

INTRODUCTION
This review serves as a primer on contemporary neuroendocrine neoplasm classification, with an emphasis on gastroenteropancreatic well-differentiated neuroendocrine tumors. Topics discussed include general features of neuroendocrine neoplasms; general neuroendocrine marker immunohistochemistry; the distinction of well-differentiated neuroendocrine tumor from pheochromocytoma/paraganglioma and other diagnostic mimics; poorly differentiated neuroendocrine carcinoma from diagnostic mimics; the concepts of differentiation and grade and the application of Ki-67 immunohistochemistry to determine the latter; the various World Health Organization (WHO) classifications of neuroendocrine neoplasms, including the 2019 WHO classification of gastroenteropancreatic tumors; organ-specific considerations for gastroenteropancreatic well-differentiated neuroendocrine tumors; immunohistochemistry to determine site of origin in metastatic well-differentiated neuroendocrine tumor of occult origin; immunohistochemistry in the distinction of well-differentiated neuroendocrine tumor G3 from large cell neuroendocrine carcinoma; and finally, required and recommended reporting elements for biopsies and resections of gastroenteropancreatic neuroendocrine epithelial neoplasms.
GENERAL FEATURES OF NEUROENDOCRINE NEOPLASMS
Neuroendocrine neoplasms include well-differentiated neuroendocrine tumors, poorly differentiated neuroendocrine carcinomas, pheochromocytoma, and paraganglioma (Fig. 1). These tumor types express general neuroendocrine markers and produce peptide hormones and/or biogenic amines. Well-differentiated neuroendocrine tumors and poorly differentiated neuroendocrine carcinomas (including small cell neuroendocrine carcinoma and large cell neuroendocrine carcinoma) are distinguished from pheochromocytoma/paraganglioma by their epithelial nature, as demonstrated by expression of the intermediate filament keratin. Neuroendocrine neoplasms, especially well-differentiated examples, characteristically express one or more somatostatin receptors, which is the physiologic basis of somatostatin receptor imaging (ie, OctreoScan or NETSPOT) and somatostatin receptor-based peptide receptor radionuclide therapy (eg, Lutathera).
Fig. 1.
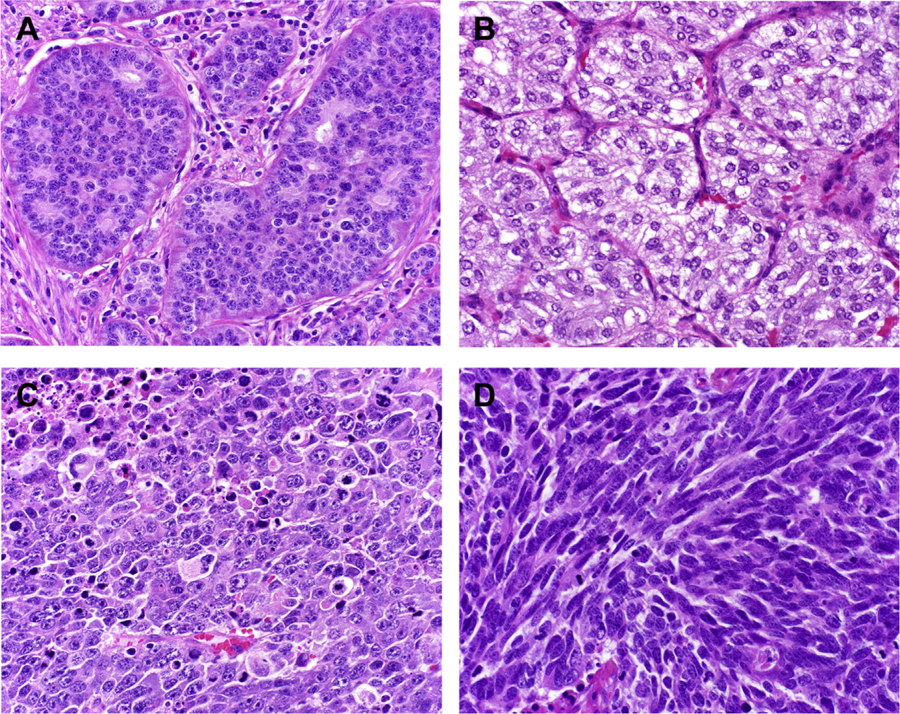
Neuroendocrine neoplasms. (A) Well-differentiated neuroendocrine tumor. (B) Paraganglioma. (C) Large cell neuroendocrine carcinoma. (D) Small cell neuroendocrine carcinoma.
Morphologically, well-differentiated neuroendocrine neoplasms, including well-differentiated neuroendocrine tumor and pheochromocytoma/paraganglioma typically demonstrate “organoid” architecture. Although the latter typically grow in a nested pattern (also known as “zellballen” in these tumor types), the former may assume one or more architectural patterns, including nested, trabecular, and pseudoglandular, either singly or in combination. Small cell neuroendocrine carcinoma typically demonstrates diffuse architecture, while large cell neuroendocrine carcinoma may be organoid or diffuse. Both well- and poorly differentiated neuroendocrine neoplasms tend to demonstrate finely granular chromatin (also known as “salt and pepper” chromatin).
GENERAL NEUROENDOCRINE MARKER IMMUNOHISTOCHEMISTRY
Immunohistochemistry for general neuroendocrine markers is essentially mandatory to confirm the neuroendocrine nature of a tumor. Traditional neuroendocrine markers include synaptophysin (component of synaptic-like vesicles) and chromogranin A (component of dense core and chromaffin granules). Synaptophysin is generally more sensitive (approaching 100% in well-differentiated tumors), while chromogranin A is more specific. Well-differentiated neuroendocrine neoplasms typically demonstrate diffuse, strong staining for one or both of these markers. Although jejunoileal and appendiceal enterochromaffin (EC)-cell neuroendocrine tumors are nearly always chromogranin A-positive, only 80% to 90% of pancreatic tumors are positive and rectal and appendiceal L-cell tumors are often weak-to-negative. CD56 (also known as neural cell adhesion molecule) is often used as a general neuroendocrine marker (it is especially popular among pulmonary pathologists), but its use is discouraged due to lack of specificity.
Poorly differentiated neuroendocrine carcinomas are less likely to be synaptophysin (~60%) and chromogranin A (≤50%)-positive and are less likely yet to be strongly expressing. I will not even consider synaptophysin and/or chromogranin A positivity in support of a diagnosis of poorly differentiated neuroendocrine carcinoma if it is seen in less than 30% of tumor cells (and even then I am wary; the stronger and more diffuse the expression, the better) (Fig. 2A, B). The reason for my trepidation—10% to 20% of nonneuroendocrine carcinomas from virtually every anatomic site demonstrate some degree of general neuroendocrine marker expression (a phenomenon I refer to as “occult” neuroendocrine differentiation) (Fig. 2C, D).1–3 It is more commonly seen with synaptophysin than chromogranin A (although it can be seen with either or both markers), generally in scattered cells (although occasionally diffuse), and is 3 to 4 times more likely in adenocarcinoma than squamous cell carcinoma. Pathologists are generally unaware of this phenomenon and may render an interpretation of “poorly differentiated carcinoma with neuroendocrine differentiation” (or “features”), which is a description rather than a diagnosis and creates confusion among treating clinicians.
Fig. 2.

Poorly differentiated neuroendocrine carcinoma versus nonneuroendocrine carcinoma with “occult” neuroendocrine differentiation. (A) This small cell lung cancer demonstrates (B) diffuse, strong synaptophysin expression. Poorly differentiated neuroendocrine carcinomas are often general neuroendocrine marker weak-to-negative, in which case dot-like keratin positivity, TTF-1 expression, and Rb loss may serve as surrogate markers for the presence of neuroendocrine differentiation. (C) This poorly differentiated lung adenocarcinoma was initially diagnosed as “poorly differentiated carcinoma with neuroendocrine features” based on (D) this synaptophysin immunostain. Ten to twenty percent of non-neuroendocrine carcinomas express general neuroendocrine markers, usually in weak, patchy fashion (as in this case), but occasionally quite strongly. This finding has no clear bearing on prognosis or therapy.
