with aging [46]. Aging itself is the biggest risk factor for AD and PD. Therefore, it is possible that part of the increased risk for AD and PD associated with aging is due to the decline of lysosomal activity and the consequential accumulation of toxic materials including damaged organelles and proteins. Thus, aging and genetic defects converge on the lysosomes, whose dysfunction may participate in not only the initiation but also the acceleration of diverse neurodegenerative processes.
The involvement of lysosomes in various neuronal processes requires and depends on their ability to move throughout the cytoplasm [47]. Indeed, it has long been recognized that lysosomes in neurons are not distributed evenly but in a polarized manner. They are particularly abundant in neuronal cell body. Fewer are present in dendrites and they are even more rare in axons [48–50]. This indicates that lysosomes in neurons are separated into different compartments to match the degradative needs of various compartments and a large portion of the final degradation mainly occurs in the cell body. Neurons are challenged to deliver unwanted components from the periphery compartments to the cell body along the long distance of axons or dendrites. In support of this, disruption of the transportation system in neurons with microtubules depolymerization compounds leads clearly to rapid autophagic vacuoles accumulation [34]. Such polarized distribution of lysosomes suggests that neurons have different mechanisms to control the movement of lysosomes in neurons [51–54].
V-ATPase and its structure
There are two related families of (H+) ATPases: the family of F-type proton-translocating ATPases (F-ATPases) and the family of vacuolar (H+) ATPase (V-ATPase). The two families of ATPase are evolutionarily related and share structural similarities [55]. One enzymatic difference between the two families of ATPase is that F-ATPase is involved in synthesizing as well as hydrolyzing ATP [56] while V-ATPase seems to function mainly in proton pumping by consuming ATP [57]. The F-ATPases are found in bacterial plasma membrane, mitochondrial inner membrane and in chloroplast thylakoid membrane [58, 59]. In addition to being present on the plasma membrane of certain eukaryotic cells, V-ATPase is widely distributed on the membrane of various subcellular organelles, such as lysosome, Golgi, and secretory granules. Each organelle maintains a characteristic internal pH, which is essential for facilitating organelle function, such as endocytosis, exocytosis, membrane flow and substance transport. The V-ATPase is the main driving force of the acid pH of the vacuolar system in eukaryotic cells. It pumps protons into the lumen of an organelle using the energy generated by ATP hydrolysis.
The V-ATPases are large multi-subunit complexes composed of two domains: a membrane integral V0 sector involved in proton translocation and a peripheral V1 domain catalyzing ATP hydrolysis. As shown at Fig. 1, the integral V0 domain is a 260 kDa complex containing six different subunits (a, c, c’, c”, d, and e) whereas the V1 domain is a 640 kDa complex including eight different subunits (A, B, C, D, E, F, G, and H) [60, 61]. In higher eukaryotes, several different H+-ATPase subunits have multiple isoforms encoded by separate genes that are located throughout the genome with differing tissue expression patterns [62–64]. For example, there are two isoforms for the B, E, d, and e subunits; three for the C and G subunits; and four for the a subunit. Some of the isoforms have different expression patterns in various tissues. For example, the d1 subunit is ubiquitously expressed while the d2 homolog is expressed only in the kidney, osteoclast and lung [65]. Similarly, the G1 isoform is expressed ubiquitously while G2 and G3 isoforms are found mainly in neuronal tissue and kidney [66]. Some subunits are encoded through splice variants, such as the a, d, e, C, G and H subunits [67–71]. This complexity leads to different possible permutations of subunit structure in individual proton pumps and unique subunit identities of pumps at different locations, which may allow the regulation of V-ATPase in a cell type and subcellular compartment specific manner. The peripheral domain and the integral domain are assembled separately and brought together into a functional proton pump at the required organelles [72]. In neuronal and neuroendocrine cells, the V-ATPase is equipped with the accessory subunits ATP6AP1 and ATP6AP2. ATP6AP1, also known as Ac45, was first identified as the accessory subunit of V-ATPase in chromaffin granules [73]. ATP6AP1 functions to guide the V-ATPase to certain subcellular compartments such as neuroendocrine regulated secretory vesicles and regulates their activity [74], the intragranular pH and Ca2+-dependent exocytotic membrane fusion [75]. ATP6AP2 was first identified as the C-terminal fragment of the (pro) renin receptor (PRR) for renin and prorenin [76]. Ablation of PRR in cardiomyocytes reveals that PRR is an integral component for the stability and assembly of V0 subunits [77]. ATP6AP2 is a key accessory protein for V-ATPase functions in the CNS and essential for stem cell self-renewal and neuronal survival [78].
Fig. 1.
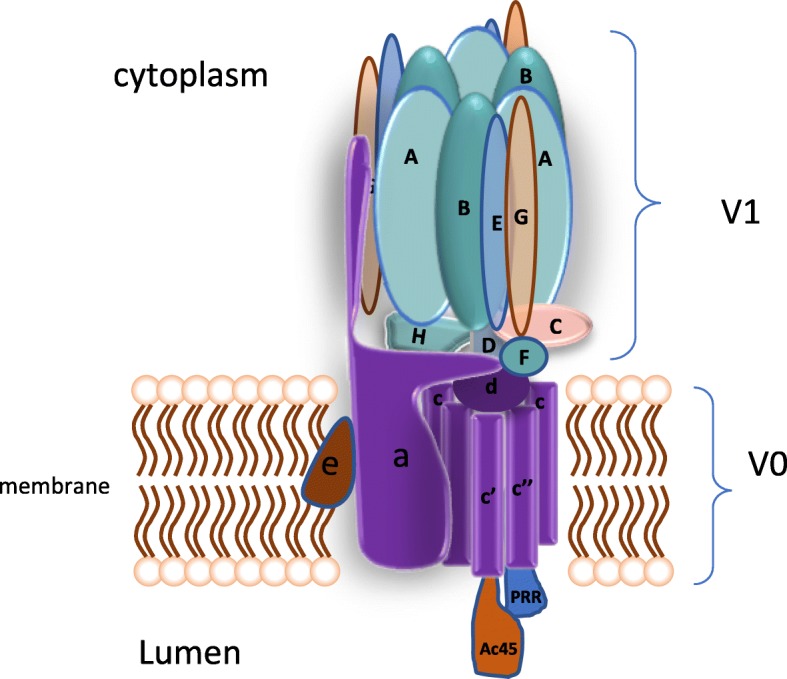
Structure of V-ATPases. V-ATPase is composed of multiple core subunits and two accessory proteins. The cytosolic V1 domain includes eight different subunits (A, B, C, D, E, F, G, and H) and the integral V0 domain contains six different subunits (a, c, c’, c”, d, and e). The hypothetic positions of the ATP6AP1/Ac45 and ATP6AP2/PRR are included
Regulation of V-ATPase
V-ATPase’s proton pumping activity plays a vital role in numerous essential cellular processes such as pH and ion homeostasis, protein trafficking, autophagy, endocytosis, signaling, and neurotransmitter release [17, 79–82]. The activity of V-ATPase is regulated by multiple mechanisms. First, the vesicle V-ATPase activity is regulated by the formation of intramolecular disulfide bonds and this is modulated by ATP or high ionic strength [83]. Studies have shown that formation and cleavage of a disulfide bond between cysteine residues at or near the catalytic site of the enzyme [84], such as the intramolecular disulfide bond formed between cysteine 254 and cysteine 532 in subunit A of the bovine V-ATPase, result in the inactivation of the enzyme [85]. A study of plant V-ATPase shows that E subunit can also form intramolecular disulfide bonds that change its conformation [86].
Regulation of V-ATPase can be achieved by the control of the abundance of V-ATPase or some of its subunits as well as their localization on the membrane [87]. For example, the level of E1 subunit increases in the medulla of kidneys from acetazolamide-treated rats [88]. The cytoplasmic membrane and intracellular vesicles containing high-density V-ATPase membrane may fuse, which leads to the adjustment of proton pump density [89]. Several specialized cells such as interdental cells of the ear, epithelial cells of the nose, vision or proximal tubule of kidney also express V-ATPase at their plasma membrane, which is involved in extracellular pH regulation [90–93]. The localization of V-ATPase to plasma membrane is controlled through reversible exocytosis and endocytosis of vesicles that contain a high density of V-ATPase [94].
