

Abstract
Background
Gliomas are the most common primary brain tumors. High-Grade Gliomas have a median survival (MS) of 18 months, while Low-Grade Gliomas (LGGs) have an MS of approximately 7.3 years. Seventy-six percent of patients with LGG express mutated isocitrate dehydrogenase (mIDH) enzyme. Survival of these patients ranges from 1 to 15 years, and tumor mutational burden ranges from 0.28 to 3.85 somatic mutations/megabase per tumor. We tested the hypothesis that the tumor mutational burden would predict the survival of patients with tumors bearing mIDH.
Methods
We analyzed the effect of tumor mutational burden on patients’ survival using clinical and genomic data of 1199 glioma patients from The Cancer Genome Atlas and validated our results using the Glioma Longitudinal AnalySiS consortium.
Results
High tumor mutational burden negatively correlates with the survival of patients with LGG harboring mIDH (P = .005). This effect was significant for both Oligodendroglioma (LGG-mIDH-O; MS = 2379 vs 4459 days in high vs low, respectively; P = .005) and Astrocytoma (LGG-mIDH-A; MS = 2286 vs 4412 days in high vs low respectively; P = .005). There was no differential representation of frequently mutated genes (eg, TP53, ATRX, CIC, and FUBP) in either group. Gene set enrichment analysis revealed an enrichment in Gene Ontologies related to cell cycle, DNA-damage response in high versus low tumor mutational burden. Finally, we identified 6 gene sets that predict survival for LGG-mIDH-A and LGG-mIDH-O.
Conclusions
we demonstrate that tumor mutational burden is a powerful, robust, and clinically relevant prognostic factor of MS in mIDH patients.
Keywords: clinical prognosis, DNA-repair prognostic signature, glioma, IDH, tumor mutational burden
Key Points.
Tumor mutational burden predicts survival in mIDH but not wtIDH tumors.
High mutational burden is associated with enrichment in DNA repair-related genes.
We developed 6 high-risk gene sets that are associated with poor prognosis in Astrocytoma and Oligodendroglioma.
Importance of the Study.
Mutations in IDH occur in approximately 75% of low-grade gliomas (LGGs) and correlate with extended survival. We performed a comprehensive mutational and survival analysis of LGG patients positive for IDH mutation, using 2 publicly available datasets, TCGA and GLASS. We found that tumor mutational burden is a unique and independent prognostic factor that negatively impacts LGG-mIDH, but not LGG-wtIDH, or glioblastoma patients. Gene expression in patients with high tumor mutational burden was associated with enrichment in DNA repair and cell cycle-related genes. LASSO analysis revealed 6 gene sets that are associated with poor prognosis in mIDH tumor. We propose that tumor mutational burden represents a clinically simple and powerful prognostic factor to predict overall survival in LGG-mIDH patients. This may potentially improve the stratification of LGG patients and provide a more accurate assessment of personalized treatment in the clinic.
Low-grade gliomas (LGGs) are slow-growing brain tumors that occur in early adult life and can progress to high-grade gliomas (HGGs).1 Molecular characterizations coupled with the histological classification revealed that a mutation in isocitrate dehydrogenase (mIDH), IDHR132H, is the main genetic lesion in LGG patients.2–5 Somatic mutation in IDH1, and far less common IDH2, results in excessive production of 2 hydroxyglutarate (2HG).6–8 2HG is a potent and competitive inhibitor of α-ketoglutarate-dependent dioxygenases, which are responsible for demethylation of DNA and histones.4,9 The ensuing hypermethylation phenotype triggers epigenetic reprogramming of the glioma cells’ transcriptome.3,10–13
The consequences of mIDH in glioma cells contribute to cancer development and progression not only by disrupting cell metabolism but also by altering the epigenetic landscape. Metabolically, IDH is one of the enzymes that encodes an irreversible reaction in the tricyclic acid cycle. Disruption of the IDH reaction results in defective mitochondrial oxidative phosphorylation, glutamine metabolism, lipogenesis, glucose sensing, and altering of cellular redox status.8,14–18 IDH also inhibits glioma stem cell differentiation,4,8 upregulates vascular endothelial growth factor to promote tumor microenvironment formation,19,20 and produces high levels of hypoxia-inducible factor-1α to promote glioma invasion,18,21,22 which ultimately leads to glioma progression. We have acquired a tremendous insight on the molecular mechanisms of genetic alterations associated with malignant transformation of IDH mutations. This includes activation of NOTCH1, RTK–RAS–PI3K, and Myc-RB1 signaling and/or deleted region of the CDKN2A/2B locus, all of which were found to be altered in progressed mIDH glioblastoma (GBM) samples compared to their lower grade counterparts.2,23–25 Nevertheless, the exact mechanism that impacts mIDH patient survival is still under debate.
In this regard, a significant advance in recent years has identified a set of genetic lesions that are characteristic of mIDH and correlate with clinical outcome. This was done in hope for better prediction of tumor behavior and outcome, including identification of secondary mutations, genetic alterations, methylation patterns, and multivariate prognostic models.26–29 Within the group of IDH-mutant gliomas, the presence of 1p/19q co-deletion (IDH-mutant–codel glioma) may present an additional prognostic marker separate from IDH-mutant glioma with intact 1p/19q chromosome arms (IDH-mutant–non-codel glioma). Other genetic alterations that affect LGG patient’s survival include mutation of PIK3CA and PIK3R1, and deletion of CDKN2A in Astrocytoma mIDH, and 10q25.2 region deletions.30–32
Given the high level of inter-tumor heterogeneity inferred from the presence of a variety of genetic lesions, and the fact that mIDH tumors are more resistant to radiotherapy than the wtIDH,33,34 it is tempting to speculate that tumor mutational burden may be a strong predictor for mIDH patient prognosis. In most solid cancers, the mutational load negatively impacts patient survival35 and it is used as an indicator of rapid tumor progression.
In the present study, we analyzed the publicly available The Cancer Genome Atlas (TCGA) and Glioma Longitudinal AnalySiS (GLASS) datasets in order to correlate the tumor mutational burden with overall survival (OS) in mIDH LGG subtypes. We found that increasing tumor mutational burden negatively impacts the survival of LGG but not GBM. Further analysis of LGG patients revealed that the effect of tumor mutational burden on survival is unique for patients with mIDH but not wtIDH. This effect is prominent for both Astrocytoma and Oligodendroglioma mIDH subgroup. Moreover, based on the prognosis, we constructed high-risk genes that predict poor prognosis in patients with IDH mutation. These data suggest that tumor mutational burden is an independent prognostic factor for glioma patients with IDH mutation and can be used as a predictor of patient survival.
Methods
Source of Data and Analyses
All clinical, RNAseq, copy number variations (CNVs), and mutational TCGA data were downloaded from the Broad Institute firebrowse36: http://firebrowse.org/?cohort=GBMLGG&download_dialog=true#. GLASS data were downloaded from https://www.glass-consortium.org/. Chinese Glioma Genome Atlas (CGGA) data were downloaded from the CGGA website: http://www.cgga.org.cn/. Patients were categorized according to their mIDH status, tumor grade, and tumor mutational load (high vs low). For mutations and CNVs frequency analysis, data from all patients were converted into matrix files and an in-house developed R-script was used to determine the frequency of each mutation in all the analyzed groups.
