

SUMMARY
Enhanced host protection against re-infection requires generation of memory T cells of sufficient quantity and functional quality. Unlike well-studied inbred mice, T cell responses of diverse size and quality are generated following infection of humans and outbred mice. Thus, additional models are needed that accurately reflect variation in immune outcomes in genetically diverse populations and to uncover underlying genetic causes. The Collaborative Cross (CC), a large recombinant inbred panel of mice, is an ideal model in this pursuit for the high degree of genetic variation present, because it allows for assessment of genetic factors underlying unique phenotypes. Here, we advance the utility of the CC as a tool to analyze the immune response to viral infection. We describe variability in resting immune cell composition and adaptive immune responses generated among CC strains following systemic virus infection and reveal quantitative trait loci responsible for generation of CD62L+ memory CD8 T cells.
Graphical Abstract
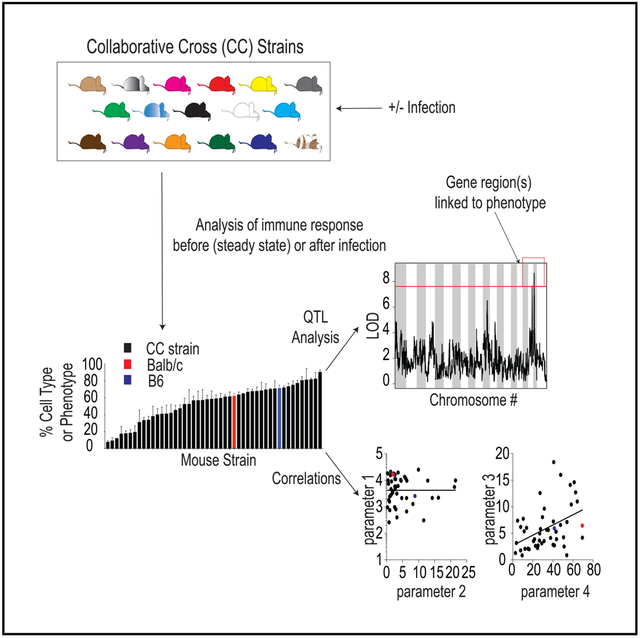
In Brief
Martin et al. advance the use of the Collaborative Cross (CC) for studying adaptive immune responses. They demonstrate that the CC better models variation in T cell responses seen in outbred mice and humans and that it can uncover genes linked to generation of qualitatively distinct memory cells following infection.
INTRODUCTION
CD8 T cells play an important role in mediating protection against cancer and bacterial, viral, and parasitic infections, and hosts containing memory CD8 T cells are often better protected against tumors or pathogenic re-infection (Epstein et al., 2011; Brown and Kelso, 2009; Duan and Thomas, 2016; Masopust, 2009; Pamer, 2004; Schmidt and Varga, 2018; Sahin et al., 2017). Therefore, research utilizing laboratory mice has focused on understanding factors influencing memory CD8 T cell generation and characteristics of memory CD8 T cell responses that confer protection against re-infection. This research has led to the understanding that, along with location, size of the memory pool and phenotypic/functional qualities of memory CD8 T cells dictate the level of host protection against re-infection (Seder et al., 2013; Schmidt et al., 2008; Mackay et al., 2012; Nolz and Harty, 2011; Wherry et al., 2003; Bachmann et al., 2005; Olson et al., 2013; Slütter et al., 2013, 2017; Martin et al., 2015; Eberlein et al., 2016; Wu et al., 2014). However, the translational value of mouse immunology studies depends on how faithfully those models reflect human immunology, and recent studies have noted areas in which mouse models fail to accurately reflect the human condition. Studies using so-called “dirty mice” have documented that the composition of immune cells present in mice housed in specific-pathogen-free (SPF) facilities is more similar to infants than adult humans and that CD8 T cell responses generated following infection are qualitatively different in SPF mice compared to dirty mice (Beura et al., 2016; Reese et al., 2016; Masopust et al., 2017). Similarly, mouse studies, conducted using 1 or 2 strains of inbred mice, fail to fully capture the array of immune responses and outcomes following infection that can be observed in genetically diverse humans (Graham et al., 2015, 2016; Ferris et al., 2013).
Using a previously described surrogate activation marker approach that can be used to track CD8 T cell responses in any mouse strain (Rai et al., 2009), we recently described that (1) the magnitude of effector and memory CD8 T cell responses generated following infection, (2) the rate of phenotypic progression of memory CD8 T cells following infection, and (3) the degree of CD8 T-cell-mediated protection against re-infection vary significantly in genetically unique outbred hosts (Martin et al., 2017). Additional studies in humans with experimental vaccination against yellow fever virus have also documented variation in the magnitude of CD8 T cell responses and changes in memory CD8 T cell phenotype and function over time after infection (Akondy et al., 2017). Taken together, these studies suggest that underlying host genetic factors influence quantitative and qualitative aspects of memory CD8 T cell development following infection, parameters that directly influence the degree of host protection against re-infection. However, due to a lack of tools available in either humans or outbred mice, determining specific genetic factors underlying diverse immune outcomes would be extremely costly and time consuming.
The Collaborative Cross (CC) model circumvents these difficulties and presents an opportunity to examine the biological networks and genetic factors regulating divergent CD8 T cell outcomes following infection in a genetically diverse population. The CC was conceptualized by the complex trait consortium in 2002 as a resource for investigation of biomolecular networks and systems level phenotypes underlying complex traits (Churchill et al., 2004). CC mice are a recombinant inbred panel of mice derived using a funnel breeding strategy with eight founder strains of mice—5 classical inbred strains (A/J, C57BL/6J, 129S1/SvImJ, non-obese diabetic [NOD]/ShiLtJ, and NZO/HILtJ) and 3 wild-derived strains (CAST/EiJ, PWK/PhJ, and WSB/EiJ; Threadgill et al., 2011). Founder strains contain representatives from the three major Mus musculus subspecies (M.m. musculus, M.m. domesticus, and M.m. castaneous) and capture almost 90% of the known genetic variation present in laboratory mice originating from M. musculus, and the variation is randomly distributed across the genome (Threadgill and Churchill, 2012). Existing CC lines contain millions of single-nucleotide polymorphisms (SNPs) and insertions or deletions (indels) that result in vast genetic diversity between lines. However, progeny within lines are inbred, genetic clones allowing for precise analysis, reproducibility, and comparative studies across different laboratories (Phillippi et al., 2014; Collaborative Cross Consortium, 2012). Efforts to characterize the genomes of CC strains facilitate the use of genetic analytical tools, such as quantitative trait linking (QTL), that allow for dissection of the genetic factors underlying complex phenotypic traits (Collaborative Cross Consortium, 2012; Srivastava et al., 2017; Graham et al., 2017), such as quantity and quality of memory CD8 T cells generated following infection.
Recent studies utilizing CC mice have documented significant variability in immune subset composition in individual strains at steady state, and QTL analysis was able to link chromosomal regions driving the diverse phenotypes observed (Graham et al., 2017; Collin et al., 2019). Further studies have analyzed disease outcomes following infection with a spectrum of micro-organisms, including influenza, Ebola, SARS, and West Nile virus (Ferris et al., 2013; Rasmussen et al., 2014; Gralinski et al., 2015; Graham et al., 2015, 2016; Elbahesh and Schughart, 2016; Lorè et al., 2015; Leist et al., 2016). Select CC strains captured disease outcomes seen in humans that are not observed in traditional inbred mice, supporting the potential translational value of the CC system. However, thus far, there has been relatively little analysis of the immune response following infection in CC mice that may contribute to differences in disease outcome or of host genetic factors that may underlie potential differences in immune response following infection.
To address this, we analyzed the T cell response following acute lymphocytic choriomeningitis virus (LCMV) Armstrong infection in 47 strains of CC mice. We corroborate previous findings that documented wide diversity in pre-infection immune compartments in CC strains (Graham et al., 2017; Collin et al., 2019). Importantly, and similarly to what can be observed in individual outbred mice (Rai et al., 2009; Martin et al., 2017) and in humans (Akondy et al., 2017), we observed a wide range in magnitude of CD8 T cell responses generated in CC strains following infection. Size of the memory CD8 T cell pool generated correlated with size of the effector CD8 T cell pool, and effector CD8 T cell pool size correlated with levels of systemic cytokines elicited following infection. Subset composition of effector and memory CD8 T cells was also highly variable among CC strains, and rates of memory CD8 T cell phenotypic progression following infection varied among strains to a similar extent to that seen in outbred mice. QTL analysis revealed significant linkages to chromosomal regions associated with the development of CD62L+ memory CD8 T cells following infection and allowed identification of genes potentially driving this phenotype.
Our results support the use of the CC to model diversity in immune responses observed in genetically diverse organisms and to uncover regulatory networks and host genetic factors underlying diverse immune outcomes following infection. This study and future studies utilizing the CC have the potential to improve translational efforts for the generation of vaccines to stimulate protective immune responses against cancer and infections of global importance.
RESULTS
Immune Subset Composition prior to Infection Is Variable among CC Strains
Orchestration of an immune response results from the interplay of cells within innate and adaptive arms of the immune system. Previous studies utilizing the CC have documented differences in immune subset composition among strains at steady state and have identified genetic linkages to observed differences (Graham et al., 2017; Collin et al., 2019). To corroborate previous studies and to document steady-state immune composition for the 47 CC strains utilized in this study, we bled all mice prior to infection and stained peripheral blood leukocytes (PBLs) using 5 flow cytometry panels to identify CD4 and CD8 T cells, Foxp3+ regulatory CD4 T cells, B220+/CD3− B cells, NKp46+/CD3− natural killer (NK) cells, and SSChi/CD11bhi granulocytes or SSClo/CD11bhi monocytes (example dot plots shown in Figure S1). All mice used in this study were female, and CC strains analyzed as well as number of mice analyzed per strain can be found in Table S1. We also analyzed commonly used inbred C57BL/6 (B6) and BALB/c strains for comparison. In agreement with previous studies, we found wide variation in representation of immune cell subsets, including CD4 T cells, CD8 T cells, Foxp3+ CD4 T cells, B cells, NK cells, granulocytes, and monocytes, and ratio of CD4 to CD8 T cells at steady state among CC strains examined in this study (Figures 1A–1H; Table S2). Of note, in all cases, subset representation was observed outside of ranges seen between B6 and BALB/c mice. Thus, immune cell composition at steady state among genetically diverse CC strains is variable, suggesting that diverse immune responses may be generated following infection.
Figure 1. CC Mice Display Variation in Immune Subset Composition prior to Infection.