In response to various stimuli, V-ATPase complexes can reversibly dissociate into V1 and V0 domains and thus close ATP-dependent proton transport. In yeast, reversible dissociation occurs rapidly in response to glucose depletion. Glucose depletion even for as little as 5 min causes dissociation of approximately 70% of the assembled V-ATPase enzyme complex into separate V1 and V0 subcomplexes. Restoration of glucose induces rapid and efficient reassembly of the enzyme from the previously synthesized subcomplexes [95]. Assembly requires the glycolytic enzyme aldolase and RAVE complex (regulator of H+-ATPase of vacuolar and endosomal membranes) [96, 97], while dissociation, which is independent of new protein synthesis, requires intact microtubules [98]. Rather than leading to disassembly, studies in mammalian cells have demonstrated that acute glucose depletion can increase V-ATPase assembly and activity through AMPK and PI3K-Akt signaling pathway [99, 100]. Amino acid starvation has been shown to promote V-ATPase assembly and re-addition of amino acid reverses this effect [101]. These findings highlight the importance of reversible dissociation of V1-V0 complexes as a common mechanism in efficiently regulating V-ATPase in response to signals.
V-ATPase acidifies the newly formed synaptic vesicles to generate a proton electrochemical gradient that drives neurotransmitter loading and clathrin-mediated endocytosis [102]. Recently, Milosevic et al. proposed a model of regulation of synaptic vesicle V-ATPase. It is thought that the activity of V-ATPase in the clathrin-coated vesicles (CCVs) is inhibited by the clathrin coat. When the coat is removed from CCVs, V-ATPase becomes functional and acidifies the uncoated vesicles [103]. This regulation of V-ATPase activity may be an important mechanism for the proper timing of synaptic vesicles refilling.
Role of V-ATPase in the nervous system
V-ATPases have multiple cellular functions at different intracellular sites. The main functions of V-ATPase rely on its H+-pumping ability in multiple membrane organelles such as lysosome, endosome, and synaptic vesicles. Through this, V-ATPase modulates a range of cellular activities including endocytic traffic, protein processing and degradation, synaptic vesicles loading and coupled transport [82, 104–106]. There are also other non-canonical effects of the V-ATPase that are not readily attributable to its proton-moving activity. This includes membrane fusion, pH sensing, amino-acid-induced activation of mammalian target of rapamycin complex1 (mTORC1), and scaffold for protein-protein interactions, etc. [5, 107, 108]. Some of the main functions of V-ATPase in neurons are summarized in Fig. 2.
Fig. 2.

Function of V-ATPase in the nervous system. In presynaptic bouton, V-ATPase is responsible for generating the H+-electrochemical gradient in synaptic vesicles, which drives the refilling of newly formed synaptic vesicles with neurotransmitter. Synaptic vesicle V-ATPase also participates in the step of fusion. Relying on its H+-pumping ability, V-ATPase modulates multiple cellular activities including endosome maturation and trafficking, protein processing and degradation via different autophagic pathways in multiple vesicle organelles such as lysosome and endosome. The acidic environment of the lysosomes is critical for not only the function of lysosomes but also many cellular processes related to lysosomes. V-ATPase is also involved in pH sensing, nutrient signaling, and scaffold for protein-protein interactions
Synaptic vesicle loading and coupled transport
It is known that V-ATPase generates the H+-electrochemical gradient in synaptic vesicles and this gradient is necessary to drive the refilling of newly formed synaptic vesicles with neurotransmitter [109]. Some vesicular transporters such as monoamine transporters utilize the H+-concentration gradient (ΔpH) whereas glutamate transporters rely primarily on the electrical potential (Δψ) generated by the V-ATPase [110]. The uptake of the positively charged acetylcholine and γ-aminobutyric acid (GABA) rely on both ΔpH and Δψ. The synaptic vesicles with neurotransmitter loaded undergo reversible tethering at the plasma membrane where they fuse and release the neurotransmitters into the synaptic space. Synaptic vesicle V-ATPase also participates in the step of fusion (discussed below).
Endosome maturation and trafficking
Defects in membrane trafficking and degradation result in the accumulation of undegraded proteins due to aberrant endosomal sorting, lysosomal degradation, or autophagy. Alterations of membrane trafficking are known to occur in AD, other tauopathies, PD, peripheral neuropathies, lysosomal storage disorders and PolyQ diseases [111]. The luminal pH is an important aspect of endosome maturation and function. Blocking V-ATPase function by pharmacological inhibitors or by siRNA leads to the accumulation of materials in the early endosomes and inhibits subsequent endocytosis [104, 105]. V-ATPase deficiency results in the formation of large hybrid organelles containing markers of immature lysosomes, granules, and autophagy, affecting the pathways of degradation and secretion [112]. These studies suggest that normal pH, which is dependent on V-ATPase-mediated acidification, is essential for sorting function of the endosomal system and subsequent endosome-lysosome processing.
pH acidification and sensing
Tight control of pH homeostasis is essential for a vast number of physiological processes of several organelles. For example, the pH of early endosomes (EEs) is between 6.8–6.1 range and the pH of late endosomes (LEs) is around 5.5. pH inside lysosome reaches the values of 4.5–4.7. The acidic pH of endocytic organelles not only provides the environment for the activation of the degradative enzymes but also is essential for membrane trafficking and sorting endosomes to promote the dissociation of internalized ligand–receptor complexes during receptor-mediated endocytosis. This process also plays a role in the inactivation of internalized pathogens [60, 113]. Recently, V-ATPase has been reported to serve as a pH sensor of endosomes in renal tubule cells [107]. The study shows that the small GTPase Arf6 and its cognate GDP/GTP exchange factor (GEF) ARNO (ADP-ribosylation factor nucleotide site opener) directly interact with the c-subunit and the a2-subunit of V-ATPase, respectively. The interaction of a2-isoform and ARNO is dependent on the intra-endosomal acidification, and disruption of this interaction inhibits endocytosis. These results suggest that the subunits of V-ATPase may perform additional functions in a luminal pH dependent manner.
Membrane fusion
Recent studies suggest that the transmembrane V0 domain of V-ATPase promotes membrane fusion independent of its role in acidification. The direct involvement of V0 domain of the V-ATPase in the process of membrane fusion comes from the studies of fusion in yeast [114]. Mutation of genes that encode subunits of the V0 domain or using inhibitory antibodies has a more significant effect on yeast vacuolar fusion than inhibition of V1 domain [115]. SNARE (soluble N-ethylmaleimide- sensitive factor attachment protein receptor) mediates the attachment step in the membrane fusion of vesicles. The V0 is involved in forming complexes between opposing membranes. After the trans-SNARE pairing, V0 trans-complex forms dependent on both Rab-GTPase Ypt7 and calmodulin [114]. Several studies from the nervous system and cells also reveal that V-ATPase is required for membrane fusion during exocytosis of synaptic and secretory vesicles [108, 116, 117]. In Drosophila melanogaster, a neuronal isoform of a subunit of V0 component, vha 100–1, is shown to colocalize with synaptic vesicles and active zones. Mutation of vha 100–1 results in accumulation of synaptic vesicles in synaptic terminals with normal levels of neurotransmitter loading [108], indicating the blocking of synaptic vesicle fusion with the presynaptic membrane. Mutation in the V0a subunit of Caenorhabditis elegans blocks the secretion of Hedgehod-related proteins through apical secretion without affecting vesicle acidification [118]. Similarly, deletion of the a3 isoform of V-ATPase impairs secretion of insulin from pancreatic β-cell without significant alternation in the pH of the secretory vesicle [117]. It should be noted that the role of V0 domain in membrane fusion has not been completely resolved. Although the above results suggest that V-ATPase has a role in membrane fusion independent of acidification, the exact mechanism by which V-ATPase promotes membrane fusion needs further clarification.
Nutrient signaling
mTOR is a serine/threonine kinase that belongs to the phosphoinositide kinase-related family. mTOR integrates signals from growth factors and amino acid availability to control cell growth. This functional enzyme is present in two distinct complexes: mTOR complexes (mTORC1 and mTORC2), both of which are characterized by different protein partners and specific substrates [119]. V-ATPase is necessary for the activation of nutrient signaling from mTORC1 and AMPK [17, 120, 121]. Amino acids promote the translocation of mTORC1 to the lysosomal surface, where it is activated [122]. The V-ATPase is necessary for amino acids to activate mTORC1 by interacting with the Ragulator, a scaffolding complex that anchors the Rag GTPase to the lysosome [17]. V-ATPase-Ragulator complex on late endosomes or lysosomes is also required for the activation of resident AMPK present on these two organelles, thus providing a switch between catabolism and anabolism [120]. It has been shown that inhibition of V-ATPase with its inhibitors Bafilomycin A1 or Concanamycin A increases the luminal concentrations of most metabolites but has no effect on the majority of essential amino acids in the lysosomes. But nutrient starvation-mediated inhibition of mTOR reduces the lysosomal efflux of most essential amino acids [123]. These results suggest that V-ATPase- and mTOR-dependent mechanisms exist for controlling lysosomal flux of metabolites.