Additional immunohistochemical support for a diagnosis of poorly differentiated neuroendocrine carcinoma in this setting would include dot-like/perinuclear keratin positivity, thyroid transcription factor 1 (TTF-1) positivity (80%–90% of small cell lung cancers; 40% of extrapulmonary visceral poorly differentiated neuroendocrine carcinomas), and Rb loss (90% of small cell lung cancers; 50% of extrapulmonary visceral neuroendocrine carcinomas).4,5
Insulinoma-associated protein 1 (INSM1) is rapidly emerging as a general neuroendocrine marker. This zinc-finger transcription factor was initially discovered by genomic subtraction of a glucagonoma cDNA library from an insulinoma library.6 It was introduced to the diagnostic pathology community by Rosenbaum and colleagues7 in 2015, who demonstrated INSM1 positivity in 88% of 129 neuroendocrine neoplasms from diverse anatomic sites and only 1 of 24 nonneuroendocrine tumors. As well-differentiated neuroendocrine neoplasms are nearly always positive for the traditional general neuroendocrine markers, its role in these is unclear (possibly useful as a “specificity” marker in a synaptophysin+/chromogranin A− tumor). Although data are still accumulating, INSM1 seems to be very useful to confirm the neuroendocrine nature of a poorly differentiated neuroendocrine carcinoma. For example, Rooper and colleagues8 reported INSM1 positivity in 95% of 39 small cell lung cancers with an average H-score of 154 (note: H-score is the product of intensity*percent cells staining and ranges from 0 to 300), while synaptophysin and chromogranin A were positive in only 62% (average H-score 60) and 49% (average H-score 85), respectively.
IMMUNOHISTOCHEMISTRY TO DISTINGUISH EPITHELIAL FROM NONEPITHELIAL NEUROENDOCRINE NEOPLASMS
In a suspected well-differentiated neuroendocrine tumor, broad-spectrum keratin immunohistochemistry (eg, keratin AE1/AE3, OSCAR, CAM5.2) is highly recommended to confirm the epithelial nature of the tumor. In tumors presenting as metastases of unknown primary, pathologists often perform CK7 and CK20 to help assign site of origin. These keratins are usually not expressed by neuroendocrine epithelial neoplasms, which preferentially express keratins 8 and 18. Rarely, broad-spectrum keratins are negative, in which case alternative broad-spectrum epithelial markers may be used, including antibodies to EpCAM (ie, MOC-31, Ber-EP4) and EMA.
In a broad-spectrum epithelial marker-negative well-differentiated neuroendocrine neoplasm, the possibility of pheochromocytoma/paraganglioma should be strongly considered. Although it is “pathology dogma” that the “positive” marker of pheochromocytoma/paraganglioma is S-100, which highlights nonneoplastic sustentacular cells, that marker is neither adequately sensitive nor specific, as pheochromocytomas/paragangliomas may lack sustentacular cells and well-differentiated neuroendocrine tumors, especially appendiceal and bronchopulmonary examples, often possess them.9 The best positive pheochromocytoma/paraganglioma marker is GATA-3 (which is most familiar to pathologists as a breast and urothelial carcinoma marker) (Fig. 3A, B).10,11 Pheochromocytomas and sympathetic paragangliomas also express tyrosine hydroxylase. Succinate dehydrogenase subunit B (SDHB) immunohistochemistry is highly recommended in pheochromocytoma/paraganglioma to screen for SDH deficiency due to inactivation of any SDH subunit, with SDHB loss (seen in 30% of thoracoabdominal and ≥15% of head and neck paragangliomas and 5% of pheochromocytomas) suggesting the possibility of a hereditary tumor (ie, hereditary paraganglioma-pheochromocytoma syndrome, Carney-Stratakis syndrome) and associated with adverse prognosis (Fig. 3C).12 Although islet 1 is often used as a marker of pancreatic origin in a well-differentiated neuroendocrine tumor (see later discussion), it is also a sympathoadrenal lineage marker and is, thus, often positive in pheochromocytoma/paraganglioma—a significant diagnostic pitfall (that can be avoided by performing a broad-spectrum keratin in suspected well-differentiated neuroendocrine tumors).13
Fig. 3.

Paraganglioma/pheochromocytoma versus well-differentiated neuroendocrine tumor. While well-differentiated neuroendocrine tumors should be broad-spectrum keratinpositive, this (A) paraganglioma expressed (B) GATA-3, supporting the diagnosis. (C) Loss of SDHB expression (with intact signal in endothelium and sustentacular cells) raises the possibility of a hereditary tumor and is prognostically adverse.
As discussed above, some well-differentiated neuroendocrine tumors are characteristically synaptophysin+/chromogranin A–. This general neuroendocrine marker immunophenotype is also characteristic of adrenal cortical carcinoma, solid pseudopapillary neoplasm, and glomus tumor, which, unfortunately, are also histologic mimics of well-differentiated neuroendocrine tumor. These should always be considered in a “synaptophysin+ only” tumor, especially in the retroperitoneum and pancreas (the former 2), stomach (glomus tumor), and at potentially metastatic sites. The former 2 may be broad-spectrum keratin positive, although typically weakly so. Positive diagnostic markers include melan A and SF1 (adrenal cortical carcinoma), nuclear β-catenin (solid pseudopapillary neoplasm), and smooth muscle actin (glomus tumor).
In a poorly differentiated neuroendocrine carcinoma, broad-spectrum keratin positivity may be useful to support the neuroendocrine nature of the tumor (if dot-like) and to distinguish it from diagnostic mimics, including small round blue cell sarcomas (panel determined by clinical presentation, often including CD99 for Ewing sarcoma and desmin for rhabdomyosarcoma) and hematolymphoid tumors (typically CD45+).
DIFFERENTIATION
Neuroendocrine neoplasms should be dichotomized into well-differentiated and poorly differentiated examples. This assignment of “differentiation” is based solely on the hematoxylin-eosin (H&E) appearance of the tumor, usually readily performed at low-to-medium power. Well-differentiated neoplasms typically demonstrate organoid architecture and possess low nucleus:cytoplasm (N:C) ratios; necrosis is typically absent and tends to be “punctate,” if present. Most neuroendocrine tumors and pheochromocytomas/paragangliomas are easily recognized as well differentiated. Small cell neuroendocrine carcinoma is an exemplar of poor differentiation. It typically demonstrates diffuse architecture, is characterized by incredibly high N:C ratios, and may show extensive (geographic) necrosis. Large cell neuroendocrine carcinomas may be recognized as poorly differentiated based on nuclear features (large nuclei, irregular nuclear countours, coarse chromatin) and may demonstrate extensive necrosis.
The WHO classification of gastroenteropancreatic neuroendocrine tumors does NOT include a “moderately differentiated” category, although some other classifications do and a pathologist may be eager to diagnose a well-differentiated neuroendocrine tumor as moderately differentiated when it is more “atypical” then their conception of how a neuroendocrine tumor should look. If a pathology report notes the presence of a “moderately differentiated neuroendocrine tumor,” ask your pathologist to reclassify the lesion according to the relevant contemporary WHO classification (see later discussion).
All that being said, there are certainly rare cases in which differentiation is not readily assigned. In fact, well-differentiated neuroendocrine tumor and poorly differentiated neuroendocrine carcinoma exist along a morphologic continuum (with the vast majority of neoplasms segregating at the extremes of the differentiation spectrum). Higher-grade well-differentiated tumors (especially G3 examples, see later discussion) can be impossible to distinguish from large cell neuroendocrine carcinoma, especially in small biopsy samples. Rather than shoehorning them into a moderately differentiated category, which implies diagnostic certainty, it is better to diagnose these cases as “neuroendocrine epithelial neoplasm, differentiation uncertain.” In these rare, morphologically ambiguous cases, immunohistochemistry may be helpful in assigning these to the “well differentiated” or “poorly differentiated” categories (see later discussion).
