

Abstract
The liver is a complex organ performing numerous vital physiological functions. For that reason, it possesses immense regenerative potential. The capacity for repair is largely attributable to the ability of its differentiated epithelial cells, hepatocytes and biliary epithelial cells, to proliferate after injury. However, in cases of extreme acute injury or prolonged chronic insult, the liver may fail to regenerate or do so suboptimally. This often results in life-threatening end-stage liver disease for which liver transplantation is the only effective treatment. In many forms of liver injury, bipotent liver progenitor cells are theorized to be activated as an additional tier of liver repair. However, the existence, origin, fate, activation, and contribution to regeneration of liver progenitor cells is hotly debated, especially since hepatocytes and biliary epithelial cells themselves may serve as facultative stem cells for one another during severe liver injury. Here, we discuss the evidence both supporting and refuting the existence of liver progenitor cells in a variety of experimental models. We also debate the validity of developing therapies harnessing the capabilities of these cells as potential treatments for patients with severe and chronic liver diseases.
Keywords: oval cell, liver regeneration, liver cancer, liver stem cell, ductular reaction
INTRODUCTION
Adult mammals possess limited organ repair capabilities, unlike lower vertebrates, such as fish or amphibians (1, 2). During postnatal development, mammals lose most of their competence for regeneration, except for minimal levels of maintenance during tissue homeostasis (1, 3, 4). In general, adult mammalian organs produce nonfunctional connective tissues after injury instead of functional restoration of lost parenchyma (5, 6). While the acute and chronic deposition of scar tissue is a protective response to reduce organ damage caused by additional insults, it results in the progressive loss of function that underlies a majority of chronic diseases, particularly in aging populations (2, 6). However, the mammalian liver is a unique organ that maintains the ability to regenerate tissue into adulthood at comparable levels to the regenerative capacity of fetal liver and the livers of lower vertebrates. The liver can restore lost functional parenchyma after various hepatic injuries, including restoring hepatic mass after surgical removal (7, 8). This extraordinary regenerative capacity is mainly based on the self-replication ability of the two main epithelial cell types: hepatocytes (HCs) and biliary epithelial cell (BECs, also known as cholangiocytes) (7,8). These two cell populations exit quiescence and replicate to compensate for lost functional parenchyma in response to both acute and chronic injuries.
However, regenerative mechanisms following acute liver injury (such as surgical removal of liver tissue or damage from hepatotoxic drug overdoses) exhibit distinct phenotypic differences from liver regeneration in the setting of chronic liver injury. Within a healthy liver, BECs are usually confined to the interlobular bile ducts that form the biliary tree, a complex network of tubules that functions to collect HC-derived bile and eventually transport it to the common bile duct, which drains into the duodenum (9). HCs secrete bile into the bile canaliculi, which connect with the smallest proximal ductules at the ductular–hepatocellular junctions known as the canals of Hering (9, 10) (Figure 1). Anatomically, the bile ducts are located near the portal vein and hepatic artery in a structure known as the portal triad, which is separated by linear cords of HCs from the central vein (9). However, during many types of liver injury there is an expansion of cells that express BEC markers, known as reactive BECs, from the periportal region into the surrounding parenchyma (Figure 1). This phenomenon is termed the ductular reaction (DR) and defined as a reaction of ductular cells, although it does not necessarily indicate their ductular origin (10). This definition affords flexibility to the cell of origin of the DR, as there is evidence that cells besides BECs can give rise to the cells of the DR, which is discussed in the section titled The Ductular Reaction in Human Liver Disease. The DR is considered by some to stem from the activation and expansion of liver progenitor cells (LPCs), also known as oval cells in rodents, which are bipotent cells capable of giving rise to HCs or BECs (8) (Figure 2). Indeed, in various murine models of hepatobiliary injury, ductular markers such as Sox9 are expressed by the DR or by HCs (Figure 3). This has led others to theorize that there are no progenitors in the liver, but during liver injury HCs and BECs can function as facultative stem cells that can transdifferentiate into one another, and the DR is evidence of such cellular plasticity (7) (Figure 4). The DR is a complex process that has different phenotypes in different injury conditions, but in general it is associated with infiltration of inflammatory cells, activation of myofibroblasts, and matrix deposition (11). Despite the prevalence of the DR after liver injury, the origin, fate, and exact roles of LPCs in the diseased liver are heavily debated and remain largely elusive.
Figure 1.
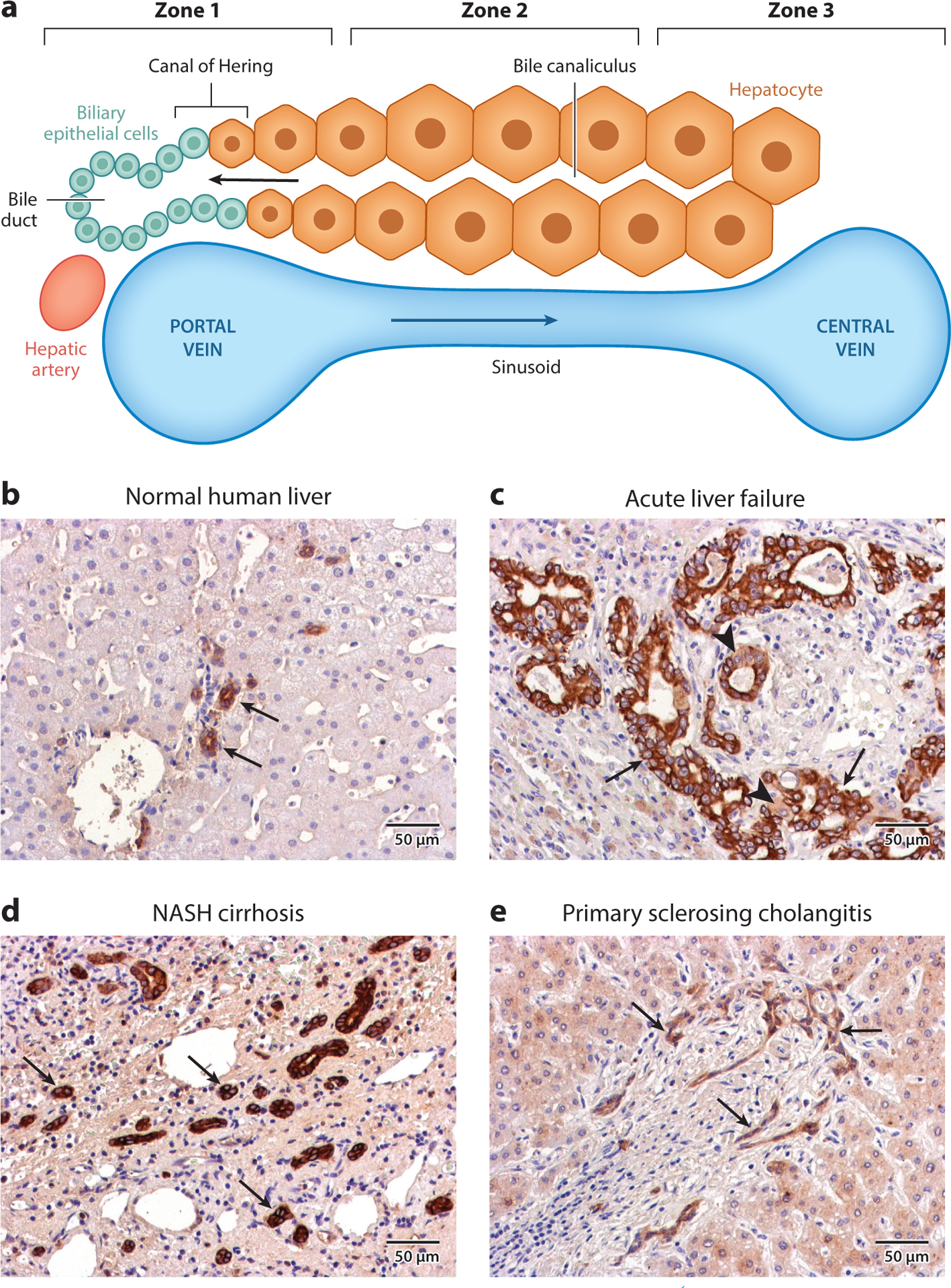
Architecture of normal and diseased liver. (a) The structure of a liver lobule consists of three zones. Zone 1 consists of the portal vein, bile ducts, and hepatic artery, which together form the portal triad. Oxygenated blood from the hepatic artery mixes with blood from the portal vein and flows through the hepatic sinusoids toward the central vein, which constitutes zone 3. The interface between the end of a bile canaliculus and the start of a bile duct is known as the canal of Hering. Cords of hepatocytes (HCs) form the bile canaliculi, which transport HC-derived bile to the bile ducts in the opposite direction of blood flow. The HCs found between zones 1 and 3 constitute zone 2, and these are also known as midzonal HCs. (b) Normal human liver stained with a pan-cytokeratin (panCK) antibody. The arrows denote bile ducts, structures with obvious lumina lined by panCK-positive cells. (c) Liver from a patient with acute liver failure stained with a panCK antibody. A ductular reaction (DR) is evidenced by a large increase in the number of panCK-positive cells (arrows). Intermediate HCs, or cells that are panCK-positive but exhibit HC morphology (arrowhead), can be observed adjacent to the cells of the DR. (d) Liver from a patient with nonalcoholic steatohepatitis (NASH)– induced cirrhosis stained with a panCK antibody. A DR is evident in the increase in the number of panCK-positive cells, which form bile duct–like structures without obvious lumina (arrows). (e) Liver from a patient with primary sclerosing cholangitis stained with a panCK antibody. A DR is evident by the increase in the number of panCK-positive cells (arrows), which have a distinctly different morphology from normal bile ducts.
Figure 2.
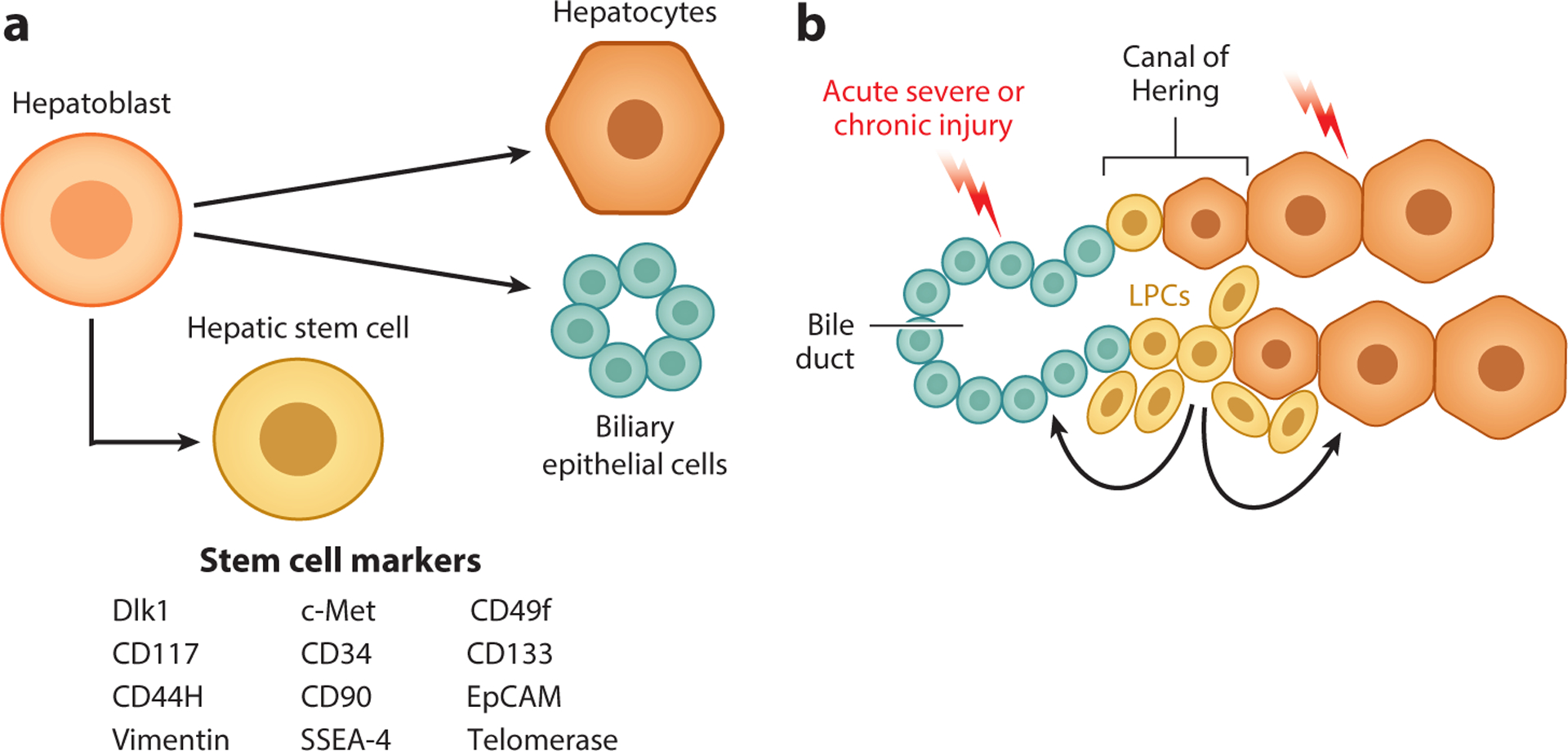
The liver progenitor cell theory. (a) During fetal liver development, hepatoblasts give rise to the two primary epithelial cell types of the mature liver: hepatocytes and biliary epithelial cells. Studies have identified potential markers of hepatoblasts with characteristics of pluripotent stem cells (shown in the figure). (b) It is theorized that quiescent liver progenitor cells (LPCs), potentially residing in the canals of Hering, are activated in the context of severe acute injury or chronic liver injury. These LPCs proliferate and are capable of giving rise to both hepatocytes and cholangiocytes, thus contributing to liver repair.
Figure 3.