Mutation Analysis
TCGA mutation analysis was done through the Broad’s Institute firebrowse stringent filtering and annotation pipeline to obtain a uniform set of mutation calls.36–39 In brief, the number of mutations and the number of covered bases for each gene were tabulated. The significant metric was calculated for each gene, using the Lawrence et al. methods (MutSigCV) which measure the significance of mutation burden.40 MutSigCV determines the P-value for observing the given quantity of non-silent mutations in the gene, given the background model determined by silent (and noncoding) mutations in the same gene, and the neighboring genes of covariate space.39 Mutational representations were plotted using comuplotter.41 GLASS mutational analysis was obtained from GLASS consortium.42 Mutation frequencies for each tumor were calculated by implementing the SQL query provided by the Jackson Lab for performing the frequency calculation within the GLASS database, published in GitHub at the following location: https://github.com/TheJacksonLaboratory/GLASS/blob/master/sql/mut_freq/mut_freq.sql. The query implements a join operation between 2 computed tables, “analysis_coverage” (id: syn18477440, last modified March 27, 2019) and “variants_ssm2_count” (id: syn21599128, last modified February 14, 2020), which were downloaded from the GLASS website location: https://www.synapse.org/#!Synapse:syn17038081/tables/. To perform the query, an SQLlite relational database instance (v 3.30.1) was instantiated locally and the downloaded tables were loaded into the instance. The provided query was performed directly in the sqlite3 command-line shell, exporting the results to a local csv file containing the coverage-adjusted mutation frequency for each aliquot barcode. Only the primary tumor sample was considered in the analysis.
For both datasets, mutation rate cutoff was determined based on the maximal log-rank cutoff test as described previously.43 This test was applied to the mutation rate in order to estimate the most appropriate cutoff values for splitting patients into groups with different OS probabilities.
Survival Analysis
Survival data were censored at the last date the patient was known to be alive. Survival functions were estimated by the Kaplan–Meier method and compared using the log-rank test. The median survival (MS) time is calculated as the smallest survival time for which the survivor function is less than or equal to 0.5. Cox proportional hazards regression was used to assess the effect of mutation numbers and IDH mutation status on patient survival. In the Cox model, an interaction between the tumor mutational burden and IDH mutation is tested to estimate the effect of mutation numbers with and without the IDH mutation. The assumptions of proportional hazard and linear form of covariates were assessed by martingale residuals plots and the Kolmogorov-type supremum test. All analyses for survival data were done using SAS 9.4 software. P < .05 was considered significant.
For regression analysis, only the deceased patients from each group were considered. Linear regression analysis was performed for the “days to death” as a dependent variable versus the number of mutated genes per patient (independent variable). The analysis was done using STATA 15.1 software. P < .05 was considered significant.
For Significance Analysis of Prognostic Signatures (SAPS), significant gene sets enriched in LGG-mIDH-Ahigh or LGG-mIDH-Ohigh were used to compute the true gene set that can predict survival in Astrocytoma and Oligodendroglioma, separately. SAPS was run using the Bioconductor R package “SAPS”: https://rdrr.io/bioc/saps/man/saps.html. All gene sets were compared in their ability to predict the survival based on the 3 P-values computed by SAPS (Ppure, Prandom, and Penrichment).44 Only gene sets that showed significance in all 3 P-values <.05 were considered significant.
Gene Set Enrichment Analysis