GRADE
In pathology, the terms differentiation and grade are generally used interchangeably. In gastroenteropancreatic neuroendocrine epithelial neoplasms, grade has a specific meaning, distinct from differentiation. Grade reflects the degree of proliferation in a tumor and is assigned based on an assessment of the mitotic rate and Ki-67 proliferation index. Well-differentiated neuroendocrine tumors may be G1 (low grade), G2 (intermediate grade), or G3 (high grade), while poorly differentiated neuroendocrine carcinomas are G3, by definition.
The mitotic rate is evaluated in 50 high-power microscopic fields (HPFs), with the result expressed as the number of mitotic figures per 10 HPF; as mitotic activity may be heterogeneously distributed in a tumor, some attempt should be made to identify the most mitotically active areas (ie, “hotspots”). The Ki-67 proliferation index should be assessed in at least 500 tumor cells, again with attention given to hotspots (which are much easier to identify on Ki-67-immunostained than H&E-stained slides) (Fig. 4). When the grades based on the mitotic rate and Ki-67 proliferation index are discrepant, the higher grade is assigned. In most of these instances, the grade based on the Ki-67 proliferation index is higher. Approximately one-third of well-differentiated neuroendocrine tumors that are G1 based on mitotic rate are G2 based on the Ki-67 proliferation index, with a similar frequency of tumors that are G2 based on mitotic rate found to be G3 based on the Ki-67 proliferation index.14–16 Therefore, Ki-67 immunohistochemistry is considered mandatory in well-differentiated neuroendocrine tumors. “Eyeball estimates” of Ki-67 proliferation indices are notoriously inaccurate, especially around grade thresholds (ie, 3% and 20%), and formal counting is generally recommended (Fig. 5).17 This usually takes the form of manual counting of a camera-captured image. Digital image analysis may also be used, if it has been successfully validated against the gold standard of a manual count.
Fig. 4.
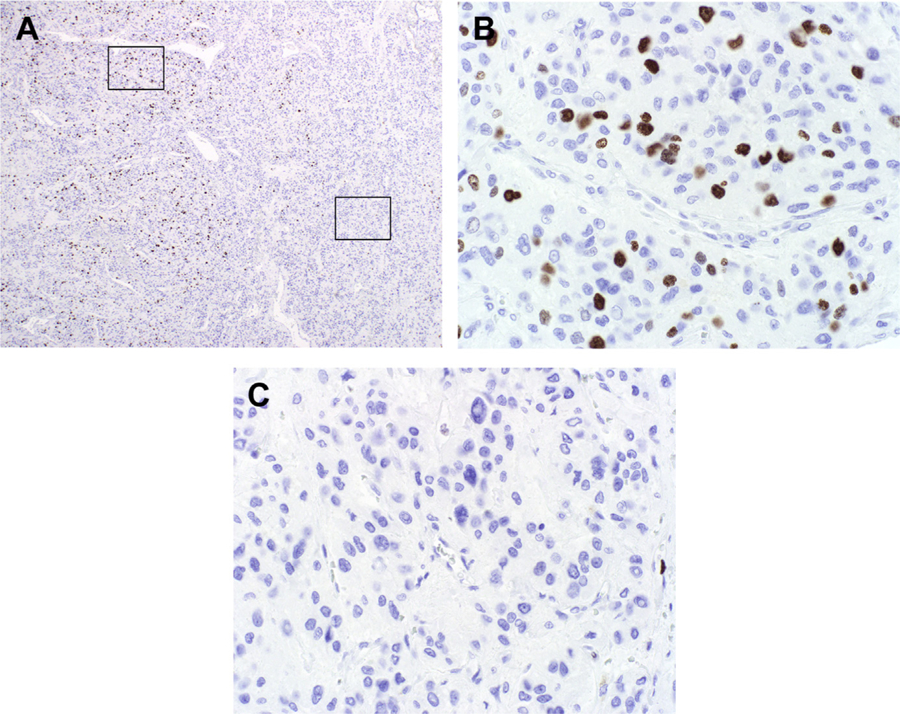
Ki-67 proliferation index heterogeneity. (A) Low-power photomicrograph of Ki-67-immunostained well-differentiated neuroendocrine neoplasm highlights areas of (B) frequent (14% in this image) and (C) absent (0%) tumor cell staining. The Ki-67 proliferation index should be based on a count of at least 500 tumor nuclei in a “hotspot.”
Fig. 5.

Ki-67 proliferation index “Eyeball Estimate” versus manual count. This Ki-67 immunostain was originally reported as “approximately 30%” by the referring pathologist. A manual count of a camera-captured image found a proliferation index of 19.9% (175 Ki-67-immunostained tumor cells/878 tumor cells counted).
Mitotic counting and Ki-67 immunohistochemistry are not required in poorly differentiated neuroendocrine carcinomas, as they are definitionally G3. I always perform Ki-67 immunohistochemistry in a suspected poorly differentiated neuroendocrine carcinoma, though, for 2 reasons: (1) crushed well-differentiated neuroendocrine tumors may be mistaken for small cell neuroendocrine carcinoma (a grave diagnostic error averted by recognition of a low Ki-67 proliferation index) and (2) the Ki-67 proliferation index may predict response to chemotherapy, with higher proliferation indices (ie, ≥55%) associated with greater likelihood of response to platinum-based chemotherapy and lower proliferation indices (ie, <60%) associated with greater likelihood of response to temozolomide.18,19
Although Ki-67 immunohistochemistry is essentially mandatory for grading, in patients with tumor at multiple sites there is little guidance on which sample to test. We were intrigued by a report from the Cedars-Sinai group several years ago of a cohort of 57 ileal well-differentiated neuroendocrine tumor patients in whom a Ki-67 proliferation index greater than 2% at either the primary or a metastatic site was the only significant predictor of progression-free survival on multivariate analysis (in a model that initially included age, presence of stage IV disease, necrosis, atypia, and grade based on mitotic rate).20 Other groups have noted a tendency for higher Ki-67 proliferation indices in liver metastases than in primary gastroenteropancreatic tumors and a relationship between tumor size and Ki-67 proliferation index.21–23 Based on these data, we perform Ki-67 immunohistochemistry on initial diagnostic biopsies as well as those taken in the setting of recurrent or progressive disease. In resection specimens, we separately test primary, regional, and distant disease, if present. In the setting of multiple primary tumors (and/or multiple lymph node metastases/tumor deposits and/or multiple distant metastases) we test tissue blocks containing the largest focus of tumor. In a cohort of 103 gastroenteropancreatic tumor patients in whom Ki-67 was performed in both primary and metastatic tumor, we recently reported a higher grade in the metastasis in 24%, a higher grade in the primary in 10%, and the same grade in 66%.24 The progression-free and overall survivals for patients with a G1 primary/G2 metastasis were superimposable on those for patients with a G2 primary. Therefore, I have taken to saying that, in terms of outcome, “any G2 trumps.”
WORLD HEALTH ORGANIZATION CLASSIFICATION OF NEUROENDOCRINE NEOPLASMS
The WHO Classification of Tumors, published as a series of “Blue Books,” represents the international gold standard for tumor classification. The 4th edition was composed of 12 organ-system-based volumes published between 2007 and 2018. The 5th edition of the WHO Classification of Tumours of the Digestive System (the first volume in the 5th series) was in press at the time I was writing this review.
Each Blue Book has “jurisdiction” over the classification of neuroendocrine neoplasms within its organ-system scope. Given the epidemiology of neuroendocrine neoplasms, the most “important” classifications are in the GI and Lung Blue Books. The WHO Classification of Tumours of the Digestive System has purview over all gastroenteropancreatic neoplasms, while the WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart has purview over bronchopulmonary (and thymic) neoplasms. The WHO Classification of Tumours of Endocrine Organs has purview over pancreatic neuroendocrine neoplasms (as well as neuroendocrine neoplasms of the pituitary, thyroid, parathyroid, adrenal, and paraganglia), but NOT other gastrointestinal (GI) neoplasms. Relevant chapters (or sections of chapters) in other Blue Books are brief. Because of the sequencing of publication and substantial overlap in authorship, the Endocrine Blue Book classification of pancreatic neuroendocrine neoplasms may serve as a “preview” of the GI Blue Book classification.