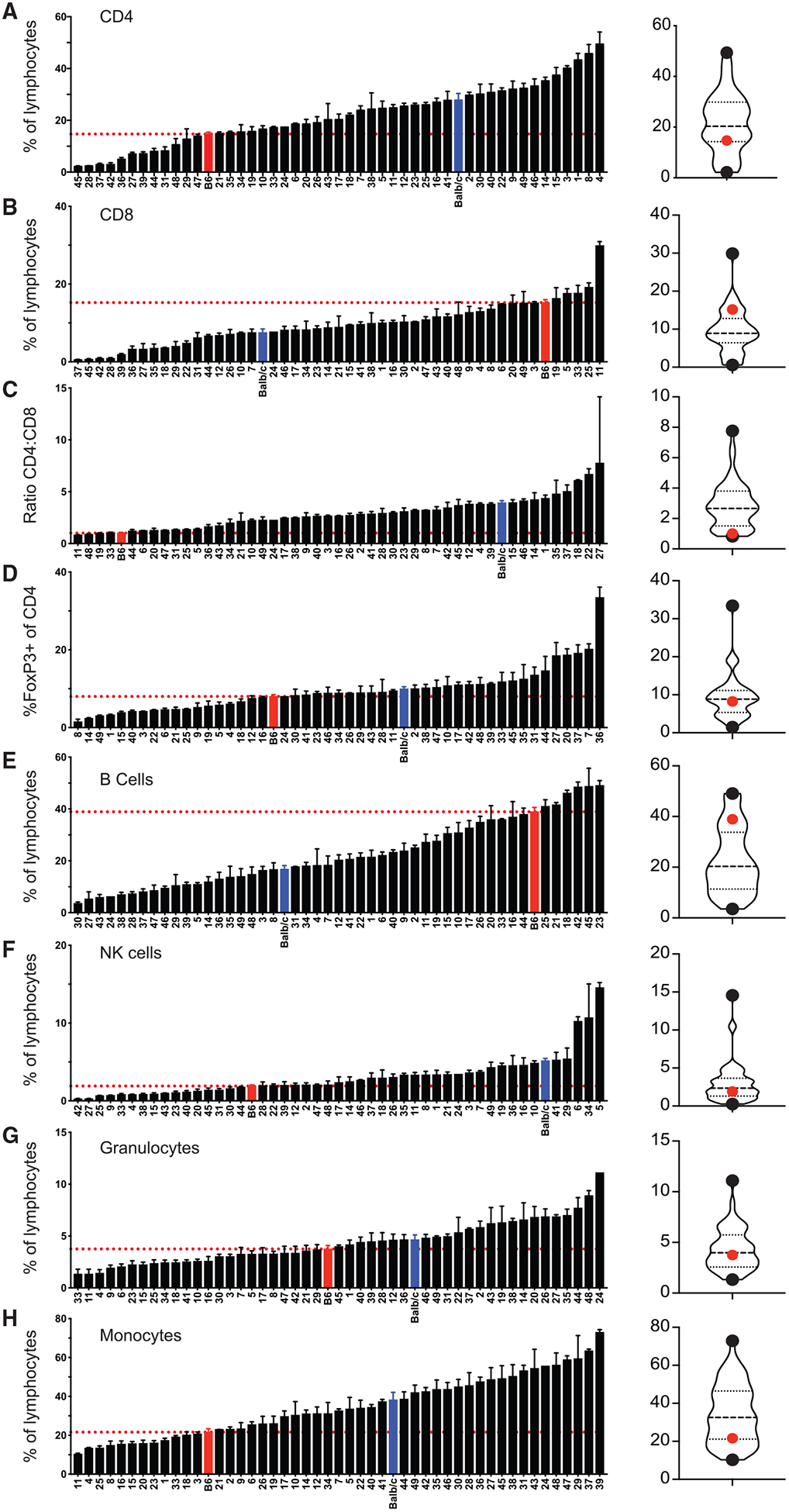
Summary graphs (left) and violin plots (right) of the percentage of CD4 (A) and CD8 (B) T cells out of total lymphocytes, ratio of CD4 to CD8 T cells (C), Foxp3+ cells of gated CD4 T cells (D), B cells (E), NK cells (F), granulocytes (G), and monocytes (H) out of total lymphocytes for uninfected B6 (red), BALB/c (blue), and CC (Hotchkiss, no. 22) strains. Data are from 1–3 individual experiments. n = 1–20 mice per group (see Table S1). Error bars for summary graphs indicate standard error of the mean and dashed red lines at percentage seen in B6 mice. For violin plots, black dots indicate collaborative cross strains with highest and lowest percentages and red dots indicate percentage in B6 mice. Dashed lines at 25th and 75th quartiles and median are shown. See also Figure S1 and Table S2.
Quantitative and Qualitative Aspects of Innate and Adaptive Effector Responses following Infection Are Variable among CC Mice
Previous studies have described a range of outcomes following infection of collaborative mice that can be observed in the human population but that are not modeled with commonly used inbred mouse strains (Graham et al., 2015, 2016). Additionally, our previous work with outbred mice has shown that quantitative and qualitative aspects of innate and adaptive immune responses following infection are variable among individual, genetically unique mice (Rai et al., 2009; Martin et al., 2017). However, at present, there has been relatively little analysis of the immune response generated following infection of CC mice. To address this, we infected B6, BALB/c, and CC strains with Armstrong strain of LCMV (LCMV Arm), as the immune response following acute infection with this virus has been well described in B6 and BALB/c mice (Zhou et al., 2012; Lapo sová et al., 2013). Following infection, we analyzed inflammatory cytokines present in serum at day 3 (d3) as a measure of the innate response, weight loss at d8 following infection, and CD4 and CD8 effector responses (CD4 Teff and CD8 Teff) at d8 as a measure of the adaptive response (Figure 2A; Table S3). Concentrations of interferon (IFN)-α and IFN-γ detected in serum 3 days after infection varied from 30- to 100-fold among strains (Figure 2B), suggesting that the magnitude of innate responses elicited was different among CC strains. Additionally, weight loss 8 days after infection was variable (Figure 2C), suggesting that the infection was experienced differently among strains. We used previously described surrogate activation marker approaches (Rai et al., 2009; McDermott and Varga, 2011) to detect effector CD4 (CD4 Teff) (CD49dhi/CD11ahi) and CD8 (CD8 Teff) (CD11ahi/CD8lo) responses at d8 (example dot plots shown in Figure S2), the peak of the adaptive response in B6 mice following LCMV infection. Magnitudes of CD4 Teff and CD8 Teff responses were strikingly different among strains, with CD4 Teff responses ranging from approximately 10% to over 90% of all CD4 T cells and CD8 Teff responses ranging from approximately 50% to close to 100% of all CD8 T cells (Figures 2D and 2E). Because CD8 T cell effector responses have been shown in B6 mice to be influenced by inflammatory cytokines (Busch et al., 2000; Curtsinger et al., 2005; Porter and Harty, 2006; Pham et al., 2009; Wirth et al., 2010), we determined whether levels of IFN-α and IFN-γ detected in the serum at d3 correlated with CD8 Teff magnitude and found statistically significant correlations for both cytokines (Figure 2F). Thus, the magnitude of innate and adaptive immune responses following infection was correlated within strains but distinct among CC strains.
Figure 2. Magnitude of Innate and Adaptive CD4 and CD8 T Cell Responses Is Variable in CC Strains.
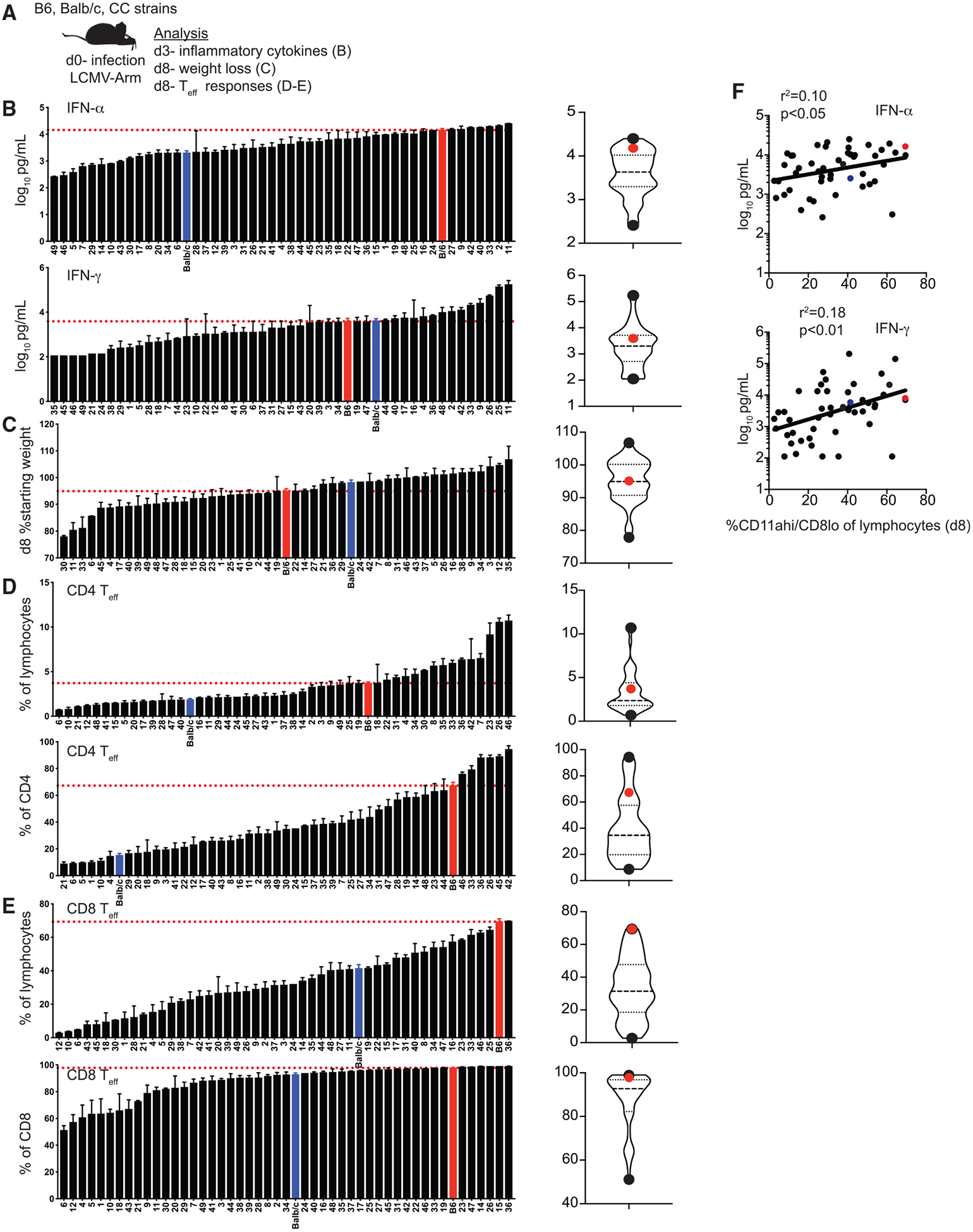
(A) Experimental design. B6, BALB/c, and CC mice were infected with LCMV-Armstrong on d0. Concentration of IFN-a and IFN-g in serum was determined on d3, and weight loss and effector T cell (Teff) responses were determined on d8. (B) Summary graphs (left) and violin plots (right) of concentration of IFN-α (top) and IFN-γ (bottom) detected in serum. (C) Summary graphs (left) and violin plots (right) of % starting weight (weight at d8 divided by weight at d0). (D) Summary graphs (left) and violin plots (right) of percentage of CD4 Teff cells (CD49dhi/CD11ahi) out of total lymphocytes (top) or gated CD4 T cells (bottom). (E) Summary graphs (left) and violin plots (right) of percentage of CD8 Teff cells (CD11ahi/CD8alo) out of total lymphocytes (top) or gated CD8 T cells (bottom). (F) Percentage of Teff CD8 T cells out of total lymphocytes (x axis) relative to concentration of IFN-a (top) or IFN-g (bottom) detected in serum (y axis).
Data from 1–3 individual experiments. n = 1–20 mice per group (see Table S1). Error bars for summary graphs indicate standard error of the mean and dashed red lines at percentage seen in B6 mice. For violin plots, black dots indicate CC strains with highest and lowest percentages and red dots indicate percentage in B6 mice. Dashed lines at 25th and 75th quartiles and median are shown. For linear correlations, red dots indicate B6 mice, blue dots indicate BALB/c mice, and black dots indicate CC strains. Statistical significance of R-squared values based on linear regression analysis is shown. See also Figure S2 and Table S3.
Effector CD8 T cells can be divided into short-lived effector cells (SLECs) (KLRG1hi/CD127lo), which are less likely to persist to memory, and memory precursor effector cells (MPECs) (KLRG1lo/CD127hi), which are more likely to persist to memory, subsets based on expression of phenotypic markers KLRG1 and CD127 (Joshi et al., 2007; example dot plots shown in Figure S3). Wide variation in CD8 Teff subset representation was observed among CC strains (Figures 3A and 3B; Table S3), suggesting that qualitatively different CD8 Teff cells are generated following infection in different strains. Because generation of SLEC and MPEC subsets has been shown to be influenced by aspects of the immune response to infection, including inflammation and CD8 T cell response magnitude (Joshi et al., 2007), we determined whether representation of SLEC and MPEC subsets could be correlated with other aspects of the immune response. However, we did not find a significant correlation between percentage of SLECs or MPECs and either levels of systemic cytokines elicited or CD8 Teff response magnitude (Figures 3C and 3D). Taken together, these data suggest that qualitatively and quantitatively different innate and effector immune responses are generated following infection in genetically distinct CC strains.