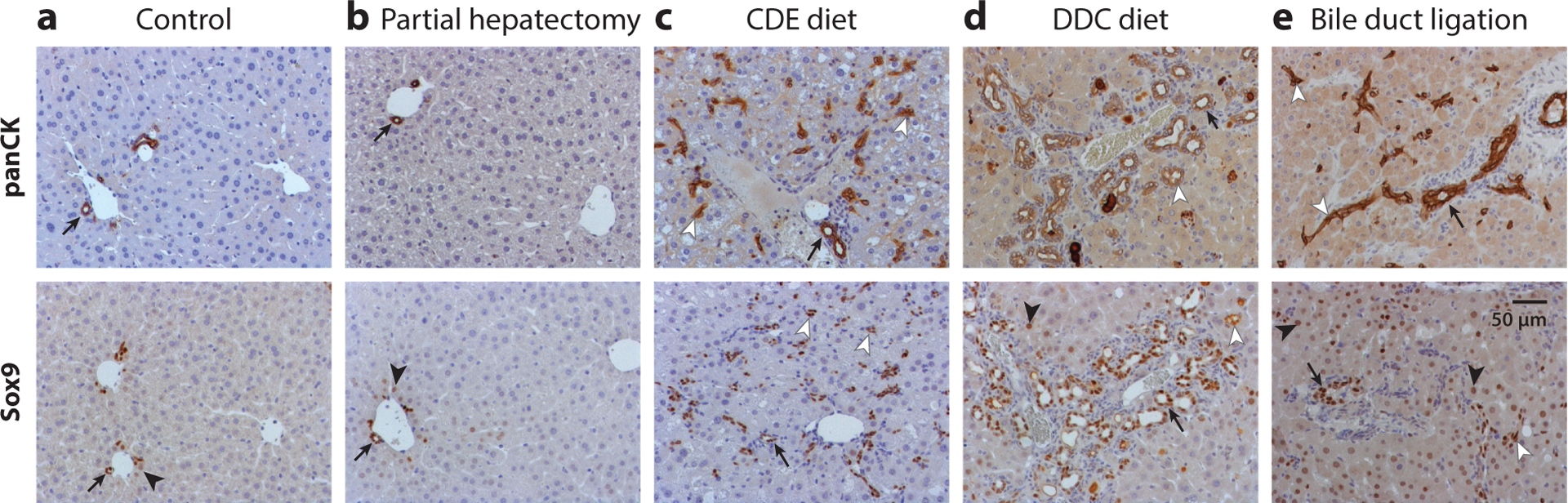
Images depict (a) normal mouse liver, and liver from a mouse (b) 1 day after two-thirds partial hepatectomy, (c) after 2 weeks of a choline-deficient, ethionine-supplemented (CDE) diet, (d) after 5 weeks of a 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet, and (e) 1 week after bile duct ligation. Liver tissues were stained with pan-cytokeratin (panCK) (top row) or Sox9 antibody (bottom row). Arrows denote bile ducts with luminal structures, lined by panCK- or Sox9-positive cells, while arrowheads denote Sox9-positive hepatocytes. (c–e) Ductular reactions are evinced by a large increase in the number of panCK- or Sox9-positive cells (white arrowheads). Dilation of luminal structures is observed after (d) the DDC diet, and (e) preexisting bile ducts are thickened, with increased numbers of panCK-positive cells, after bile duct ligation. A few Sox9-positive hepatocytes are observed adjacent to the portal vein in (a) normal liver, while this population is (d) modestly increased after administration of the DDC diet, and it is (e) hugely increased throughout the whole liver following bile duct ligation.
Figure 4.
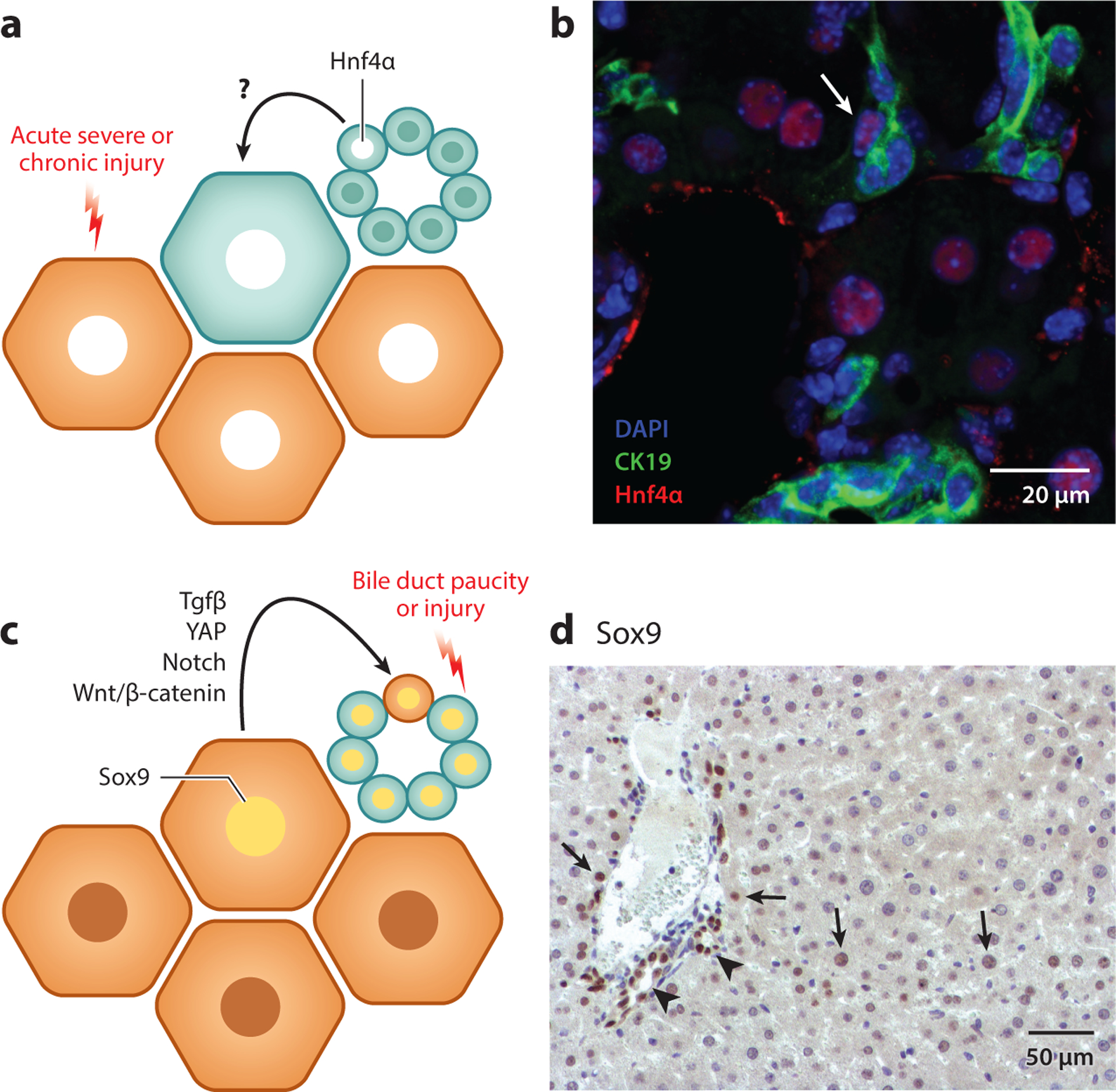
The transdifferentiation theory. (a) During acute severe or chronic liver injury in which hepatocyte (HC) function is significantly impaired, biliary epithelial cells (BECs) can directly transdifferentiate into HCs to mediate liver repair. During this process, BECs express HC markers such as Hnf4α. (b) The liver from a mouse fed a choline-deficient, ethionine-supplemented diet for 2 weeks, followed by 3 days of recovery on a normal diet, stained for DAPI (blue), cytokeratin (CK) 19 (green) and Hnf4α (red). A transdifferentiating cell that expressed both CK19 and Hnf4α is evident (white arrow). (c) Following bile duct paucity or severe injury to BECs, HCs can transdifferentiate into BECs, and during this process, HCs will express BEC markers, such as Sox9. Signaling pathways, including transforming growth factor-β (Tgfβ), yes-associated protein (YAP), Notch, and Wnt/β-catenin, have been identified as playing roles in promoting this process. (d) Liver from a mouse stained for Sox9 2 days after bile duct ligation reveals HCs that express Sox9 (arrows) in comparison to the presence of Sox9 in the BECs lining the bile ducts (arrowheads).
Currently, liver transplantation is the only reliable treatment for prolonging the lives of patients with advanced liver diseases. However, the number of donor livers is not sufficient to match an increasing demand (12). Therefore, alternative therapeutic options are desperately needed. Given the prevalence of DRs in human diseased liver and recent findings that suggest LPCs could be a reliable source of new HCs in diseased liver, promoting the differentiation of activated LPCs into parenchymal liver cells has garnered attention as a potential therapeutic option for patients with advanced liver diseases. However, the pathophysiology and molecular mechanisms underlying the DR and its subsequent differentiation remain largely unknown. This review summarizes the available models for studying LPC-driven liver regeneration and discusses updated findings from these models and their potential benefits and limitations for clinical application in end-stage liver diseases.
THE DUCTULAR REACTION IN HUMAN LIVER DISEASE
The activation of the DR is thought to be triggered by an impairment of the regenerative capacity of the differentiated epithelial cells of the liver during liver injury. In support of this theory, one study found that patients diagnosed with acute liver failure or severe liver impairment who exhibited severe loss (>50%) of HCs in combination with impaired proliferation of the remaining HCs developed a robust DR (13). This study also tracked the appearance of intermediate HCs [those that express the BEC markers cytokeratin (CK) 7 or CK19] and concluded that LPCs appear early but need approximately 1 week to differentiate into HCs (13). In line with this theory, the spontaneous recovery of patients with massive hepatic necrosis, a condition in which nearly all parenchymal cells are acutely lost, is theorized to occur partly via LPC-mediated liver regeneration (reviewed in Reference 14). There is also evidence of LPC-derived HCs in human cirrhosis patients with parenchyma extinction (15). However, even in the setting of acute liver failure, the degree of DR is positively correlated with liver stiffness and hepatic stellate cell activation (14, 16), demonstrating an intricate relationship between these various cell types (reviewed in Reference 17).
In chronic liver injury, the degree of the DR positively correlates with disease severity in a wide range of pathological settings (14, 18–21). In chronic hepatitis C virus (HCV) infection, an advanced stage of fibrosis was associated with higher numbers of activated hepatic stellate cells and LPCs, both in adult (22) and pediatric patients (23). In patients with nonalcoholic steatohepatitis (NASH), the degree of DR strongly correlated with HC replicative arrest, fibrosis stage, and extent of portal inflammation, specifically CD68+ macrophages and CD8+ lymphocytes (24,25). Furthermore, in patients with NASH, expansion of the DR reaction into the centrilobular region correlated with fibrosis stage and fibrosis progression (26). In pediatric NASH, the degree of proinflammatory macrophage polarization also correlated with disease severity, DR, and portal fibrosis (27).
Although the DR is observed in almost all forms of severe and chronic liver injury, there are thought to be disease-specific phenotypic differences (Figure 1b–e). The cells of the DR can differ in morphology, ranging from organized ductules with clear lumen to more disorganized strings of cells with no visible lumen, and the morphology varies depending on the etiology of the liver injury (reviewed in Reference 19). A recent study characterized the DR in the cholangiopathies primary sclerosing cholangitis (PSC) and primary biliary cholangitis (PBC) and found that while the DR was a prognostic marker in both conditions, the DR phenotype and activation of signaling pathways differed between PSC and PBC (28). Another study used laser capture microdissection to isolate cells of the DR from patients with HCV or PSC, and high-throughput RNA sequencing revealed numerous differences, including a neoangiogenesis signature in HCV in contrast to a profile of oxidative stress–related and proinflammatory gene expression in PSC (29).
There is evidence that LPCs themselves can secrete proinflammatory and profibrogenic cytokines (30). In alcoholic hepatitis, the expansion of the DR correlates with disease progression, and cells of the DR have been found to express chemokines and inflammatory mediators, promoting neutrophil infiltration in the periportal area (31). A recent study found that activation of noncanonical NF-κB signaling via RELB in BECs was important for BEC proliferation during the DR in a variety of pathologies, including PSC, PBC, NASH, autoimmune hepatitis, viral hepatitis, and alcoholic liver disease. In early-stage PBC and PSC, the expression of the cytokine lymphotoxin-β increased in BECs and induced RELB activation. A corresponding mouse model found that deletion of RELB reduced inflammatory cell infiltration and the expression of inflammatory cytokines (32), providing further evidence that cells of the DR can promote inflammation.
A subset of patients with chronic liver disease will go on to develop hepatocellular carcinoma (HCC). Chronic inflammation is known to be a driver of hepatic carcinogenesis (33). There is a long-standing debate about the role of LPCs in hepatic tumorigenesis (34). LPCs are thought to be a potential source of HCC due to the strong association between the DR and the progression of chronic liver disease and the fact that up to 50% of human HCCs express markers of BECs. Additionally, in humans approximately 55% of small cell dysplastic foci are composed of LPCs and intermediate HCs (35). Furthermore, proliferating peritumoral DR was found to strongly correlate with inflammation and fibrosis, and it was an independent prognostic factor for disease-free survival after surgical resection of HCC (36, 37). A proliferating peritumoral DR was also found to correlate with overall survival and disease-free survival posthepatectomy in patients with combined hepatocellular–cholangiocarcinoma (38). These data underscore the clear clinical significance of the DR in liver disease, providing the impetus to study the role of the DR in liver disease and tumorigenesis in a variety of animal models.
EVIDENCE FOR HEPATIC STEM CELLS IN FETAL AND ADULT LIVER
While the idea of an LPC population in the liver remains a controversial topic, the presence of LPCs in liver development is unequivocal (Figure 2a). The liver originates from the foregut endoderm, and during hepatic specification there is formation of the liver bud, which is composed of hepatoblasts that give rise to both HCs and BECs (for a full review of liver development see References 39, 40). A subset of hepatoblasts have been shown to have classical stem cell–like properties and have been termed hepatic stem cells (HpSCs) (39). Cells expressing c-Met and CD49f and negative for c-Kit, CD45, and TER119 isolated from embryonic day (E) 13.5 fetal mouse liver were found to self-renew in culture and give rise to HCs and BECs. In fact, during in vivo transplantation experiments, these cells were found to be multipotent by their capability of differentiating into pancreatic ductal and acinar cells or intestinal epithelial cells (41). Another group identified delta-like 1 homolog (Dlk1)–positive cell fractions isolated from E14.5 fetal mouse liver to be highly proliferative and found that it could differentiate into BECs and HCs in vitro (42). These cells gave rise to HCs when transplanted intrasplenically in vivo. In human fetal liver samples, CD117+;CD34+;CD90− cells were found to express both hepatic and biliary markers in vitro (43). Other groups identified cells that express markers such as CD133; CK8, 18, and 19; CD44H; telomerase; claudin 3; and albumin (44). Additional markers expressed included CD34, CD90, c-Kit, EpCAM, c-Met, vimentin, and SSEA-4 (45). These cells have displayed more than 100 population doublings in culture and the ability to differentiate into fat, bone, cartilage, and endothelial cells. When α-fetoprotein (AFP)+;CK19+;albumin+ HpSCs were isolated from E14 rat livers and transplanted into rats subjected to 70% partial hepatectomy (PHx), they transdifferentiated into both HCs and BECs and represented 23.5% of total liver mass after 6 months, suggesting continuous proliferation of the donor cells during the duration of the experiment (46). However, the question remains whether HpSCs are confined to fetal liver development or persist into adulthood.