Dysfunction of V-ATPase-dependent lysosomal acidification in neurodegenerative diseases
As our knowledge of the lysosome as a multifunctional organelle in cellular clearance, signaling and energy metabolism progresses, the importance of its pH homeostasis becomes increasingly recognized [124]. Dysregulation of pH and lysosomal dysfunction are being linked to the congenital CNS diseases such as Renal tubular acidosis with deafness [125, 126], early-onset CNS diseases such as X-linked Parkinson Disease with Spasticity (XPDS) [127, 128], Wolfram syndrome [129–131], and adult-onset neurodegenerative disorders such as AD, PD, and amyotrophic lateral sclerosis [132, 133]. Here we focus on the LSD, PD and AD.
Neurodegenerative lysosomal storage disorders
In eukaryotes, lysosomes are the main organelles for intracellular digestion [2]. It contains > 50 hydrolases that require an acidic pH for optimal degradation [134]. It has been reported that dysregulation of lysosomal acidification contributes to pathogenesis in virtually all LSDs [135, 136], which include neuronal ceroid lipofuscinosis (NCL), also known as Batten’s disease. This is a group of the most prevalent neurodegenerative LSDs caused by mutations in more than 13 different genes called the CLNs (ceroid lipofuscinosis neuronal) [137], Niemann-Pick type C (NPC), and mucolipidosis type IV (MLIV) [138]. It has been reported that inactivating mutations in the CLN1 gene, which encodes palmitoyl-protein thioesterase-1(PPT1) can cause infant NCL (INCL) [139, 140], a devastating NLSD. In neurons of Cln1−/− INCL model mice, lack of PPT1 activity causes V0a1 misrouted to plasma membrane, preventing its interaction with AP-3, which is required for its transport from the sorting endosome to the late endosomal/lysosomal membrane. This impairs lysosomal V-ATPase activity, thereby dysregulating lysosomal acidification [141]. These findings reveal a role of Cln1 in regulating lysosomal targeting of V0a1 and suggest that dysregulation of lysosomal acidification caused by varying factors adversely affecting V-ATPase function may be common in other LSDs and neurodegenerative diseases.
Parkinson’s disease
PD is caused by the interaction of various genetic factors, environmental factors, and the process of aging [142]. Many of the genetic factors, such as Leucine-rich repeat kinase 2 (LRRK2), α-synuclein (SNCA), Parkin RBR E3 ubiquitin protein ligase (PARK2), Parkinson protein 7 (PARK7), PTEN induced putative kinase 1 (PINK1), scavenger receptor class B, member 2 (SCARB2), etc., are involved in the autophagy-lysosome pathway [143–145]. Notably, Glucocerebrosidase (GBA) gene, which encodes a lysosomal hydrolase glucocerebrosidase (GCase) and whose mutation causes the lysosomal storage disorder Gauchers disease, has recently been shown to be a genetic risk factor for PD when mutated [146]. On the one hand, lysosomes are responsible for the elimination of several PD related toxic factors, such as misfolded proteins or aggregation-prone α-synuclein, and old or damaged mitochondria. Lysosomal failure leads to aberrant accumulation of these toxic materials, which is the molecular hallmark of pathogenic events in PD. On the other hand, many of the genetic risk factors associated with PD often adversely affect V-ATPase and lysosomes. For example, PC12 cells expressing A53T mutant α-synuclein, but not the wildtype α-synuclein, showed lysosomal de-acidification, measured by staining with the acidotropic dye lysotracker, and disruption of lysosomal function [147, 148]. Mutations of ATP6AP2 impair V-ATPase function and result in lysosomal de-acidification, causing lysosomal system failure in neurons and juvenile-onset Parkinsonism [149, 150]. Patients with exon-skipping mutations in ATP6AP2 show cognitive disorders such as XPDS [127, 128]. Another splice site mutation in ATP6AP2 is linked to X-linked intellectual disability (XLID), epilepsy and parkinsonism [151].
ATP13A2 gene (also known as PARK9) encodes a transmembrane endo−/lysosomal- associated P5-type ATPase. Recent study shows that ATP13A2 regulates endosomal and lysosomal cargo sorting and proteostasis through PI (3,5) P2-mediated scaffolding function [152]. Mutation of ATP13A2 is found in autosomal recessive forms of early-onset Parkinsonism and linked to lysosomal dysfunction [153–155]. Mechanistically, fibroblasts derived from PD patients with loss of ATP13A2 function show impaired lysosomal acidification. This is correlated with a decrease in proteolytic processing by lysosomal enzymes, in lysosome-mediated clearance of autophagosomes, and in degradation of lysosomal substrates [153, 155].
The mutation of LRRK2 is regarded as the most frequent cause of familial PD [156]. LRRK2 mutation results in lysosomal expansion and diminished lysosomal degradation of substrates. There is evidence supporting LRRK2 and V-ATPase interaction. In a C. elegans model, mutations of LRRK2 cause an increase in V-ATPase subunits as well as the level of V-ATPase modified by 4-hydroxy-2-nonenal (HNE), indicating an increase in oxidative damage to V-ATPase and other proteins as observed in the cerebral cortex in the case of sporadic PD [157, 158]. Recently, LRRK2 is found to associate directly with the a1 subunit of V-ATPase. This interaction is abolished by the LRRK2 R1441C mutation, leading to a decrease in a1 protein and its cellular mis-localization [159].
In addition to genetic factors, many environmental neurotoxic agents, such as methamphetamine, rotenone and MPP+, promote PD and PD-like symptoms via impairing lysosomal acidification and lysosomal activity [160–166]. Therefore, dysfunction of lysosomal acidification plays a general role in genetic, sporadic, and toxin-induced forms of Parkinsonism.
Alzheimer’s disease
The aggregation of Aβ, the main component of amyloid plaques in AD, is regarded as one of the key pathogenic factors contributing to AD [167]. Aβ is processed from amyloid precursor protein (APP) mainly in the late endosomes into sAPPβ and α-CTF by the action of beta secretase-1 (BACE1), whose peak activity is at pH 4.5 [168]. sAPPβ is subsequently processed into As by γ-secretase. Either an increase in Aβ production or a decrease in its degradation can lead to the accumulation of Aβ and thus leads to the pathological process of AD [169, 170]. Conditions that cause lysosomal dysfunction may prolong the residence time of APP in the acidic late endosomes and thus increase Aβ production. Glial cells participate in the regulation of Aβ level in the brain. Multiple types of brain cells participate in the regulation of Aβ level in the brain [171, 172]. For example, microglia phagocytose Aβ and degrade it via the lysosomal enzymes cathepsin B [173] or endothelin-converting enzymes-2 (ECE-2), a metalloendopeptidase [174]. Several studies suggest that the APP processing machinery may be linked to V-ATPase. Presenilin-1 (PS1) is a component of γ-secretase involved in APP processing and appears to be essential for the targeting of V-ATPase to lysosomes. It has been reported that loss-of-function of PS1 is associated with V-ATPase deficiency, contributing to abnormal cellular Ca2+ homeostasis and lysosomal/autophagy dysfunction [175]. Moreover, N-glycosylation of the V0a1 subunit, which is essential for its efficient ER-to-lysosome delivery, requires the selective binding of PS1 holoprotein to the un-glycosylated V0a1 and the Sec61α in the translocon and OST (oligosaccharyltransferase) complex [10]. Sec61αis a subunit of translocon. It is speculated that PS1 binding to translocon facilitates the presentation of the V0a1 subunit to the OST complex, thus promoting its glycosylation and export to lysosomes. In 5 × FAD mice, in which Aβ is over-produced in part due to dysfunctional PS1, the lysosomal acidification is also impaired by a decrease in N-glycoyslation of the V-ATPase subunit V0a1 and reduced cathepsin D activity. Administration of a competitive GSK-3 inhibitor, L803-mts, restores lysosomal acidification in 5 × FAD brains, enhances the clearance of Aβ, and ameliorates the cognitive dysfunction [176]. However, a subsequent study by Zhang et al. failed to observe the alterations in the N-glycosylation of V0a1 in mouse embryonic fibroblasts (MEFs) deficient in PS1 and PS2 [177]. Nor did it find evidence that the turnover of autophagic substrates, vesicle pH or lysosome function is altered in cells lacking PS1 and PS2. Thus, the precise link between V0a1 and PS1 remains to be clarified.