The classification of neuroendocrine neoplasms of the digestive system in the 2019 WHO Classification of Tumours of the Digestive System will adopt the classification from the 2017 Endocrine Blue Book (Table 1). Although technically it only had purview over pancreatic tumors, as soon as it was published I began to apply it to all gastroenteropancreatic neuroendocrine neoplasms, as it addressed 3 shortcomings of the 2010 WHO Classification of Tumours of the Digestive System (4th edition): (1) the “G1/G2 Ki-67 hole”, (2) well-differentiated neuroendocrine tumor G3, and (3) mixed tumors composed of various elements. In the 2010 classification, the Ki-67 proliferation index of a G1 tumor was defined as ≤2%, while that of a G2 tumor was defined as 3% to 20%. There was no accounting for proliferation indices greater than 2 and less than 3%. In the 2017 and 2019 classifications, the Ki-67 proliferation index of a G1 tumor is defined as less than 3%, closing the “hole.” As soon as the 2010 classification was published, investigators noted rare tumors that were morphologically well differentiated but that had Ki-67 proliferation indices greater than 20% (Fig. 6). The 2017 and 2019 classifications created the category “neuroendocrine tumor G3.”25–27 In my practice, well-differentiated neuroendocrine tumor G3 constitutes up to 5% of all neuroendocrine tumors and, among digestive tumors, is more common among pancreatic tumors. Not surprisingly, these tumors typically pursue a clinical course in-between that of well-differentiated neuroendocrine tumor G2 and poorly differentiated neuroendocrine carcinoma and are less likely than poorly differentiated neuroendocrine carcinoma to respond to chemotherapy. The 2010 classification referred to neoplasms composed of combinations of nonneuroendocrine and neuroendocrine elements as “mixed adenoneuroendocrine carcinoma”. The 2017 and 2019 classifications acknowledge that the nonneuroendocrine carcinoma component of these tumors may be squamous (eg, in the esophagus or anus) and that, rarely, the neuroendocrine component is well-differentiated, designating these mixed tumors as “mixed neuroendocrine-nonneuroendocrine neoplasms”. These rare tumors are distinguished from the nonneuroendocrine carcinomas with occult neuroendocrine differentiation discussed above in that the neuroendocrine and nonneuroendocrine components must be morphologically distinct, with the minor component representing at least 30% of the tumor.
Table 1.
2019 WHO classification of gastroenteropancreatic neuroendocrine epithelial neoplasms
| Classification/Grade | Ki-67 Proliferation Index | Mitotic Count |
|---|---|---|
| (Well-differentiated) neuroendocrine tumor: | ||
| G1 | <3% | <2 per 10 HPF |
| G2 | 3%–20% | 2–20 per 10 HPF |
| G3 | >20% | >20 per 10 HPF |
| (Poorly differentiated) neuroendocrine carcinoma: | ||
| G3 | >20% | >20 per 10 HPF |
| Mixed neuroendocrine-nonneuroendocrine neoplasm | ||
From Klimstra DS, Kloppel G, LaRosa S, Rindi G. Classification of neuroendocrine neoplasms of the digestive system. In: WHO Classification of Tumours of the Digestive System. 5th ed. Lyon: IARC; 2019.
Fig. 6.

Well-differentiated neuroendocrine tumor G3. (A) Pancreatic well-differentiated neuroendocrine tumor with (B) Ki-67 proliferation index heterogeneity readily noted at low power. (C, D) Whereas most of the tumor had a low-to-intermediate proliferation index (3.5% in D), there was a distinct clone with a high proliferation index (44%) (E, F). This tumor pursued an aggressive clinical course with the patient dying 9 months after distal pancreatectomy.
The classification of neuroendocrine neoplasms of the lung from the 2015 WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart is presented in Table 2.28 This classification includes 2 well-differentiated neuroendocrine tumor types, typical and atypical carcinoid tumor, and 2 poorly differentiated neuroendocrine carcinoma types, small cell and large cell neuroendocrine carcinoma. Although the classification does not make use of the Ki-67 proliferation index, I routinely perform Ki-67 immunohistochemistry, which is especially useful in small biopsies containing too few high-power microscopic fields for reliable mitotic counting and in crushed specimens in which morphologic assessment is difficult; it also likely provides independent prognostic information.29 Typical carcinoid tumor is roughly equivalent to gastroenteropancreatic well-differentiated neuroendocrine tumor G1, with Ki-67 proliferation indices typically less than 4%, while atypical carcinoid tumor is roughly equivalent to gastroenteropancreatic well-differentiated neuroendocrine tumor G2, with Ki-67 proliferation indices typically between 4% and 25%.30 The lung classification does not include a well-differentiated G3 category, although I have seen rare examples.31 The poorly differentiated neuroendocrine carcinoma types are analogous, although the mitotic threshold is lower in the lung (>10 per 2 mm2) than in the GI tract (>20 per 10 HPF).
Table 2.
2015 WHO classification of bronchopulmonary neuroendocrine epithelial neoplasms
| Classification | Mitotic Count | Necrosis | Other Features |
|---|---|---|---|
| Carcinoid tumor: | |||
| Typical carcinoid | <2 per 2 mm2 | Absent | Tumor must be ≥0.5 cm |
| Atypical carcinoid | 2–10 per 2 mm2 | Present | Diagnosis based on presence of either or both features |
| Poorly differentiated neuroendocrine carcinoma: | |||
| Small cell carcinoma | >10 per 2 mm2 | Frequent | Characteristic histology |
| Large cell neuroendocrine carcinoma | >10 per 2 mm2 | Frequent | Organoid morphology and expression of at least 1 general neuroendocrine marker |
Reprinted from Travis WD, Colby TV, Corrin B et al. (1999). Histological Typing of Lung and Pleural Tumours. WHO International Histological Classification of Tumours 3rd ed. Springer-Verlag: Berlin. With kind permission of Springer Science + Business Media.
SITE-SPECIFIC CONSIDERATIONS
Although gastroenteropancreatic neuroendocrine neoplasms arise throughout the GI tract and are subject to a common classification, site of origin is at least as, if not more, relevant to biology than grade is. In this section, I will thumbnail essential site-specific considerations for these well-differentiated neuroendocrine tumors.
Esophagus
Well-differentiated neuroendocrine tumors of the esophagus are so rare that I have never seen one, and, if I did, I would write a case report.
Stomach
Gastric well-differentiated neuroendocrine tumors arise in 3 main settings.32 That vast majority (60%–70%) arise in association with autoimmune atrophic gastritis (so-called type I tumors). These enterochromaffin-like (ECL) cell tumors arise in the gastric body driven by hypergastrinemia, tend to be multiple, are nearly always clinically benign, and are readily dealt with endoscopically. Type II tumors (10%) arise in the setting of combined multiple endocrine neoplasia type 1 (MEN1)/Zollinger-Ellison syndrome. Again, these are ECL-cell tumors driven by hypergastrinemia (in this case a gastrinoma) and tend to be multiple and indolent; they may even spontaneously regress with resection of the underlying gastrinoma. Type III tumors (25%) are sporadic, solitary, and aggressive, sometimes requiring gastrectomy. To assist in this typing, your pathologist should comment on the background oxyntic mucosa if it is present (atrophic in type I, possibly hypertrophic/hyperplastic in type II; ECL-cell hyperplasia in types I and II) and you should order a serum gastrin.