A potential location for HpSCs in the adult liver is in the canals of Hering. Examination of human postnatal liver samples has revealed hybrid cells in the canals of Hering that express HpSC markers (44, 47, 48), such as BECs that weakly express Hnf4α and HCs that weakly express Hnf1β(49). In normal adult mice, a small fraction of cells expressing markers, such as EpCAM, CD133, CD13, and SOX9, have been isolated and reported to be adult LPCs (50–52), although the efficiency of these cells to form colonies and differentiate into HCs in vitro was reduced with age(53). In line with these results, single SOX9-positive ductal progenitor cells formed self-renewing organoids in culture (54). More recently, ST14 was identified as a marker of clonogenic cholangiocytes that gave rise to organoids that could be serially passaged, although it was not determined whether these ST14-positive BECs were localized to the canals of Hering (55). Studies using sub-lethal acetaminophen overdose have demonstrated through bromodeoxyuridine administration that the cells in the canals of Hering are label-retaining cells, a property characteristic of stem cell niches (56, 57). Likewise, cells in the canals of Hering have been shown to be regulated by Hedgehog signaling, similar to the progenitor compartments of other organs, such as skin, bone marrow, and intestine (58). A laminin-containing basement membrane is present in the canals of Hering (57, 59). Laminin has been shown to be important for the proliferation and maintenance of LPCs in culture (60) and surrounds LPCs during liver injury in vivo (61, 62). Additional morphological studies after severe liver injury in mice have indicated that LPCs may be derived from the canals of Hering (63) (Figures 2b and 3). Collectively, these data suggest the existence of a population of LPCs in the adult liver that functions in liver regeneration in specific models of liver injury rather than contributing to homeostasis in an adult liver.
EVIDENCE FOR LIVER PROGENITOR CELLS AND BILIARY EPITHELIAL CELL TO HEPATOCYTE DIFFERENTIATION IN RODENT MODELS
Original Description of Oval Cells in Rats
The first description of what came to be known as putative adult LPCs after hepatic injury was a study of liver injury in rats published in the 1950s (64). In a study cataloguing the histological changes following the administration of chemical carcinogens in rats, Dr. Emmanuel Farber(64) noted that a very early change was the proliferation of what he termed oval cells, which first appeared in the periportal area and over time expanded throughout the entire liver lobule, a phenomenon also described as a DR. These cells were presumed to be of BEC origin (64). It was noted that the expansion of these cells preceded liver regeneration, and it was speculated that oval cells were giving rise to a subset of HCs in hyperplastic liver nodules. It was further found that oval cells expressed fetal liver markers such as AFP (65). Studies of azo dye carcinogenesis similarly reported the transitioning of oval cells into HCs (66, 67), and it was suggested that oval cells function to provide new HCs during prolonged, severe liver injury. However, the origin and fate of oval cells was intensely debated (8). Other groups performed cell-labeling experiments with tritiated thymidine in multiple models of liver injury, including chemical carcinogenesis, bile duct ligation [a surgical procedure in which the common bile duct is ligated, leading to obstructive cholestasis, proliferation of BECs, and periportal fibrosis (68)], and in PHx in combination with carbon tetrachloride administration and argued that oval cells did not give rise to HCs and instead underwent removal by cell death (69–71).
The study of oval cells was furthered by the development of new rodent models, including the Solt–Farber model of hepatocarcinogenesis in which rats were injected with 2-acetylaminofluorene (2-AAF) to block HC proliferation and then subjected to PHx, which led to a massive expansion of oval cells (72). Administration of the biliary toxin methylene dianiline (DAPM) prior to the 2-AAF–PHx protocol blocked the expansion of oval cells and prevented the expression of AFP, providing further evidence that oval cells were derived from cholangiocytes(73). Pulse-chase labeling of oval cells with tritiated thymidine (74, 75) and careful immunohistochemical analysis (76) in the 2-AAF–PHx model demonstrated the conversion of oval cells into HCs, leading to the hypothesis that oval cells give rise to HCs only when HC proliferation is impaired in the context of liver injury, an idea that remains popular to this day (Figures 2 and 4). However, due to a lack of fate-tracing studies in rat models and an inability to use 2-AAF in mice, conclusive studies demonstrating that BECs are the source of LPCs and, eventually, HCs have continued to nag the field in general.
The Model Choline-Deficient, Ethionine-Supplemented Diet
Another model used to study oval cells in rats involved the combination of the hepatocarcinogen ethionine with choline deficiency. Prolonged choline deficiency induces liver tumors in rats (77,78). Ethionine is an analog of methionine and is extremely toxic to cells. Its incorporation into proteins reduces protein translation and inhibits DNA replication (79), and prolonged administration of ethionine also promotes the development of liver tumors in rats (64). Administration of a choline-deficient, ethionine-supplemented (CDE) diet in rats led to acute immune infiltration, cell necrosis, fat accumulation, and massive oval cell proliferation (80). The CDE diet model has since been adapted for use in mice, where it also gives rise to steatohepatitis and oval cell expansion (81). The efficiency and speed of the CDE diet in promoting the expansion of putative LPCs has made it a widely used model in this field (Figure 3c). Liver intravital microscopy revealed a compromised blood–bile barrier in mice fed the CDE diet for 4 days, which was accompanied by disruption of tight junctions as well as liver injury (82). Deposition of matrix around the portal tract occurs as early as 3 days after feeding a CDE diet and increases over the course of injury, but expansion of the DR begins around day 7 of CDE diet–induced liver injury and continues to increase over time (62, 83).
It has been theorized that hepatic injury–mediated inflammatory signaling is required for the initiation of the DR. Previously it was shown that a type 1 T helper (Th1) cell–mediated cellular immune response is necessary for a DR by demonstrating impaired LPC activation in CDE diet– fed mice that were immunocompromised due to a defect in T cell–mediated immune response genes such as CD3E, Rag2, and IFNR (84, 85). In fact, bone marrow cell transplantation or the injection of recombinant tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) is sufficient to induce a DR in mice even in the absence of liver injury, supporting the role of macrophages and TWEAK in triggering the DR (86).
Although it has long been speculated that oval cells give rise to HCs in the CDE diet injury model, the advent of lineage tracing in mice has allowed researchers to directly test this hypothesis. First, it was shown that the cells of the DR observed after CDE diet feeding are of BEC origin; one group labeled ductal plate cells (an embryonic structure consisting of a single-layered sleeve of Sox9-positive cells around the periportal mesenchyme that gives rise to cholangiocytes and periportal HCs) with yellow fluorescent protein (YFP) via Sox9-CreERT2 and found that after CDE diet administration the CK19-positive oval cells also expressed YFP, indicating ductal plate origin (87). Other lineage tracing systems used to label BEC-derived oval cells in CDE diet–fed mice include osteopontin (OPN)-CreERT2 (59), Foxl1-Cre (88), Hnf1-CreERT2 (89), and Krt19-CreERT2 (90–92). While these BECs do not contribute to HCs during homeostasis or during toxic or surgical loss of liver mass (54, 59, 90), several studies demonstrated that LPCs give rise to HCs after CDE diet–induced liver injury, which is followed by recovery on normal chow, with the proportions of LPC-derived HCs ranging from 1.86% (89) to 2.45% (59) to 29% in one study in which analysis was limited to mice that lost more than 14% of their initial body weight upon exposure to the CDE diet (88). However, other groups performing lineage tracing in the CDE diet model have found that BECs do not significantly contribute to HCs. One study utilizing Sox9-CreERT2 to label BECs found no BEC-derived HCs after CDE diet and recovery (54). Another group utilized Krt19-CreERT2 to label BECs and failed to detect BEC-derived HCs after CDE diet and recovery (90). Several groups have utilized adeno-associated virus serotype 8 (AAV8), a virus that preferentially infects HCs (93), to deliver Cre recombinase driven by an HC-specific promoter. With this technique, more than 99% of HCs can be genetically labeled (90, 92–94). In these HC lineage-tracing studies, several groups found no contribution of BECs to HCs during CDE diet and recovery (90, 94).
Comparing 2-AAF–PH studies in rats with CDE studies in mice, a potential explanation for the presence of very-few-to-no BEC-derived HCs in mice is that HC proliferation is not impaired during a CDE diet in mice (54, 92, 94). One group utilized the Ah-Cre system to conditionally delete the E3 ubiquitin ligase Mdm2 in up to 98% of HCs, leading to overexpression of p21, HC senescence, HC injury, and widespread DRs. LPCs isolated from CDE diet–fed mice were transfected with a green fluorescent protein (GFP) plasmid and transplanted into Mdm2 HC null mice, and after 3 months, GFP-positive HCs and BECs represented approximately 15% of liver tissue, suggesting LPC-to-HC differentiation (95). In a follow-up study, Krt19-CreERT2 was utilized to label BECs, and animals were injected with AAV8-p21 to overexpress p21 in HCs, followed by CDE diet and recovery, which resulted in approximately 15.3% of HCs being derived from BECs. In the same study, the authors used AAV8-thyroid binding globulin (TBG)-Cre to delete β1-integrin specifically in HCs, which were simultaneously labeled with the marker tdTomato. When these animals were given a methionine- and choline-deficient (MCD) diet to induce liver injury, followed by a recovery on a normal diet, 20–30% of HCs were found to be tdTomato-negative, indicating they did not originate from a preexisting HC. These results were confirmed in Krt19-CreERT mice with tdTomato-labeled BECs given RNA interference against Itgb1 (β1-integrin) and subjected to an MCD diet followed by recovery, in which tdTomato-positive HCs were observed (91). In a recent study from our group, mice with HC-specific enhanced YFP (eYFP) labeling and simultaneous deletion of β-catenin via AAV8-TBG-Cre that were subjected to a CDE diet displayed a profound impairment of HC proliferation (92). Following recovery on a normal diet for 2 weeks, there was expansion of β-catenin-positive, eYFP-negative HCs, accounting for approximately 20% of periportal HCs. Interestingly, at between 3 and 7 days of recovery on a normal diet after a CDE diet, small β-catenin-positive HCs were observed along with β-catenin+;CK19+;Hnf4α+ cells, suggesting they were BECs in the process of differentiating into HCs. Positive lineage tracing via Krt19-CreERT2 mice with tdTomato-labeled BECs injected with Ctnnb1 RNA interference and placed on a CDE diet, followed by recovery, resulted in tdTomato-positive HCs, confirming that BECs were giving rise to HCs in this model (92). Together, these results suggest that when HC proliferation is impaired in the CDE diet model, BECs and LPCs give rise to HCs.
The DDC Diet Model
Another model that has been used to study DRs and potential BEC-to-HC differentiation is the 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet, which causes porphyrin plugs in the bile, leading to bile duct obstruction, DR, cholangitis, and periportal fibrosis (96) (Figure 3d). A notable feature of the DDC diet, in addition to the robust proliferation of HCs and BECs, is the appearance of biphenotypic HCs that express Hnf4α and biliary marker A6 (97). However, similar to work with the CDE diet, different groups have reported conflicting conclusions regarding LPC-to-HC differentiation. Groups utilizing Krt19-CreERT2 mice (90) or Sox9-CreERT2 mice (54) with labeled BECs found no BEC-derived HCs after 4 weeks of DDC diet–induced liver injury(54) or 2–3 weeks of DDC diet followed by recovery on a normal diet (90). However, when HC proliferation was impaired through the deletion of HC-specific β1-integrin or overexpression of p21 and animals were subjected to a DDC diet and recovery, significant numbers of BEC-derived HCs were observed (91). One group did lineage tracing of Lgr5+ cells and found that while Lgr5 was not readily detectable in normal liver, treatment with a DDC diet led to the labeling of both BECs and HCs. When single Lgr5+ cells were isolated, they could be cultured into self-renewing organoids expressing markers of both BECs and HCs. These organoids could differentiate into HCs when transplanted in vivo, suggesting that these Lgr5+ cells marked damage-induced LPCs(98). In a recent study, Krt19-CreERT2 mice with labeled BECs were fed a DDC diet for 24 weeks, and that led to 9.1% of HCs being derived from BECs (99). This study provides proof of concept that extended liver insult may yield BEC-derived HCs even in the absence of any genetic manipulation, and, thus, it supports such phenomena occurring during chronic liver injury in humans.
It has also been reported that epigenetic chromatin remodeling plays important roles in LPC activation in models such as the DDC diet. Polycomb repressive complexes (PRCs) have been reported to be crucial regulators of the proliferation of hepatoblasts (100, 101) and LPCs (102) via repression of Bim1 expression (100). Jalan-Sakrikar et al. (102) suggested that Hedgehog signaling is an upstream regulator of enhancer of zeste homolog 2 (EZH2)–PRC during proliferation of LPCs in DDC-fed mice. Recently, it was reported in both zebrafish and mice that chromatin readers of the bromodomain and extraterminal (BET) protein family regulate LPC activation in a myca-dependent manner (103). Altogether, chromatin modification appears necessary for activation of a quiescent LPC, although further studies are needed. Likewise, the distinct roles of chromatin remodeling in the reprogramming or dedifferentiation of parenchymal cells during regeneration require further investigation.