Figure 3. Representation of Effector CD8 T Cell Subsets Generated following Infection Is Variable in CC Strains.
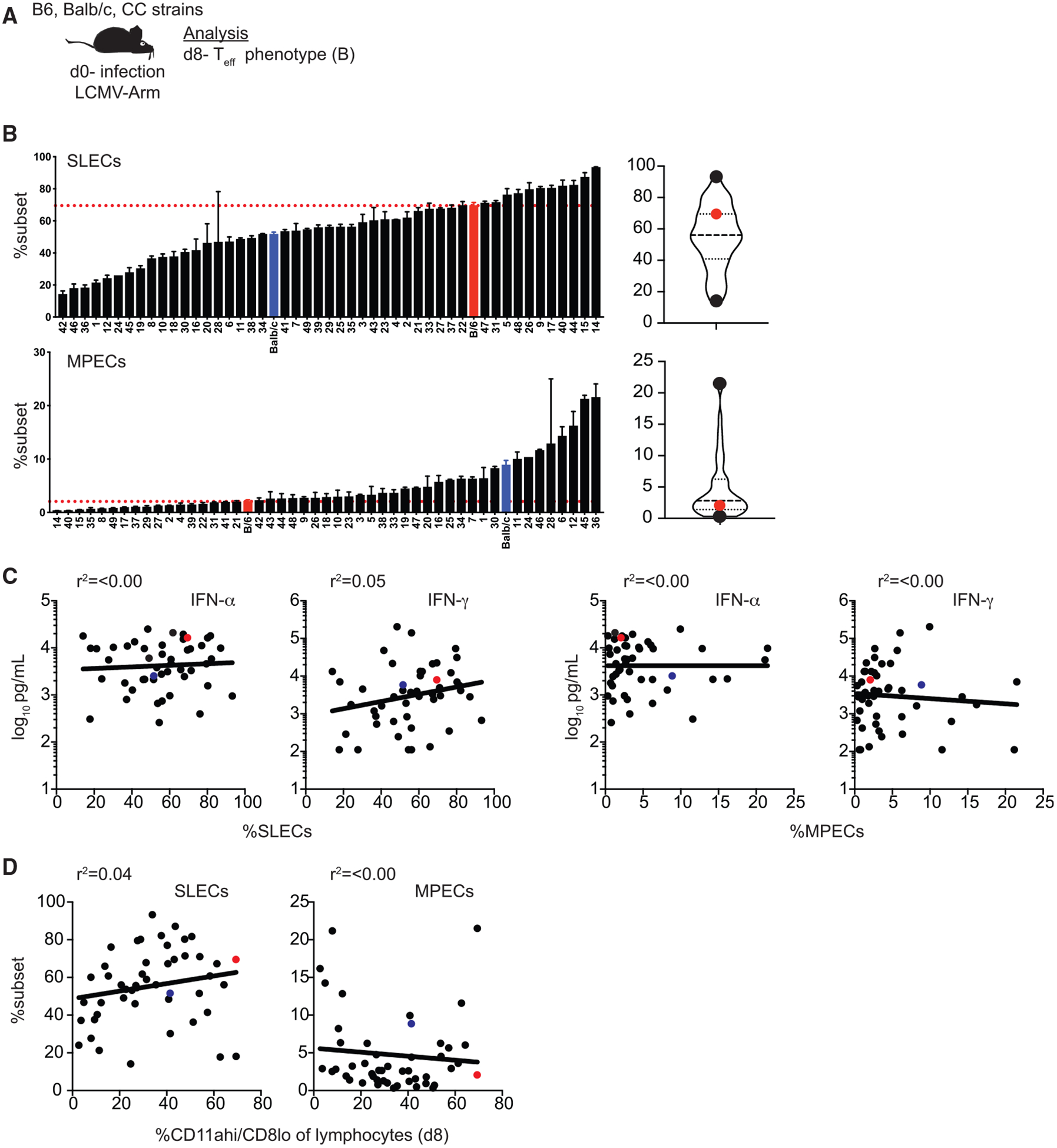
(A) Experimental design. B6, BALB/c, and CC mice were infected with LCMV-Armstrong on d0. Phenotype of Teff cells (CD11ahi/CD8alo) was determined on d8. (B) Summary graphs (left) and violin plots (right) of percentage of CD8 Teff cells displaying a short-lived effector cell (SLEC) (KLRG1hi/CD127lo; top) or memory precursor effector cell (MPEC) (KLRG1lo/CD127hi; bottom) phenotype. (C) Percentage of SLECs (left 2 graphs) out of CD8 Teff cells (x axis) relative to concentration of IFN-α (left) or IFN-γ (right) detected in serum (y axis) or percentage of MPECs (right 2 graphs) out of CD8 Teff cells (x axis) relative to concentration of IFN-α (left) or IFN-γ (right) detected in serum (y axis). (D) Percentage of SLECs (left) or MPECs (right) out of CD8 Teff cells relative to percentage of CD8 Teff cells out of total lymphocytes (x axis).
Data from 1–3 individual experiments. n = 1–20 mice per group (see Table S1). Error bars for summary plots indicate standard error of the mean and dashed red lines at percentage seen in B6 mice. For violin plots, black dots indicate CC strains with highest and lowest percentages and red dots indicate percentage in B6 mice. Dashed lines at 25th and 75th quartiles and median are shown. For linear correlations, red dots indicate B6 mice, blue dots indicate BALB/c mice, and black dots indicate CC strains. R-squared values were all not significant based on linear regression analysis. See also Figure S3 and Table S3.
Size and Phenotype of the Memory CD8 T Cell Pool Generated following Infection Are Highly Variable among CC Mice
Following contraction, CD4 and CD8 T cells persist as memory cells that are capable of providing the host with increased protection against re-infection. Degree of memory CD8 T-cell-mediated protection against re-infection has been correlated with size of the memory CD8 T cell pool (Seder et al., 2013; Schmidt et al., 2008, 2010), and we previously noted that magnitude of the memory CD8 T cell pool generated following infection is highly variable in outbred mice (Rai et al., 2009; Martin et al., 2017). Additional studies in humans have documented variation in size of the memory CD8 T cell pool generated in response to experimental vaccination (Akondy et al., 2017), suggesting that memory CD8 T cell response magnitude is determined at least in part by host genetic makeup. To determine whether memory T cell pools of different sizes are generated in genetically distinct CC strains, we identified memory CD4 (CD4 TM) (CD49dhi/CD11ahi) and CD8 (CD8 TM) (CD11ahi/CD8lo) T cells 75 days after LCMV infection (Figure 4A; Table S4; example dot plots shown in Figure S4). We found strikingly different sizes of CD4 and CD8 TM cell pools ranging from 1% to 8% (CD4 TM) and 1% to 20% (CD8 TM) of all lymphocytes in different CC strains (Figures 4B–4D). There was a statistically significant correlation with the size of the memory CD8 T cell pool generated and size of the effector response (Figure 4E), suggesting that the relative size of the memory pool generated could be predicted based on size of the CD8 Teff pool. Thus, TM pools of distinct sizes are generated in genetically diverse CC strains, which could impact degree of memory-mediated protection against re-infection, and the best predictor of CD8 TM cell pool size was size of the CD8 Teff pool generated.
Figure 4. Magnitude of Memory CD4 and CD8 T Cell Responses Is Variable in CC Mice.
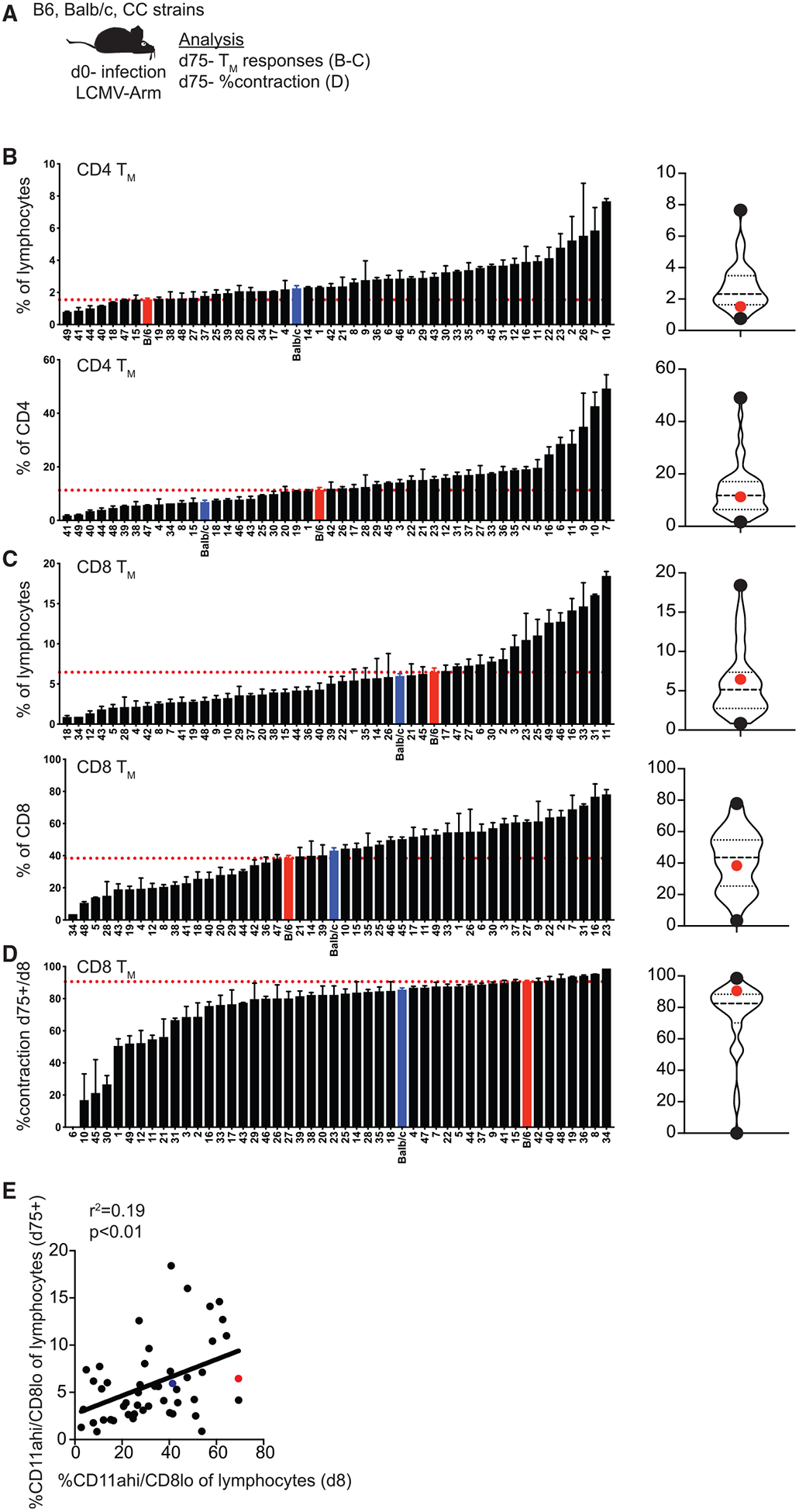
(A) Experimental design. B6, BALB/c, and CC mice were infected with LCMV-Armstrong on d0. Memory T cell (TM) responses and % contraction of CD8 T cells (% of d75 TM/% of d8 TEff) were determined on d75. (B) Summary graphs (left) and violin plots (right) of percentage of CD4 TM cells (CD49dhi/CD11ahi) out of total lymphocytes (top) or gated CD4 T cells (bottom). (C) Summary graphs (left) and violin plots (right) of percentage of CD8 TM cells (CD11ahi/CD8alo) out of total lymphocytes (top) or gated CD8 T cells (bottom). (D) Summary graphs (left) and violin plots (right) of percent contraction of CD8 T cell responses. (E) Percentage of CD8 Teff cells (d8) out of total lymphocytes (x axis) relative to percentage of CD8 TM cells (d75) out of total lymphocytes (y axis).