All patients were screened for the tumor grade, mutations load, and IDH status. Rank file was created by implementing R script from Bader lab (https://github.com/BaderLab/EM-tutorials-docker/blob/master/R_scripts/supplemental_protocol2_rnaseq.R). The gmt file downloaded from Broad Institute website (http://software.broadinstitute.org/gsea/index.jsp) and it contains all gene ontology (GO) sets to be included in the gene set enrichment analysis (GSEA). Files for positively and negatively enriched groups were used as input files to create the enrichment map in Cytoscape.
Results
Tumor Mutational Burden Unexpectedly Predicts Increased Aggressive Clinical Course Only in LGG-mIDH
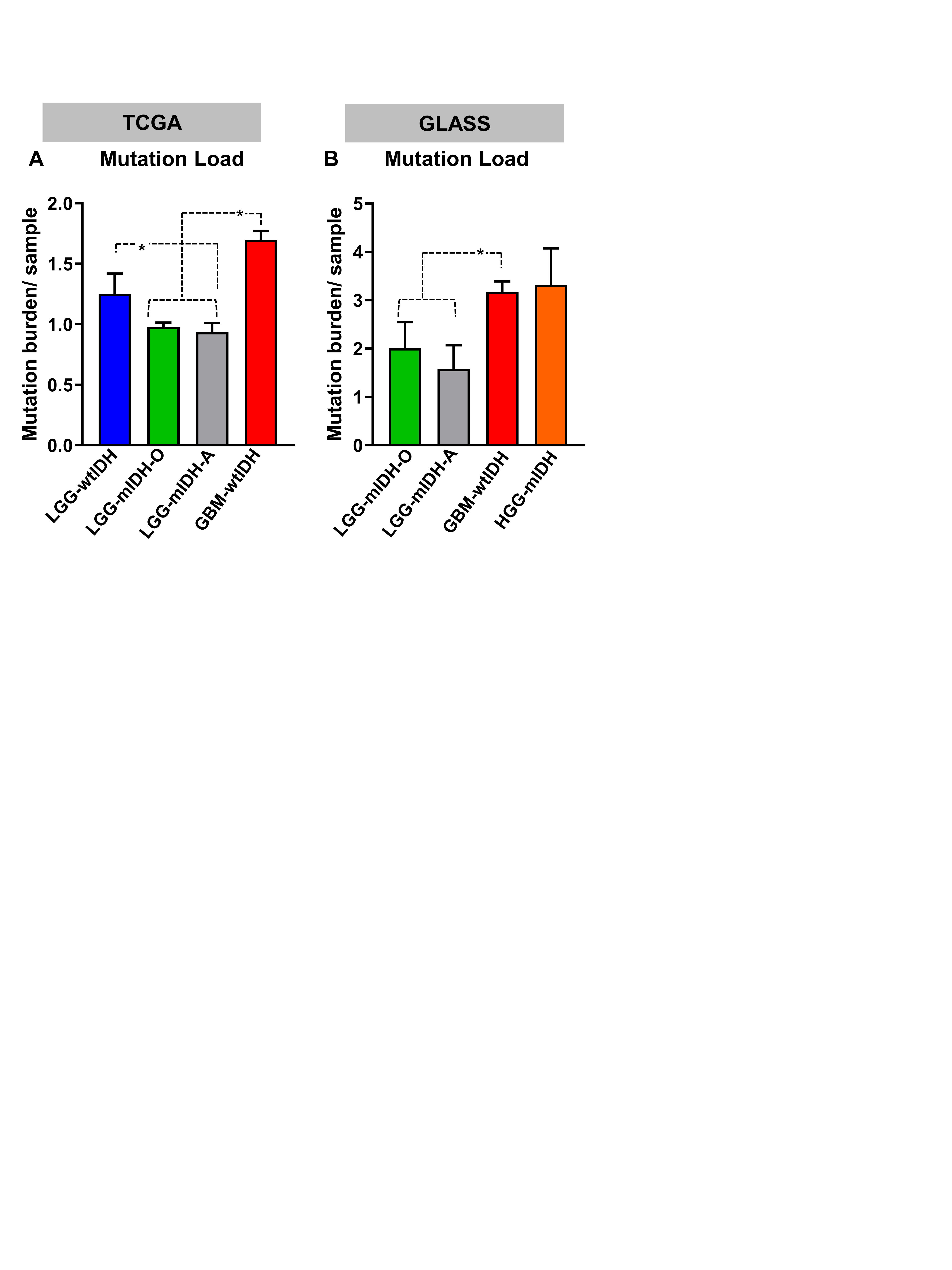
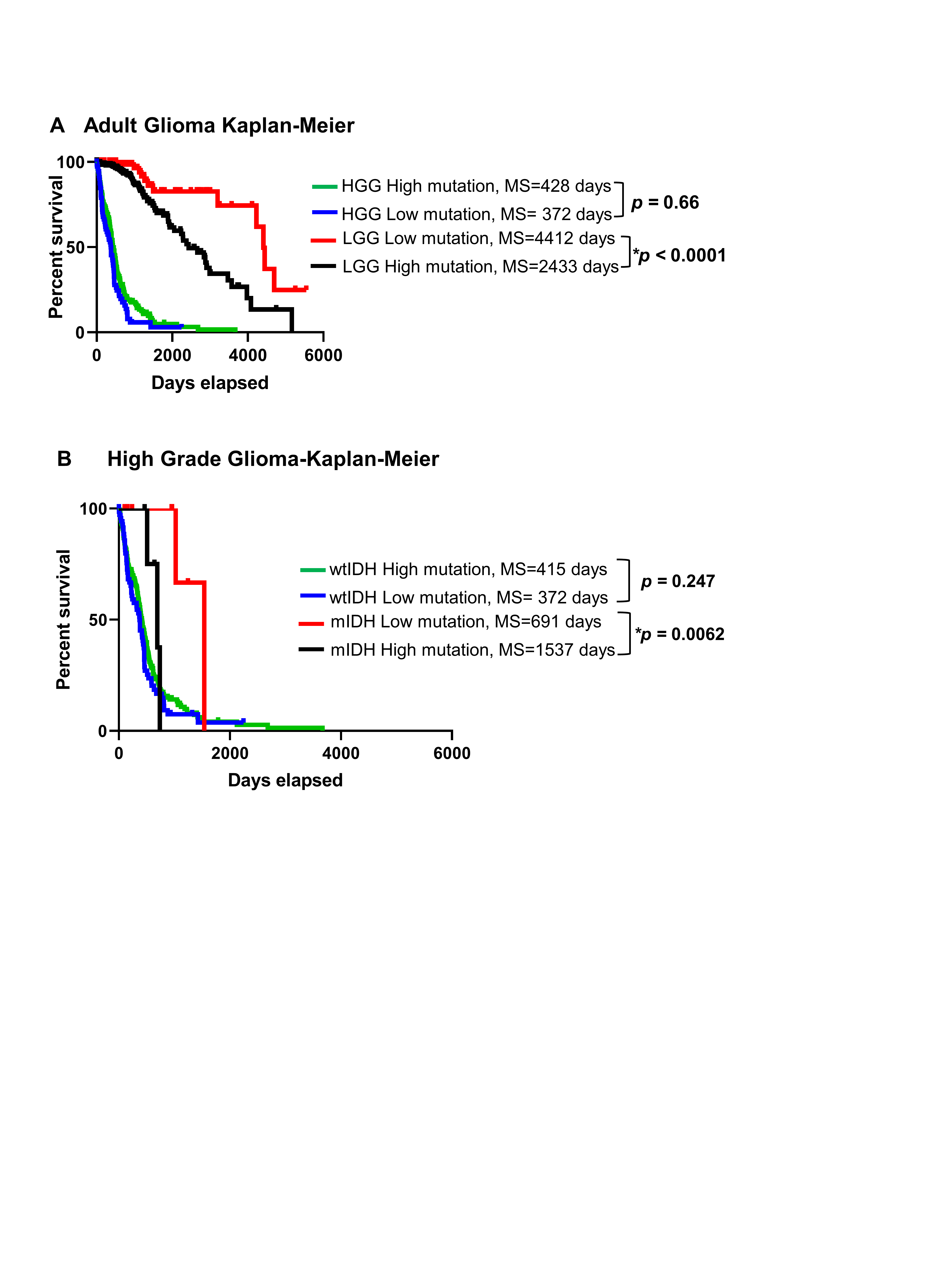
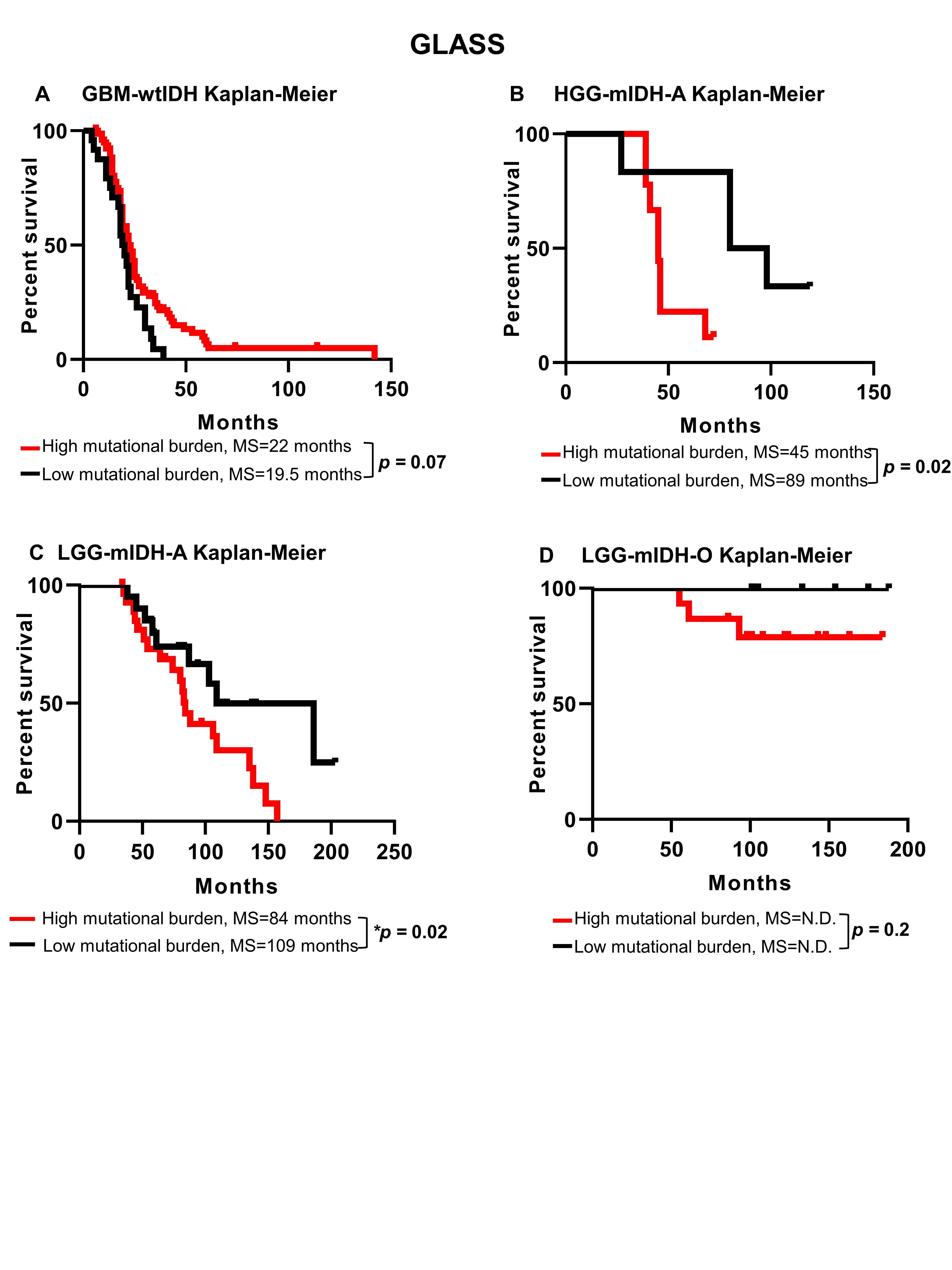
We analyzed the clinical data of 1199 patients from TCGA. Among the TCGA patients, we used 799 patients for whom there are available complete mutational data. We examined the tumor mutational burden (#Mutations/Mb) in HGG and LGG in TCGA and validated our results utilizing 196 patients from the GLASS consortium.42 All patients’ clinical and mutational data are summarized in Supplementary Tables S1 and S2. To assess the impact of tumor mutational burden on the patient’s survival, we classified all patients according to the tumor mutational burden into 2 groups (high vs low), based on the maximal log-rank cutoff test as described before43 (Figure 1A). We found that overall mutational load was higher in HGG patients compared to the LGG patients, regardless of the IDH mutation (Supplementary Figure S1A and B). We then applied the “life test” procedure to compare the survival probabilities between patients with high versus low tumor mutational burden. Surprisingly, there was no difference in survival between HGG with high tumor mutational burden as compared to HGG with low tumor mutational burden (Supplementary Figure S2A). In contrast, LGG patients with high tumor mutational burden have a significant decrease in MS as compared to LGG patients with low tumor mutational burden (Supplementary Figure S2A). We stratified patients in terms of the IDH mutation and found no significant differences in MS between HGG patients with wild-type IDH (wtIDH) who have a high mutation load (GBM-wtIDHhigh) as compared to those with low mutation load (GBM-wtIDHlow) (Figure 1B). Similar results were obtained when comparing LGG patients with tumors expressing wtIDH; there were no differences in MS between high mutation versus low mutation group (Figure 1D). However, high tumor mutational burden was negatively correlated with patients’ prognosis in mIDH LGG patients with no 1p/19q codeletion (ie, Astrocytoma; LGG-mIDH-A), as well as in mIDH LGG patients with 1p/19q codeletion (ie, Oligodendroglioma; LGG-mIDH-O) (Figure 1F and H). In HGG patients with mIDH tumors, tumor mutational burden was also negatively correlated with survival, even though the number of patients available to be studied was low (n = 14) (Supplementary Figure S2B). Except for patient age which impacts the prognosis of LGG-mIDH-O, there was no effect of patient’s gender, radiation therapy, or TMZ treatment on OS between the analyzed groups (Supplementary Table S3). We validated the impact of tumor mutational burden on the survival of LGG-mIDH using two approaches. First, we tested if there was a correlation between the survival rate and tumor burden in all tumor groups. We applied a linear regression model to see if we could predict the “days to death” (dependent variable) of glioma patients based on the tumor mutational burden (independent variable). Since an endpoint is required for linear regression, we only considered the deceased patients in this analysis. Linear regression modeling showed a significant dependency of patients “days to death” on tumor mutational burden only in patients with LGG-mIDH tumors (Figure 1C, E, G, and I). Second, we validated the TCGA results by studying 196 patients from the GLASS database to further