Duodenum
A recent large series of duodenal well-differentiated neuroendocrine tumors highlighted 4 different types: (1) nonfunctioning (60% of 176), (2) ampullary somatostatin-expressing (21%), (3) gastrinoma (11%), and (4) gangliocytic paraganglioma (7%).33 Tumor type had no bearing on survival, although ampullary somatostatin-expressing tumors and gastrinomas were more likely to present with lymph node metastasis than nonfunctioning tumors. Most nonfunctioning tumors expressed one or more hormones immunohistochemically (gastrin >> somatostatin > serotonin >> other) and, as such, hormone immunohistochemistry need not be routinely used (ie, functionality is defined as presentation with a hypersecretion syndrome). Ampullary somatostatin-expressing tumors typically demonstrate pseudoglandular architecture and are, thus, apt to be mistaken for adenocarcinoma; although there is a disease association with NF1 (10% in this series), most tumors arise sporadically. Thirty-five percent of the gastrinomas in this series arose in association with MEN1. They have a propensity for regional lymph node metastasis, occasionally in the setting of a minute (and thus occult) primary. Gangliocytic paragangliomas consist of an admixture of neuroendocrine epithelial and neural elements (Schwann cells, axons, ganglion cells) and nearly always arise in the ampullary region.
Pancreas
Most pancreatic well-differentiated neuroendocrine tumors arise sporadically and are nonfunctioning. Tumors measuring less than 0.5 cm are classified as “microadenomas” and are presumed benign. Insulinomas account for up to 20% of resected tumors, while other functional tumors are rare. Pathologists should take care to distinguish pancreatic well-differentiated neuroendocrine tumors from morphologic mimics, including acinar cell carcinoma (trypsin+) and solid pseudopapillary neoplasm (nuclear β-catenin+). The presence of multiple pancreatic well-differentiated neuroendocrine tumors is typically seen with MEN1 and, less commonly, with von Hippel-Lindau syndrome. In the latter instance, tumors may demonstrate microvesicular cytoplasm, and patients often manifest concurrent pancreatic serous cystadenomas.
Jejunoileum
Jejunoileal well-differentiated neuroendocrine tumors are nearly always composed of serotonin-producing EC cells, are often multifocal (50%), and have a predilection for the distal small intestine.34 Unlike the situation in the pancreas, multifocality does not imply a genetic basis. Although 5% of patients have an affected first-degree relative, a germline mutation is only exceptionally identified.35 Small primary tumors are notorious for metastasis (eg, in the Armed Forces Institute of Pathology [AFIP] series of 159 tumors, 21% of tumors <1 cm involved regional lymph nodes and 29% of tumors measuring 1 to 2 cm demonstrated distant metastasis).36 Serotonin and substance P production by the tumors causes mesenteric vascular elastosis and resulting ischemia, while patients with large volume hepatic metastasis may manifest carcinoid syndrome (5%).
Appendix
The vast majority of appendiceal well-differentiated neuroendocrine tumors are incidentally discovered, and appendectomy is generally sufficient. Most are EC-cell tumors (similar to jejunoileal primaries) with 10% to 20% representing L-cell (enteroglucagon-expressing) tumors. The latter demonstrate trabecular or tubular architecture, are nearly always miniscule (up to a few millimeters in size), and are categorically benign. The North American Neuroendocrine Tumor Society (NANETS) and the European Neuroendocrine Tumor Society (ENETS) recommend right hemicolectomy in appendiceal well-differentiated neuroendocrine tumor given one or more of the following: size greater than 2 cm, location at the base of the appendix, positive margin, mesoappendiceal (NANETS) or deep (>3 mm) mesoappendiceal invasion (ENETS), and gross nodal involvement (NANETS).37,38 Although NANETS recommends right hemicolectomy given lymph-vascular space invasion or G2, ENETS is more circumspect, recommending a discussion about (rather than mandating) a right hemicolectomy. Finally, despite the name, goblet cell carcinoid tumor is an adenocarcinoma variant and should be managed accordingly.
Colorectum
Most colorectal neuroendocrine tumors are incidentally discovered and benign. They have a special predilection for the rectum, are typically composed of L-cells, and characteristically demonstrate trabecular architecture. In the AFIP series of colorectal tumors 81 arose in the rectum and 3 in the distal sigmoid colon.39 Rare cecal tumors have more in common with jejunoileal EC-cell tumors. The ENETS recently published a colorectal management guideline.40 Most tumors are effectively managed endoscopically. Surgery is indicated for tumors greater than 2 cm, tumors 1 to 2 cm invasive into the muscularis propria, rare tumors less than 1 cm invasive into the muscularis propria in which a local resection has failed, and in G3 patients considered candidates for resection (many of these latter patients will have distant metastases).
IMMUNOHISTOCHEMISTRY FOR SITE OF ORIGIN ASSIGNMENT IN METASTASIS OF OCCULT ORIGIN
Ten to 20% of well-differentiated neuroendocrine tumors present as metastases of occult origin, typically from the jejunoileum (especially) and pancreas, and less commonly from the lung or other sites.4,41–43 Most jejunoileal primaries are difficultto-impossible to reach with conventional endoscopy, are difficult-to-impossible to detect on cross-sectional imaging, and, as mentioned previously, have a tendency to metastasize even at small sizes. Determination of site of origin is prognostically and therapeutically significant. For example, the median survival in patients with distant metastases from tumors of jejunoileal, pancreatic, and lung origin is 65, 27, and 17 months, respectively; resection of the primary tumor may be considered even in the face of widespread metastatic disease and is associated with prolonged survival and, in midgut primaries, protects patients from significant bleeding and obstruction risks; capecitabine/temozolomide and mTOR and receptor tyrosine kinase inhibitors are much more likely to be used in foregut than midgut tumors.41 Immunohistochemistry is useful to assign site of origin.
The University of Iowa immunohistochemical algorithm to assign site of origin in a well-differentiated neuroendocrine tumor is presented in Fig. 7 (see also Fig. 8). I originally developed it in the setting of a comprehensive review on this topic published in 2013 but have updated it extensively since, as superior markers of lung and rectal and complementary markers of pancreatic and midgut neuroendocrine tumors have emerged.4 Given the epidemiology of metastatic well-differentiated neuroendocrine tumors of occult origin, its “first round” emphasizes identifying tumors of jejunoileal and pancreatic origin. CDX2 is expressed by 90% of jejunoileal tumors, although it is also expressed by 15% of pancreatic tumors. Islet 1 is the most sensitive pancreas marker (70% of metastases in the aggregated published literature, although 85% in my anecdotal experience). To boost the sensitivity of the classifier to identify pancreatic tumors, I had originally intended to add polyclonal PAX8 (55% sensitive in the aggregated published literature). When I tested polyclonal PAX8 in my laboratory, pancreatic tumors were nonreactive and I substituted PAX6, which is the main PAX-family transcription factor expressed by normal islets and to which polyclonal PAX8 cross-reacts.44 Islet 1 and PAX6 are never significantly expressed by midgut tumors, although both are usually expressed by rectal tumors (ie, the same transcriptional machinery regulates islet and L-cell development). SATB2, which is typically used in diagnostic pathology as a marker of adenocarcinoma of lower GI origin, has emerged as the preferred marker of lower GI well-differentiated neuroendocrine tumors; I recently found moderate-tostrong expression in 92% of 25 rectosigmoid, 55% of 33 appendical, and 0% of 331 other well-differentiated neuroendocrine neoplasms from diverse anatomic sites.45 Islet 1 may also be expressed by pheochromocytoma/paraganglioma (broad-spectrum keratin–), medullary thyroid carcinoma and pituitary tumors (unlikely to present as metastasis of occult origin), and, rarely, by bronchopulmonary tumors.46
Fig. 7.

University of Iowa immunohistochemical algorithm for well-differentiated neuroendocrine tumor site of origin.
Fig. 8.

Metastatic well-differentiated neuroendocrine tumor of unknown primary—site of origin immunohistochemistry. (A) This well-differentiated neuroendocrine tumor metastatic to liver expresses (B) CDX2 and not (C) islet 1 (depicted) or PAX6, supporting a midgut (ie, jejunoileal) origin. Subsequent Ga 68-DOTATATE scan demonstrated focal uptake in the distal ileum.