The Thioacetamide Model of Liver Injury
Another model that has been used to study BEC-to-HC differentiation is thioacetamide (TAA)-induced liver injury. TAA is processed into a toxic metabolite in HCs and induces centrilobular necrosis (104). Chronic administration of TAA in drinking water leads to the development of cirrhosis (105). A recent study utilized AAV8-TBG-Cre to label HCs with tdTomato and simultaneously deleted β1-integrin in HCs. When these mice were exposed to TAA for 3 weeks, followed by recovery on normal diet, 20–30% of HCs were tdTomato-negative (91). A more recent study subjected Krt19-CreERT2 mice with labeled BECs to TAA treatment for 24 weeks and found that approximately 10% of HCs were BEC derived (99). Excitingly, the authors of this study identified biphenotypic cells with BEC morphology that expressed CK19 and Hnf4α, and these accounted for approximately 3.87% of the ductular cells. These cells had reduced expression of mature BEC markers such as protein kinase Cζ and primary cilia, and they did not express Lgr5 or AFP, which led the authors to claim that these cells were BECs directly converting into HCs as opposed to undergoing differentiation through an LPC intermediate stage (99). However, the expression of more widely accepted markers of LPCs was not assessed in this study, and the renewal capacity of these cells was not tested in vitro. Hence, the question remains whether these biphenotypic cells represent the differentiation of mature BECs or activation of LPCs (Figures 2b and 4).
TRANSDIFFERENTIATION OF HEPATOCYTES INTO BILIARY EPITHELIAL CELLS: EVIDENCE FOR THE FACULTATIVE STEM CELL HYPOTHESIS
An alternative theory to that of LPCs is that there is no stem cell population in the liver and, instead, HCs and cholangiocytes serve as facultative stem cells (7, 106), meaning that the normal cell population dedifferentiates in response to injury. By this theory, cholangiocytes directly transdifferentiate into HCs and vice versa to mediate liver regeneration when normal proliferation of one of these populations is impaired (Figures 3c,d and 4). In the case of HC-to-cholangiocyte transdifferentiation, this theory is supported both in vivo and in organoid cultures. Rats lacking the enzyme dipeptidyl peptidase IV (DPPIV) were given retrorsine, an agent that blocks HC proliferation, and then transplanted with DPPIV-positive HCs, which resulted in livers with colonies of donor-derived DPPIV-positive HCs. When organoid cultures were derived from these hybrid livers, cells resembling BECs on the surface of the organoid culture were found to be DPPIV-positive, indicating HC origin (107). This finding was further confirmed when DPPIV-negative rats were given retrorsine and subjected to PHx followed by transplantation with DPPIV-positive HCs. When these chimeric livers were pretreated with the biliary toxin DAPM and subjected to bile duct ligation, the appearance of DPPIV-positive ductules was observed (108). A similar study found that GFP-labeled HCs transplanted into rats after bile duct ligation adopted a cholangiocyte phenotype, indicating HC-to-BEC transdifferentiation (109).
The DDC diet has also been used to study HC-to-cholangiocyte differentiation. When mice were given AAV8-TBG-Cre to label HCs with YFP and subjected to the DDC diet or bile duct ligation, YFP-positive cells with biliary morphology that expressed BEC markers were detected(97). A different study found that Sox9+;EpCAM− cells isolated from DDC diet–fed mice could differentiate into HCs or BECs in vitro (110). Yet another study utilized β-galactosidase-labeled HCs transplanted into mice subjected to retrorsine treatment, PHx, and carbon tetrachloride (CCl4) or DDC injury and observed β-galactosidase-positive BECs (111). A recent study identified tdTomato-positive BECs following DDC diet–induced liver injury in mice expressing tdTomato only in telomerase reverse transcriptase–positive random HCs (112).
In terms of the signaling pathways that drive HC-to-BEC transdifferentiation, the crucial role of Notch signaling has been confirmed by numerous groups. HC-specific overexpression of the Notch intracellular domain (NICD) in the absence of injury was sufficient to induce HC-to-BEC conversion, and HC-specific deletion of Rbpj (the principal effector of Notch signaling) in mice fed a DDC diet significantly reduced the number of HC-derived BECs, suggesting that Notch signaling is required for HC reprogramming (97). Another group found that in chronic DDC diet–induced liver injury, the majority of cholangiocytes were derived from HCs, and the DR in these mice was significantly increased by NICD overexpression and significantly reduced by deletion of Hes1, a Notch target gene (113). Notch signaling was also shown to be required for in vitro differentiation of LPCs into BECs, and in vivo blockage of Notch receptor cleavage during administration of a DDC diet reduced the extent of the DR, which was attributed to impaired HC dedifferentiation (114). Importantly, the overexpression of YAP in HCs was sufficient to dedifferentiate HCs into ductal-like cells, and Notch signaling was found to be a downstream target of YAP signaling during the dedifferentiation process (115). Similarly, another group described periportal HCs that express Sox9 and differentiate into ductal cells after DDC diet–induced liver injury (116). Another signaling pathway that may be involved in HC-to-BEC transdifferentiation is the Wnt/β-catenin signaling pathway. Mice overexpressing a stabilized form of β-catenin showed increased HC expression of BEC markers after DDC diet–induced liver injury, along with improvement in cholestasis (117), and blocking Wnt secretion from BECs via deletion of Wntless reduced the number of HCs expressing BEC markers following administration of a DDC diet (118).
Intriguingly, some lineage-tracing studies performed on mice fed a DDC diet or subjected to bile duct ligation detected no evidence of HC-derived BECs (93). Other groups showed that HCs that adopt a biliary phenotype during injury revert back to the HC fate upon the cessation of injury, arguing for the process to be metaplasia rather than transdifferentiation (119). Interestingly, a recent paper demonstrated the transdifferentiation of HCs into BECs in a mouse model of human Alagille syndrome. Mice with Alb-Cre–mediated deletion of Rbpj and Hnf6 both in HCs and cholangiocytes are born lacking peripheral bile ducts, but they spontaneously form intrahepatic bile ducts by approximately 4 months of age. Labeling HCs in these mice with GFP via injection of AAV8 encoding flippase under an HC-specific promoter led to the demonstration of newly derived GFP-positive BECs, indicating their HC origin (120). The study further proved that this transdifferentiation of HCs into BECs required transforming growth factor-β (Tgfβ) signaling, as Alb-Cre+/−;Rbpjf/f;Hnf6f/f;Tgfbr2f/f mice failed to form de novo peripheral bile ducts. These HC-derived bile ducts were stable throughout life, indicating true HC transdifferentiation (120).
LIVER PROGENITOR CELLS IN LIVER CANCER
In addition to determining their role in liver regeneration, the role of LPCs in the development of liver cancer has been an area of interest (Figure 5). The extent of expansion of the DR during liver injury correlates with the severity of liver injury, and LPCs are known to secrete a host of profibrogenic and proinflammatory factors (30). Combined with the putative stem cell–like properties of LPCs, this has led to the speculation that LPCs during prolonged chronic liver injury can give rise to cancer (Figure 5). Recently, the theory of cancer stem cells (CSCs)—a small fraction of tumor cells that are capable of tumor formation and growth, self-renewal, and differentiation into mature cells—has gained support, including from the identification of CSCs in liver cancer (121–123). Most HCCs and intrahepatic cholangiocarcinomas (ICCs) arise in conditions of chronic injury and inflammation, such as viral hepatitis, nonalcoholic fatty liver disease, alcoholic liver disease, and PSC. The expansion of LPCs during chronic liver injuries may potentiate their transformation and subsequent development into liver cancers; indeed, many HCCs express markers of LPCs, such as AFP and CK19 (35). One of the first studies to address whether LPCs could function as CSCs involved the isolation, purification, and generation of oval cell lines from rats fed a CDE diet. These cell lines were found to be nontransforming and nontumorigenic when injected into nude mice; however, when transfected with the rasH oncogene and injected into nude mice, they gave rise to tumors resembling well-differentiated trabecular HCC (124).
Figure 5.
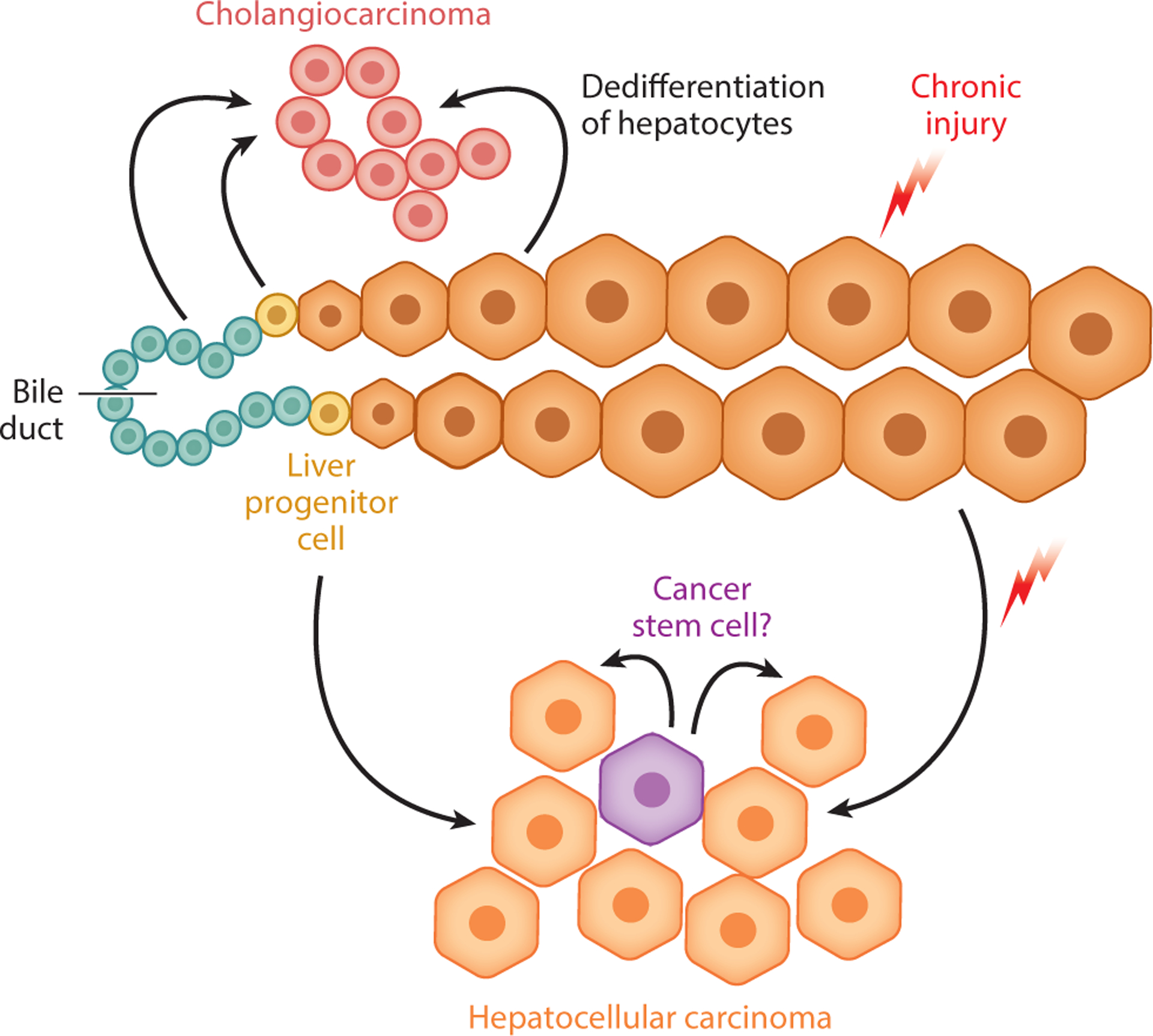
Liver progenitor cells (LPCs) in liver cancer. Generally, the two major forms of adult liver cancer are thought to arise from their corresponding epithelial cell counterparts, often in the setting of chronic injury and inflammation: hepatocellular carcinoma from hepatocytes and cholangiocarcinoma from biliary epithelial cells. At the same time, the question remains whether LPCs serve as another source of both hepatocellular carcinoma and cholangiocarcinoma due to their stem cell–like properties and their long-term activation in the setting of liver injury. In addition, studies have identified potential cancer stem cells in liver cancer, which may or may not be related in origin to LPCs.
Analysis of human and mouse tumors has provided some evidence that LPCs may be the origin of certain liver cancers. Analysis of an EpCAM+;AFP+ HCC subtype revealed that these cells exhibited properties of CSCs and gave rise to invasive HCC when injected into nonobese diabetic, severe combined immunodeficient (NOD-SCID) mice (125). Another group identified a subgroup of human HCC that expressed markers of fetal hepatoblasts and oval cells and indicated a poor prognosis (126). Hepatic stem cells isolated from E13.5 mice, transfected with Bmi1 and constitutively active β-catenin, and transplanted into NOD-SCID mice, gave rise to tumors with combined features of HCC and cholangiocarcinoma (127). Mutations in isocitrate dehydrogenase 1 (IDH1) or IDH2 are common in ICC, and it was shown that mutations in IDH1 or IDH2 prevented LPCs from differentiating into HCs through suppression of Hnf4α. Furthermore, liver-specific expression of IDH2R172K and KrasG12D led to the development of ICC with simultaneous oval cell expansion, although it was unclear whether these tumors developed from LPCs or HCs (128). Another study identified human HCC cells that expressed stem cell markers and lacked expression of TGFβ receptor type II and embryonic liver fodrin (ELF), a molecule crucial in TGFβ signaling. Mice heterozygous for Elf developed spontaneous HCC driven by interleukin (IL)-6 signaling (129). IL-6 signaling was also identified as an autocrine signaling pathway in HCC progenitor cells isolated from mice treated with diethylnitrosamine (DEN) followed by retrorsine and CCl4, and these cells were found to be transcriptomically similar to oval cells. However, the authors demonstrated that their HCC progenitor cells gave rise to HCC after transplantation into major urinary protein (MUP)–urokinase-type plasminogen activator (uPA) mice, whereas oval cells did not, leading the authors to theorize that their HCC progenitor cells originated from dedifferentiated HCs (130).