corroborate our hypothesis using a separate dataset.42 In agreement with the results from the TCGA, no difference in survival was seen between GBM-wtIDHhigh and GBM-wtIDHlow (Supplementary Figure S3A). However, the high mutational burden negatively impacts patient survival in both HGG and LGG astrocytoma patients with mIDH (Supplementary Figure S3B and C). Despite the low number of patients in the LGG-mIDH-O group, all patients within LGG-mIDH-Olow were alive, whereas, 4 patients within the LGG-mIDH-Ohigh were deceased (P = .2, NS, Supplementary Figure S3D). These data confirm the effect of tumor mutational burden on the survival of patients with LGG expressing the IDH mutation, in two unrelated databases.
Figure 1.

Poor prognosis in mIDH patients from TCGA with a high mutational burden. (A) Classification of glioma patients according to the subtype and mutational burden used in this study. (B) Kaplan–Meier survival of GBM-wtIDH patients with high mutational burden (red; N = 71) or low mutational burden (black; N = 208) from TCGA. There was no significant difference in survival between groups (hazard ratio [HR] = 1.203, CI [1.644–0.87]). (C) Linear regression model of patients’ “days to death” versus mutational burden/patient in GBM-wtIDH (N = 218). There was no correlation between patients’ “days to death” and mutational load. (D) Kaplan–Meier curves of LGG-wtIDH patients classified according to the mutational burden in TCGA. There is no difference in survival between LGG-wtIDHhigh (N = 72) and LGG-wtIDHlow (N = 22) (HR = 0.58, CI [0.29–1.125]). (E) Linear regression model of patients’ “days to death” versus mutation burden/patient in LGG-wtIDH (N = 51). There was no correlation between patients’ “days to death” and mutational load. (F) LGG-mIDH-Ahigh (N = 189) has statistically significant decreased median survival as compared to LGG-mIDHlow (N = 63) (HR = 0.3891, CI [0.2193–0.6905]). (G) Linear regression model of patients’ “days to death” versus mutation burden/patient in LGG-mIDH-A (N = 51). There is a significant correlation between patients’ days to death and mutation load/patient in LGG-mIDH-A (R = −0.31, P < .001). (H) LGG-mIDH-Ohigh (N = 74) has statistically significantly decreased median survival as compared to LGG-mIDHlow (N = 95) (HR = 0.3198, CI [0.1343–0.7616]). (I) Linear regression model of patients’ “days to death” versus mutation burden/patient in LGG-mIDH-O (N = 23). There is a significant correlation between patients’ days to death and mutation load/patient in LGG-mIDH-O (R = −0.54, P < .001).
Association of Genetic Alterations With OS
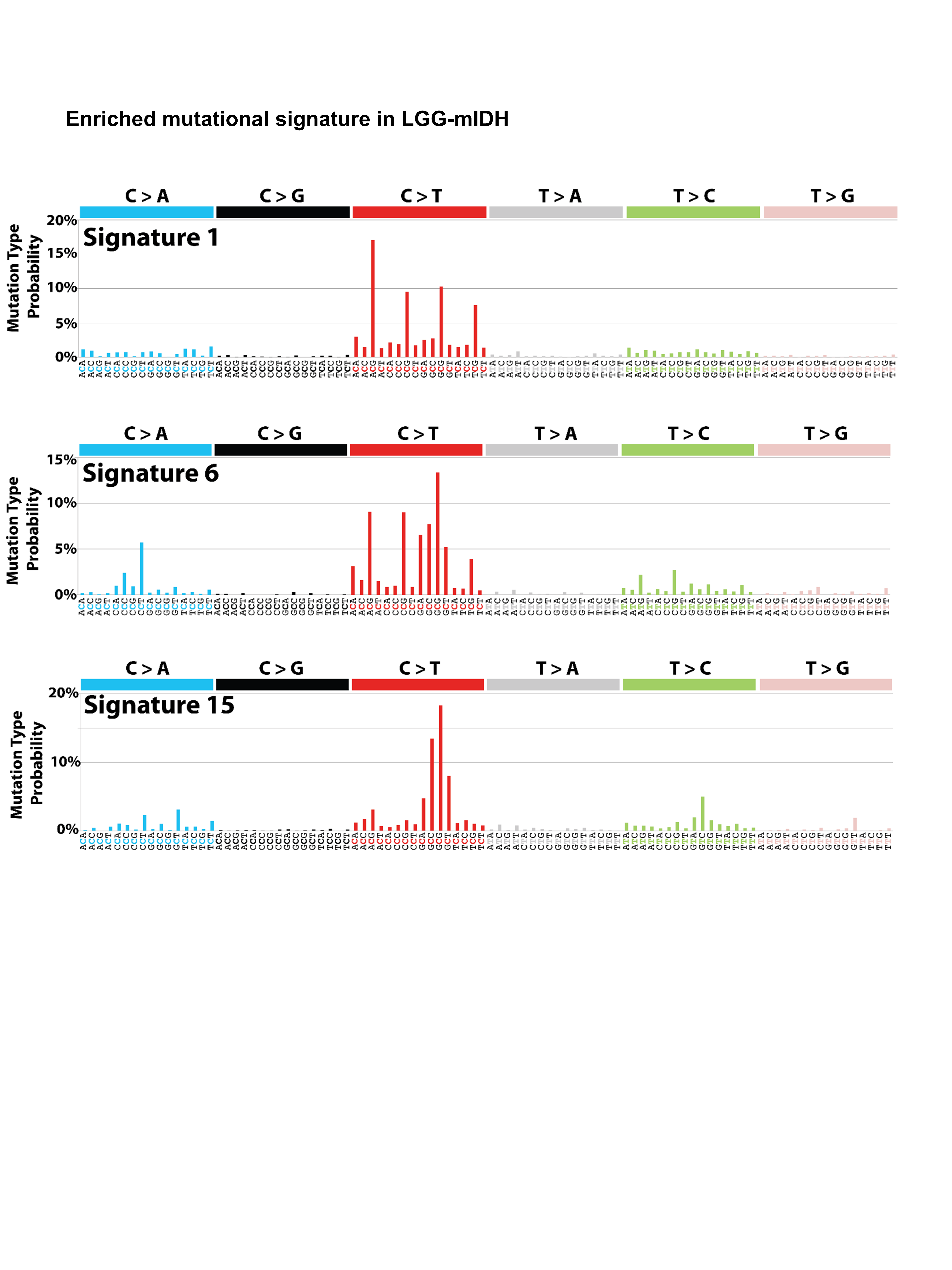
Next we tested if the differences in tumor mutational load could be explained by changes in the frequency of individual mutations, mutational signatures (COSMIC), or CNV. In LGG-mIDH-A, we found that 4 out of the top 10 commonly mutated genes are shared in both high and low mutation groups. These genes are IDH, TP53, ATRX, and TTN (Figure 2A). None of these mutations have a significant impact on OS between LGG-mIDH-Ahigh and LGG-mIDH-Alow (Figure 4A). Mutated genes that are unique to the LGG-mIDH-Ahigh are MUC16, SMARCA4, MUC17, CSMD3, APOB, and NOTCH1 (Figure 2A). With the exception of SMARCA4, none of these mutations have an impact on patient’s survival (Figure 4A). Notably, the frequency of SMARCA4 is less than 8% of total patients within the LGG-mIDH-Ahigh. Mutations in RYR2, PCDH19, CIC, C3, BCOR, and ACSM5 genes are unique in the LGG-mIDH-Alow (Figure 2B). ACSM5 was the only mutation associated with survival in LGG-mIDH-Alow (Figure 2B). The COSMIC database has categorized 30 reference mutation spectra signatures based on the analysis of 40 distinct types of human cancer.45 The genetic mutation signatures across all patients within the LGG-mIDH-Ahigh and LGG-mIDH-Alow groups were identical (Figure 2A and B, bottom plots). The most common signatures in either group were 1, 6, and 15 (Figure 2A and B, Supplementary Figure S4).