Along with SATB2, other “second round” markers include OTP, ATRX, clusterin, serotonin, progesterone receptor (PR), and prostatic acid phosphatase (PrAP). Although SATB2 may be used in islet 1/PAX6-positive tumors to interrogate a rectal origin, the other markers typically come into play with CDX2/PAX6/Islet 1 “triple-negative” tumors. Although TTF-1 is most commonly applied (although aggregated published sensitivity is only 31%), OTP is the clear first choice lung well-differentiated neuroendocrine tumor marker, at least twice as sensitive without sacrificing specificity.47 I recently found OTP positivity in 82% of 77 typical and 50% of 12 atypical carcinoids and only 1 of 603 gastroenteropancreatic well-differentiated neuroendocrine tumors (a pancreatic tumor that also expressed islet 1 and PAX6).48 Frequent ATRX inactivation (10%–20%) was noted in recent studies defining the molecular genetic landscape of pancreatic neuroendocrine tumors and not in jejunoileal or bronchopulmonary tumors.49–51 In addition to using it as a pancreatic site of origin marker, I also apply it as a prognostic marker once a pancreatic origin is established (inactivation is associated with unfavorable prognosis overall, although more favorable prognosis in the subset of patients presenting with metastatic disease).52,53 Of note, inactivation was also recently found in 3% of 103 pheochromocytomas/paragangliomas.54 Clusterin is usually strongly expressed by well-differentiated neuroendocrine tumors of diverse anatomic sites with the exception of jejunoileal tumors, in which it is rarely, weakly expressed. I found clusterin positivity in 82% of 148 nonjejunoileal tumors (average H-score 183) and only 8% of 107 jejunoileal tumors (average H-score 31).55 Since CDX2 is only 90% sensitive for tumors of jejunoileal origin, I recently added serotonin as an additional midgut marker. I found serotonin positivity in 75% of 256 metastatic jejunoileal, 3% of 63 metastatic pancreatic, and 0% of 44 bronchopulmonary well-differentiated neuroendocrine tumors.56 Adding serotonin to CDX2 increased the sensitivity for detecting jejunoileal origin from 90% to 96%, and all serotonin-positive pancreatic tumors expressed islet 1 and/or PAX6. Although data are limited, the largest published study of PR reported expression in 58% of 96 pancreatic, 0% of 29 tubal gut, and 7% of 15 lung tumors.57 PrAP, although typically used as a prostatic adenocarcinoma marker, is usually (>80%) expressed by rectal well-differentiated neuroendocrine tumors. When I formally evaluated it, I found expression in 88% of 17 rectal but also 37% of 41 metastatic midgut and only 8% of 13 metastatic pancreatic and 0% of 20 lung well-differentiated neuroendocrine tumors.58 Before I had SATB2 and serotonin in my laboratory, I used PrAP as my primary rectal and as a secondary midgut marker.
A simplified site of origin algorithm is presented in Fig. 9. All the markers are performed simultaneously. It substitutes PR and polyclonal PAX8 for the more sensitive combination of islet 1 and PAX6 and TTF-1 for the more sensitive OTP. All of the markers used in the simplified algorithm are in widespread clinical use by pathologists for alternative diagnostic applications. As a note of caution, many pathology laboratories have shifted to performing monoclonal PAX8 immunohistochemistry (a fact your pathologist may be unaware of), which is negative in pancreatic well-differentiated neuroendocrine tumors.
Fig. 9.

Simplified immunohisochemical algorithm for well-differentiated neuroendocrine tumor site of origin.
In poorly differentiated neuroendocrine carcinomas, beyond the use of TTF-1 and CK20 to distinguish tumors of visceral from cutaneous origin, immunohistochemistry has almost no role in site of origin assignment. In the aggregated published literature, TTF-1 is expressed by 83% of small cell lung cancers, 36% of large cell neuroendocrine lung cancers, 36% of extrapulmonary visceral poorly differentiated neuroendocrine carcinomas, and only 0.8% of Merkel cell carcinomas.4 CK20 is expressed by 88% of Merkel cell carcinomas, 63% of poorly differentiated neuroendocrine carcinomas of the major salivary glands (usually parotid), 6% of other extrapulmonary visceral poorly differentiated neuroendocrine carcinomas, and 5% of small cell lung cancers. Beyond TTF-1 and CK20, any neurofilament and strong SATB2 expression are more common in Merkel cell than visceral poorly differentiated neuroendocrine carcinomas, while achaete-scute homolog 1 (ASCL1; also known as MASH1) positivity favors a visceral over a cutaneous origin.45,59 Up to 80% of Merkel cell carcinomas are driven by a polyomavirus (ie, Merkel cell polyomavirus), and I was initially excited about immunohistochemistry to the virus’s large T antigen (clone CM2B4) to increase the accuracy of the TTF-1/CK20 classifier.60 It has subsequently been found that CK20-negative Merkel cell carcinomas are also apt to be CM2B4-negative.61 The immunostain is useful, though, in distinguishing Merkel cell carcinoma (CM2B4+) from poorly differentiated neuroendocrine carcinoma of major salivary gland origin (CM2B4–).
Unlike the situation in well-differentiated neuroendocrine tumors, other than TTF-1, ASCL1, and SATB2, transcription factor immunohistochemistry is not useful to assign site of origin in poorly differentiated neuroendocrine carcinomas. In fact, these tumors have a tendency to express multiple transcription factors independent of site of origin, generally in weak, patchy fashion but occasionally more strongly. I have dubbed this phenomenon “marked transcription factor lineage infidelity.”62
IMMUNOHISTOCHEMISTRY TO DISTINGUISH WELL-DIFFERENTIATED NEUROENDOCRINE TUMOR G3 FROM POORLY DIFFERENTIATED NEUROENDOCRINE CARCINOMA
As mentioned previously, up to 5% of well-differentiated neuroendocrine tumors are G3 (ie, possess Ki-67 proliferation indices >20% and/or mitotic counts >20 per 10 HPF) and these can be very difficult to distinguish from large cell neuroendocrine carcinoma, especially in small biopsies—typically from metastatic sites. Fig. 10 presents the University of Iowa immunohistochemical algorithm for distinguishing morphologically ambiguous well-differentiated neuroendocrine tumor G3 from poorly differentiated neuroendocrine carcinoma. The molecular genetic hallmark of small cell lung cancer is biallelic inactivation of TP53 and RB1.63 Although not as extensively studied, large cell neuroendocrine carcinoma of lung origin and extrapulmonary visceral neuroendocrine carcinomas often, although not always, share this molecular genetic fingerprint.26,64,65 p53 and Rb immunohistochemistry are, thus, critical elements of this algorithm (Fig. 11). I recently found mutant-pattern p53 staining (missense or null patterns) in 71% of 31 small cell lung and 76% of extrapulmonary visceral, Rb loss in 85% of small cell lung and 52% of extrapulmonary visceral, and mutant-patten p53 and/or Rb loss in 97% and 81% of small cell lung cancers and extrapulmonary visceral poorly differentiated neuroendocrine carcinomas, respectively.5
Fig. 10.
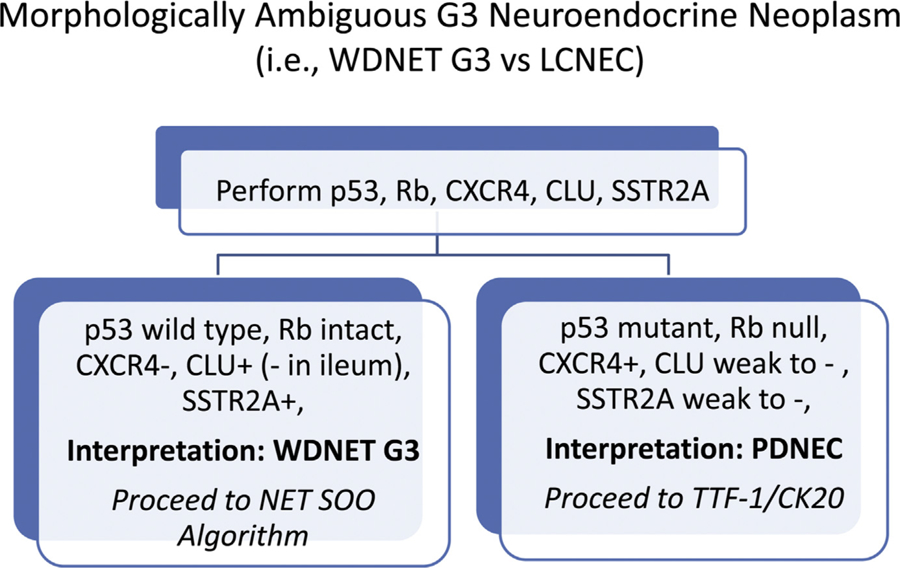
Immunohistochemical algorithm for morphologically ambiguous G3 neuroendocrine epithelial neoplasms.