Along these lines, although many cases of human or rodent HCC exhibit markers of LPCs, there is a debate over whether the cell that gave rise to the cancer was actually an LPC or a dedifferentiated HC. More recent lineage-tracing experiments have led to mixed results about whether LPCs are the origin of HCC. One group utilized Sox9-CreERT2 to label BECs with eYFP in mice with HC-specific expression of human unconventional prefoldin RPB5 interactor, which leads to NAD+-deficit-induced DNA damage and tumorigenesis. By 65 weeks of age, 46% of tumors in mice were heterogeneously positive, while 8% were completely positive, for eYFP. It was determined that 54% of the lesions that were positive for eYFP were benign dysplasia, regenerative nodules, or hepatocellular adenomas, and only 14% of HCCs originated from LPCs. However, when the authors performed lineage tracing in other models of HCC, including injection with DEN followed by CCl4 injections and in mice with HC-specific loss of Mdr2, they found that all HCCs originated from HCs (131). Similarly, Sox9-CreERT2 was used to label BECs with YFP and in tumorigenesis models including DEN injections, and no YFP-positive tumors were detected in MUP-uPA mice fed a high-fat diet or in mice with streptozotocin-induced diabetes fed a high-fat diet (116). In mice fed TAA for 52 weeks, all tumors were found to be derived from endogenous HCs, not BECs or BEC-derived HCs (99). Finally, when OPN-CreERT2 was used to label BECs with YFP and mice were subjected to repeated injections of DEN, CCl4, or a combination of the two, no YFP-positive HCCs were detected. Furthermore, when AAV8-TBG-Cre was used to label HCs with YFP, the LPCs within the tumors induced by DEN or CCl4 were found to be YFP-positive, indicating their derivation from HCs (132). The apparent lack of LPC-derived tumors may signify that LPCs are more resistant to oncogenic transformation or that HCs are more prone to accumulating DNA damage, as they are the cell type that express enzymes that metabolize chemicals such as DEN and CCl4 into toxic by-products. The former hypothesis is not supported by the observation that LPCs, hepatoblasts, and HCs transfected with H-Ras and SV40LT all acquired CSC properties and initiated fast-growing tumors (133).
Furthermore, multiple experimental mouse models have described the dedifferentiation of HCs into LPCs following activation of Notch signaling or YAP1 through inactivation of the Hippo pathway kinases MST1 and MST2 or the cell junction regulator Merlin (Nf2) (115, 134, 135). These models eventually give rise to liver cancer, including HCC and ICC, originating from HC-derived LPCs expressing bipotential progenitor cell markers. In another study, HCs were labeled with β-galactosidase through Alb-CreERT2, while in a separate cohort of mice cholangiocytes were labeled through Krt19-CreERT2. These mice were injected with TAA to induce ICC, and it was found that only in Alb-CreERT2 mice were the ICC cells positive for β-galactosidase, indicating HC origin (136). Another group labeled HCs via AAV8-Ttr-Cre injection into Rosa-eYFP mice and induced ICC via hydrodynamic tail vein injection of an NICD plasmid and a human AKT overexpression plasmid. ICC tumor cells were found to express eYFP in this model, indicating they originated from HCs (137). However, the contribution of HCs to ICC appears to be context dependent. A study utilized Alb-CreERT2 mice to express oncogenic KrasG12D with simultaneous homozygous deletion of Pten to induce carcinogenesis. Administration of tamoxifen at postnatal day 10 resulted in Cre expression in BECs and HCs and led to the development of ICC. However, tamoxifen administration at 8 weeks of age activated Cre expression only in HCs and led to the development of HCC. Finally, tamoxifen administration to Krt19-CreERT2;LSL:KrasG12D/+;Ptenflox/flox mice led to the development of ICC, leading the authors to conclude that the expression of oncogenic KrasG12D combined with Pten loss only in BECs, not HCs, led to the development of ICC (138). Similar results were seen during Alb-Cre-driven expression of Notch-2 intracellular domain in both HCs and BECs, which led to the development of HCC. When tumorigenesis was enhanced by DEN, a subset of mice also developed ICCs, which were determined to be of BEC origin (139). In contrast, a different group found that in the TAA model of ICC, Kupffer cells express Jagged 1 to activate Notch signaling in HCs, inducing their transdifferentiation into BECs (140). Together, these results indicate that ICC can result from multiple cell types, and under certain conditions HCs may transdifferentiate to BECs to give rise to ICC (Figure 5).
BILIARY EPITHELIAL CELL–DERIVED LIVER PROGENITOR CELL–MEDIATED REGENERATION IN THE ZEBRAFISH MODEL SYSTEM
In 2014, prior to LPC-to-HC conversion being verified in the mouse system through positive lineage tracing (91), three groups reported robust conversion of BEC-derived LPCs into functional HCs in zebrafish by pharmacogenetic pan-HC ablation (141–143). Using a model of acute zebrafish injury employing transgenic lines expressing bacterial nitroreductase fused with a fluorescent protein under the HC-specific fabp10a promoter, treatment with metronidazole allowed for selective ablation of more than 98% of nitroreductase-positive HCs. Remnant Notch-positive BECs were shown to proliferate and to express HC-specific genes, such as fabp10a and hnf4a. Subsequently, this population lost expression of BEC-specific genes to express more mature HC markers, indicating the conversion of BECs to mature HCs (Figure 6b). The transition of BECs into HCs was verified by lineage tracing (144). Interestingly, during HC ablation, the intrahepatic tubular biliary structure completely collapsed and was subsequently reestablished during the regeneration process (141, 145). Therefore, this zebrafish model is amenable as an in vivo system for studying bipotent LPC differentiation into BECs as well as into HCs (Figure 6b). Because following HC ablation more than 95% of HCs are derived from LPCs during repopulation, the larval zebrafish model provides a homogeneous, large-scale, fate-changing in vivo system to examine the molecular mechanisms of LPC generation (i.e., DR) and differentiation using pharmacogenetic approaches, and thus has distinct advantages over rodent models.
Figure 6.
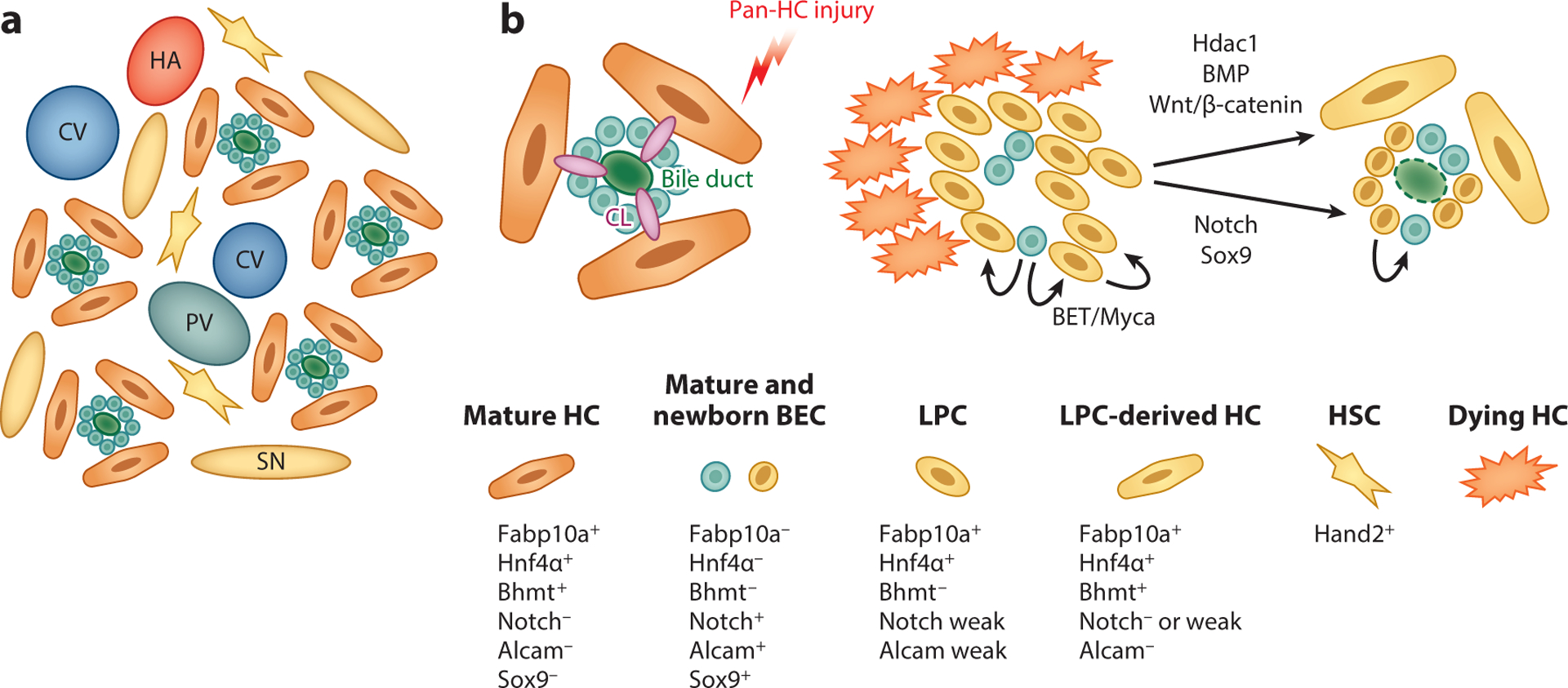
Schematic of zebrafish liver anatomy and liver progenitor cell (LPC)–driven liver regeneration. (a) Schematic of zebrafish liver architecture. Unlike the hepatic zonation found in the mammalian liver, portal veins (PVs), central veins (CVs), and hepatic arteries (HAs) are randomly distributed in the zebrafish liver. Hepatocytes (HCs) are arranged in tubules around bile ducts interspersed with hepatic stellate cells (HSCs) and surrounded by fenestrated sinusoidal endothelium (SN). (b) During near-total HC ablation, bile canaliculi (CL) and biliary networks completely collapse, and remnant biliary epithelial cells (BECs) are packed without a central lumen. Subsequently, BECs rapidly dedifferentiate into LPCs, which coexpress Notch signaling targets, Alcam, Fabp10a, and Hnf4α. This is followed by redifferentiation of LPCs into functional HCs and BECs. Using this model, signaling pathways, including BET/Myca, Hdac1/Sox9, BMP, and Wnt/β-catenin have been identified as regulators of LPC-mediated liver regeneration.
Making use of this model, several studies have described the identification of small molecules regulating LPC-to-HC conversion during regeneration. He et al. (142) reported that pretreatment with a γ-secretase inhibitor combined with homozygous sox9b mutation impairs BEC-derived HC formation, suggesting that activation of the Notch/Sox9b axis is required for this transition. These data are consistent with impaired DR after Notch inhibition in rodent fibrosis models since treatment with a Notch inhibitor occurred before ablation, which may affect biliary homeostasis and injury-mediated biliary proliferation, as shown in rodents (146–148). However, given that Notch or Sox9 inhibition promotes LPC-to-HC differentiation in vitro (149), it needs to be further clarified whether Notch/Sox9 is indispensable for the dedifferentiation (i.e., proliferation) of BECs into LPCs or if Notch activity is actually required for LPC-to-HC differentiation in vivo. It has additionally been reported that BMP signaling is required for maturation of BEC-derived HCs and transient proliferation of BECs to reconstitute the biliary network during regeneration (150) (Figure 6b). More recently, by using targeted chemical screening in zebrafish, Hdac1 was identified as a crucial regulator of the differentiation of LPCs into HCs and BECs through the repression of, respectively, Sox9b and the Cdk8/Fbxw7/Notch3 signaling axis (Figure 6b). In addition, this novel zebrafish finding was further confirmed in a mouse system as well as in samples of diseased human liver, demonstrating the potential of zebrafish models of LPC-driven liver regeneration to be relevant systems for studying human LPC pathophysiology in diseased liver (145).
However, there are caveats to using a zebrafish system of LPC-driven liver regeneration as a preclinical model for end-stage human liver disease with DR. First, anatomically, in the zebrafish liver all HCs are directly adjacent to vascular sinusoids through the apical membrane, and there is no hepatic zonation (151–153) (Figure 6a). In contrast, the mammalian liver is composed of three zones based on the location within the porto-central axis in each hepatic lobule, with zone 1 consisting of HCs around the portal triad, zone 3 consisting of HCs around the central vein, and zone 2 consisting of the HCs between zones 1 and 3 (154) (Figure 1a). Such compartmentalization not only allows for the functional partition of labor but also restricts particular toxicant-mediated injuries to specific zones, allowing surviving cells in other zones to repair the liver (154). Moreover, to maintain systemic metabolic homeostasis, mammalian hepatic zonation and the heterogeneity of HC gene expression are tightly regulated by various physiological factors and signaling pathways, such as the Wnt/β-catenin pathway, which is considered one of the master regulators of hepatic zonation (92, 155, 156). The lack of hepatic zonation in the zebrafish liver means that findings relevant to understanding zebrafish liver regeneration may not be able to be extrapolated to higher vertebrates. Second, the zebrafish embryonic liver is still developing during the analysis used in the HC ablation model. Although zebrafish liver is functional at 3.5 days post-fertilization when metronidazole is administered for HC ablation, HCs and BECs in a non-injured liver are highly proliferative, and the overall liver size is still growing at this time (157). This aspect of developing and regenerating zebrafish liver needs to be carefully considered when translating the results to adult mammalian liver regeneration. Third, thymus development is completed at approximately 3 weeks post-fertilization in zebrafish, and, therefore, prior to this point, zebrafish lack a competent adaptive immune system (158, 159). Thus, the adaptive immune response is not fully mature and functional in the zebrafish HC ablation model. Given the critical importance of the Th1-mediated adaptive immune response in the mammalian DR and cell fate conversion during the regeneration of various organs (84, 85, 160), this larval model needs to be carefully extrapolated when interpreting LPC-driven regeneration that is especially relevant to immune cells. The contribution of LPCs to the zebrafish model is minimal compared with repopulation by preexisting BECs and HCs during mammalian liver injury and regeneration. Furthermore, the mammalian liver contains distinct regenerating microenvironments, including dissimilar cell populations to those of the zebrafish model (161, 162). Altogether, the established zebrafish model of LPC-driven liver regeneration may provide a powerful platform and synergizes well with mammalian regenerative models to examine the fundamental pathophysiological mechanisms of LPCs in diseased liver.
CONCLUSIONS AND FUTURE DIRECTIONS
The literature indicates the existence of remarkable cellular plasticity within the epithelial cells of the liver during hepatic injury, such that under specific circumstances the HCs and BECs can change their cellular fate to mediate liver regeneration (Table 1). Whether this cell type conversion is direct transdifferentiation or goes through a progenitor cell intermediate remains to be definitively elucidated. It also remains to be proven whether there are only small subsets of HCs and BECs primed to undergo cell fate changes, and thus these subsets represent facultative stem cells or a progenitor cell compartment. Based on their diverse origins and their dynamic cellular fate in diseased liver, LPC-specific markers (or a marker that is absent in mature BECs and HCs but is expressed in a cell undergoing a cell fate conversion) are not currently available. Therefore, it is not feasible to distinguish between LPC activation and BEC proliferation or hyperplasia initiated to repair damage to biliary structures. Although several markers have been suggested as LPC markers, such as Foxl1 (88), Lgr5 (163), Trop2 (51), and Ncam (164), none of these are universally sensitive or specific for detecting all LPC populations regardless of etiology. There is no doubt that the identification of LPC-specific markers would greatly advance this field; however, we also need to acknowledge that such concrete and common LPC markers may not exist.