Figure 2.

Somatic genetic alterations identified in LGG-mIDH-A according to the tumor mutational burden. (A) Upper plot shows the mutation rate for each tumor sample in LGG-mIDH-Ahigh. Middle plot: Heatmap of the most frequent somatic mutations identified in LGG-mIDH-Ahigh. Genes are sorted according to the FDR q-value. Lower plot: Mutational signature analysis in LGG-mIDH-Ahigh samples. (B) Upper plot shows the mutation rate for each tumor sample in LGG-mIDH-Alow. Middle plot: Heatmap of the most frequent somatic mutations identified in LGG-mIDH-Alow. Genes are sorted according to the FDR q-value. Lower plot: Mutational signature analysis in LGG-mIDH-Alow samples. Color codes represent the mutation type or mutation signature as indicated in the left panel.
Figure 4.

CNVs frequency and hazard ratios (HRs) for OS in the Cox regression model in LGG-mIDH-A and LGG-mIDH-O. (A) HRs for OS in the Cox regression model according to the presence or absence of the frequently mutated genes among the LGG-mIDH-A. (B) HRs for OS in the Cox regression model according to the presence or absence of the frequently mutated genes among the LGG-mIDH-O. (C and D) The most frequent CNVs in high versus low mutation in LGG-mIDH-A and LGG-mIDH-O, respectively. (E and F) HRs for OS in the Cox regression model according to the presence or absence of the most frequent CNVs among the groups.
Similar to the Astrocytoma, LGG-mIDH-Ohigh and LGG-mIDH-Olow shared 6 of the top 10 frequent mutations: IDH, CIC, FUBP1, NOTCH1, PIK3CA, and ZBRB20 (Figure 3A and B). Except for a mutation in PIK3CA (which have a negative impact on the survival of both groups), none of these mutations have a differential impact on the OS of LGG-mIDH-Ohigh or LGG-mIDH-Olow (Figure 4B). Moreover, the most common COSMIC signatures were similar to the signatures found in LGG-mIDH-A groups (ie, 1, 6, and 15) (Figure 3A and B, Supplementary Figure S4).
Figure 3.

Somatic genetic alterations identified in LGG-mIDH-O according to the tumor mutational burden. (A) Upper plot shows the mutation rate for each tumor sample in LGG-mIDH-Ohigh. Middle plot: Heatmap of the most frequent somatic mutations identified in LGG-mIDH-Ohigh. Genes are sorted according to the FDR q-value. Lower plot: Mutational signature analysis in LGG-mIDH-Ohigh samples. (B) Upper plot shows the mutation rate for each tumor sample in LGG-mIDH-Olow. Middle plot: Heatmap of the most frequent somatic mutations identified in LGG-mIDH-Olow. Genes are sorted according to the FDR q-value. Lower plot: Mutational signature analysis in LGG-mIDH-Olow samples. Color codes represent the mutation type or mutation signature as indicated in the left panel.
We then investigated the effect of the most frequent CNVs on both LGG-mIDH subgroups. Overall, the percentage of the most frequently altered CNVs was approximately 11% in LGG-mIDH-A and approximately 10% in LGG-mIDH-O (Figure 4C and D). In all LGG-mIDH-A, the most frequent CNVs that have an impact on patient’s survival are found in the following genes: CCND2, PTPN6, FGF6, FGF23, CHD4, and ZNF384 (Figure 4E). Interestingly, all of these CNVs are shared in the same patients within the LGG-mIDH-Ahigh group (Figure 4C and E). In LGG-mIDH-O, none of the most frequent CNVs had a differential effect on OS between LGG-mIDH-Ohigh and LGG-mIDH-Olow (Figure 4D and F). Overall, these data suggest that tumor mutational burden is a unique and independent prognostic factor that negatively impacts LGG-mIDH patient survival.
Unique GO Groups Are Enriched in the LGG-mIDHhigh
Since the high mutational burden impacts LGG-mIDH but not the LGG-wtIDH, we hypothesized that LGG-mIDHhigh is associated with a differential gene expression profile, which could be used to predict patients’ outcome. We performed the differential gene analysis based on tumors with either high or low mutational burden. Using an FDR of 0.05 as the lower limit of significance, we identified 8321 genes that were upregulated and 6527 genes that were downregulated in the LGG-mIDH-Ahigh. In LGG-mIDH-Ohigh, there were 7345 upregulated and 5298 downregulated genes. We then performed GSEA to evaluate the functional aspects of the differentially expressed genes.