Data from 1–3 individual experiments. n = 2–20 mice per group (see Table S1). Error bars for summary plots indicate standard error of the mean and dashed red lines at percentage seen in B6 mice. For violin plots, black dots indicate CC strains with highest and lowest percentages and red dots indicate percentage in B6 mice. Dashed lines at 25th and 75th quartiles and median are shown. For linear correlations, red dots indicate B6 mice, blue dots indicate BALB/c mice, and black dots indicate CC strains. Statistical significance of R-squared values based on linear regression analysis is shown. See also Figure S4 and Table S4.
In addition to the number of memory CD8 T cells, their functional abilities (quality) impact degree of memory CD8 T-cell-mediated protection against re-infection (Mackay et al., 2012; Nolz and Harty, 2011; Wherry et al., 2003; Bachmann et al., 2005; Olson et al., 2013; Slütter et al., 2013, 2017; Wu et al., 2014). Circulating memory CD8 T cells were originally divided into subsets described as T effector memory (Tem), which do not express CD62L, are less proliferative, and more cytotoxic, and T central memory (Tcm), which express CD62L, home to secondary lymphoid organs, are more proliferative, and are better protective against chronic infections (Sallusto et al., 1999; Wherry et al., 2003). More recently, Tem (Cx3Cr1hi/CD27lo) and Tcm (Cx3Cr1lo/CD27hi) subsets have been identified based on expression of Cx3Cr1 and CD27, which allows for identification of an additional memory subset described as peripheral memory (Tpm) (Cx3Cr1int/CD27hi) that can circulate among peripheral tissues (Gerlach et al., 2016). We and others have shown that the phenotype of memory CD8 T cells in inbred mice changes with time after infection such that the memory pool becomes highly represented by CD62L+ Tcm cells (Martin et al., 2015; Eberlein et al., 2016). However, the rate of memory CD8 T cell phenotypic changes was variable in individual outbred mice (Martin et al., 2017), suggesting that memory CD8 T cells generated in genetically unique organisms will differ qualitatively and in the ability to provide protection against re-infection.
To determine whether development of phenotypically distinct memory CD8 T cells in CC mice occurs at different rates, we stained TM cells at d75 after infection for expression of Tcm-associated markers CD127, CD62L, and CD27hi and Tem-associated markers KLRG1 and Cx3Cr1 (Table S4; example dot plots shown in Figure S5A). We also identified Tem (Cx3Cr1hi/CD27lo), Tpm (Cx3Cr1int/CD27hi), and Tcm (Cx3Cr1lo/CD27hi) subsets based on expression of Cx3Cr1 and CD27 (Table S4; example dot plots shown in Figure S5B). We found that the phenotype of CD8 TM cells at this point following infection could be strikingly different, with some strains possessing as few as 10% or almost 90% of CD8 TM cells expressing Tcm-associated markers and with some strains possessing as few as 10% or as many as 90% of CD8 TM cells expressing Tem-associated markers (Figure 5A). We were unable to find any strong correlations between expression of Tem- or Tcm-associated markers and the magnitude of the effector or memory CD8 T cell populations generated or the levels of systemic cytokines elicited early in the response (Figure S6).
Figure 5. Phenotype of Memory CD8 T Cells Generated following Infection Is Diverse in CC Strains.
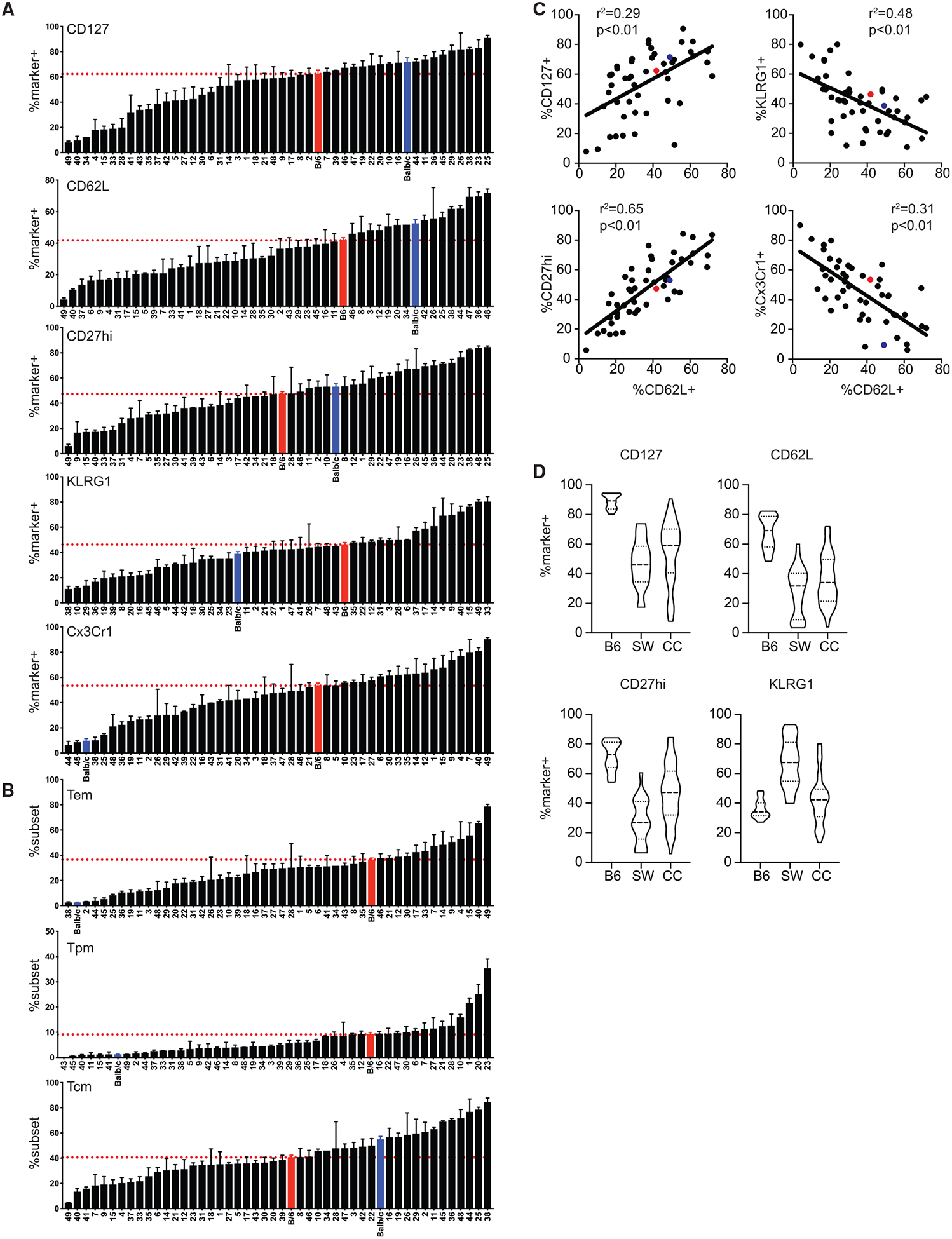
B6, BALB/c, and CC mice were infected with LCMV-Armstrong on d0. Phenotype of TM cells (CD11ahi/CD8alo) was determined on d75. (A) Summary graphs of percentage of CD8 TM cells expressing CD127, CD62L, CD27hi, KLRG1, or Cx3Cr1. (B) Summary graphs of percentage of CD8 TM cells displaying an effector memory (Tem) (Cx3Cr1hi/CD27lo; top), peripheral memory (Tpm) (Cx3Cr1int/CD27hi; middle), or central memory (Tcm) (Cx3Cr1lo/CD27hi; bottom) phenotype. (C) Percentage of CD8 TM cells (d75) expressing CD62L (x axis) relative to percentage expressing CD127, CD27hi, KLRG1, or Cx3Cr1 (y axis). (D) B6, NIH Swiss (SW), and CC mice were infected with LCMV-Armstrong on d0. Phenotype of TM cells (CD11ahi/CD8alo) was determined on d75+. Violin plots of the percentage of TM cells expressing CD127, CD62L, CD27hi, and KLRG1 for B6 or SW mice or CC strains are shown.
Data from 1–3 individual experiments. n = 2–20 mice per group (see Table 1). Error bars for summary plots indicate standard error of the mean and dashed red lines at percentage seen in B6 mice. For linear correlations, red dots indicate B6 mice, blue dots indicate BALB/c mice, and black dots indicate CC strains. Statistical significance of R-squared values based on linear regression analysis. For violin plots, solid lines are at the 25th and 75th quartiles and dashed line indicates the median. See also Figures S5 and S6 and Table S4.
We also found that subset representation of memory CD8 T cells was highly variable between strains, ranging from 5% to 80% Tem cells, 0% to 40% Tpm cells, and 5% to 80% Tcm cells (Figure 5B). Expression of CD62L correlated with expression of other Tcm-associated markers CD127 and CD27hi, and expression of CD62L was inversely correlated with Tem-associated markers KLRG1 and Cx3Cr1 (Figure 5C), suggesting that expression of CD62L is a good surrogate for true Tcm cells in any host. Interestingly, phenotypic diversity of memory CD8 T cells was greater among CC strains than B6 mice and similar to that seen in individual outbred NIH Swiss mice (Figure 5D), validating use of the CC as a model of diverse CD8 T cell memory outcomes following infection of genetically heterogeneous organisms. Additionally, these data suggest that heritable genetic factors may underlie development of T cell responses of distinct magnitude and/or quality following infection. To determine the heritability of CD4 and CD8 Teff and TM size and phenotypes of CD8 Teff and TM generated following infection that were quantified in Figures 2, 3, 4, and 5, we used a broad sense heritability method to estimate the proportion of total variance contributed by genetic variance. This analysis showed that the genetic contribution was high among all phenotypes examined (Table S5), which further suggested that genetic factors may underlie development of T cell responses of distinct magnitude and/or quality following infection.
QTL Mapping Reveals Genetic Associations Influencing Development of CD62L+ CD8 TM Cells
One of the powerful aspects of the CC model is that it allows for genetic mapping studies to identify genetic factors underlying complex phenotypes. In an attempt to identify gene regions associated with quantitative measurements of the CD8 T cell response to infection, we performed QTL mapping on size of the CD8 Teff response (Figure 2E) and size of the CD8 TM pool generated (Figure 4C). For QTL scans, 1,000 permutations were run, and log of the odds ratio (LOD) scores above the 95th percentile of the distribution were selected as significant QTLs. QTL intervals were then identified based on the LOD scores and effect of founder alleles in Diversity Outbred (DO) mice strains observed at those regions (Gatti et al., 2014). We did not find significant QTLs associated with size of the CD8 Teff or TM pool (data not shown). However, linear correlation analysis revealed that size of the CD8 TM pool generated correlated with size of the TM pool (Figure 4E) and that size of the Teff pool generated correlated with levels of systemic cytokines elicited early during the response (Figure 2F). This suggests that host factors regulating the innate response to infection may play an important role in determining the magnitude of effector and memory CD8 T cells generated in hosts of diverse genetic backgrounds.