In addition to AD, V-ATPase has also been linked to cognitive function. For example, altered splicing and conditional knockout of the ATP6AP2 gene in mouse and fly have been shown to cause cognitive impairment and other congenital disorders associated with neurodegeneration [178, 179]. Pathologically, ATP6AP2 conditional knockdown in mouse and fly results in presynaptic transmission defects and abnormal changes of both the number and morphology in synapses. Molecularly, lack of ATP6AP2 also triggers autophagy defects and axonal transport alterations. Consistent with these animal studies, patients with ATP6AP1/Ac45 deficiency display neurocognitive abnormalities as well as immunodeficiency phenotype associated with hypogammaglobulinemia and hepatopathy [180].
We summarize the V-ATPase subunits and their related genes dysfunction in congenital, early-onset, and late-onset CNS diseases in Table 1.
Table 1.
Summary of changes of V-ATPase and neurodegenerative diseases
| Gene | Diseases | Species/model | Pathological Mechanisms | Reference |
|---|---|---|---|---|
| V0a1 | Alzheimer Disease | 5 × FAD mice | decrease of N-glycosylation of V0a1 | [176] |
| PS/APP mice | decrease of mature V0a1 in the lysosomal fraction | [181] | ||
| V0a2 | Autosomal recessive cutis laxa typeII/Wrinkly Skin Syndrome | human | abnormal glycosylation of serum proteins (CDG-II) and impairment of Golgi trafficking by V0a2 mutation | [182–185] |
| V0a3 | Autosomal recessive osteopetrosis with neurodegeneration | R444L mutant mice | endoplasmic reticulum retention and misprocessing of V0a3 due to R444L mutation | [186] |
| human | loss of V0a3 function due to truncation or impaired splicing caused by mutations | [187, 188] | ||
| V0a4 | Renal Tubular Acidosis with hearing loss | human | mutations | [189] |
| V0a4−/− mice | proximal tubule dysfunction with defective endocytic trafficking and accumulation of lysosomal material with V0a4 knockout | [190, 191] | ||
| V1B1 | Renal Tubular Acidosis with hearing loss | human | mutations | [189] |
| V1B2 | Zimmermann-Laband syndrome | human | impaired complex assembly due to missense mutation | [192] |
| Dominant Deafness-Onychodystrophy syndrome | human | c.1516C > T mutation | [193] | |
| cognitive deficits | ATPV1B2 mutant mice | weaker interaction with the V1E2E and abnormal brain development | [194] | |
| ATP6AP2 | X-linked mental retardation and epilepsy | human | impairment of ERK1/2 activation | [127] |
| X-linked Parkinson Disease | human | overexpression of a minor splice isoform due to mutation | [128] | |
| cognitive impairment | ATP6AP2 conditional knockout Drosophila/mice | defects in presynaptic transmission and synapses abnormal caused by conditional knockout | [179] | |
| ATP13A2 | Neuronal ceroid lipofuscinosis | human | mutation | [195] |
| ATP13A2 ko mice | increased insoluble α-synuclein in the hippocampus | [196] | ||
| Kufor-Rakeb syndrome | ATP13A2 ko mice | increase in gliosis, lipofuscinosis and lysosomal markers; protein aggregation but no α-synuclein abnormalities; selective defects in autophagy | [197] | |
| Hereditary parkinsonism | human/in vitro | retaintion in the endoplasmic reticulum and degradation by the proteasome due to truncation | [198] | |
| WFS1 | Wolfram Syndrome | human | mutation | [130] |
| WFS1−/− mice | V1A/V1B instability | [199] | ||
| CLN1 | Neuronal ceroid lipofuscinoses | human | mutation | [139] |
| CLN1−/− mice | misrouting of V-ATPase subunit V0a | [141] |
Conclusion
Although V-ATPase complex is a universal proton pump and plays important roles in lysosome acidification for all cell types, the most profound impact of V-ATPase dysfunction appears to manifest in the central nervous system as neurodegeneration. There is evidence that targeting lysosomal acidification and V-ATPase offers therapeutic benefits [200–203]. For example, studies have demonstrated that FK506 exerts neuroprotective effects by binding with ATP6V1A and induces autophagy. This provides a new and attractive strategy for treating neurodegenerative diseases [204]. Dendrobium nobile Lindl Alkaloids (DNLA) has been used in APP/PS1 mice, which exhibit impaired lysosomal function, and shown to increase the level of V-ATPase A1 subunit and improve learning and memory function in this AD model [205]. In further support of this strategy, low-dose bafilomycin A1 has also been proved to be cytoprotective in part through its maintenance of the autophagy-lysosome pathway [206]. Similarly, the bafilomycin A1-binding subunit ATP6V0C may also serve as a therapeutic target to enhance substrate degradation in age-related neurodegenerative disease [207]. In this regard, it is particularly worth noting that the diversity of subunit isoforms existed for the V-ATPases in different organs and organelles should offer the opportunities for selectively targeting particular V-ATPase complexes that are involved in certain disease processes. Further understanding the molecular details that regulate V-ATPase in the CNS may identify additional and attractive targets for treating neurodegenerative diseases.
Acknowledgements
None.
Abbreviations
- Aβ
amyloid beta
- AD
Alzheimer’s Disease
- ARNO
ADP- ribosylation factor nucleotide site opener
- ALPs
autophagy-lysosomal pathways
- ALS
amyotrophic lateral sclerosis
- AP
autophagosomes
- APP
amyloid precursor protein
- BACE1
beta secretase-1
- CLNs
ceroid lipofuscinosis neuronal
- CMA
chaperone-mediated autophagy
- CI-M6PR
cation-independent mannose-6-phosphate receptor
- DNLA
Dendrobium nobile Lindl Alkaloids
- ECE-2
endothelin-converting enzymes-2
- EEs
early endosomes
- HD
Huntington’s disease
- HEK-293 T
human embryonic kidney-293 T
- HNE
4-hydroxy-2-nonenal
- INCL
infant NCL
- GBA
Glucocerebrosidase
- LSDs
lysosomal storage disorders
- LEs
late endosomes
- LAMP-2A
lysosome-associated membrane protein type 2A
- LRRK2
Leucine-rich repeat kinase 2
- MEFs
mouse embryonic fibroblasts
- MLIV
Mucolipidosis type IV
- mTOR
Mammalian target of rapamycin
- mTORC1
mechanistic target of rapamycin complex 1
- MVBs
multivesicular bodies
- NCLs
neuronal ceroid lipofuscinoses
- NLSD
Neurodegenerative lysosomal storage disorders
- NPC
Niemann-Pick type C
- NTs
Neurotransmitters
- OST
oligosaccharyltransferase
- PARK2
Parkin RBR E3 ubiquitin protein ligase
- PARK7
Parkinson protein 7
- PD
Parkinson’s disease
- PINK1
PTEN induced putative kinase 1
- PPT1
palmitoyl-protein thioesterase-1
- PS1
presenilin 1
- PS2
presenilin 2
- RAVE
regulator of H+-ATPase of vacuolar and endosomal membranes
- SCARB2
scavenger receptor class B, member 2
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- SNCA
α-synuclein
- V-ATPase
vacuolar-type ATPase
- XLID
X-linked intellectual disability
- XPDS
X-linked Parkinson’s disease with sputum
Authors’ contributions
All the three authors critically reviewing the literature, drafting and revising the manuscript; All authors read and approved the final manuscript.