Table 1.
Summary of hepatic injury models of transdifferentiation
| Type of transdifferentiation | Species | Stimulus (injury) | Model or type of lineage tracing | Transdifferentiation? | Percentage of liver repopulated with transdifferentiated cells | Reference(s) |
|---|---|---|---|---|---|---|
| Bile duct ligation | Tritiated thymidine tracing | No | 0 | 69 | ||
| PHx + BDL; PHx + CDE; PHx + CC14 | Tritiated thymidine tracing | No | 0 | 70 | ||
| 2-AAF + PHx | Tritiated thymidine tracing | No | NA | 71 | ||
| 2-AAF + PHx | Tritiated thymidine tracing + histological analysis | Yes | NA | 74–76 | ||
| 3’-methyl-4-dimethylaminoazobenzene | Glucose 6-phosphatase activity + histological analysis | Yes | NA | 66, 67 | ||
| PHx | No | 0 | 54 | |||
| AAV8-Ttr:Cre;Rosa26:tdTom,ato | ||||||
| PHx | Opn-CreERT2;Rosa26-eYFP | No | 0 | 59 | ||
| PHx | No | 0 | 90, 93, 94 | |||
| AAV8-TBG-Cre;Rosa26-eYFP | ||||||
| PHx + CDE | No | <1 | 54 | |||
| AAV8-Ttr-Cre;Rosa26-tdTomato | ||||||
| CDE | No | <1 | 90, 92–94 | |||
| AAV8-TBG-Cre;Rosa26-eYFP | ||||||
| CDE | OPN-CreERT2;Rosa26-eYFP | Yes | <2.45 | 59 | ||
| CDE | Hnf1βl-CreERT2;Rosa26-YFP or LacZ | Yes | <1.86 | 89 | ||
| CDE (5% sucrose in drinking water) | Foxl1-Cre;Rosa26-eYFP | Yes | <26 | 88 | ||
| AAV8-TBG:p21 + CDE | Krt19-Cre;Rosa26-tdTomato | Yes | <15.3 | 91 | ||
| AAV8-TBG-Cre; Cmnb1(f/f) + CDE | Yes | <20 | 92 | |||
| AAV8-TBG-Cre;Rosa26-eYFP | ||||||
| AAVS-TBG-Cre; Itgb1f(f/f) + MCD | Krt19-Cre;Rosa26-tdTomato | Yes | 20–30 | 91 | ||
| DDC | No | 0 | 54 | |||
| AAV8-Ttr-Cre;Rosa26-tdTomato | ||||||
| DDC | No | <1 | 90 | |||
| AAV8-TBG-Cre;Rosa26-eYFP | ||||||
| DDC | Hnf1β-CreERT2;Rosa26-eYFP or LacZ | No | 0 | 89 | ||
| DDC (24 weeks) | Krt19-Cre;Rosa26-mGFP | Yes | ~20 | 99 | ||
| AAV8-TBG:Cre; hgb1(f/f) + DDC | Krtl9-Cre;Rosa26-tdTomato | Yes | ~25 | 91 | ||
| Ah-Cre-Mdm2(f/f) | LPC transplantation into Ah-Cre;Mdm2(f’f) mice | Yes | <15 | 95 | ||
| PHx + CC14 | No | <1 | 54 | |||
| AAV8-Ttr-Cre;Rosa26-tdTomato | ||||||
| CCl4 | Krt19-Cre;Rosa26-tdTomato | No | 0 | 59 | ||
| CCl4 | No | <1 | 90, 93, 94 | |||
| AAV8-TEG-Cre;Rosa26-eYFP | ||||||
| TAA (24 weeks) | Krt19-Cre;Rosa26-mGFP | Yes | ~10 | 99 | ||
| Rat | DAPM + BDL | Dipeptidyl peptidase IV + HC tracing | Yes | 30–60 | 108, 109 | |
| BDL | AAV8-TBG-Cre;Rosa26-eYFP | Yes | 2–62 | 97 | ||
| DDC | AAV8-TBG-Cre;Rosa26-eYFP | Yes | 14–48 | 97 | ||
| PHx | AAV8-TBG-Cre;Rosa26-eYFP | No | 0 | 97 | ||
| DDC | Yes | <1.9 | 111 | |||
| Mx1-Cre;Rosa26R + polyinosinic-polycytidylic acid | ||||||
| DDC + DAPM | Yes | <4.7 | 111 | |||
| Mx1-Cre;Rom26R + polyinosinic-polycytidylic acid | ||||||
| CC14 (20 weeks) | Yes | ~20 | 111 | |||
| Mx1-Cre;Rom26R + polyinosinic-polycytidylic acid | ||||||
| TAA | Yes | <4.3 | 111 | |||
| Mx1-Cre;Rosa26R + polyinosinic-polycytidylic acid | ||||||
| TERThigh HC ablation + DDC | Tert-CreERT2;Rosa26-LSL-Tomato | Yes | 5–7 | 112 | ||
| DDC | CK19+/Hnf4α+ co-staining | Yes | ~10 | 118 | ||
| DDC | Yes | 30–40 | 113 | |||
| Krtl9-Cre;Rosa26-eYFP | ||||||
| PHx | No | 0 | 113 | |||
| Krt19-Cre;Rosa26-eYFP | ||||||
| CCl4 | No | <0.14 | 113 | |||
| Krt19-Cre;Rosa26-eYFP | ||||||
| PHx | AAV8-Ttr-Cre;Rosa26-eYFP | No | 0 | 93 | ||
| DDC | AAV8-Ttr-Cre;Rosa26-eYFP | No | 0 | 93 | ||
| BDL | AAV8-Ttr-Cre;Rosa26-eYFP | No | 0 | 93 | ||
| CC14 | AAV8-Ttr-Cre; Rosa26-eYFP | No | 0 | 93 | ||
| Alb-Cre;Rbpj(f/f);Hnf6(f/f) | Alb-Cre;Rbpj(f/f);Hnf6(f/f);Rosa26-ZG; AAV8-Ttr-Flp | Yes | ~100 | 120 |
Abbreviations: 2-AAF, 2-acetylaminofluorene; AAV8, adeno-associated virus serotype 8; BDL, bile duct ligation; BEC, biliary epithelial cell; CCl4, carbon tetrachloride; CDE, choline-deficient ethionine-supplemented; DAPM, methylene dianiline; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; HC, hepatocyte; LPC, liver progenitor cell; MCD, methionine- and choline-deficient; NA, not applicable; PHx, partial hepatectomy; TAA, thioacetamide.
It also remains to be seen whether targeting LPCs for differentiation into HCs represents a viable therapeutic strategy for chronic liver disease. Only a subset of patients with chronic liver disease will progress to end-stage liver disease and liver cancer, and the role or function of LPCs in these processes remains less understood. Most studies have focused on proving the phenomenon of cell fate conversion and sought to identify the molecular mechanisms underlying this process. While these experiments are essential for developing druggable targets, few studies have examined the long-term consequences of promoting LPC differentiation. Since the DR is known to promote fibrogenesis and inflammation, will targeting the DR to induce LPC differentiation into HCs be sufficient to restore the hepatic microenvironment? The technology for targeting BECs in the DR is limited and, hence, a major roadblock in developing new therapeutics. Many studies involved in promoting LPC differentiation into HCs have removed the underlying liver injury to allow recovery and enhance LPC differentiation. While new therapies for several hepatic diseases are being discovered and may eventually remove the stimulus for the DR, facilitating LPC differentiation into a useful cell type may be yet another viable alternative.
In the context of liver cancer, the question of whether HCs dedifferentiate into LPCs that subsequently give rise to tumors is an area of debate. It remains unclear whether HCs first dedifferentiate into LPCs that subsequently differentiate into BECs or whether they undergo a gradual reprogramming to complete transdifferentiation, since both theories are congruent with the presence of cells expressing both HC and BEC markers. This question is muddled by the very nature of many genetic models of liver cancer, which specifically rely on direct injury or genetic manipulation of HCs or BECs. More studies are necessary to determine the cell of origin in models of liver cancer that induce tumorigenesis without direct genetic manipulation of a specific cell type in the liver. There is also the question of whether promoting LPC-to-HC differentiation as a treatment for chronic liver disease will impact LPC-mediated tumorigenesis.
In conclusion, the liver is a remarkable organ with a diverse array of regenerative responses to hepatic injury. It can exhibit activation of LPCs and mutual transdifferentiation of BECs and HCs to serve as facultative stem cells to ensure liver regeneration, but these changes can be associated with alterations in the hepatic microenvironment. Advances in the field of hepatic cellular plasticity may hold great promise for developing therapies for patients with chronic liver disease.
ACKNOWLEDGMENTS
This work is supported by grants from the US National Institutes of Health to S.P.M. (DK62277, DK100287, and CA204586) and J.O.R. (T32EB0010216 and 1F31DK115017). S.P.M is the Endowed Chair for Experimental Pathology, University of Pittsburgh School of Medicine.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Footnotes
The Annual Review of Pathology: Mechanisms of Disease is online at pathol.annualreviews.org
LITERATURE CITED
- 1.Stoick-Cooper CL, Moon RT, Weidinger G. 2007. Advances in signaling in vertebrate regeneration as a prelude to regenerative medicine. Genes Dev. 21:1292–315 [DOI] [PubMed] [Google Scholar]
- 2.Cordero-Espinoza L, Huch M. 2018. The balancing act of the liver: tissue regeneration versus fibrosis. J. Clin. Investig 128:85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vivien CJ, Hudson JE, Porrello ER. 2016. Evolution, comparative biology and ontogeny of vertebrate heart regeneration. NPJ Regen. Med 1:16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferretti P, Zhang F, O’Neill P. 2003. Changes in spinal cord regenerative ability through phylogenesis and development: lessons to be learnt. Dev. Dyn 226:245–56 [DOI] [PubMed] [Google Scholar]
- 5.Wynn TA. 2008. Cellular and molecular mechanisms of fibrosis. J. Pathol 214:199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forbes SJ, Newsome PN. 2016. Liver regeneration—mechanisms and models to clinical application. Nat. Rev. Gastroenterol. Hepatol 13:473–85 [DOI] [PubMed] [Google Scholar]
- 7.Michalopoulos GK, Khan Z. 2015. Liver stem cells: experimental findings and implications for human liver disease. Gastroenterology 149:876–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fausto N, Campbell JS. 2003. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech. Dev 120:117–30 [DOI] [PubMed] [Google Scholar]
- 9.Tabibian JH, Masyuk AI, Masyuk TV, O’Hara SP, LaRusso NF. 2013. Physiology of cholangiocytes. Compr. Physiol 3:541–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roskams TA, Theise ND, Balabaud C, Bhagat G, Bhathal PS, et al. 2004. Nomenclature of the finer branches of the biliary tree: canals, ductules, and ductular reactions in human livers. Hepatology 39:1739– 45 [DOI] [PubMed] [Google Scholar]
- 11.Sato K, Marzioni M, Meng F, Francis H, Glaser S, Alpini G. 2019. Ductular reaction in liver diseases: pathological mechanisms and translational significances. Hepatology 69:420–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim WR, Lake JR, Smith JM, Schladt DP, Skeans MA, et al. 2018. OPTN/SRTR 2016 annual data report: liver. Am. J. Transplant 18(Suppl. 1):172–253 [DOI] [PubMed] [Google Scholar]
- 13.Katoonizadeh A, Nevens F, Verslype C, Pirenne J, Roskams T. 2006. Liver regeneration in acute severe liver impairment: a clinicopathological correlation study. Liver Int. 26:1225–33 [DOI] [PubMed] [Google Scholar]
- 14.Weng HL, Cai X, Yuan X, Liebe R, Dooley S, Li H, Wang TL. 2015. Two sides of one coin: massive hepatic necrosis and progenitor cell–mediated regeneration in acute liver failure. Front. Physiol 6:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stueck AE, Wanless IR. 2015. Hepatocyte buds derived from progenitor cells repopulate regions of parenchymal extinction in human cirrhosis. Hepatology 61:1696–707 [DOI] [PubMed] [Google Scholar]
- 16.Dechêne A, Sowa JP, Gieseler RK, Jochum C, Bechmann LP, et al. 2010. Acute liver failure is associated with elevated liver stiffness and hepatic stellate cell activation. Hepatology 52:1008–16 [DOI] [PubMed] [Google Scholar]
- 17.Williams MJ, Clouston AD, Forbes SJ. 2014. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology 146:349–56 [DOI] [PubMed] [Google Scholar]
- 18.Lowes KN, Brennan BA, Yeoh GC, Olynyk JK. 1999. Oval cell numbers in human chronic liver diseases are directly related to disease severity. Am. J. Pathol 154:537–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gouw AS, Clouston AD, Theise ND. 2011. Ductular reactions in human liver: diversity at the interface. Hepatology 54:1853–63 [DOI] [PubMed] [Google Scholar]
- 20.Machado MV, Michelotti GA, Pereira TA, Xie G, Premont R, et al. 2015. Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J. Hepatol 63:962–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guldiken N, Kobazi Ensari G, Lahiri P, Couchy G, Preisinger C, et al. 2016. Keratin 23 is a stress-inducible marker of mouse and human ductular reaction in liver disease. J. Hepatol 65:552–59 [DOI] [PubMed] [Google Scholar]
- 22.Helal TESA Ehsan NA, Radwan NA Abdelsameea E. 2018. Relationship between hepatic progenitor cells and stellate cells in chronic hepatitis C genotype 4. APMIS 126:14–20 [DOI] [PubMed] [Google Scholar]
- 23.El-Araby HA, Ehsan NA, Konsowa HA, Abd-Elaati BM, Sira AM. 2015. Hepatic progenitor cells in children with chronic hepatitis C: correlation with histopathology, viremia, and treatment response. Eur. J. Gastroenterol. Hepatol 27:561–69 [DOI] [PubMed] [Google Scholar]
- 24.Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander-Tetri BA, et al. 2007. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology 133:80–90 [DOI] [PubMed] [Google Scholar]
- 25.Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, et al. 2014. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 59:1393–405 [DOI] [PubMed] [Google Scholar]
- 26.Zhao L, Westerhoff M, Pai RK, Choi WT, Gao ZH, Hart J. 2018. Centrilobular ductular reaction correlates with fibrosis stage and fibrosis progression in non-alcoholic steatohepatitis. Mod. Pathol 31:150–59 [DOI] [PubMed] [Google Scholar]
- 27.Carpino G, Nobili V, Renzi A, De Stefanis C, Stronati L, et al. 2016. Macrophage activation in pediatric nonalcoholic fatty liver disease (NAFLD) correlates with hepatic progenitor cell response via Wnt3a pathway. PLOS ONE 11:e0157246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carpino G, Cardinale V, Folseraas T, Overi D, Floreani A, et al. 2018. Hepatic stem/progenitor cell activation differs between primary sclerosing and primary biliary cholangitis. Am. J. Pathol 188:627– 39 [DOI] [PubMed] [Google Scholar]
- 29.Govaere O, Cockell S, Van Haele M, Wouters J, Van Delm W, et al. 2019. High-throughput sequencing identifies aetiology-dependent differences in ductular reaction in human chronic liver disease. J. Pathol 248:66–76 [DOI] [PubMed] [Google Scholar]
- 30.Svegliati-Baroni G, De Minicis S, Marzioni M. 2008. Hepatic fibrogenesis in response to chronic liver injury: novel insights on the role of cell-to-cell interaction and transition. Liver Int. 28:1052–64 [DOI] [PubMed] [Google Scholar]
- 31.Aguilar-Bravo B, Rodrigo-Torres D, Ariño S, Coll M, Pose E, et al. 2019. Ductular reaction cells display an inflammatory profile and recruit neutrophils in alcoholic hepatitis. Hepatology 69:2180–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elßner C, Goeppert B, Longerich T, Scherr AL, Stindt J, et al. 2019. Nuclear translocation of RELB is increased in diseased human liver and promotes ductular reaction and biliary fibrosis in mice. Gastroenterology 156:1190–205.e14 [DOI] [PubMed] [Google Scholar]