We also performed QTL mapping to show the contribution of each of the founder alleles and identify genes associated with generation of qualitatively distinct memory CD8 T cells based on expression of CD8 TM phenotypic markers (Figure 5A) and CD8 TM subset representation (Figure 5B). Here, we did find significant QTLs associated with development of CD62L+ CD8 TM cells with LOD scores corresponding to p < 0.05 within chromosome 18 between 60 and 80 Mb-telomere regions and within chromosome 19 between 10 and 20 Mb-telomere regions (Figure 6A). The QTL intervals were determined using the Bayesian credible interval and represents the region most likely to contain the causative polymorphism(s) (https://www.ncbi.nlm.nih.gov/pubmed/11560912). Our analysis indicated that QTL regions with max LOD scores explained 61% of variance for generation of CD62L+ CD8 TM cells. Analysis of founder effects did not reveal clear founder effects for QTLs associated with development of CD62L+ CD8 TM cells. However, NOD/ShiLtJ, CAST/EiJ, and NZO/HILtJ haplotypes between 60 and 80 Mb-telomere regions of chromosome 18 were associated with high frequency of CD62L+ CD8 TM cells although inheritance from A/J and PWK/PhJ was associated with low frequency (Figure 6B). Additionally, 129S1/SvImJ and NZO/HILtJ haplotypes between 10 and 20 Mb-telomere regions of chromosome 19 were associated with high frequency of CD62L+ CD8 TM cells although inheritance from A/J, CAST EiJ, and PWK/PhJ was associated with low frequency (Figure 6B). CD62L+ CD8 T cells have classically been described as Tcm cells (Sallusto et al., 1999; Wherry et al., 2003), but more recent subsetting strategies have also identified Tcm cells as Cx3Cr1lo/CD27hi (Gerlach et al., 2016). Using this strategy to subset Tcm cells, we found a trending QTL associated with development of Tcm cells at the same region within chromosome 18 associated with development of CD62L+ TM cells (Figure 6C) that accounted for 50% of variance for generation of Tcm cells, providing further evidence that this chromosomal region is associated with the development of qualitatively distinct Tcm CD8 T cells.
Figure 6. Significant QTLs within Chromo somes 18 and 19 Drive the Frequency of CD62L+ CD8 TM Cells.
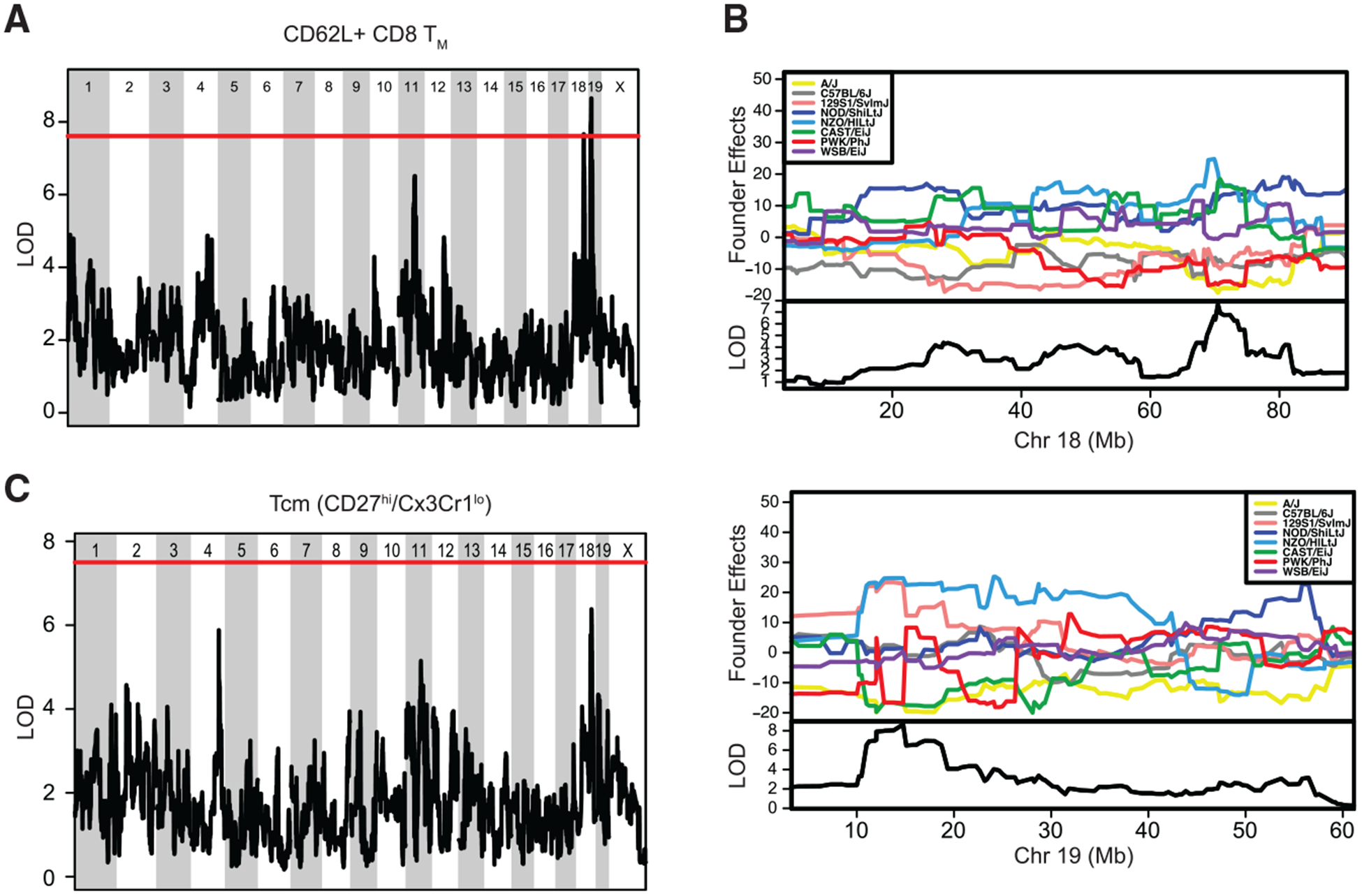
(A) QTL analysis for chromosomal regions associated with frequency of CD62L+ CD8 TM cells. Significant QTLs were found within chromosome 18 at position 60–80 Mb and within chromosome 19 at position 10–20 Mb. (B) Analysis of founder effects associated with the significant QTL within chromosome 18 revealed a correlation with inheritance from NOD/ShiLtJ, CAST/EiJ, and NZO/HILtJ and high frequency of CD62L+ CD8 TM cells and inheritance from A/J and PWK/PhJ with low frequency of CD62L+ CD8 TM cells (top). Analysis of founder effects associated with the significant QTL within chromosome 19 revealed a correlation with inheritance from 129S1/SvImJ and NZO/HILtJ with high frequency of CD62L+ CD8 TM cells and inheritance from A/J, CAST EiJ, and PWK/PhJ with low frequency of CD62L+ CD8 TM cells (bottom). (C) QTL analysis for chromosomal regions associated with Tcm (Cx3Cr1lo/CD27hi) CD8 TM subset representation revealed a trending QTL at the same region within chromosome 18, driving frequency of CD62L+ CD8 TM cells.
Data from 1–3 individual experiments. In (A) and (C), the solid red line indicates the threshold for an LOD score with p < 0.05. See also Figure S7 and Tables S5, S6, and S7.
To identify SNPs and the candidate genes that may be driving development of CD62L+ TM cells, we performed association mapping of the QTL interval regions in chromosomes 18 and 19 with SNPs from the DO founder strains using Mouse Genome Project data from the Sanger Institute (ftp://ftp.jax.org/SNPtools/variants/mgp.v5.merged.snps_all.dbSNP142.vcf.gz). SNPs with LOD score greater than 3 were selected for the analysis of candidate genes. Further, we surveyed the significant SNPs to see whether those were present only in causal haplotypes or were commonly shared across all haplotypes. Within the QTL region on chromosome 18, we identified 209 SNPs that were shared in all haplotypes (Figure S7A; Table S6) and determined that SNPs within two candidate genes, Mbd2 and Dcc (Table 1), could affect protein structure or function. Of 7 mutations near the gene region encoding for Mbd2, 3 were intronic, 2 were upstream, and 2 were downstream of the gene, although all SNPs associated with Dcc were intronic. SNPs identified in chromosome 19 QTL regions were present only in the haplotypes A/J, CAST/EiJ, and PWK/PhJ, which were associated with low-fre quency CD62L+ CD8 TM cells (Figure S7B; Table S7). Candidate genes within the QTL regions of chromosome 19 that contained missense mutations included Ms4a3, Patl1, Gm22272, Olfr235, Olfr1434, Olfr1436, and Pfpl (Table 1). Therefore, we have been able to characterize diverse adaptive immune responses within the CC reflective of outbred mice and the human population and identify gene regions and candidate genes driving this diversity. This further validates the usefulness of the CC as a translatable model that can identify complex interactions underlying unique immune responses following infection.
Table 1.
Candidate Genes Driving Observed Phenotypes
| Phenotype | OTL Region | Founder Effects | Candidate Genes in Region |
|---|---|---|---|
| CD62L+ CD8 TM | Chr 18: 60–80 MB | NOD, CAST, WSB high A/J, PWK low | Mbd2, Dcc |
| Chr 19: 10–20 MB | 129S1, NZO high A/J, CAST, PWK low | Ms4a3, Patll, Gm22272, Olfr235, Olfr1434, Olfr1436, Pfpl |
DISCUSSION
Previous studies have demonstrated the translational value of the collaborative cross due to its ability to model variation in immune cell composition and function at steady state and reveal disease outcomes observed in humans, but not in traditionally used inbred mouse strains (Graham et al., 2015, 2016, 2017; Collin et al., 2019; Elbahesh and Schughart, 2016; Ferris et al., 2013; Gralinski et al., 2015; Leist et al., 2016; Lorè et al., 2015; Rasmussen et al., 2014). These studies have also demonstrated the power of the CC as a tool to identify genetic factors underlying complex traits by using QTL mapping to identify gene regions and gene candidates driving differences in immune cell composition at steady state. Here, we further advance the utility of the CC by providing detailed analysis of the immune response following acute virus infection in 47 strains of CC mice. We corroborate previous findings of variation in immune subset composition among strains at steady state. Additionally, we describe wide variation in the magnitude of cytokines elicited during the adaptive immune response, size of the effector and memory T cell response, and phenotype and subset representation of effector and memory CD8 T cells generated following infection. This variation is similar to that seen in outbred mice (Rai et al., 2009; Martin et al., 2017) and that can be observed in the human population (Akondy et al., 2017) but that is not reflected in inbred B6 and BALB/c mice. We were also able to identify factors underlying qualitative and quantitative differences in the immune response following infection. Using linear correlation analysis, we demonstrate that magnitude of the memory CD8 T cell pool generated correlates with size of the effector pool generated and that effector pool size is correlated with levels of systemic cytokines elicited during the innate response. We also performed QTL mapping to identify gene regions and candidate genes driving the generation of CD62L+ Tcm CD8 T cells.
Size of the memory CD8 T cell pool has been correlated with host ability to provide protection against re-infection (Seder et al., 2013; Schmidt et al., 2008, 2010). Consequently, immunologists have sought to determine factors influencing size of the CD8 T cell pool generated following infection and have demonstrated that inflammation can affect CD8 T cell response magnitude (Busch et al., 2000; Curtsinger et al., 2005; Porter and Harty, 2006; Pham et al., 2009; Wirth et al., 2010). Here, we have shown that the degree of inflammation elicited during the innate immune response to infection correlated with magnitude of the effector CD8 T cell response and that size of the effector response was a good predictor of memory CD8 T cell pool size. Importantly, levels of inflammation elicited during the innate response differed among CC strains. This suggests that the CC could be used to dissect factors influencing generation of inflammatory cytokines by cells of the innate arm of the immune system and how this impacts generation of memory CD8 T cells in genetically diverse organisms. Exploration of this kind could lead to improved knowledge of ways to improve the adaptive immune response following infection and/or vaccination in order to increase size of the memory CD8 T cell pool generated in hosts that do not generate sufficiently robust innate and/or adaptive immune responses.