Funding
This work was supported in part by grants R01NS107505 and R01NS095269 to Z.M.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–635. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- 2.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 3.Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007;8:622–632. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- 4.Sasaki K, Yoshida H. Organelle autoregulation-stress responses in the ER, Golgi, mitochondria and lysosome. J Biochem. 2015;157:185–195. doi: 10.1093/jb/mvv010. [DOI] [PubMed] [Google Scholar]
- 5.Zhang CS, et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014;20:526–540. doi: 10.1016/j.cmet.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 6.Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74:69–86. doi: 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- 7.Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–734. doi: 10.1083/jcb.201208152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793:684–696. doi: 10.1016/j.bbamcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Hu YB, Dammer EB, Ren RJ, Wang G. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl Neurodegener. 2015;4:18. doi: 10.1186/s40035-015-0041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee J-H, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 2020;21:101–118. doi: 10.1038/s41580-019-0185-4. [DOI] [PubMed] [Google Scholar]
- 12.Lie PPY, Nixon RA. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol Dis. 2019;122:94–105. doi: 10.1016/j.nbd.2018.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marzella L, Ahlberg J, Glaumann H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch B Cell Pathol Incl Mol Pathol. 1981;36:219–234. doi: 10.1007/BF02912068. [DOI] [PubMed] [Google Scholar]
- 14.Weckman A, et al. Autophagy in the endocrine glands. J Mol Endocrinol. 2014;52:R151–R163. doi: 10.1530/JME-13-0241. [DOI] [PubMed] [Google Scholar]
- 15.Csizmadia T, et al. Molecular mechanisms of developmentally programmed crinophagy in. J Cell Biol. 2018;217:361–374. doi: 10.1083/jcb.201702145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 17.Zoncu R, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 19.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19:365–381. doi: 10.1038/s41580-018-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sahu R, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:131–139. doi: 10.1016/j.devcel.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- 24.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 25.Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci. 2011;68:749–763. doi: 10.1007/s00018-010-0565-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gary-Bobo M, Nirde P, Jeanjean A, Morere A, Garcia M. Mannose 6-phosphate receptor targeting and its applications in human diseases. Curr Med Chem. 2007;14:2945–2953. doi: 10.2174/092986707782794005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richo GR, Conner GE. Structural requirements of procathepsin D activation and maturation. J Biol Chem. 1994;269:14806–14812. [PubMed] [Google Scholar]
- 28.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of mammalian iron metabolism. Cell. 2010;142:24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 30.Asano T, et al. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol. 2011;31:2040–2052. doi: 10.1128/MCB.01437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yambire KF, et al. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. Elife. 2019;8. [DOI] [PMC free article] [PubMed]
- 32.Diering GH, Numata M. Endosomal pH in neuronal signaling and synaptic transmission: role of Na(+)/H(+) exchanger NHE5. Front Physiol. 2014;4:412. doi: 10.3389/fphys.2013.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carstea ED, et al. Niemann-pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 34.Boland B, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hara T, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 36.Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 37.Ferguson SM. Neuronal lysosomes. Neurosci Lett. 2019;697:1–9. doi: 10.1016/j.neulet.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nixon RA, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 39.Felbor U, et al. Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc Natl Acad Sci U S A. 2002;99:7883–7888. doi: 10.1073/pnas.112632299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivy GO, et al. Lipofuscin-like substances accumulate rapidly in brain, retina and internal organs with cysteine protease inhibition. Adv Exp Med Biol. 1989;266:31–45. doi: 10.1007/978-1-4899-5339-1_3. [DOI] [PubMed] [Google Scholar]
- 41.Rubinsztein DC, et al. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11–22. doi: 10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- 42.Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- 43.Dehay B, et al. Lysosomal impairment in Parkinson's disease. Mov Disord. 2013;28:725–732. doi: 10.1002/mds.25462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nixon RA, Yang DS. Autophagy failure in Alzheimer's disease--locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015;38:26–35. doi: 10.1016/j.tins.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 46.Carmona-Gutierrez D, Hughes AL, Madeo F, Ruckenstuhl C. The crucial impact of lysosomes in aging and longevity. Ageing Res Rev. 2016;32:2–12. doi: 10.1016/j.arr.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pu J, Guardia CM, Keren-Kaplan T, Bonifacino JS. Mechanisms and functions of lysosome positioning. J Cell Sci. 2016;129:4329–4339. doi: 10.1242/jcs.196287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becker NH, Goldfischer S, Shin WY, Novikoff AB. The localization of enzyme activities in the rat brain. J Biophys Biochem Cytol. 1960;8:649–663. doi: 10.1083/jcb.8.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parton RG, Simons K, Dotti CG. Axonal and dendritic endocytic pathways in cultured neurons. J Cell Biol. 1992;119:123–137. doi: 10.1083/jcb.119.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gorenstein C, Bundman MC, Lew PJ, Olds JL, Ribak CE. Dendritic transport. I Colchicine stimulates the transport of lysosomal enzymes from cell bodies to dendrites. J Neurosci. 1985;5:2009–2017. doi: 10.1523/JNEUROSCI.05-08-02009.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin EJ, et al. Live Observation of Two Parallel Membrane Degradation Pathways at Axon Terminals. Curr Biol. 2018;28:1027–1038.e1024. doi: 10.1016/j.cub.2018.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maday S, Holzbaur EL. Compartment-specific regulation of autophagy in primary neurons. J Neurosci. 2016;36:5933–5945. doi: 10.1523/JNEUROSCI.4401-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maday S, Wallace KE, Holzbaur EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196:407–417. doi: 10.1083/jcb.201106120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maday S, Holzbaur EL. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell. 2014;30:71–85. doi: 10.1016/j.devcel.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nelson N. Evolution of organellar proton-ATPases. Biochim Biophys Acta. 1992;1100:109–124. doi: 10.1016/0005-2728(92)90072-A. [DOI] [PubMed] [Google Scholar]
- 56.Dittrich M, Hayashi S, Schulten K. On the mechanism of ATP hydrolysis in F1-ATPase. Biophys J. 2003;85:2253–2266. doi: 10.1016/S0006-3495(03)74650-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grüber G, Wieczorek H, Harvey WR, Müller V. Structure-function relationships of A-, F- and V-ATPases. J Exp Biol. 2001;204:2597–2605. doi: 10.1242/jeb.204.15.2597. [DOI] [PubMed] [Google Scholar]
- 58.Futai M. Our research on proton pumping ATPases over three decades: their biochemistry, molecular biology and cell biology. Proc Jpn Acad Ser B Phys Biol Sci. 2007;82:416–438. doi: 10.2183/pjab.82.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song J, Pfanner N, Becker T. Assembling the mitochondrial ATP synthase. Proc Natl Acad Sci U S A. 2018;115:2850–2852. doi: 10.1073/pnas.1801697115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 61.Arata Y, et al. Structure, subunit function and regulation of the coated vesicle and yeast vacuolar (H(+))-ATPases. Biochim Biophys Acta. 2002;1555:71–74. doi: 10.1016/S0005-2728(02)00257-8. [DOI] [PubMed] [Google Scholar]
- 62.Pérez-Sayáns M, et al. An update in the structure, function, and regulation of V-ATPases: the role of the C subunit. Braz J Biol. 2012;72:189–198. doi: 10.1590/S1519-69842012000100023. [DOI] [PubMed] [Google Scholar]
- 63.Miranda KC, Karet FE, Brown D. An extended nomenclature for mammalian V-ATPase subunit genes and splice variants. PLoS One. 2010;5:e9531. doi: 10.1371/journal.pone.0009531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Toei M, Saum R, Forgac M. Regulation and isoform function of the V-ATPases. Biochemistry. 2010;49:4715–4723. doi: 10.1021/bi100397s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith AN, Borthwick KJ, Karet FE. Molecular cloning and characterization of novel tissue-specific isoforms of the human vacuolar H(+)-ATPase C, G and d subunits, and their evaluation in autosomal recessive distal renal tubular acidosis. Gene. 2002;297:169–177. doi: 10.1016/S0378-1119(02)00884-3. [DOI] [PubMed] [Google Scholar]