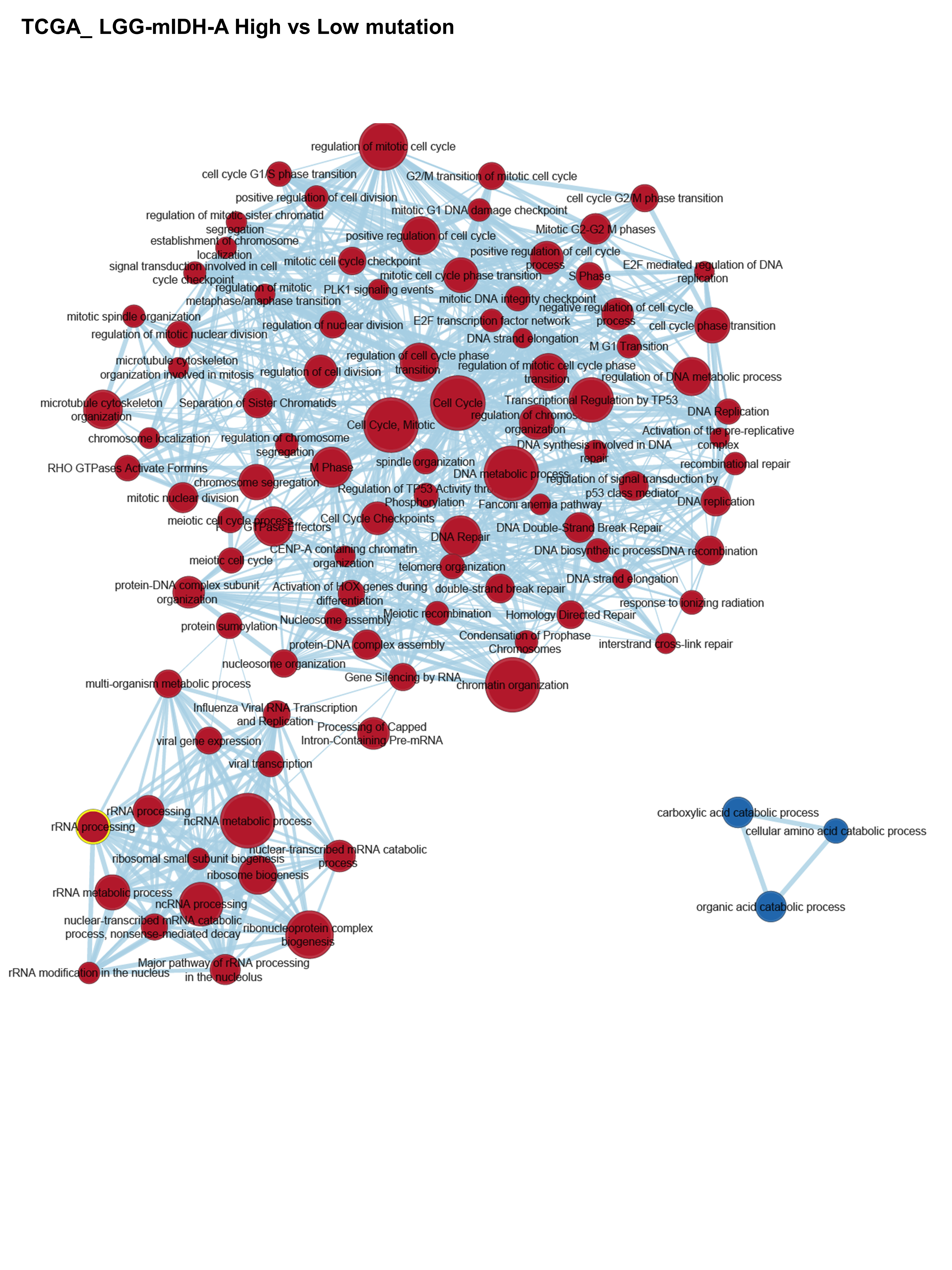
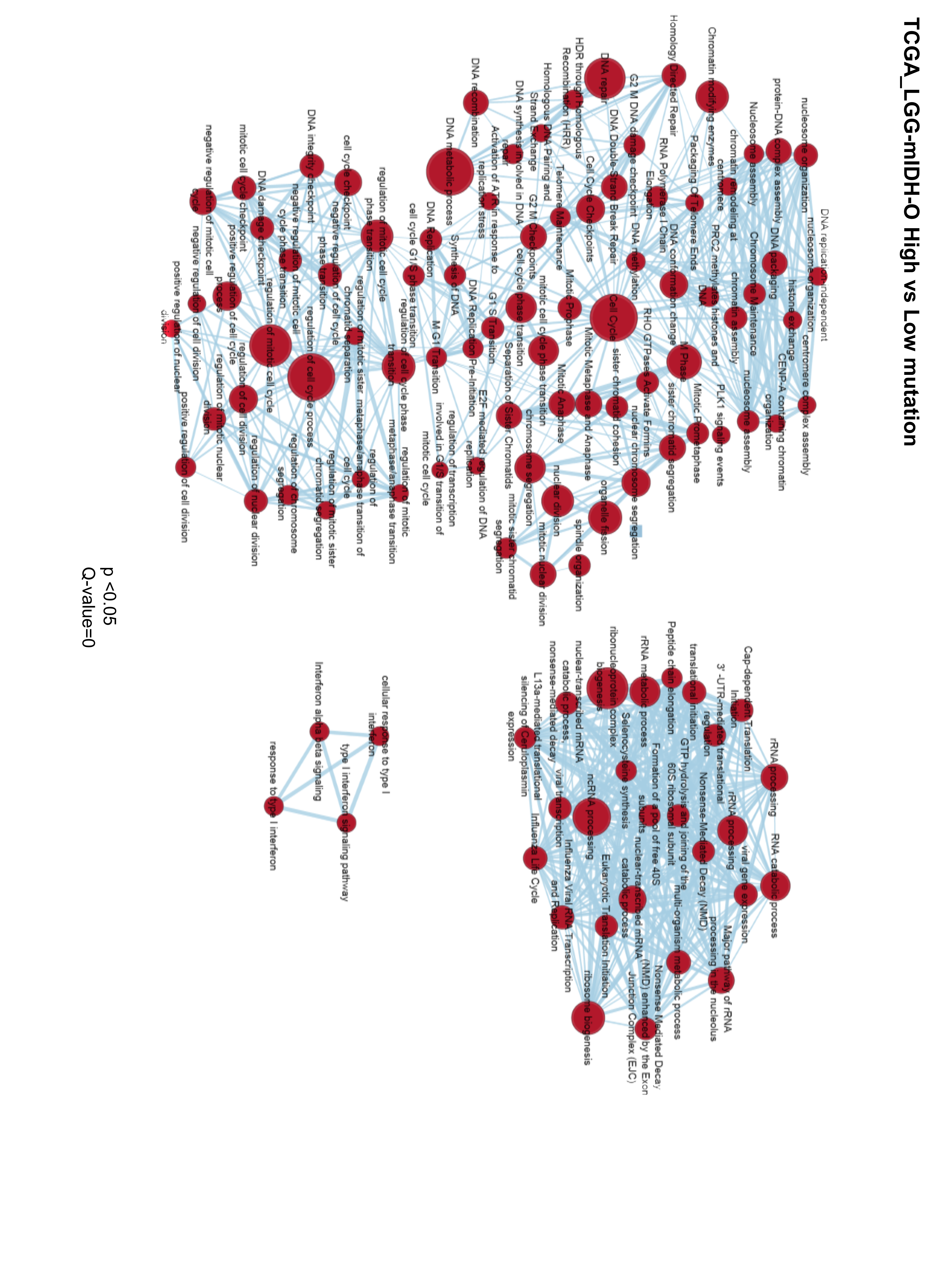
We compared the differences between significant GOs in LGG-mIDHhigh vs low in both astrocytoma and oligodendroglioma. Results suggested that high tumor mutational burden was associated with the upregulation of DNA repair, cell cycle-related processes, DNA mismatch repair, and chromosomal remodeling in LGG-mIDHhigh vs low (Figure 5A–D, Supplementary Figure S5). The enriched GO groups were prominent in the high mutated groups of both LGG-mIDH-A and LGG-mIDH-O (Figure 5A–D). Interestingly, there was a positive enrichment of GO families that belong to RNA and pre-RNA processing in LGG-mIDHhigh vs low (Figure 5C and D). This is consistent with a recent report which highlights the effect of enrichment in RNA processing associated GO groups in LGG patients’ survival.46 Unique GOs related to the amino acids catabolism were downregulated in LGG-mIDH-Ahigh (Figure 5A and B), whereas unique GOs belonged to type-I interferon signaling were enriched in LGG-mIDH-Ohigh (Figure 5C and D). These data elucidate the molecular differences in the high tumor mutational burden within LGG-mIDH tumors and suggest that GOs belonging to cell cycle regulation and RNA processing may be negatively correlated with mIDH patient survival.