In addition to memory CD8 T cell pool size, quality of memory CD8 T cells, which encompasses functional abilities that are different among phenotypically distinct subsets of memory CD8 T cells, influences protective capacity (Mackay et al., 2012; Nolz and Harty, 2011; Wherry et al., 2003; Bachmann et al., 2005; Olson et al., 2013; Slütter et al., 2013, 2017; Wu et al., 2014). We found that phenotype and subset representation of memory CD8 T cells generated differed among collaborative cross strains, suggesting that qualitatively different memory CD8 T cells are generated within strains. CD62L+ Tcm cells are highly proliferative, become the dominant subset within the memory pool with time after infection, and provide enhanced protection compared to Tem cells following chronic viral infection (Sallusto et al., 1999; Wherry et al., 2003; Martin et al., 2015; Eberlein et al., 2016). Through mapping analysis, we identified QTLs within chromosomes 18 and 19 and several candidate genes within these regions underlying the generation of CD62L+ memory CD8 T cells (Figure 6; Table 1). Genes listed in Table 1 require further studies to determine whether and how their products may regulate development of CD62L+ memory as well as their mode of action. These genes could be acting intrinsically within CD8 T cells to influence development of CD62L+ memory CD8 T cells. However, Mbd2 is known to impact function of antigen-presenting cells and differentiation of CD4 T cells (Cook et al., 2015; Jia et al., 2017), suggesting that genes of interest may act extrinsically by modulating function of other cell types that regulate priming of CD8 T cells and/or the cytokine milieu during the response. Therefore,examination of the impact of these genes of interest on development of CD62L+ Tcm cells will need to include careful analysis of CD8 T cells themselves as well as other cell types that may impact CD8 T cell differentiation.
In this study, we focused primarily on CD8 T cells found in circulation, but additional immune cell types shape the primary and memory-mediated responses to infection. Our analysis of the CD4 T cell response indicated that representation of CD4 T cells and of regulatory CD4 T cells at steady state differed among strains, as did magnitude of effector and memory CD4 T cell responses following infection. CD4 T cells differentiate into functionally distinct subsets following infection (DuPage and Bluestone, 2016), and differences in CD4 T cell differentiation following infection among strains, which was not examined here, have the potential to shape the inflammatory environment and to influence the development of CD8 T cell and B cell responses (Laidlaw et al., 2016; Crotty, 2015). Furthermore, in addition to Tem, Tpm, and Tcm cells found in the circulation, memory CD8 T cells can persist as tissue resident memory (Trm) cells that provide protection against infections originating at barrier tissues (Mackay et al., 2012; Slütter et al., 2013; Schenkel et al., 2013). Trm cells differentiate from phenotypically and transcriptionally unique precursor cells within the circulation (Mackay and Kallies, 2017), and data here that describe the generation of phenotypically distinct effector CD8 T cells among collaborative cross strains following LCMV infection suggest that strains may differ in their ability to form Trm populations. Therefore, the CC may be useful for modeling development of qualitatively unique adaptive immune responses following infection and for identification of factors driving differentiation of distinct B cell and CD4 and CD8 T cell subsets.
Research utilizing the CC has examined disease outcomes following infection with pathogens of human interest, including influenza, Ebola, SARS, and West Nile virus (Ferris et al., 2013; Rasmussen et al., 2014; Gralinski et al., 2015; Graham et al., 2015, 2016; Elbahesh and Schughart, 2016; Lorè et al., 2015; Leist et al., 2016), but little in- depth analysis of how underlying differences in immune responses influence divergent disease outcomes has been conducted. Our analysis of responses following LCMV Armstrong infection described here can serve as a blueprint for examination of immune responses following infection with additional pathogens of human interest, including those that cause localized and/or chronic infections.
As is the case with all experimental science, the study that we describe here does possess limitations that will be important to examine in future studies. Due to the number of mice required for this study, and because there is a limited amount of tissue that can be collected from a mouse for cellular analysis and the need to keep mice alive for an extended period of time to examine memory T cell responses, all CC strains in this study received the same infectious dose of virus and T cell responses were all analyzed at the same effector time point, which has been described as the peak of the response in B6 mice. Differences in kinetics and magnitude of early viral infection and clearance are known to affect the magnitude of T cell responses, and we were unable to determine whether there were differences in the course of infection among CC strains. Future studies should determine this and what role any differences in initial infection have on the CD8 T cell response. Additionally, because the CC strains possess different major histocompatibility complex (MHC) haplotypes, we were unable to analyze T cell responses of known epitope specificity. Future studies should examine T cell responses against bona fide epitopes, but this may require generation of recombinant inbred cross (RIX) lines that contain fixed MHC haplotypes capable of generating T cells recognizing known epitopes, such as the well-studied CD8 T cell response to the GP33 epitope of LCMV. Additionally, differences in memory CD8 T cell response quantity and quality observed in this study suggest that CC strains may possess a differential ability to provide protection against secondary infection. This was not examined in this study but will be important to follow up on in future studies. However, this examination may also require adjustments to the model, as secondary LCMV infection is rapidly cleared due to the magnitude of the primary response. In this case, RIX mice may also be useful, as it would allow secondary infection with recombinant bacteria that have been engineered to express epitopes present in the organism used for the primary infection. Lastly, as was previously mentioned in the discussion, it will be important to isolate and test candidate genes driving generation of memory CD8 T cells of distinct quality identified in this study.
In summary, we have advanced the utility of the CC as a tool to study the immune response to infection in a model that reflects diversity of responses seen in the human population. We have shown how the CC can reveal genetic factors influencing generation of qualitatively and quantitatively distinct adaptive immune responses following infection. Selection of CC strains with unique responses following acute, systemic infection revealed in this study may prove valuable in understanding factors regulating generation of memory CD8 T cells of unique phenotypes required to provide protection against infection with pathogens of human interest.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Vladimir Badovinac (vladimir-badovinac@uiowa.edu). This study did not generate unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Inbred female C57BL/6 and BALB/c mice were purchased from the National Cancer Institute (Frederick, MD) and bred at the University of Iowa. Outbred female NIH Swiss mice were obtained from Charles River Laboratories. Female CC mice were obtained from the Systems Genetics Core Facility at the University of North Carolina, Chapel Hill (Keane, #79) (Welsh et al., 2012). Prior to their relocation to UNC, CC lines were generated and bred at Tel Aviv University in Israel (Iraqi et al., 2008), Geniad in Australia (Morahan et al., 2008), and Oak Ridge National Laboratory in the United States (Chesler et al., 2008). All mice were housed at the University of Iowa under specific pathogen-free conditions at the appropriate biosafety level and used at 6–20 weeks of age. All animal experiments were approved by the IACUC of the University of Iowa and met stipulations of the Guide for Care and Use of Laboratory Animals (National Institutes of Health.
METHOD DETAILS
Infections
All LCMV Armstrong infections were performed intraperitoneally with 2×105 plaque forming units per mouse. All mice were weighed prior to infection and on d8 post infection.
Flow Cytometry
Prior to infection and at indicated days post infection, blood was collected and red blood cells were lysed with ACK. Prior to infection, cells were stained for surface expression of CD8, CD4, CD11a, and CD49d; CD3, NKp46, Ly49H, and CD122; CD11b, CD11c, and Gr1; or CD3 and B220; or for surface expression of CD4 and intracellular expression of Foxp3 using Foxp3 staining kit protocols (eBioscience). Following infection, cells were stained for surface expression of CD8, CD4, CD11a, CD49d, and PD1; CD8, CD11a, KLRG1, and CD127; CD8, CD11a, CD62L, and CD27; or CD8, CD11a, Cx3Cr1, and CD27. Ag-experienced CD4 T cells were detected based on increased expression of CD11a and CD49d as previously described (McDermott and Varga, 2011). Ag-experienced CD8 T cells were detected based on increased expression of CD11a and decreased expression of CD8 as previously described (Rai et al., 2009). Short-lived effector cells (SLECs- KLRG1hi/CD127lo) and memory precursor effector cells (MPECs- KLRG1lo/CD127hi) were detected on d8 post infection based on expression of KLRG1 and CD127 as previously described (Joshi et al., 2007). Effector memory (Tem - Cx3Cr1hi/CD27lo), peripheral memory (Tpm- Cx3Cr1/CD27hi), and central memory (Tcm - Cx3Cr1lo/CD27hi) CD8 T cells were detected on d75 post infection based on expression of Cx3Cr1 and CD27 as previously described (Gerlach et al., 2016).
ELISAs
Blood was collected from mice at d3 post infection and serum was separated and collected by centrifugation of samples at 13,300xg for 3 minutes. IFN-α was measured using a mouse IFN-a platinum ELISA kit (eBioscience). For detection of IFN-γ, purified IFN-γ mAb (eBioscience) was diluted to 2 μg/mL and 50 μL/well was added to a flat bottom 96 well MaxiSorp ELISA plate and incubated overnight at 4°C. The following day, the plate was washed with PBS/Tween, and 200 μL/well of RP10 was added and plates were incubated at room temperature for 2 hours. Plates were then washed with PBS/Tween, and 25 μL of serum sample was added to wells along with 25 μL of PBS, and standards were prepared and plated with a range of 156.2–80,000 pg/mL, and plates were incubated overnight at 4°C. The following day, plates were washed with PBS/Tween. Biotinylated anti-IFN-γ detecting mAb (eBioscience) was diluted to 1 μg/mL in PBS, and 100 μL was added per well, and plates were incubated at room temperature for 2 hours. Plates were then washed with PBS/Tween. Avidin-peroxidase was diluted to 2.5 μg/mL in PBS, and 100 μL was added per well and plates were incubated at room temperature for 30 minutes. Plates were then washed with PBS/Tween. 100 μL of TMB substrate containing 0.2 μL/mL of hydrogen peroxide was added per well and plates were incubated for 10 minutes at room temperature. The reaction was then stopped by adding 25 μL/well of 2 M sulfuric acid. Absorbance values (450 nM) were measured and assessed for all plates using Gen5 software (BioTek).
QTL Mapping
QTL mapping was performed with select phenotypes observed as CD8 T cell response to infection. Phenotype data were imported into the R software environment (http://www.r-project.org) and DOQTL version 1.19.0 Bioconductor package (Gatti et al., 2014) was used for mapping. 1000 permutations were run for QTL scans. Genotype markers in the CC mice were obtained from the Jackson Laboratory (ftp://ftp.jax.org/MUGA/muga_snps) and the haplotype probabilities from the Systems Genetics Core Facility at the University of North Carolina, Chapel Hill (Keane, #79). DOQTL implements regression analysis to calculate Log of the odds ratio (LOD) score comparing the phenotype observations with and without the founder genotype probabilities at each locus (Gatti et al., 2014). The statistical significance of LOD scores is determined via a permutation test and the genomic loci with scores above the threshold p value of 0.05 were selected as associated QTLs with founder effects. The most likely causative SNPs and genes in significantly associated QTL regions were determined by mapping the with range of SNPs in the Sanger Mouse Genomes obtained from the Jackson Laboratory (ftp://ftp.jax.org/SNPtools/variants/mgp.v5.merged.snps_all.dbSNP142.vcf.gz). SNPs with potential effect on protein function or gene regulation were selected as higher-priority candidate genes.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data for summary graphs are presented as mean values ± SEM. For violin plots, black dots indicate collaborative cross strains with highest and lowest percentages and red dots indicate percentage in B6 mice. Dashed lines at 25th and 75th quartiles and median. R-squared values and statistical significance were calculated from linear regression analysis using GraphPad Prism software version 8 (GraphPad Software Inc., San Diego, CA). Broad sense heritability method was used to estimate the proportion of genetic variance that contributed to total variance of observed parameters (Wray and Visscher, 2008). LOD values for QTL mapping were determined using DOQTL version 1.19.0 Biocunductor package (Gatti et al., 2014), and statistical significance of LOD scores was determined by a permutation test where genomic loci with scores above the threshold p value of 0.05 were selected as associated with QTLs with founder effects.