Fig. 11.

p53 and Rb immunohistochemistry to distinguish well-differentiated neuroendocrine tumor G3 versus large cell neuroendocrine carcinoma. (A) This intermediate- to high-grade neuroendocrine epithelial neoplasm demonstrates (B) wild-type pattern p53 staining and (C) intact Rb expression, supporting the diagnosis of well-differentiated neuroendocrine tumor. (D) This high-grade neuroendocrine epithelial neoplasm demonstrates (E) missensemutation pattern p53 staining (ie, diffuse, strong staining obscuring nuclear detail) and (F) loss of Rb expression, supporting the diagnosis of large cell neuroendocrine carcinoma.
We supplement these with clusterin, SSTR2A, and CXCR4. As discussed previously, clusterin is usually strongly expressed by nonjejunoileal well-differentiated neuroendocrine tumors and is only occasionally, weakly expressed by poorly differentiated neuroendocrine carcinomas (19%, average H-score 36).55 SSTR2A is ubiquitously (>99%) and very frequently (>85%) strongly expressed by well-differentiated neuroendocrine tumors of jejunoileal and pancreatic origin, respectively; we have observed less frequent expression by bronchopulmonary tumors (40%).66 Poorly differentiated neuroendocrine carcinomas are less frequently positive (30%–40%), and even when positive they are typically less strongly expressing (H-scores in the 100–150 range).67 Our group is investigating the chemokine receptor CXCR4 as a theranostic target in poorly differentiated neuroendocrine carcinomas. I recently found frequent, moderately strong (84%; average H-score 104) expression in 95 poorly differentiated neuroendocrine carcinomas and only rare, weak expression (4.5%; average H-score 3) in 66 well-differentiated gastroenteropancreatic tumors.68 More recently we have found that, among well-differentiated neuroendocrine tumors, CXCR4 positivity seems to be largely confined to atypical carcinoid tumor of lung origin (8 of 10 cases; average H-score 130).
REPORTING OF BIOPSY AND RESECTION SPECIMENS
Table 3 summarizes required and recommended reporting elements for biopsies and resections of gastroenteropancreatic neuroendocrine epithelial neoplasms. Reports of resection specimens should include all of the required data elements from the relevant College of American Pathologists Cancer Protocol.69 These Cancer Protocols have been adapted from current staging criteria promulgated by the American Joint Committee on Cancer.70 Of note, because of their especially poor prognosis, poorly differentiated neuroendocrine carcinomas are reported/staged based on protocols/criteria for nonneuroendocrine carcinomas.
Table 3.
Required and recommended reporting elements for biopsies and resections of gastroenteropancreatic neuroendocrine epithelial neoplasms
| Associated Required or Recommended Immunohistochemistry | |
|---|---|
| Required Data Element: | |
| Diagnosis: well-differentiated neuroendocrine tumor or poorly differentiated neuroendocrine carcinoma | ● Synaptophysin and chromogranin A to establish neuroendocrine nature (required) ● Broad-spectrum keratin to confirm epithelial nature (highly recommended in primary and regional disease and required in distant metastasis) ● p53 and Rb are recommended in the distinction of well-differentiated neuroendocrine tumor G3 from poorly differentiated neuroendocrine carcinoma |
| Ki-67 proliferation index (proliferation index >20% is implied for poorly differentiated neuroendocrine carcinoma and performance is not mandatory) | ● Ki-67 on at least 1 block of tumor (required) ● Ki-67 on at least1 block of primary tumor and matched metastasis (recommended) |
| Mitotic count per 10 HPF (in biopsies with <50 HPF to assess it is reasonable to express the total number of mitotic figures in the total number of microscopic fields; for poorly differentiated neuroendocrine carcinoma a mitotic count >20 per 10 HPF is implied and performance is not mandatory) | |
| Grade: G1, G2, or G3 (G3 is implied for poorly differentiated neuroendocrine carcinoma and need not be explicitly stated) | |
| Data elements in College of American Pathologists Cancer Protocol: for resection specimens | |
| Recommend Data Element: | |
| Comment on site of origin (for metastasis of occult origin) | ● Panel in a well-differentiated neuroendocrine tumor may include some combination of CDX2 for midgut origin; polyclonal PAX8 and/or PR (or islet 1 and PAX6) for pancreatic origin; TTF-1 (or OTP) for bronchopulmonary origin; and SATB2 for rectal origin ● Panel in a poorly differentiated neuroendocrine carcinoma to include TTF-1 for visceral origin and CK20 for cutaneous origin; neurofilament and strong SATB2 expression also support a cutaneous origin |
KEY POINTS.
Neuroendocrine neoplasms include well-differentiated neuroendocrine tumor, pheochromocytoma/paraganglioma, and poorly differentiated neuroendocrine carcinoma, which are characterized by general neuroendocrine marker expression and the production of peptide hormones and/or biogenic amines.
“Differentiation” refers to the morphologic appearance of a neoplasm and is dichotomized into well and poorly differentiated categories and “grade” takes mitotic count and Ki-67 proliferation index into account with well-differentiated neuroendocrine tumor stratified into low- (G1), intermediate- (G2), and high-grade (G3) groups, and poorly differentiated neuroendocrine carcinoma considered G3, by definition.
The 2019 WHO Classification of Digestive System Tumours (“WHO GI Blue Book”) contains the current gold standard classification of gastroenteropancreatic neuroendocrine neoplasms; it includes the category “neuroendocrine tumor G3,” which was absent from the 2010 WHO classification.
Immunohistochemistry is useful for determining the site of origin of metastatic well-differentiated neuroendocrine tumor of occult origin, most of which arise from the jejunoileum (CDX2+) or pancreas (islet 1+), and for distinguishing morphologically ambiguous well-differentiated neuroendocrine tumor G3 (p53 wild-type pattern, Rb intact) from large cell neuroendocrine carcinoma (p53 mutant pattern and/or Rb lost).
Acknowledgments
DISCLOSURE
This work was supported by NIH grant P50 CA174521-01A1 (AMB).