Figure 5.

Gene set enrichment analysis (GSEA) of high versus low mutation load in LGG-mIDH-A and LGG-mIDH-O. (A and C) Cytoscape map visualization of the positive (red) and negative (blue) enriched GO groups in high versus low mutation load in LGG-mIDH-A (LGG-mIDH-AhighN = 189, LGG-mIDH-AlowN = 63) (A) and LGG-mIDH-O (LGG-mIDH-OhighN = 74, LGG-mIDH-OlowN = 95) (C). (B and D) Enrichment plots of the top significantly altered GO in the high versus low mutation load in LGG-mIDH-A and LGG-mIDH-O.
Identification of High-Risk Gene Sets Associated With Survival in LGG-mIDH Patients
We aimed to construct gene sets that could be used to predict survival in LGG-mIDH. To do so, we obtained the differentially enriched GOs between the high mutation and low mutation groups in both LGG-mIDH subtypes. The resulting enriched gene sets include genes related to DNA repair, cell cycle-related processes, and chromosomal remodeling. To determine if these gene sets correlate with survival, we used the SAPS test.44 SAPS is a powerful tool that computes 3 P-values (Ppure, Prandom, and Penrichment) for candidate prognostic gene sets and integrates the 3 P-values in the form of the SAPS q-value. Out of 53 gene sets that were enriched in LGG-mIDH, only 8 gene sets had a significant SAPS score in LGG-mIDH-A, and 9 gene sets had significant SAPS score in LGG-mIDH-O (Q-value <0.01). We performed the “least absolute shrinkage and selection operator” (LASSO) (Figure 6A) to identify genes within each gene set most associated with survival. LASSO selected 12 genes from 3 gene sets, set1, set2, and set3, associated with the survival of patients with LGG-mIDH-A tumors, and 18 genes from 3 gene sets, set4, set5, and set6, that impact survival of LGG-mIDH-O (Figure 6B and C, Supplementary Table S4). We then validated our model using an independent dataset from the CGGA. Within the CGGA dataset, all 6 gene sets predicted survival of LGG-mIDH tumors (Figure 6B and C). Therefore, we propose that set1, set2, and set3 could be utilized as a prognostic marker for survival of patients with LGG-mIDH-A, whereas set4, set5, and set6 could predict prognosis in LGG-mIDH-O. All genes are listed in Supplementary Table S4.
Figure 6.

Kaplan–Meier survival analysis of LGG-mIDH patients with high-risk versus a low-risk score of each gene set. (A) Flow chart illustrating the construction of the high-risk gene set in LGG-mIDH-A and LGG-mIDH-O. Significant gene sets in LGG-mIDH were selected based on GSEA. SAPS analysis was done to test the significant prognostic gene sets. Finally, LASSO was performed to predict the genes that mostly impact survival. (B) Kaplan–Meier analysis of patients with high-risk versus a low-risk score based on the 3 genes sets that predict survival in LGG-mIDH-A of TCGA (training) and CGGA (validation) datasets. (C) Kaplan–Meier analysis of patients with high-risk versus low-risk score based on the 3 genes sets that predict survival in LGG-mIDH-O of TCGA (training) and CGGA (validation) dataset (TCGA: LGG-mIDH-A [N = 245], TCGA: LGG-mIDH-O [N = 149], CGGA: LGG-mIDH-A [N = 108], CGGA: LGG-mIDH-O [N = 80]).
Discussion
Expression of mIDH in LGG results in a hypermethylation phenotype and enhanced patients’ survival. In this study, we analyzed the tumor mutational burden, CNVs, mRNA expression, and clinical outcomes of adult glioma utilizing the TCGA database and validated the results using the GLASS consortium and the CGGA database. We propose that tumor mutational burden is a predictor of OS in patients with LGG-mIDH tumors. Furthermore, we constructed a list of “high-risk” gene sets which we propose could be used as a prognostic marker of OS of mIDH glioma.
Although the total number of mutations in GBM is higher than in LGG-mIDH tumor, the correlation between tumor mutational burden and patient outcome was specific to LGG-mIDH. This suggests that the mutational burden is an important contributor to survival in LGG-mIDH patients, albeit, the role of IDHR132H on the mutation rate in LGG tumors remains to be determined.
Tumor mutational burden has been associated with better response to immunotherapy in many, but not all, tumor types.47 However, the tumor mutational burden can also give rise to intratumor heterogeneity which increases treatment resistance, including resistance to immune therapy.48 As recurrent tumors contain an increased tumor mutational burden, and are also more resistant to treatment, and more aggressive than the primary tumor,2,23,24 there exists a general correlation between tumor mutational burden and tumor progression and aggressiveness. Recently, Barthel et al.42 have shown that upon recurrence, hypermutation in all glioma patients was not associated with prognosis. Future studies will elucidate the effect of the mutational burden in recurrent tumors on prognosis.
Previous studies have shown that mutation in PIK3CA and deletion of CDKN2A are associated with poor clinical outcome30,31 in LGG. We found that CDKN2A is only present in LGG-wtIDH (less than 1% of LGG-mIDH), and it was associated with poor clinical outcome in both LGG-wtIDHhigh and LGG-wtIDHlow. Mutation in PIK3CA was present in approximately 13% of LGG-mIDH-O patient, and it did drive unfavorable prognosis in both LGG-mIDH-Ohigh and LGG-mIDH-Olow. Moreover, mutational analysis revealed that the main COSMIC signatures enriched in the high and low mutated tumors were signatures 1, 6, and 15. Signature 1 is common in most tumor type and is found in most cancer samples. Both signature 6 and signature 15 are resulted from high numbers of small (shorter than 3 bp) insertions and deletions at mono/polynucleotide repeats and are associated with DNA mismatch repair pathway.45 The exact contribution of these mutational signatures on glioma patient’s prognosis outcome is yet to be discovered.
GSEA suggests that LGG-mIDHhigh tumors have positive enrichments in cell cycle regulation and DNA repair GO groups as compared to LGG-mIDHlow. This suggests that activation of the DNA repair pathway results in worse prognosis in mIDH glioma. These data are consistent with our recent study which showed that the mIDH tumor is associated with epigenetic regulation of genes involved in DNA repair pathway such as ATM.33 It is possible to speculate that activation of DNA repair mechanisms in tumors with high mutational burden could be a cause of increased mutation or its consequence. Future experimental studies will need to address this issue. Another GO which was unique in LGG-mIDHhigh versus LGG-mIDHlow was “RNA and non-coding RNA processing”.49–52 The role of changes in RNA processing, whether causal or effect of high levels of mutation, will also need to be evaluated in forthcoming studies.
In summary, we propose that the tumor mutational burden could be used as a potentially highly reliable marker of OS in patients with LGG-mIDH tumors. Furthermore, we identified 6 gene sets whose expression levels correlate significantly with the OS in this group of patients. As our data were established using the TCGA database and validated using 2 independent databases (GLASS and CGGA), we propose that our results have high clinical statistical significance. We believe our results are of clinical relevance for therapeutic decisions and the stratification of patients for clinical trials.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Padma Kadiyala for reading the manuscript. We thank the “Consulting for Statistics, Computing and Analytics Research” (CSCAR) at the University of Michigan for the helpful input. We thank the support and academic leadership of Dr. Karin Muraszko, and the administrative and technical support of Angela Collada, and Marta Edwards, respectively. The content is solely the responsibility of the authors.