- 66.Murata Y, et al. Differential localization of the vacuolar H+ pump with G subunit isoforms (G1 and G2) in mouse neurons. J Biol Chem. 2002;277:36296–36303. doi: 10.1074/jbc.M200586200. [DOI] [PubMed] [Google Scholar]
- 67.Poëa-Guyon S, Amar M, Fossier P, Morel N. Alternative splicing controls neuronal expression of v-ATPase subunit a1 and sorting to nerve terminals. J Biol Chem. 2006;281:17164–17172. doi: 10.1074/jbc.M600927200. [DOI] [PubMed] [Google Scholar]
- 68.Kawasaki-Nishi S, Yamaguchi A, Forgac M, Nishi T. Tissue specific expression of the splice variants of the mouse vacuolar proton-translocating ATPase a4 subunit. Biochem Biophys Res Commun. 2007;364:1032–1036. doi: 10.1016/j.bbrc.2007.10.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crider BP, et al. Subunit G of the vacuolar proton pump. Molecular characterization and functional expression. J Biol Chem. 1997;272:10721–10728. doi: 10.1074/jbc.272.16.10721. [DOI] [PubMed] [Google Scholar]
- 70.Zhou Z, et al. Molecular characterization of the 50- and 57-kDa subunits of the bovine vacuolar proton pump. J Biol Chem. 1998;273:5878–5884. doi: 10.1074/jbc.273.10.5878. [DOI] [PubMed] [Google Scholar]
- 71.Blake-Palmer KG, Su Y, Smith AN, Karet FE. Molecular cloning and characterization of a novel form of the human vacuolar H+-ATPase e-subunit: an essential proton pump component. Gene. 2007;393:94–100. doi: 10.1016/j.gene.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 72.Cotter K, Stransky L, McGuire C, Forgac M. Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem Sci. 2015;40:611–622. doi: 10.1016/j.tibs.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Supek F, et al. A novel accessory subunit for vacuolar H(+)-ATPase from chromaffin granules. J Biol Chem. 1994;269:24102–24106. [PubMed] [Google Scholar]
- 74.Jansen EJ, Scheenen WJ, Hafmans TG, Martens GJ. Accessory subunit Ac45 controls the V-ATPase in the regulated secretory pathway. Biochim Biophys Acta. 2008;1783:2301–2310. doi: 10.1016/j.bbamcr.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 75.Jansen EJ, et al. Identification of domains within the V-ATPase accessory subunit Ac45 involved in V-ATPase transport and Ca2+−dependent exocytosis. J Biol Chem. 2012;287:27537–27546. doi: 10.1074/jbc.M112.356105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ludwig J, et al. Identification and characterization of a novel 9.2-kDa membrane sector-associated protein of vacuolar proton-ATPase from chromaffin granules. J Biol Chem. 1998;273:10939–10947. doi: 10.1074/jbc.273.18.10939. [DOI] [PubMed] [Google Scholar]
- 77.Kinouchi K, et al. The (pro) renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ Res. 2010;107:30–34. doi: 10.1161/CIRCRESAHA.110.224667. [DOI] [PubMed] [Google Scholar]
- 78.Hirose T, et al. ATP6AP2 variant impairs CNS development and neuronal survival to cause fulminant neurodegeneration. J Clin Invest. 2019;129. [DOI] [PMC free article] [PubMed]
- 79.Xu Y, et al. Epidermal growth factor-induced vacuolar (H+)-ATPase assembly a role in signaling via mTORC1 activation. J Biol Chem. 2012;287:26409–26422. doi: 10.1074/jbc.M112.352229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yan Y, Denef N, Schüpbach T. The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev Cell. 2009;17:387–402. doi: 10.1016/j.devcel.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sun-Wada G-H, et al. The a3 isoform of V-ATPase regulates insulin secretion from pancreatic β-cells. J Cell Sci. 2006;119:4531–4540. doi: 10.1242/jcs.03234. [DOI] [PubMed] [Google Scholar]
- 82.Vavassori S, Mayer A. A new life for an old pump: V-ATPase and neurotransmitter release. J Cell Biol. 2014;205:7–9. doi: 10.1083/jcb.201403040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Feng Y, Forgac M. Cysteine 254 of the 73-kDa a subunit is responsible for inhibition of the coated vesicle (H+)-ATPase upon modification by sulfhydryl reagents. J Biol Chem. 1992;267:5817–5822. [PubMed] [Google Scholar]
- 84.Feng Y, Forgac M. A novel mechanism for regulation of vacuolar acidification. J Biol Chem. 1992;267:19769–19772. [PubMed] [Google Scholar]
- 85.Feng Y, Forgac M. Inhibition of vacuolar H(+)-ATPase by disulfide bond formation between cysteine 254 and cysteine 532 in subunit a. J Biol Chem. 1994;269:13224–13230. [PubMed] [Google Scholar]
- 86.Kawamura Y, Arakawa K, Maeshima M, Yoshida S. ATP analogue binding to the a subunit induces conformational changes in the E subunit that involves a disulfide bond formation in plant V-ATPase. Eur J Biochem. 2001;268:2801–2809. doi: 10.1046/j.1432-1327.2001.02139.x. [DOI] [PubMed] [Google Scholar]
- 87.Toyomura T, et al. From lysosomes to the plasma membrane: localization of vacuolar-type H+ −ATPase with the a3 isoform during osteoclast differentiation. J Biol Chem. 2003;278:22023–22030. doi: 10.1074/jbc.M302436200. [DOI] [PubMed] [Google Scholar]
- 88.Bagnis C, Marshansky V, Breton S, Brown D. Remodeling the cellular profile of collecting ducts by chronic carbonic anhydrase inhibition. Am J Physiol Renal Physiol. 2001;280:F437–F448. doi: 10.1152/ajprenal.2001.280.3.F437. [DOI] [PubMed] [Google Scholar]
- 89.Hinton A, Bond S, Forgac M. V-ATPase functions in normal and disease processes. Pflügers Arch Eur J Physiol. 2009;457:589–598. doi: 10.1007/s00424-007-0382-4. [DOI] [PubMed] [Google Scholar]
- 90.Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 91.Nishi T, Forgac M. The vacuolar (H+)-ATPases--nature's most versatile proton pumps. Nat Rev Mol Cell Biol. 2002;3:94–103. doi: 10.1038/nrm729. [DOI] [PubMed] [Google Scholar]
- 92.Smith AN, et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26:71–75. doi: 10.1038/79208. [DOI] [PubMed] [Google Scholar]
- 93.Shine L, Kilty C, Gross J, Kennedy B. Vacuolar ATPases and their role in vision. Adv Exp Med Biol. 2014;801:97–103. doi: 10.1007/978-1-4614-3209-8_13. [DOI] [PubMed] [Google Scholar]
- 94.Wagner CA, et al. Renal vacuolar H+-ATPase. Physiol Rev. 2004;84:1263–1314. doi: 10.1152/physrev.00045.2003. [DOI] [PubMed] [Google Scholar]
- 95.Kane PM. Disassembly and reassembly of the yeast vacuolar H+-ATPase in vivo. J Biol Chem. 1995;270:17025–17032. [PubMed] [Google Scholar]
- 96.Lu M, Ammar D, Ives H, Albrecht F, Gluck SL. Physical interaction between aldolase and vacuolar H+-ATPase is essential for the assembly and activity of the proton pump. J Biol Chem. 2007;282:24495–24503. doi: 10.1074/jbc.M702598200. [DOI] [PubMed] [Google Scholar]
- 97.Smardon AM, Tarsio M, Kane PM. The RAVE complex is essential for stable assembly of the yeast V-ATPase. J Biol Chem. 2002;277:13831–13839. doi: 10.1074/jbc.M200682200. [DOI] [PubMed] [Google Scholar]
- 98.Xu T, Forgac M. Microtubules are involved in glucose-dependent dissociation of the yeast vacuolar [H+]-ATPase in vivo. J Biol Chem. 2001;276:24855–24861. doi: 10.1074/jbc.M100637200. [DOI] [PubMed] [Google Scholar]
- 99.McGuire CM, Forgac M. Glucose starvation increases V-ATPase assembly and activity in mammalian cells through AMP kinase and phosphatidylinositide 3-kinase/Akt signaling. J Biol Chem. 2018;293:9113–9123. doi: 10.1074/jbc.RA117.001327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Parra KJ, Hayek SR. A lysosomal proton pump turns on when glucose runs out. J Biol Chem. 2018;293:9124–9125. doi: 10.1074/jbc.H118.002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stransky LA, Forgac M. Amino acid availability modulates vacuolar H+-ATPase assembly. J Biol Chem. 2015;290:27360–27369. doi: 10.1074/jbc.M115.659128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Milosevic I. Revisiting the role of Clathrin-mediated Endoytosis in synaptic vesicle recycling. Front Cell Neurosci. 2018;12:27. doi: 10.3389/fncel.2018.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Farsi Z, et al. Clathrin coat controls synaptic vesicle acidification by blocking vacuolar ATPase activity. Elife. 2018;7. [DOI] [PMC free article] [PubMed]
- 104.Peña-Llopis S, et al. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011;30:3242–3258. doi: 10.1038/emboj.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Smith GA, et al. Extracellular and luminal pH regulation by vacuolar H+-ATPase isoform expression and targeting to the plasma membrane and endosomes. J Biol Chem. 2016;291:8500–8515. doi: 10.1074/jbc.M116.723395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morel N. Neurotransmitter release: the dark side of the vacuolar-H+ATPase. Biol Cell. 2003;95:453–457. doi: 10.1016/S0248-4900(03)00075-3. [DOI] [PubMed] [Google Scholar]
- 107.Hurtado-Lorenzo A, et al. V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat Cell Biol. 2006;8:124–136. doi: 10.1038/ncb1348. [DOI] [PubMed] [Google Scholar]
- 108.Hiesinger PR, et al. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell. 2005;121:607–620. doi: 10.1016/j.cell.2005.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hnasko TS, Edwards RH. Neurotransmitter corelease: mechanism and physiological role. Annu Rev Physiol. 2012;74:225–243. doi: 10.1146/annurev-physiol-020911-153315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.El Mestikawy S, Wallén-Mackenzie A, Fortin GM, Descarries L, Trudeau LE. From glutamate co-release to vesicular synergy: vesicular glutamate transporters. Nat Rev Neurosci. 2011;12:204–216. doi: 10.1038/nrn2969. [DOI] [PubMed] [Google Scholar]