- 33.Yu LX, Ling Y, Wang HY. 2018. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol 2:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sia D, Villanueva A, Friedman SL, Llovet JM. 2017. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 152:745–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roskams T 2006. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene 25:3818–22 [DOI] [PubMed] [Google Scholar]
- 36.Ye F, Jing YY, Guo SW, Yu GF, Fan QM, et al. 2014. Proliferative ductular reactions correlate with hepatic progenitor cell and predict recurrence in HCC patients after curative resection. Cell Biosci. 4:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu M, Xie F, Qian G, Jing Y, Zhang S, et al. 2014. Peritumoral ductular reaction: a poor postoperative prognostic factor for hepatocellular carcinoma. BMC Cancer 14:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai X, Zhai J, Kaplan DE, Zhang Y, Zhou L, et al. 2012. Background progenitor activation is associated with recurrence after hepatectomy of combined hepatocellular–cholangiocarcinoma. Hepatology 56:1804–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gordillo M, Evans T, Gouon-Evans V. 2015. Orchestrating liver development. Development 142:2094– 108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gérard C, Tys J, Lemaigre FP. 2017. Gene regulatory networks in differentiation and direct reprogramming of hepatic cells. Semin. Cell Dev. Biol 66:43–50 [DOI] [PubMed] [Google Scholar]
- 41.Suzuki A, Zheng YW, Kaneko S, Onodera M, Fukao K, et al. 2002. Clonal identification and characterization of self-renewing pluripotent stem cells in the developing liver. J. Cell Biol 156:173–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanimizu N, Nishikawa M, Saito H, Tsujimura T, Miyajima A. 2003. Isolation of hepatoblasts based on the expression of Dlk/Pref-1. J. Cell Sci 116:1775–86 [DOI] [PubMed] [Google Scholar]
- 43.Nava S, Westgren M, Jaksch M, Tibell A, Broomé U, et al. 2005. Characterization of cells in the developing human liver. Differentiation 73:249–60 [DOI] [PubMed] [Google Scholar]
- 44.Schmelzer E, Zhang L, Bruce A, Wauthier E, Ludlow J, et al. 2007. Human hepatic stem cells from fetal and postnatal donors. J. Exp. Med 204:1973–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dan YY, Riehle KJ, Lazaro C, Teoh N, Haque J, et al. 2006. Isolation of multipotent progenitor cells from human fetal liver capable of differentiating into liver and mesenchymal lineages. PNAS 103:9912– 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oertel M, Menthena A, Dabeva MD, Shafritz DA. 2006. Cell competition leads to a high level of normal liver reconstitution by transplanted fetal liver stem/progenitor cells. Gastroenterology 130:507–20 [DOI] [PubMed] [Google Scholar]
- 47.Turner R, Lozoya O, Wang Y, Cardinale V, Gaudio E, et al. 2011. Human hepatic stem cell and maturational liver lineage biology. Hepatology 53:1035–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardinale V, Wang Y, Carpino G, Cui CB, Gatto M, et al. 2011. Multipotent stem/progenitor cells in human biliary tree give rise to hepatocytes, cholangiocytes, and pancreatic islets. Hepatology 54:2159–72 [DOI] [PubMed] [Google Scholar]
- 49.Isse K, Lesniak A, Grama K, Maier J, Specht S, et al. 2013. Preexisting epithelial diversity in normal human livers: a tissue-tethered cytometric analysis in portal/periportal epithelial cells. Hepatology 57:1632– 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamiya A, Kakinuma S, Yamazaki Y, Nakauchi H. 2009. Enrichment and clonal culture of progenitor cells during mouse postnatal liver development in mice. Gastroenterology 137:1114–26.e14 [DOI] [PubMed] [Google Scholar]
- 51.Okabe M, Tsukahara Y, Tanaka M, Suzuki K, Saito S, et al. 2009. Potential hepatic stem cells reside in EpCAM+ cells of normal and injured mouse liver. Development 136:1951–60 [DOI] [PubMed] [Google Scholar]
- 52.Dorrell C, Erker L, Schug J, Kopp JL, Canaday PS, et al. 2011. Prospective isolation of a bipotential clonogenic liver progenitor cell in adult mice. Genes Dev. 25:1193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanimizu N, Kobayashi S, Ichinohe N, Mitaka T. 2014. Downregulation of miR122 by grainyhead-like 2 restricts the hepatocytic differentiation potential of adult liver progenitor cells. Development 141:4448– 56 [DOI] [PubMed] [Google Scholar]
- 54.Tarlow BD, Finegold MJ, Grompe M. 2014. Clonal tracing of Sox9+ liver progenitors in mouse oval cell injury. Hepatology 60:278–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li B, Dorrell C, Canaday PS, Pelz C, Haft A, et al. 2017. Adult mouse liver contains two distinct populations of cholangiocytes. Stem Cell Rep. 9:478–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuwahara R, Kofman AV, Landis CS, Swenson ES, Barendswaard E, Theise ND. 2008. The hepatic stem cell niche: identification by label-retaining cell assay. Hepatology 47:1994–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kordes C, Häussinger D. 2013. Hepatic stem cell niches. J. Clin. Investig 123:1874–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sicklick JK, Li YX, Melhem A, Schmelzer E, Zdanowicz M, et al. 2006. Hedgehog signaling maintains resident hepatic progenitors throughout life. Am. J. Physiol. Gastrointest. Liver Physiol 290:G859– 70 [DOI] [PubMed] [Google Scholar]
- 59.Español-Suñer R, Carpentier R, Van Hul N, Legry V, Achouri Y, et al. 2012. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology 143:1564–75.e7 [DOI] [PubMed] [Google Scholar]
- 60.Clayton E, Forbes SJ. 2009. The isolation and in vitro expansion of hepatic Sca-1 progenitor cells. Biochem. Biophys. Res. Commun 381:549–53 [DOI] [PubMed] [Google Scholar]
- 61.Lorenzini S, Bird TG, Boulter L, Bellamy C, Samuel K, et al. 2010. Characterisation of a stereotypical cellular and extracellular adult liver progenitor cell niche in rodents and diseased human liver. Gut 59:645–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van Hul NK, Abarca-Quinones J, Sempoux C, Horsmans Y, Leclercq IA. 2009. Relation between liver progenitor cell expansion and extracellular matrix deposition in a CDE-induced murine model of chronic liver injury. Hepatology 49:1625–35 [DOI] [PubMed] [Google Scholar]
- 63.Factor VM, Radaeva SA, Thorgeirsson SS. 1994. Origin and fate of oval cells in Dipin-induced hepatocarcinogenesis in the mouse. Am. J. Pathol 145:409–22 [PMC free article] [PubMed] [Google Scholar]
- 64.Farber E 1956. Similarities in the sequence of early histological changes induced in the liver of the rat by ethionine, 2-acetylamino-fluorene, and 3′-methyl-4-dimethylaminoazobenzene. Cancer Res. 16:142–48 [PubMed] [Google Scholar]
- 65.Dempo K, Chisaka N, Yoshida Y, Kaneko A, Onoé T. 1975. Immunofluorescent study on α-fetoprotein-producing cells in the early stage of 3′-methyl-4-dimethylaminoazobenzene carcinogenesis. Cancer Res. 35:1282–87 [PubMed] [Google Scholar]
- 66.Inaoka Y 1967. Significance of the so-called oval cell proliferation during azo-dye hepatocarcinogenesis. Gan 58:355–66 [PubMed] [Google Scholar]
- 67.Ogawa K, Minase T, Onhoe T. 1974. Demonstration of glucose 6-phosphatase activity in the oval cells of rat liver and the significance of the oval cells in azo dye carcinogenesis. Cancer Res. 34:3379–86 [PubMed] [Google Scholar]
- 68.Tag CG, Sauer-Lehnen S, Weiskirchen S, Borkham-Kamphorst E, Tolba RH, et al. 2015. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J. Vis. Exp 10:e52438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rubin E 1964. The origin and fate of proliferated bile ductular cells. Exp. Mol. Pathol 3:279–86 [DOI] [PubMed] [Google Scholar]
- 70.Grisham JW, Porta EA. 1964. Origin and fate of proliferated hepatic ductal cells in the rat: electron microscopic and autoradiographic studies. Exp. Mol. Pathol 3:242–61 [DOI] [PubMed] [Google Scholar]
- 71.Tatematsu M, Ho RH, Kaku T, Ekem JK, Farber E. 1984. Studies on the proliferation and fate of oval cells in the liver of rats treated with 2-acetylaminofluorene and partial hepatectomy. Am. J. Pathol 114:418–30 [PMC free article] [PubMed] [Google Scholar]
- 72.Solt D, Farber E. 1976. New principle for the analysis of chemical carcinogenesis. Nature 263:701–3 [Google Scholar]
- 73.Petersen BE, Zajac VF, Michalopoulos GK. 1997. Bile ductular damage induced by methylene dianiline inhibits oval cell activation. Am. J. Pathol 151:905–9 [PMC free article] [PubMed] [Google Scholar]
- 74.Evarts RP, Nagy P, Marsden E, Thorgeirsson SS. 1987. A precursor-product relationship exists between oval cells and hepatocytes in rat liver. Carcinogenesis 8:1737–40 [DOI] [PubMed] [Google Scholar]
- 75.Evarts RP, Nagy P, Nakatsukasa H, Marsden E, Thorgeirsson SS. 1989. In vivo differentiation of rat liver oval cells into hepatocytes. Cancer Res. 49:1541–47 [PubMed] [Google Scholar]
- 76.Alison M, Golding M, Lalani EN, Nagy P, Thorgeirsson S, Sarraf C. 1997. Wholesale hepatocytic differentiation in the rat from ductular oval cells, the progeny of biliary stem cells. J. Hepatol 26:343– 52 [DOI] [PubMed] [Google Scholar]
- 77.Copeland DH, Salmon WD. 1946. The occurrence of neoplasms in the liver, lungs, and other tissues of rats as a result of prolonged choline deficiency. Am. J. Pathol 22:1059–79 [PubMed] [Google Scholar]
- 78.Newberne PM, Camargo JL, Clark AJ. 1982. Choline deficiency, partial hepatectomy, and liver tumors in rats and mice. Toxicol. Pathol 10:95–106 [DOI] [PubMed] [Google Scholar]
- 79.Alix JH. 1982. Molecular aspects of the in vivo and in vitro effects of ethionine, an analog of methionine. Microbiol. Rev 46:281–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shinozuka H, Lombardi B, Sell S, Iammarino RM. 1978. Early histological and functional alterations of ethionine liver carcinogenesis in rats fed a choline-deficient diet. Cancer Res. 38:1092–98 [PubMed] [Google Scholar]
- 81.Akhurst B, Croager EJ, Farley-Roche CA, Ong JK, Dumble ML, et al. 2001. A modified choline-deficient, ethionine-supplemented diet protocol effectively induces oval cells in mouse liver. Hepatology 34:519–22 [DOI] [PubMed] [Google Scholar]
- 82.Pradhan-Sundd T, Vats R, Russell JM, Singh S, Michael AA, et al. 2018. Dysregulated bile transporters and impaired tight junctions during chronic liver injury in mice. Gastroenterology 155:1218–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Knight B, Matthews VB, Akhurst B, Croager EJ, Klinken E, et al. 2005. Liver inflammation and cytokine production, but not acute phase protein synthesis, accompany the adult liver progenitor (oval) cell response to chronic liver injury. Immunol. Cell Biol 83:364–74 [DOI] [PubMed] [Google Scholar]
- 84.Knight B, Akhurst B, Matthews VB, Ruddell RG, Ramm GA, et al. 2007. Attenuated liver progenitor (oval) cell and fibrogenic responses to the choline deficient, ethionine supplemented diet in the BALB/c inbred strain of mice. J. Hepatol 46:134–41 [DOI] [PubMed] [Google Scholar]
- 85.Strick-Marchand H, Masse GX, Weiss MC, Di Santo JP. 2008. Lymphocytes support oval cell– dependent liver regeneration. J. Immunol 181:2764–71 [DOI] [PubMed] [Google Scholar]
- 86.Bird TG, Lu WY, Boulter L, Gordon-Keylock S, Ridgway RA, et al. 2013. Bone marrow injection stimulates hepatic ductular reactions in the absence of injury via macrophage-mediated TWEAK signaling. PNAS 110:6542–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carpentier R, Suñer RE, van Hul N, Kopp JL, Beaudry JB, et al. 2011. Embryonic ductal plate cells give rise to cholangiocytes, periportal hepatocytes, and adult liver progenitor cells. Gastroenterology 141:1432–38.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shin S, Upadhyay N, Greenbaum LE, Kaestner KH. 2015. Ablation of Foxl1-Cre–labeled hepatic progenitor cells and their descendants impairs recovery of mice from liver injury. Gastroenterology 148:192– 202.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rodrigo-Torres D, Affò S, Coll M, Morales-Ibanez O, Millán C, et al. 2014. The biliary epithelium gives rise to liver progenitor cells. Hepatology 60:1367–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yanger K, Knigin D, Zong Y, Maggs L, Gu G, et al. 2014. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell 15:340–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Raven A, Lu WY, Man TY, Ferreira-Gonzalez S, O’Duibhir E, et al. 2017. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 547:350–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Russell JO, Lu WY, Okabe H, Abrams M, Oertel M, et al. 2019. Hepatocyte-specific β-catenin deletion during severe liver injury provokes cholangiocytes to differentiate into hepatocytes. Hepatology 69:742– 59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Malato Y, Naqvi S, Schürmann N, Ng R, Wang B, et al. 2011. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J. Clin. Investig 121:4850–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schaub JR, Malato Y, Gormond C, Willenbring H. 2014. Evidence against a stem cell origin of new hepatocytes in a common mouse model of chronic liver injury. Cell Rep. 8:933–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lu WY, Bird TG, Boulter L, Tsuchiya A, Cole AM, et al. 2015. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat. Cell Biol 17:971–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fickert P, Stöger U, Fuchsbichler A, Moustafa T, Marschall HU, et al. 2007. A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am. J. Pathol 171:525–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, et al. 2013. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 27:719–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huch M, Dorrell C, Boj SF, van Es JH, Li VS, et al. 2013. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 494:247–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Deng X, Zhang X, Li W, Feng RX, Li L, et al. 2018. Chronic liver injury induces conversion of biliary epithelial cells into hepatocytes. Cell Stem Cell 23:114–22.e3 [DOI] [PubMed] [Google Scholar]