Funding
This work was supported by National Institutes of Health/National Institute of Neurological Disorders & Stroke (NIH/NINDS) Grants [R37-NS094804, R01-NS105556, R21-NS107894 and Rogel Cancer Center Scholar Award to M.G.C.]; NIH/NINDS Grants [R01-NS076991, R01-NS082311, and R01-NS096756 to P.R.L.]; NIH/NIBIB [R01-EB022563 and NCI/UO1-CA-224160 to M.G.C. and P.R.L.]; the Department of Neurosurgery; Leah’s Happy Hearts Foundation, ChadTough Foundation, Pediatric Brain Tumor Foundation, and Smiles for Sophie Forever Foundation [to M.G.C. and P.R.L]; RNA Biomedicine Grant [F046166 to M.G.C.] NIH/NCI [T32-CA009676 Post-Doctoral Fellowship to M.S.A].
Conflict of interest statement. The authors declare no conflict of interest.
Authorship Statement. M.S.A., M.G.C., and P.R.L. designed the research study and experiments. M.S.A., R.T., R.A., L.Z., A.D., A.T., P.J.U., and P.R.L. analyzed data. L.Z. performed the statistical analysis. M.S.A., M.G.C., and P.R.L. wrote the manuscript. All authors read and commented on the manuscript.
References
- 1. Ceccarelli M, Barthel FP, Malta TM, et al. TCGA Research Network: Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell Rep. 2016;164(3):550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bai H, Harmancı AS, Erson-Omay EZ, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet. 2016;48(1):59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dominik Sturm HW, Hovestadt V, Khuong-Quang D-A, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–437. [DOI] [PubMed] [Google Scholar]
- 6. Jin G, Reitman ZJ, Duncan CG, et al. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013;73(2):496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu XY, Gerges N, Korshunov A, et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012;124(5):615–625. [DOI] [PubMed] [Google Scholar]
- 11. Jiao Y, Killela PJ, Reitman ZJ, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3(7):709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lai A, Kharbanda S, Pope WB, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol. 2011;29(34):4482–4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Verhaak RG, Hoadley KA, Purdom E, et al. ; Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Izquierdo-Garcia JL, Viswanath P, Eriksson P, et al. Metabolic reprogramming in mutant IDH1 glioma cells. PLoS One. 2015;10(2):e0118781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karpel-Massler G, Ishida CT, Bianchetti E, et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat Commun. 2017;8(1):1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ronnebaum SM, Ilkayeva O, Burgess SC, et al. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J Biol Chem. 2006;281(41):30593–30602. [DOI] [PubMed] [Google Scholar]
- 17. Seltzer MJ, Bennett BD, Joshi AD, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70(22):8981–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reitman ZJ, Duncan CG, Poteet E, et al. Cancer-associated isocitrate dehydrogenase 1 (IDH1) R132H mutation and d-2-hydroxyglutarate stimulate glutamine metabolism under hypoxia. J Biol Chem. 2014;289(34):23318–23328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saetta AA, Levidou G, El-Habr EA, et al. Expression of pERK and pAKT in human astrocytomas: correlation with IDH1-R132H presence, vascular endothelial growth factor, microvascular characteristics and clinical outcome. Virchows Arch. 2011;458(6):749–759. [DOI] [PubMed] [Google Scholar]
- 20. Yalaza C, Ak H, Cagli MS, Ozgiray E, Atay S, Aydin HH. R132H mutation in IDH1 gene is associated with increased tumor HIF1-alpha and serum VEGF levels in primary glioblastoma multiforme. Ann Clin Lab Sci. 2017;47(3):362–364. [PubMed] [Google Scholar]
- 21. Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hiromichi Suzuki KA, Chiba K, Sato Y, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 47(5):458–468. [DOI] [PubMed] [Google Scholar]
- 24. Johnson BE, Mazor T, Hong C, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343(6167):189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mur P, Mollejo M, Ruano Y, et al. Codeletion of 1p and 19q determines distinct gene methylation and expression profiles in IDH-mutated oligodendroglial tumors. Acta neuropathologica. 2013;126(2):277–289. [DOI] [PubMed] [Google Scholar]
- 26. Li Z-H, Guan Y-L, Liu Q, Wang Y, Cui R, Wang Y-J. Astrocytoma progression scoring system based on the WHO 2016 criteria. Sci Rep. 2019;9(1):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu X, Martinez-Ledesma E, Zheng S, et al. Multigene signature for predicting prognosis of patients with 1p19q co-deletion diffuse glioma. Neuro Oncol. 2017;19(6):786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reis GF, Pekmezci M, Hansen HM, et al. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization grades II–III) astrocytomas. J Neuropathol Exp Neurol. 2015;74(5):442–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cimino PJ, Zager M, McFerrin L, et al. Multidimensional scaling of diffuse gliomas: application to the 2016 World Health Organization classification system with prognostically relevant molecular subtype discovery. Acta Neuropathol Commun. 2017;5(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aoki K, Nakamura H, Suzuki H, et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 2017;20(1):66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Draaisma K, Wijnenga MM, Weenink B, et al. PI3 kinase mutations and mutational load as poor prognostic markers in diffuse glioma patients. Acta Neuropathol Commun. 2015;3(1):88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Thuijl HF, Scheinin I, Sie D, et al. Spatial and temporal evolution of distal 10q deletion, a prognostically unfavorable event in diffuse low-grade gliomas. Genome Biol. 2014;15(9):471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nunez FJ, Mendez FM, Kadiyala P, et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci Transl Med. 2019;11(479):eaaq1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wahl DR, Dresser J, Wilder-Romans K, et al. Glioblastoma therapy can be augmented by targeting IDH1-mediated NADPH biosynthesis. Cancer Res. 2017;77(4):960–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klebanov N, Artomov M, Goggins WB, Daly E, Daly MJ, Tsao H. Burden of unique and low prevalence somatic mutations correlates with cancer survival. Sci Rep. 2019;9(1):4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. The Cancer Genome Atlas Research N, Chang K, Creighton CJ, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. The Cancer Genome Atlas Research. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas, N Engl J Med. 2015;372(26):2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Center BITGDA. Mutation Analysis (MutSig 2CV v3.1). Broad Institute of MIT and Harvard; 2016. doi: 10.7908/C1D21X2X. [DOI] [Google Scholar]
- 40. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang PJ, Lin HH, Lee CC, et al. CoMutPlotter: a web tool for visual summary of mutations in cancer cohorts. BMC Med Genomics. 2019;12(Suppl 5):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barthel FP, Johnson KC, Varn FS, et al. ; GLASS Consortium Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576(7785):112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hothorn T, Lausen B. On the exact distribution of maximally selected rank statistics. Comput Stat Data Anal. 2003;43(2):121–137. [Google Scholar]
- 44. Beck AH, Knoblauch NW, Hefti MM, et al. Significance analysis of prognostic signatures. PLoS Comput Biol. 2013;9(1):e1002875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tate JG, Bamford S, Jubb HC, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018;47(D1):D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chai RC, Li YM, Zhang KN, et al. RNA processing genes characterize RNA splicing and further stratify lower-grade glioma. JCI Insight. 2019;4(17):e130591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–334. [DOI] [PubMed] [Google Scholar]
- 49. Wang ZH, Guo XQ, Zhang QS, et al. Long non-coding RNA CCAT1 promotes glioma cell proliferation via inhibiting microRNA-410. Biochem Biophys Res Commun. 2016;480(4):715–720. [DOI] [PubMed] [Google Scholar]
- 50. Wang R, Li Y, Zhu G, et al. Long noncoding RNA CASC2 predicts the prognosis of glioma patients and functions as a suppressor for gliomas by suppressing Wnt/β-catenin signaling pathway. Neuropsychiatr Dis Treat. 2017;13(1):1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu L, Lv QL, Chen SH, et al. Up-regulation of long non-coding RNA AB073614 predicts a poor prognosis in patients with glioma. Int J Environ Res Public Health. 2016;13(4):433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li J, Wang YM, Song YL. Knockdown of long noncoding RNA AB073614 inhibits glioma cell proliferation and migration via affecting epithelial-mesenchymal transition. Eur Rev Med Pharmacol Sci. 2016;20(19):3997–4002. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.