- 111.Wang D, Chan CC, Cherry S, Hiesinger PR. Membrane trafficking in neuronal maintenance and degeneration. Cell Mol Life Sci. 2013;70:2919–2934. doi: 10.1007/s00018-012-1201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sobota JA, Bäck N, Eipper BA, Mains RE. Inhibitors of the V0 subunit of the vacuolar H+-ATPase prevent segregation of lysosomal-and secretory-pathway proteins. J Cell Sci. 2009;122:3542–3553. doi: 10.1242/jcs.034298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Marshansky V, Futai M. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr Opin Cell Biol. 2008;20:415–426. doi: 10.1016/j.ceb.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Peters C, et al. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature. 2001;409:581–588. doi: 10.1038/35054500. [DOI] [PubMed] [Google Scholar]
- 115.Bayer MJ, Reese C, Buhler S, Peters C, Mayer A. Vacuole membrane fusion: V0 functions after trans-SNARE pairing and is coupled to the Ca2+−releasing channel. J Cell Biol. 2003;162:211–222. doi: 10.1083/jcb.200212004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Poëa-Guyon S, et al. The V-ATPase membrane domain is a sensor of granular pH that controls the exocytotic machinery. J Cell Biol. 2013;203:283–298. doi: 10.1083/jcb.201303104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sun-Wada GH, et al. The a3 isoform of V-ATPase regulates insulin secretion from pancreatic beta-cells. J Cell Sci. 2006;119:4531–4540. doi: 10.1242/jcs.03234. [DOI] [PubMed] [Google Scholar]
- 118.Liegeois S, Benedetto A, Garnier JM, Schwab Y, Labouesse M. The V0-ATPase mediates apical secretion of exosomes containing hedgehog-related proteins in Caenorhabditis elegans. J Cell Biol. 2006;173:949–961. doi: 10.1083/jcb.200511072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 120.Zhang C-S, et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014;20:526–540. doi: 10.1016/j.cmet.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 121.Efeyan A, et al. Regulation of mTORC1 by the rag GTPases is necessary for neonatal autophagy and survival. Nature. 2013;493:679. doi: 10.1038/nature11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sancak Y, et al. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Abu-Remaileh M, et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science. 2017;358:807–813. doi: 10.1126/science.aan6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Settembre C, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Blake-Palmer KG, Karet FE. Cellular physiology of the renal H+ATPase. Curr Opin Nephrol Hypertens. 2009;18:433–438. doi: 10.1097/MNH.0b013e32832e9c58. [DOI] [PubMed] [Google Scholar]
- 126.Karet FE, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- 127.Ramser J, et al. A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Hum Mol Genet. 2005;14:1019–1027. doi: 10.1093/hmg/ddi094. [DOI] [PubMed] [Google Scholar]
- 128.Korvatska O, et al. Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS) Hum Mol Genet. 2013;22:3259–3268. doi: 10.1093/hmg/ddt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Strom TM, et al. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet. 1998;7:2021–2028. doi: 10.1093/hmg/7.13.2021. [DOI] [PubMed] [Google Scholar]
- 130.Inoue H, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome) Nat Genet. 1998;20:143. doi: 10.1038/2441. [DOI] [PubMed] [Google Scholar]
- 131.Hatanaka M, et al. Wolfram syndrome 1 gene (WFS1) product localizes to secretory granules and determines granule acidification in pancreatic beta-cells. Hum Mol Genet. 2011;20:1274–1284. doi: 10.1093/hmg/ddq568. [DOI] [PubMed] [Google Scholar]
- 132.Şentürk M, et al. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat Cell Biol. 2019;21:384–396. doi: 10.1038/s41556-019-0281-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yang Y, Klionsky DJ. A novel role of UBQLNs (ubiquilins) in regulating autophagy, MTOR signaling and v-ATPase function. Autophagy. 2020;16:1–2. doi: 10.1080/15548627.2019.1665293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mellman I, Fuchs R, Helenius A. Acidification of the endocytic and exocytic pathways. Annu Rev Biochem. 1986;55:663–700. doi: 10.1146/annurev.bi.55.070186.003311. [DOI] [PubMed] [Google Scholar]
- 135.Platt FM, Boland B, van der Spoel AC. Lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–734. doi: 10.1083/jcb.201208152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Futerman AH, Van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- 137.Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease) Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2015;1852:2237–2241. doi: 10.1016/j.bbadis.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Holopainen JM, Saarikoski J, Kinnunen PK, Järvelä I. Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs) Eur J Biochem. 2001;268:5851–5856. doi: 10.1046/j.0014-2956.2001.02530.x. [DOI] [PubMed] [Google Scholar]
- 139.Vesa J, et al. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. 1995;376:584. doi: 10.1038/376584a0. [DOI] [PubMed] [Google Scholar]
- 140.Camp LA, Verkruyse LA, Afendis SJ, Slaughter CA, Hofmann SL. Molecular cloning and expression of palmitoyl-protein thioesterase. J Biol Chem. 1994;269:23212–23219. [PubMed] [Google Scholar]
- 141.Bagh MB, et al. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat Commun. 2017;8:14612. doi: 10.1038/ncomms14612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tanner CM, et al. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119:866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bras J, et al. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Human molecular genetics. 2014;23:6139–6146. doi: 10.1093/hmg/ddu334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gan-Or Z, Dion PA, Rouleau GA. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy. 2015;11:1443–1457. doi: 10.1080/15548627.2015.1067364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kalinderi K, Bostantjopoulou S, Fidani L. The genetic background of Parkinson's disease: current progress and future prospects. Acta Neurol Scand. 2016;134:314–326. doi: 10.1111/ane.12563. [DOI] [PubMed] [Google Scholar]
- 146.Almeida MR. Glucocerebrosidase involvement in Parkinson disease and other synucleinopathies. Front Neurol. 2012;3:65. doi: 10.3389/fneur.2012.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 149.Kinouchi K, Ichihara A, Itoh H. Functional characterization of (pro) renin receptor in association with V-ATPase. Front Biosci (Landmark Ed) 2011;16:3216–3223. doi: 10.2741/3907. [DOI] [PubMed] [Google Scholar]
- 150.Rujano MA, et al. Mutations in the X-linked. J Exp Med. 2017;214:3707–3729. doi: 10.1084/jem.20170453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gupta HV, Vengoechea J, Sahaya K, Virmani T. A splice site mutation in ATP6AP2 causes X-linked intellectual disability, epilepsy, and parkinsonism. Parkinsonism Relat Disord. 2015;21:1473–1475. doi: 10.1016/j.parkreldis.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 152.Demirsoy S, et al. ATP13A2/PARK9 regulates endo−/lysosomal cargo sorting and proteostasis through a novel PI(3, 5)P2-mediated scaffolding function. Hum Mol Genet. 2017;26:1656–1669. doi: 10.1093/hmg/ddx070. [DOI] [PubMed] [Google Scholar]
- 153.Dehay B, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci. 2012;109:9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Park JS, et al. Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor-Rakeb syndrome, a form of early-onset parkinsonism. Hum Mutat. 2011;32:956–964. doi: 10.1002/humu.21527. [DOI] [PubMed] [Google Scholar]
- 155.Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32:4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson's disease: progress and therapeutic implications. Mov Disord. 2013;28:14–23. doi: 10.1002/mds.25249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Di Domenico F, et al. Redox proteomics analyses of the influence of co-expression of wild-type or mutated LRRK2 and Tau on C. elegans protein expression and oxidative modification: relevance to Parkinson disease. Antioxid Redox Signal. 2012;17:1490–1506. doi: 10.1089/ars.2011.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Choi J, et al. Oxidative modifications and aggregation of cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J Biol Chem. 2005;280:11648–11655. doi: 10.1074/jbc.M414327200. [DOI] [PubMed] [Google Scholar]