DATA AND CODE AVAILABILITY
The accession number for unproccessed FACS Files pertaining to Figures 1–5 and raw QTL mapping pertaining to Figure 6 are available at Mendeley Data https://dx.doi.org/10.17632/r7gfn2brw4.1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD8α (53–6.7) PerCP-Cy5.5 | eBioscience | Cat # 45-0081-82; RRID:AB_1107004 |
| Anti-mouse CD8α (53–6.7) APC-Cy7 | Biolegend | Cat # 100714; RRID:AB_312753 |
| Anti-mouse CD8α (53–6.7) APC | eBioscience | Cat #17-0081-82; RRID:AB_469335 |
| Anti-mouse CD4 (GK1.5) APC | eBioscience | Cat # 17-0041-82; RRID:AB_469320 |
| Anti-mouse CD11a (M17/4) FITC | Biolegend | Cat# 101106; RRID:AB_312779 |
| Anti-mouse CD49d (R1–2) PE | eBioscience | Cat # 12-0492-82; RRID:AB_465697 |
| Anti-mouse CD3 (145–2C11) PerCP-Cy5.5 | eBioscience | Cat # 45-0031-82; RRID:AB_1107000 |
| Anti-mouse NKp46 (29A1.4) FITC | Biolegend | Cat # 137606; RRID:AB_2298210 |
| Anti-mouse Ly49H (3D10) APC | eBioscience | Cat # 14-5886-82; RRID:AB_906245 |
| Anti-mouse CD122 (5H4) PE | eBioscience | Cat # 12-1221-82; RRID:AB_465833 |
| Anti-mouse CD11b (M17/0) FITC | eBioscience | Cat #11-0112-82; RRID:AB_464935 |
| Anti-mouse CD11c (N418) PE | eBioscience | Cat # 12-0114-82; RRID:AB_465552 |
| Anti-mouse Gr1 (RB6–8C5) APC | eBioscience | Cat # 17-5931-82; RRID:AB_469476 |
| Anti-mouse B220 (RA3–6B2) APC | eBioscience | Cat # 17-0452-82; RRID:AB_469395 |
| Anti-mouse Foxp3 (FJK-16S) PE | eBioscience | Cat # 12-5773-82; RRID:AB_465936 |
| Anti-mouse PD1 (J43) PerCP-eF710 | eBioscience | Cat # 46-9985-82; RRID:AB_11150055 |
| Anti-mouse KLRG1 (2F1) APC | eBioscience | Cat # 17-5893-82; RRID:AB_469469 |
| Anti-mouse CD127 (A7R34) PE | eBioscience | Cat # 12-1271-82; RRID:AB_465844 |
| Anti-mouse CD62L (MEL-14) APC-eF780 | eBioscience | Cat # 47-0621-82; RRID:AB_1603256 |
| Anti-mouse CD62L (MEL-14) APC | eBioscience | Cat # 17-0621-82; RRID:AB_469410 |
| Anti-mouse CD27 (LG.7F9) PE | eBioscience | Cat # 12-0271-82; RRID:AB_465614 |
| Anti-mouse Cx3Cr1 (SA011F11) PerCP-Cy5.5 | Biolegend | Cat# 149010; RRID:AB_2564494 |
| IFN gamma monoclonal antibody (R4–6A2) unconjugated | eBioscience | Cat # MM701; RRID:AB_223538 |
| IFN gamma monoclonal antibody (XMG1.2) Biotin | eBioscience | Cat # MM700; RRID:AB_223608 |
| Bacterial and Virus Strains | ||
| LCMV-Armstrong | Badovinac Lab | NA |
| Critical Commercial Assays | ||
| Mouse IFN alpha Platinum ELISA 96 tests Kit | eBioscience | Cat #: BMS6027; RRID:AB_2575643 |
| Deposited Data | ||
| Unprocessed FACS Files and raw QTL mapping | Mendeley Data | https://dx.doi.org/10.17632/r7gfn2brw4.1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | National Cancer Institute | #556 |
| Mouse: BALB/c | National Cancer Institute | #555 |
| Mouse: Swiss Webster (CFW) | Charles River | #550 |
| Mouse: CC003/UNC | University of North Carolina | IMSR Cat# UNC102; RRID:IMSR_UNC:102 |
| Mouse: CC002/UNC | University of North Carolina | IMSR Cat # UNC44; RRID:IMSR_UNC:44 |
| Mouse: CC019/TAUUNC | University of North Carolina | IMSR Cat# UNC:114; RRID:IMSR_UNC:114 |
| Mouse: CC037/TAUUNC | University of North Carolina | IMSR Cat# UNC:159; RRID:IMSR_UNC:159 |
| Mouse: CC001/UNC | University of North Carolina | IMSR Cat# UNC:28; RRID:IMSR_UNC:28 |
| Mouse: CC041/TAUUNC | University of North Carolina | IMSR Cat# UNC:140; RRID:IMSR_UNC:140 |
| Mouse: CC068/TAUUNC | University of North Carolina | IMSR Cat# UNC:163; RRID:IMSR_UNC:163 |
| Mouse: CC055/TAUUNC | University of North Carolina | IMSR Cat# UNC:154; RRID:IMSR_UNC:154 |
| Mouse: CC006/TAUUNC | University of North Carolina | IMSR Cat# UNC:123; RRID:IMSR_UNC:123 |
| Mouse: CC071/TAUUNC | University of North Carolina | IMSR Cat# UNC:167; RRID:IMSR_UNC:167 |
| Mouse: CC051/TAUUNC | University of North Carolina | IMSR Cat# UNC:138; RRID:IMSR_UNC:138 |
| Mouse: CC041/TAUUNC | University of North Carolina | IMSR Cat# UNC:140; RRID:IMSR_UNC:140 |
| Mouse: CC011/UNC | University of North Carolina | IMSR Cat# UNC:11; RRID:IMSR_UNC:11 |
| Mouse: CC057/UNC | University of North Carolina | IMSR Cat# UNC:155; RRID:IMSR_UNC:155 |
| Mouse: CC036/UNC | University of North Carolina | IMSR Cat# UNC:131; RRID:IMSR_UNC:131 |
| Mouse: CC035/UNC | University of North Carolina | IMSR Cat# UNC:143; RRID:IMSR_UNC:143 |
| Mouse: CC023/GENIUNC | University of North Carolina | IMSR Cat# UNC:122; RRID:IMSR_UNC:122 |
| Mouse: CC053/UNC | University of North Carolina | IMSR Cat# UNC:149; RRID:IMSR_UNC:149 |
| Mouse: CC031/GENIUNC | University of North Carolina | IMSR Cat# UNC:96; RRID:IMSR_UNC:96 |
| Mouse: CC008/GENIUNC | University of North Carolina | IMSR Cat# UNC:94; RRID:IMSR_UNC:94 |
| Mouse: CC032/GENIUNC | University of North Carolina | IMSR Cat# UNC:31; RRID:IMSR_UNC:31 |
| Mouse: CC030/GENIUNC | University of North Carolina | IMSR Cat# UNC:25; RRID:IMSR_UNC:25 |
| Mouse: CC025/GENIUNC | University of North Carolina | IMSR Cat# UNC:126; RRID:IMSR_UNC:126 |
| Mouse: CC012/GENIUNC | University of North Carolina | IMSR Cat# UNC:127; RRID:IMSR_UNC:127 |
| Mouse: CC027/GENIUNC | University of North Carolina | IMSR Cat# UNC:152; RRID:IMSR_UNC:152 |
| Mouse: CC079/TAUUNC | University of North Carolina | IMSR Cat# UNC:172; RRID:IMSR_UNC:172 |
| Mouse: CC065/UNC | University of North Carolina | IMSR Cat# UNC:42; RRID:IMSR_UNC:42 |
| Mouse: CC072/TAUUNC | University of North Carolina | IMSR Cat# UNC:119; RRID:IMSR_UNC:119 |
| Mouse: CC004/TAUUNC | University of North Carolina | IMSR Cat# UNC:16; RRID:IMSR_UNC:16 |
| Mouse: CC005/TAUUNC | University of North Carolina | IMSR Cat# UNC:15; RRID:IMSR_UNC:15 |
| Mouse: CC059/TAUUNC | University of North Carolina | IMSR Cat# UNC:153; RRID:IMSR_UNC:153 |
| Mouse: CC013/GENIUNC | University of North Carolina | IMSR Cat# UNC:108; RRID:IMSR_UNC:108 |
| Mouse: CC015/UNC | University of North Carolina | IMSR Cat# UNC:10; RRID:IMSR_UNC:10 |
| Mouse: CC024/GENIUNC | University of North Carolina | IMSR Cat# UNC:125; RRID:IMSR_UNC:125 |
| Mouse: CC017/UNC | University of North Carolina | IMSR Cat# UNC:112; RRID:IMSR_UNC:112 |
| Mouse: CC021/UNC | University of North Carolina | IMSR Cat# UNC:117; RRID:IMSR_UNC:117 |
| Mouse: CC046/UNC | University of North Carolina | IMSR Cat# UNC:156; RRID:IMSR_UNC:156 |
| Mouse: CC056/GENIUNC | University of North Carolina | IMSR Cat# UNC:134; RRID:IMSR_UNC:134 |
| Mouse: CC043/GENIUNC | University of North Carolina | IMSR Cat# UNC:35; RRID:IMSR_UNC:35 |
| Mouse: CC044/UNC | University of North Carolina | IMSR Cat# UNC:158; RRID:IMSR_UNC:158 |
| Mouse: CC050/UNC | University of North Carolina | IMSR Cat# UNC:3; RRID:IMSR_UNC:3 |
| Mouse: CC052/GENIUNC | University of North Carolina | IMSR Cat# UNC:151; RRID:IMSR_UNC:151 |
| Mouse: CC058/UNC | University of North Carolina | IMSR Cat# UNC:168; RRID:IMSR_UNC:168 |
| Mouse: CC060/UNC | University of North Carolina | IMSR Cat# UNC:165; RRID:IMSR_UNC:165 |
| Mouse: CC061/GENIUNC | University of North Carolina | IMSR Cat# UNC:137; RRID:IMSR_UNC:137 |
| Mouse: CC063/UNC | University of North Carolina | IMSR Cat# UNC:141; RRID:IMSR_UNC:141 |
| Mouse: CC078/TAUUNC | University of North Carolina | IMSR Cat# UNC:171; RRID:IMSR_UNC:171 |
| Software and Algorithms | ||
| FACSCanto | BD Biosciences | https://www.bdbiosciences.com/en-us |
| Gen5 microplate reader and imager software | BioTek | https://www.biotek.com/products/software-robotics-software/gen5-microplate-reader-and-imager-software/ |
| R software environment | The R Project for Statistical Computing | https://www.r-project.org/ |
| DOQTL version 1.19.0 Bioconductor package | Daniel Gatti, Karl Broman, Andrey Shabalin, Petr Simecek | https://rdrr.io/bioc/DOQTL/ |
| Prism 8.0 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo | BD Biosciences | https://www.flowjo.com/ |
Highlights.