- 100.Aoki R, Chiba T, Miyagi S, Negishi M, Konuma T, et al. 2010. The polycomb group gene product Ezh2 regulates proliferation and differentiation of murine hepatic stem/progenitor cells. J. Hepatol 52:854–63 [DOI] [PubMed] [Google Scholar]
- 101.Koike H, Ouchi R, Ueno Y, Nakata S, Obana Y, et al. 2014. Polycomb group protein Ezh2 regulates hepatic progenitor cell proliferation and differentiation in murine embryonic liver. PLOS ONE 9:e104776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jalan-Sakrikar N, De Assuncao TM, Lu J, Almada LL, Lomberk G, et al. 2016. Hedgehog signaling overcomes an EZH2-dependent epigenetic barrier to promote cholangiocyte expansion. PLOS ONE 11:e0168266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ko S, Choi TY, Russell JO, So J, Monga SPS, Shin D. 2016. Bromodomain and extraterminal (BET) proteins regulate biliary-driven liver regeneration. J. Hepatol 64:316–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hunter AL, Holscher MA, Neal RA. 1977. Thioacetamide-induced hepatic necrosis. I. Involvement of the mixed-function oxidase enzyme system. J. Pharmacol. Exp. Ther 200:439–48 [PubMed] [Google Scholar]
- 105.Müller A, Machnik F, Zimmermann T, Schubert H. 1988. Thioacetamide-induced cirrhosis-like liver lesions in rats—usefulness and reliability of this animal model. Exp. Pathol 34:229–36 [DOI] [PubMed] [Google Scholar]
- 106.Gracz AD, Fuller MK, Wang F, Li L, Stelzner M, et al. 2013. CD24 and CD44 mark human intestinal epithelial cell populations with characteristics of active and facultative stem cells. Stem Cells 31:2024–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Michalopoulos GK, Bowen WC, Mulè K, Lopez-Talavera JC, Mars W. 2002. Hepatocytes undergo phenotypic transformation to biliary epithelium in organoid cultures. Hepatology 36:278–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Michalopoulos GK, Barua L, Bowen WC. 2005. Transdifferentiation of rat hepatocytes into biliary cells after bile duct ligation and toxic biliary injury. Hepatology 41:535–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yovchev MI, Locker J, Oertel M. 2016. Biliary fibrosis drives liver repopulation and phenotype transition of transplanted hepatocytes. J. Hepatol 64:1348–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tanimizu N, Nishikawa Y, Ichinohe N, Akiyama H, Mitaka T. 2014. Sry HMG box protein 9-positive (Sox9+) epithelial cell adhesion molecule-negative (EpCAM−) biphenotypic cells derived from hepatocytes are involved in mouse liver regeneration. J. Biol. Chem 289:7589–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nagahama Y, Sone M, Chen X, Okada Y, Yamamoto M, et al. 2014. Contributions of hepatocytes and bile ductular cells in ductular reactions and remodeling of the biliary system after chronic liver injury. Am. J. Pathol 184:3001–12 [DOI] [PubMed] [Google Scholar]
- 112.Lin S, Nascimento EM, Gajera CR, Chen L, Neuhöfer P, et al. 2018. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature 556:244–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sekiya S, Suzuki A. 2014. Hepatocytes, rather than cholangiocytes, can be the major source of primitive ductules in the chronically injured mouse liver. Am. J. Pathol 184:1468–78 [DOI] [PubMed] [Google Scholar]
- 114.Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, et al. 2012. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat. Med 18:572– 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, et al. 2014. Hippo pathway activity influences liver cell fate. Cell 157:1324–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Font-Burgada J, Shalapour S, Ramaswamy S, Hsueh B, Rossell D, et al. 2015. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell 162:766–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Thompson MD, Awuah P, Singh S, Monga SP. 2010. Disparate cellular basis of improved liver repair in β-catenin-overexpressing mice after long-term exposure to 3,5-diethoxycarbonyl-1,4-dihydrocollidine. Am. J. Pathol 2010. 177:1812–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Okabe H, Yang J, Sylakowski K, Yovchev M, Miyagawa Y, et al. 2016. Wnt signaling regulates hepatobiliary repair following cholestatic liver injury in mice. Hepatology 64:1652–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tarlow BD, Pelz C, Naugler WE, Wakefield L, Wilson EM, et al. 2014. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 15:605–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schaub JR, Huppert KA, Kurial SNT, Hsu BY, Cast AE, et al. 2018. De novo formation of the biliary system by TGFβ-mediated hepatocyte transdifferentiation. Nature 557:247–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, et al. 2006. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell–like properties. Hepatology 44:240–51 [DOI] [PubMed] [Google Scholar]
- 122.Ma S, Chan KW, Hu L, Lee TK, Wo JY, et al. 2007. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 132:2542–56 [DOI] [PubMed] [Google Scholar]
- 123.Marquardt JU, Raggi C, Andersen JB, Seo D, Avital I, et al. 2011. Human hepatic cancer stem cells are characterized by common stemness traits and diverse oncogenic pathways. Hepatology 54:1031–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Braun L, Goyette M, Yaswen P, Thompson NL, Fausto N. 1987. Growth in culture and tumorigenicity after transfection with the ras oncogene of liver epithelial cells from carcinogen-treated rats. Cancer Res. 47:4116–24 [PubMed] [Google Scholar]
- 125.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, et al. 2009. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 136:1012–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, et al. 2006. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med 12:410–16 [DOI] [PubMed] [Google Scholar]
- 127.Chiba T, Zheng YW, Kita K, Yokosuka O, Saisho H, et al. 2007. Enhanced self-renewal capability in hepatic stem/progenitor cells drives cancer initiation. Gastroenterology 133:937–50 [DOI] [PubMed] [Google Scholar]
- 128.Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, et al. 2014. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 513:110–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tang Y, Kitisin K, Jogunoori W, Li C, Deng CX, et al. 2008. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-β and IL-6 signaling. PNAS 105:2445–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, et al. 2013. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 155:384–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tummala KS, Brandt M, Teijeiro A, Graña O, Schwabe RF, et al. 2017. Hepatocellular carcinomas originate predominantly from hepatocytes and benign lesions from hepatic progenitor cells. Cell Rep. 19:584– 600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mu X, Español-Suñer R, Mederacke I, Affò S, Manco R, et al. 2015. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J. Clin. Investig 125:3891–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Holczbauer Á, Factor VM, Andersen JB, Marquardt JU, Kleiner DE, et al. 2013. Modeling pathogenesis of primary liver cancer in lineage-specific mouse cell types. Gastroenterology 145:221–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Benhamouche S, Curto M, Saotome I, Gladden AB, Liu CH, et al. 2010. Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev. 24:1718–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fitamant J, Kottakis F, Benhamouche S, Tian HS, Chuvin N, et al. 2015. YAP inhibition restores hepatocyte differentiation in advanced HCC, leading to tumor regression. Cell Rep. 17:1692–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sekiya S, Suzuki A. 2012. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J. Clin. Investig 122:3914–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, et al. 2012. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Investig 122:2911–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ikenoue T, Terakado Y, Nakagawa H, Hikiba Y, Fujii T, et al. 2016. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci. Rep 6:23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dill MT, Tornillo L, Fritzius T, Terracciano L, Semela D, et al. 2013. Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology 57:1607–19 [DOI] [PubMed] [Google Scholar]
- 140.Terada M, Horisawa K, Miura S, Takashima Y, Ohkawa Y, et al. 2016. Kupffer cells induce Notch-mediated hepatocyte conversion in a common mouse model of intrahepatic cholangiocarcinoma. Sci. Rep 6:34691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Choi TY, Ninov N, Stainier DY, Shin D. 2014. Extensive conversion of hepatic biliary epithelial cells to hepatocytes after near total loss of hepatocytes in zebrafish. Gastroenterology 146:776–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.He J, Lu H, Zou Q, Luo L. 2014. Regeneration of liver after extreme hepatocyte loss occurs mainly via biliary transdifferentiation in zebrafish. Gastroenterology 146:789–800.e8 [DOI] [PubMed] [Google Scholar]
- 143.Huang M, Chang A, Choi M, Zhou D, Anania FA, Shin CH. 2014. Antagonistic interaction between Wnt and Notch activity modulates the regenerative capacity of a zebrafish fibrotic liver model. Hepatology 60:1753–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ninov N, Hesselson D, Gut P, Zhou A, Fidelin K, Stainier DY. 2013. Metabolic regulation of cellular plasticity in the pancreas. Curr. Biol 23:1242–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ko S, Russell JO, Tian J, Gao C, Kobayashi M, et al. 2019. Hdac1 regulates differentiation of bipotent liver progenitor cells during regeneration via Sox9b and Cdk8. Gastroenterology 156:187–202.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang X, Du G, Xu Y, Li X, Fan W, et al. 2016. Inhibition of Notch signaling pathway prevents cholestatic liver fibrosis by decreasing the differentiation of hepatic progenitor cells into cholangiocytes. Lab Investig. 96:350–60 [DOI] [PubMed] [Google Scholar]
- 147.Yongping M, Zhang X, Xuewei L, Fan W, Chen J, et al. 2015. Astragaloside prevents BDL-induced liver fibrosis through inhibition of Notch signaling activation. J. Ethnopharmacol 169:200–9 [DOI] [PubMed] [Google Scholar]
- 148.Fiorotto R, Raizner A, Morell CM, Torsello B, Scirpo R, et al. 2013. Notch signaling regulates tubular morphogenesis during repair from biliary damage in mice. J. Hepatol 59:124–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Paganelli M, Nyabi O, Sid B, Evraerts J, El Malmi I, et al. 2014. Downregulation of Sox9 expression associates with hepatogenic differentiation of human liver mesenchymal stem/progenitor cells. Stem Cells Dev. 23:1377–91 [DOI] [PubMed] [Google Scholar]
- 150.Choi TY, Khaliq M, Tsurusaki S, Ninov N, Stainier DYR, et al. 2017. Bone morphogenetic protein signaling governs biliary-driven liver regeneration in zebrafish through tbx2b and id2a. Hepatology 66:1616– 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hardman RC, Volz DC, Kullman SW, Hinton DE. 2007. An in vivo look at vertebrate liver architecture: three-dimensional reconstructions from medaka (Oryzias latipes). Anat. Rec 290:770–82 [DOI] [PubMed] [Google Scholar]
- 152.Lorent K, Moore JC, Siekmann AF, Lawson N, Pack M. 2010. Reiterative use of the Notch signal during zebrafish intrahepatic biliary development. Dev. Dyn 239:855–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Goessling W, Sadler KC. 2015. Zebrafish: an important tool for liver disease research. Gastroenterology 149:1361–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Russell JO, Monga SP. 2018. Wnt/β-catenin signaling in liver development, homeostasis, and pathobiology. Annu. Rev. Pathol. Mech. Dis 13:351–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Soto-Gutierrez A, Gough A, Vernetti LA, Taylor DL, Monga SP. 2017. Pre-clinical and clinical investigations of metabolic zonation in liver diseases: the potential of microphysiology systems. Exp. Biol. Med 242:1605–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ko S, Monga SP. 2018. Hepatic zonation now on hormones! Hepatology 69:1339–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chu J, Sadler KC. 2009. New school in liver development: lessons from zebrafish. Hepatology 50:1656–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kulkeaw K, Sugiyama D. 2012. Zebrafish erythropoiesis and the utility of fish as models of anemia. Stem Cell Res. Ther 3:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Langenau DM, Zon LI. 2005. The zebrafish: a new model of T-cell and thymic development. Nat. Rev. Immunol 5:307–17 [DOI] [PubMed] [Google Scholar]
- 160.Iismaa SE, Kaidonis X, Nicks AM, Bogush N, Kikuchi K, et al. 2018. Comparative regenerative mechanisms across different mammalian tissues. NPJ Regen. Med 3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hernandez-Martinez JM, Forrest CM, Darlington LG, Smith RA, Stone TW. 2017. Quinolinic acid induces neuritogenesis in SH-SY5Y neuroblastoma cells independently of NMDA receptor activation. Eur. J. Neurosci 45:700–11 [DOI] [PubMed] [Google Scholar]
- 162.Robinson MA, Graham DJ, Morrish F, Hockenbery D, Gamble LJ. 2015. Lipid analysis of eight human breast cancer cell lines with ToF-SIMS. Biointerphases 11:02A303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lin Y, Fang ZP, Liu HJ, Wang LJ, Cheng Z, et al. 2017. HGF/R-spondin1 rescues liver dysfunction through the induction of Lgr5. Nat. Commun 8:1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Xu J, Tan Y, Shao X, Zhang C, He Y, Wang J, Xi Y. 2018. Evaluation of NCAM and c-Kit as hepatic progenitor cell markers for intrahepatic cholangiocarcinomas. Pathol. Res. Pract 214:2011–17 [DOI] [PubMed] [Google Scholar]