- 159.Wallings R, Connor-Robson N, Wade-Martins R. LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum Mol Genet. 2019;28:2696–2710. doi: 10.1093/hmg/ddz088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Callaghan RC, Cunningham JK, Sykes J, Kish SJ. Increased risk of Parkinson's disease in individuals hospitalized with conditions related to the use of methamphetamine or other amphetamine-type drugs. Drug Alcohol Depend. 2012;120:35–40. doi: 10.1016/j.drugalcdep.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 161.Betarbet R, et al. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 162.Pal R, et al. NADPH oxidase promotes Parkinsonian phenotypes by impairing autophagic flux in an mTORC1-independent fashion in a cellular model of Parkinson's disease. Sci Rep. 2016;6:22866. doi: 10.1038/srep22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bourdenx M, et al. Nanoparticles restore lysosomal acidification defects: implications for Parkinson and other lysosomal-related diseases. Autophagy. 2016;12:472–483. doi: 10.1080/15548627.2015.1136769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dehay B, et al. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci. 2010;30:12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Prévot G, et al. Harnessing Lysosomal pH through PLGA Nanoemulsion as a treatment of Lysosomal-related neurodegenerative diseases. Bioconjug Chem. 2018;29:4083–4089. doi: 10.1021/acs.bioconjchem.8b00697. [DOI] [PubMed] [Google Scholar]
- 166.Boland B, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2018;17:660–688. doi: 10.1038/nrd.2018.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 168.Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer's disease beta-secretase. J Biol Chem. 2000;275:33729–33737. doi: 10.1074/jbc.M004175200. [DOI] [PubMed] [Google Scholar]
- 169.Cataldo AM, Paskevich PA, Kominami E, Nixon RA. Lysosomal hydrolases of different classes are abnormally distributed in brains of patients with Alzheimer disease. Proc Natl Acad Sci. 1991;88:10998–11002. doi: 10.1073/pnas.88.24.10998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cataldo AM, et al. Gene expression and cellular content of cathepsin D in Alzheimer's disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 1995;14:671–680. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 171.Ries M, Sastre M. Mechanisms of Aβ clearance and degradation by glial cells. Front Aging Neurosci. 2016;8:160. doi: 10.3389/fnagi.2016.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Nalivaeva NN, Beckett C, Belyaev ND, Turner AJ. Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer's disease? J Neurochem. 2012;120(Suppl 1):167–185. doi: 10.1111/j.1471-4159.2011.07510.x. [DOI] [PubMed] [Google Scholar]
- 173.Nakanishi H. Microglial functions and proteases. Mol Neurobiol. 2003;27:163–176. doi: 10.1385/MN:27:2:163. [DOI] [PubMed] [Google Scholar]
- 174.Pacheco-Quinto J, Eckman EA. Endothelin-converting enzymes degrade intracellular β-amyloid produced within the endosomal/lysosomal pathway and autophagosomes. J Biol Chem. 2013;288:5606–5615. doi: 10.1074/jbc.M112.422964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lee J-H, et al. Presenilin 1 maintains lysosomal Ca2+ homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 2015;12:1430–1444. doi: 10.1016/j.celrep.2015.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Avrahami L, et al. Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013;288:1295–1306. doi: 10.1074/jbc.M112.409250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang X, et al. A role for presenilins in autophagy revisited: normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J Neurosci. 2012;32:8633–8648. doi: 10.1523/JNEUROSCI.0556-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Goldstein B, Speth RC, Trivedi M. Renin–angiotensin system gene expression and neurodegenerative diseases. J Renin-Angiotensin-Aldosterone Syst. 2016;17:1470320316666750. doi: 10.1177/1470320316666750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dubos A, et al. Conditional depletion of intellectual disability and parkinsonism candidate gene ATP6AP2 in fly and mouse induces cognitive impairment and neurodegeneration. Hum Mol Genet. 2015;24:6736–6755. doi: 10.1093/hmg/ddv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jansen EJ, et al. ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nat Commun. 2016;7:11600. doi: 10.1038/ncomms11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wolfe DM, et al. Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013;37:1949–1961. doi: 10.1111/ejn.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kornak U, et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet. 2008;40:32. doi: 10.1038/ng.2007.45. [DOI] [PubMed] [Google Scholar]
- 183.Morava E, et al. Defining the phenotype in an autosomal recessive cutis laxa syndrome with a combined congenital defect of glycosylation. Eur J Hum Genet. 2008;16:28. doi: 10.1038/sj.ejhg.5201947. [DOI] [PubMed] [Google Scholar]
- 184.Guillard M, et al. Vacuolar H+-ATPase meets glycosylation in patients with cutis laxa. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2009;1792:903–914. doi: 10.1016/j.bbadis.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 185.Beyens A, et al. ATP 6V0A2-related cutis laxa in 10 novel patients: focus on clinical variability and expansion of the phenotype. Exp Dermatol. 2018. [DOI] [PubMed]
- 186.Bhargava A, et al. Osteopetrosis mutation R444L causes endoplasmic reticulum retention and misprocessing of vacuolar H+-ATPase a3 subunit. J Biol Chem. 2012;287:26829–26839. doi: 10.1074/jbc.M112.345702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sobacchi C, et al. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum Mol Genet. 2001;10:1767–1773. doi: 10.1093/hmg/10.17.1767. [DOI] [PubMed] [Google Scholar]
- 188.Kornak U, et al. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet. 2000;9:2059–2063. doi: 10.1093/hmg/9.13.2059. [DOI] [PubMed] [Google Scholar]
- 189.Stover E, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet. 2002;39:796–803. doi: 10.1136/jmg.39.11.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hennings JC, et al. A mouse model for distal renal tubular acidosis reveals a previously unrecognized role of the V-ATPase a4 subunit in the proximal tubule. EMBO Mol Med. 2012;4:1057–1071. doi: 10.1002/emmm.201201527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lorente-Cánovas B, et al. Mice deficient in H+-ATPase a4 subunit have severe hearing impairment associated with enlarged endolymphatic compartments within the inner ear. Dis Model Mech. 2013;6:434–442. doi: 10.1242/dmm.010645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kortüm F, et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat Genet. 2015;47:661–667. doi: 10.1038/ng.3282. [DOI] [PubMed] [Google Scholar]
- 193.Menendez I, et al. Dominant deafness-onychodystrophy syndrome caused by an. Clin Case Rep. 2017;5:376–379. doi: 10.1002/ccr3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhao W, et al. A subunit of V-ATPases, ATP6V1B2, underlies the pathology of intellectual disability. EBioMedicine. 2019;45:408–421. doi: 10.1016/j.ebiom.2019.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet. 2012;21:2646–2650. doi: 10.1093/hmg/dds089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Schultheis PJ, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. 2013;22:2067–2082. doi: 10.1093/hmg/ddt057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kett LR, et al. α-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J Neurosci. 2015;35:5724–5742. doi: 10.1523/JNEUROSCI.0632-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ramirez A, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 199.Gharanei S, et al. Vacuolar-type H+-ATPase V1A subunit is a molecular partner of Wolfram syndrome 1 (WFS1) protein, which regulates its expression and stability. Hum Mol Genet. 2013;22:203–217. doi: 10.1093/hmg/dds400. [DOI] [PubMed] [Google Scholar]
- 200.Kane PM. Targeting reversible disassembly as a mechanism of controlling V-ATPase activity. Curr Protein Peptide Sci. 2012;13:117–123. doi: 10.2174/138920312800493142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kartner N, Manolson MF. Novel techniques in the development of osteoporosis drug therapy: the osteoclast ruffled-border vacuolar H+-ATPase as an emerging target. Expert Opin Drug Discovery. 2014;9:505–522. doi: 10.1517/17460441.2014.902155. [DOI] [PubMed] [Google Scholar]
- 202.Spugnini EP, Citro G, Fais S. Proton pump inhibitors as anti vacuolar-ATPases drugs: a novel anticancer strategy. J Exp Clin Cancer Res. 2010;29:44. doi: 10.1186/1756-9966-29-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bowman EJ, Bowman BJ. V-ATPases as drug targets. J Bioenerg Biomembr. 2005;37:431–435. doi: 10.1007/s10863-005-9485-9. [DOI] [PubMed] [Google Scholar]
- 204.Kim D, et al. FK506, an Immunosuppressive Drug, Induces Autophagy by Binding to the V-ATPase Catalytic Subunit A in Neuronal Cells. J Proteome Res. 2017;16:55–64. doi: 10.1021/acs.jproteome.6b00638. [DOI] [PubMed] [Google Scholar]
- 205.Nie J, Jiang L, Lu Y, Shi J. Dendrobium nobile Lindl. alkaloids decreases the level of intracellular β-amyloid by improving impaired autolysosomal proteolysis in APP/PS1 mice. FASEB J. 2019;33:806.817. doi: 10.1096/fj.201900253RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pivtoraiko VN, et al. Low-dose bafilomycin attenuates neuronal cell death associated with autophagy-lysosome pathway dysfunction. J Neurochem. 2010;114:1193–1204. doi: 10.1111/j.1471-4159.2010.06838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Mangieri LR, et al. ATP6V0C knockdown in neuroblastoma cells alters autophagy-lysosome pathway function and metabolism of proteins that accumulate in neurodegenerative disease. PLoS One. 2014;9:e93257. doi: 10.1371/journal.pone.0093257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.