The Collaborative Cross (CC) models generation of T cell responses of various quantity
The CC models generation of qualitatively diverse memory CD8 T cells in response to LCMV
QTL mapping reveals candidate genes underlying generation of CD8 T central memory
ACKNOWLEDGMENTS
Funding for this study was provided from NIH grants AI42767, AI85515, and AI100527 (J.T.H.); AI114543 (J.T.H. and V.P.B.); and AI147064, GM113961, and GM134880 (V.P.B.). We wish to thank Dr. Anne Kwitek for initial help with QTL analysis and Ginger Shaw for help with procurement of Collaborative Cross mice used in this study.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.03.072.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, Youngblood BA, Abdelsamed HA, McGuire DJ, Cohen KW, et al. (2017). Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552, 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann MF, Wolint P, Schwarz K, Jäger P, and Oxenius A (2005). Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor alpha and CD62L. J. Immunol 175, 4686–4696. [DOI] [PubMed] [Google Scholar]
- Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, et al. (2016). Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LE, and Kelso A (2009). Prospects for an influenza vaccine that induces cross-protective cytotoxic T lymphocytes. Immunol. Cell Biol 87, 300–308. [DOI] [PubMed] [Google Scholar]
- Busch DH, Kerksiek KM, and Pamer EG (2000). Differing roles of inflammation and antigen in T cell proliferation and memory generation. J. Immunol 164, 4063–4070. [DOI] [PubMed] [Google Scholar]
- Chesler EJ, Miller DR, Branstetter LR, Galloway LD, Jackson BL, Philip VM, Voy BH, Culiat CT, Threadgill DW, Williams RW, et al. (2008). The Collaborative Cross at Oak Ridge National Laboratory: developing a powerful resource for systems genetics. Mamm. Genome 19, 382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, et al. ; Complex Trait Consortium (2004). The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet 36, 1133–1137. [DOI] [PubMed] [Google Scholar]
- Collaborative Cross Consortium (2012). The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics 190, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin R, Balmer L, Morahan G, and Lesage S (2019). Common heritable immunological variations revealed in genetically diverse inbred mouse strains of the Collaborative Cross. J. Immunol 202, 777–786. [DOI] [PubMed] [Google Scholar]
- Cook PC, Owen H, Deaton AM, Borger JG, Brown SL, Clouaire T, Jones GR, Jones LH, Lundie RJ, Marley AK, et al. (2015). A dominant role for the methyl-CpG-binding protein Mbd2 in controlling Th2 induction by dendritic cells. Nat. Commun 6, 6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S (2015). A brief history of T cell help to B cells. Nat. Rev. Immunol 15, 185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, and Mescher MF (2005). Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol 174, 4465–4469. [DOI] [PubMed] [Google Scholar]
- Duan S, and Thomas PG (2016). Balancing immune protection and immune pathology by CD8(+) T-cell responses to influenza infection. Front. Immunol 7, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, and Bluestone JA (2016). Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat. Rev. Immunol 16, 149–163. [DOI] [PubMed] [Google Scholar]
- Eberlein J, Davenport B, Nguyen T, Victorino F, Haist K, Jhun K, Karimpour-Fard A, Hunter L, Kedl R, Clambey ET, and Homann D (2016). Aging promotes acquisition of naive-like CD8+ memory T cell traits and enhanced functionalities. J. Clin. Invest 126, 3942–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbahesh H, and Schughart K (2016). Genetically diverse CC-founder mouse strains replicate the human influenza gene expression signature. Sci. Rep 6, 26437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein JE, Tewari K, Lyke KE, Sim BK, Billingsley PF, Laurens MB, Gunasekera A, Chakravarty S, James ER, Sedegah M, et al. (2011). Live attenuated malaria vaccine designed to protect through hepatic CD8+ T cell immunity. Science 334, 475–480. [DOI] [PubMed] [Google Scholar]
- Ferris MT, Aylor DL, Bottomly D, Whitmore AC, Aicher LD, Bell TA, Bradel-Tretheway B, Bryan JT, Buus RJ, Gralinski LE, et al. (2013). Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog. 9, e1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti DM, Svenson KL, Shabalin A, Wu LY, Valdar W, Simecek P, Goodwin N, Cheng R, Pomp D, Palmer A, et al. (2014). Quantitative trait locus mapping methods for diversity outbred mice. G3 (Bethesda) 4, 1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, de la Torre JC, and von Andrian UH (2016). The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity 45, 1270–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JB, Thomas S, Swarts J, McMillan AA, Ferris MT, Suthar MS, Treuting PM, Ireton R, Gale M Jr., and Lund JM (2015). Genetic diversity in the collaborative cross model recapitulates human West Nile virus disease outcomes. MBio 6, e00493–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JB, Swarts JL, Wilkins C, Thomas S, Green R, Sekine A, Voss KM, Ireton RC, Mooney M, Choonoo G, et al. (2016). A mouse model of chronic West Nile virus disease. PLoS Pathog. 12, e1005996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JB, Swarts JL, Mooney M, Choonoo G, Jeng S, Miller DR, Ferris MT, McWeeney S, and Lund JM (2017). Extensive homeostatic T cell phenotypic variation within the Collaborative Cross. Cell Rep. 21, 2313–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralinski LE, Ferris MT, Aylor DL, Whitmore AC, Green R, Frieman MB, Deming D, Menachery VD, Miller DR, Buus RJ, et al. (2015). Genome wide identification of SARS-CoV susceptibility loci using the Collaborative Cross. PLoS Genet. 11, e1005504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iraqi FA, Churchill G, and Mott R (2008). The Collaborative Cross, developing a resource for mammalian systems genetics: a status report of the Well-come Trust cohort. Mamm. Genome 19, 379–381. [DOI] [PubMed] [Google Scholar]
- Jia A, Wang Y, Sun W, Xiao B, Wei Y, Qiu L, Mu L, Xu L, Li J, Zhang X, et al. (2017). MBD2 regulates Th17 cell differentiation and experimental severe asthma by affecting IRF4 expression. Mediators Inflamm. 2017, 6249685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM (2007). Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidlaw BJ, Craft JE, and Kaech SM (2016). The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol 16, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapo sová K, Pastoreková S, and Tomá sková J (2013). Lymphocytic choriomeningitis virus: invisible but not innocent. Acta Virol. 57, 160–170. [DOI] [PubMed] [Google Scholar]
- Leist SR, Pilzner C, van den Brand JM, Dengler L, Geffers R, Kuiken T, Balling R, Kollmus H, and Schughart K (2016). Influenza H3N2 infection of the collaborative cross founder strains reveals highly divergent host responses and identifies a unique phenotype in CAST/EiJ mice. BMC Genomics 17, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorè NI, Iraqi FA, and Bragonzi A (2015). Host genetic diversity influences the severity of Pseudomonas aeruginosa pneumonia in the Collaborative Cross mice. BMC Genet. 16, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, and Kallies A (2017). Transcriptional regulation of tissue-resident lymphocytes. Trends Immunol. 38, 94–103. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, and Gebhardt T (2012). Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl. Acad. Sci. USA 109, 7037–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MD, Kim MT, Shan Q, Sompallae R, Xue HH, Harty JT, and Badovinac VP (2015). Phenotypic and functional alterations in circulating memory CD8 T cells with time after primary infection. PLoS Pathog. 11, e1005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MD, Danahy DB, Hartwig SM, Harty JT, and Badovinac VP (2017). Revealing the complexity in CD8 T cell responses to infection in inbred C57B/6 versus outbred Swiss mice. Front. Immunol 8, 1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masopust D (2009). Developing an HIV cytotoxic T-lymphocyte vaccine: issues of CD8 T-cell quantity, quality and location. J. Intern. Med 265, 125–137. [DOI] [PubMed] [Google Scholar]
- Masopust D, Sivula CP, and Jameson SC (2017). Of mice, dirty mice, and men: using mice to understand human immunology. J. Immunol 199, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott DS, and Varga SM (2011). Quantifying antigen-specific CD4 T cells during a viral infection: CD4 T cell responses are larger than we think. J. Immunol 187, 5568–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morahan G, Balmer L, and Monley D (2008). Establishment of “The Gene Mine”: a resource for rapid identification of complex trait genes. Mamm. Genome 19, 390–393. [DOI] [PubMed] [Google Scholar]
- Nolz JC, and Harty JT (2011). Protective capacity of memory CD8+ T cells is dictated by antigen exposure history and nature of the infection. Immunity 34, 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JA, McDonald-Hyman C, Jameson SC, and Hamilton SE (2013). Effector-like CD8+ T cells in the memory population mediate potent protective immunity. Immunity 38, 1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamer EG (2004). Immune responses to Listeria monocytogenes. Nat. Rev. Immunol 4, 812–823. [DOI] [PubMed] [Google Scholar]
- Pham NL, Badovinac VP, and Harty JT (2009). A default pathway of memory CD8 T cell differentiation after dendritic cell immunization is deflected by encounter with inflammatory cytokines during antigen-driven proliferation. J. Immunol 183, 2337–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillippi J, Xie Y, Miller DR, Bell TA, Zhang Z, Lenarcic AB, Aylor DL, Krovi SH, Threadgill DW, de Villena FP, et al. (2014). Using the emerging Collaborative Cross to probe the immune system. Genes Immun. 15, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter BB, and Harty JT (2006). The onset of CD8+-T-cell contraction is influenced by the peak of Listeria monocytogenes infection and antigen display. Infect. Immun 74, 1528–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai D, Pham NL, Harty JT, and Badovinac VP (2009). Tracking the total CD8 T cell response to infection reveals substantial discordance in magnitude and kinetics between inbred and outbred hosts. J. Immunol 183, 7672–7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen AL, Okumura A, Ferris MT, Green R, Feldmann F, Kelly SM, Scott DP, Safronetz D, Haddock E, LaCasse R, et al. (2014). Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science 346, 987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese TA, Bi K, Kambal A, Filali-Mouhim A, Beura LK, Bürger MC, Pulendran B, Sekaly RP, Jameson SC, Masopust D, et al. (2016). Sequential infection with common pathogens promotes human-like immune gene expression and altered vaccine response. Cell Host Microbe 19, 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. (2017). Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Förster R, Lipp M, and Lanzavecchia A (1999). Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712. [DOI] [PubMed] [Google Scholar]
- Schenkel JM, Fraser KA, Vezys V, and Masopust D (2013). Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol 14, 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ME, and Varga SM (2018). The CD8 T cell response to respiratory virus infections. Front. Immunol 9, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt NW, Podyminogin RL, Butler NS, Badovinac VP, Tucker BJ, Bahjat KS, Lauer P, Reyes-Sandoval A, Hutchings CL, Moore AC, et al. (2008). Memory CD8 T cell responses exceeding a large but definable threshold provide long-term immunity to malaria. Proc. Natl. Acad. Sci. USA 105, 14017–14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt NW, Butler NS, Badovinac VP, and Harty JT (2010). Extreme CD8 T cell requirements for anti-malarial liver-stage immunity following immunization with radiation attenuated sporozoites. PLoS Pathog. 6, e1000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seder RA, Chang LJ, Enama ME, Zephir KL, Sarwar UN, Gordon IJ, Holman LA, James ER, Billingsley PF, Gunasekera A, et al. ; VRC 312 Study Team (2013). Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science 341, 1359–1365. [DOI] [PubMed] [Google Scholar]
- Slütter B, Pewe LL, Kaech SM, and Harty JT (2013). Lung airway-surveilling CXCR3(hi) memory CD8(+) T cells are critical for protection against influenza A virus. Immunity 39, 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slütter B, Van Braeckel-Budimir N, Abboud G, Varga SM, Salek-Ardakani S, and Harty JT (2017). Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci. Immunol 2, eaag2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Morgan AP, Najarian ML, Sarsani VK, Sigmon JS, Shorter JR, Kashfeen A, McMullan RC, Williams LH, Giusti-Rodríguez P, et al. (2017). Genomes of the mouse Collaborative Cross. Genetics 206, 537–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threadgill DW, and Churchill GA (2012). Ten years of the collaborative cross. G3 (Bethesda) 2, 153–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threadgill DW, Miller DR, Churchill GA, and de Villena FP (2011). The collaborative cross: a recombinant inbred mouse population for the systems genetic era. ILAR J. 52, 24–31. [DOI] [PubMed] [Google Scholar]
- Welsh CE, Miller DR, Manly KF, Wang J, McMillan L, Morahan G, Mott R, Iraqi FA, Threadgill DW, and de Villena FP (2012). Status and access to the Collaborative Cross population. Mamm. Genome 23, 706–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, and Ahmed R (2003). Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol 4, 225–234. [DOI] [PubMed] [Google Scholar]
- Wirth TC, Harty JT, and Badovinac VP (2010). Modulating numbers and phenotype of CD8+ T cells in secondary immune responses. Eur. J. Immunol 40, 1916–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray NR, and Visscher PM (2008). Estimating trait heritability. Nature Education 1, 29. [Google Scholar]
- Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, and Cauley LS (2014). Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol 95, 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Ramachandran S, Mann M, and Popkin DL (2012). Role of lymphocytic choriomeningitis virus (LCMV) in understanding viral immunology: past, present and future. Viruses 4, 2650–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for unproccessed FACS Files pertaining to Figures 1–5 and raw QTL mapping pertaining to Figure 6 are available at Mendeley Data https://dx.doi.org/10.17632/r7gfn2brw4.1.