REFERENCES
- 1.Ionescu DN, Treaba D, Gilks CB, et al. Nonsmall cell lung carcinoma with neuroendocrine differentiation–an entity of no clinical or prognostic significance. Am J Surg Pathol 2007;31(1):26–32. [DOI] [PubMed] [Google Scholar]
- 2.Howe MC, Chapman A, Kerr K, et al. Neuroendocrine differentiation in non-small cell lung cancer and its relation to prognosis and therapy. Histopathology 2005; 46(2):195–201. [DOI] [PubMed] [Google Scholar]
- 3.Graziano SL, Tatum AH, Newman NB, et al. The prognostic significance of neuroendocrine markers and carcinoembryonic antigen in patients with resected stage I and II non-small cell lung cancer. Cancer Res 1994;54(11):2908–13. [PubMed] [Google Scholar]
- 4.Bellizzi AM. Assigning site of origin in metastatic neuroendocrine neoplasms: a clinically significant application of diagnostic immunohistochemistry. Adv Anat Pathol 2013;20(5):285–314. [DOI] [PubMed] [Google Scholar]
- 5.Bellizzi A p53 and Rb immunohistochemistry as molecular surrogates show distinctive patterns in visceral and cutaneous poorly differentiated neuroendocrine carcinomas. Mod Pathol 2019;32(Supplement 2):540A. [Google Scholar]
- 6.Goto Y, De Silva MG, Toscani A, et al. A novel human insulinoma-associated cDNA, IA-1, encodes a protein with “zinc-finger” DNA-binding motifs. J Biol Chem 1992;267(21):15252–7. [PubMed] [Google Scholar]
- 7.Rosenbaum JN, Guo Z, Baus RM, et al. INSM1: a novel immunohistochemical and molecular marker for neuroendocrine and neuroepithelial neoplasms. Am J Clin Pathol 2015;144(4):579–91. [DOI] [PubMed] [Google Scholar]
- 8.Rooper LM, Sharma R, Li QK, et al. INSM1 demonstrates superior performance to the individual and combined use of synaptophysin, chromogranin and CD56 for diagnosing neuroendocrine tumors of the thoracic cavity. Am J Surg Pathol 2017; 41(11):1561–9. [DOI] [PubMed] [Google Scholar]
- 9.Al-Khafaji B, Noffsinger AE, Miller MA, et al. Immunohistologic analysis of gastrointestinal and pulmonary carcinoid tumors. Hum Pathol 1998;29(9):992–9. [DOI] [PubMed] [Google Scholar]
- 10.So JS, Epstein JI. GATA3 expression in paragangliomas: a pitfall potentially leading to misdiagnosis of urothelial carcinoma. Mod Pathol 2013;26(10):1365–70. [DOI] [PubMed] [Google Scholar]
- 11.Perrino CM, Ho A, Dall CP, et al. Utility of GATA3 in the differential diagnosis of pheochromocytoma. Histopathology 2017;71(3):475–9. [DOI] [PubMed] [Google Scholar]
- 12.Barletta JA, Hornick JL. Succinate dehydrogenase-deficient tumors: diagnostic advances and clinical implications. Adv Anat Pathol 2012;19(4):193–203. [DOI] [PubMed] [Google Scholar]
- 13.Huber K, Narasimhan P, Shtukmaster S, et al. The LIM-homeodomain transcription factor Islet-1 is required for the development of sympathetic neurons and adrenal chromaffin cells. Dev Biol 2013;380(2):286–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rege TA, King EE, Barletta JA, et al. Ki-67 proliferation index in pancreatic endocrine tumors: comparison with mitotic count, interobserver variability, and impact on grading. Mod Pathol 2011;24(1S):372A. [Google Scholar]
- 15.McCall CM, Shi C, Cornish TC, et al. Grading of well-differentiated pancreatic neuroendocrine tumors is improved by the inclusion of both Ki67 proliferative index and mitotic rate. Am J Surg Pathol 2013;37(11):1671–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Velthuysen ML, Groen EJ, van der Noort V, et al. Grading of neuroendocrine neoplasms: mitoses and Ki-67 are both essential. Neuroendocrinology 2014; 100(2–3):221–7. [DOI] [PubMed] [Google Scholar]
- 17.Tang LH, Gonen M, Hedvat C, et al. Objective quantification of the Ki67 proliferative index in neuroendocrine tumors of the gastroenteropancreatic system: a comparison of digital image analysis with manual methods. Am J Surg Pathol 2012;36(12):1761–70. [DOI] [PubMed] [Google Scholar]
- 18.Pelosi G, Rodriguez J, Viale G, et al. Typical and atypical pulmonary carcinoid tumor overdiagnosed as small-cell carcinoma on biopsy specimens: a major pitfall in the management of lung cancer patients. Am J Surg Pathol 2005;29(2):179–87. [DOI] [PubMed] [Google Scholar]
- 19.Sorbye H, Welin S, Langer SW, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): the NORDIC NEC study. Ann Oncol 2013;24(1):152–60. [DOI] [PubMed] [Google Scholar]
- 20.Dhall D, Mertens R, Bresee C, et al. Ki-67 proliferative index predicts progression-free survival of patients with well-differentiated ileal neuroendocrine tumors. Hum Pathol 2012;43(4):489–95. [DOI] [PubMed] [Google Scholar]
- 21.Zen Y, Heaton N. Elevated Ki-67 labeling index in ‘synchronous liver metastases’ of well differentiated enteropancreatic neuroendocrine tumor. Pathol Int 2013; 63(11):532–8. [DOI] [PubMed] [Google Scholar]
- 22.Grillo F, Albertelli M, Brisigotti MP, et al. Grade increases in gastroenteropancreatic neuroendocrine tumor metastases compared to the primary tumor. Neuroendocrinology 2016;103(5):452–9. [DOI] [PubMed] [Google Scholar]
- 23.Shi C, Gonzalez RS, Zhao Z, et al. Liver metastases of small intestine neuroendocrine tumors: Ki-67 heterogeneity and World Health Organization grade discordance with primary tumors. Am J Clin Pathol 2015;143(3):398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keck KJ, Choi A, Maxwell JE, et al. Increased grade in neuroendocrine tumor metastases negatively impacts survival. Ann Surg Oncol 2017;24(8):2206–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basturk O, Yang Z, Tang LH, et al. The high-grade (WHO G3) pancreatic neuroendocrine tumor category is morphologically and biologically heterogenous and includes both well differentiated and poorly differentiated neoplasms. Am J Surg Pathol 2015;39(5):683–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang LH, Untch BR, Reidy DL, et al. Well-differentiated neuroendocrine tumors with a morphologically apparent high-grade component: a pathway distinct from poorly differentiated neuroendocrine carcinomas. Clin Cancer Res 2016; 22(4):1011–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milione M, Maisonneuve P, Spada F, et al. The clinicopathologic heterogeneity of grade 3 gastroenteropancreatic neuroendocrine neoplasms: morphological differentiation and proliferation identify different prognostic categories. Neuroendocrinology 2017;104(1):85–93. [DOI] [PubMed] [Google Scholar]
- 28.Travis WD, Brambilla E, Burke AP, et al. , editors. WHO classification of Tumours of the lung, Pleura, Thymus and Heart. 4th edition. Lyon (France): IARC; 2015. [DOI] [PubMed] [Google Scholar]
- 29.Pelosi G, Rindi G, Travis WD, et al. Ki-67 antigen in lung neuroendocrine tumors: unraveling a role in clinical practice. J Thorac Oncol 2014;9(3):273–84. [DOI] [PubMed] [Google Scholar]
- 30.Rindi G, Klersy C, Inzani F, et al. Grading the neuroendocrine tumors of the lung: an evidence-based proposal. Endocr Relat Cancer 2014;21(1):1–16. [DOI] [PubMed] [Google Scholar]
- 31.Quinn AM, Chaturvedi A, Nonaka D. High-grade neuroendocrine carcinoma of the lung with carcinoid morphology: a study of 12 cases. Am J Surg Pathol 2017;41(2):263–70. [DOI] [PubMed] [Google Scholar]
- 32.La Rosa S, Inzani F, Vanoli A, et al. Histologic characterization and improved prognostic evaluation of 209 gastric neuroendocrine neoplasms. Hum Pathol 2011;42(10):1373–84. [DOI] [PubMed] [Google Scholar]
- 33.Vanoli A, La Rosa S, Klersy C, et al. Four neuroendocrine tumor types and neuroendocrine carcinoma of the duodenum: analysis of 203 cases. Neuroendocrinology 2017;104(2):112–25. [DOI] [PubMed] [Google Scholar]
- 34.Keck KJ, Maxwell JE, Utria AF, et al. The distal predilection of small bowel neuroendocrine tumors. Ann Surg Oncol 2018;25(11):3207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sei Y, Zhao X, Forbes J, et al. A hereditary form of small intestinal carcinoid associated with a germline mutation in inositol polyphosphate multikinase. Gastroenterology 2015;149(1):67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burke AP, Thomas RM, Elsayed AM, et al. Carcinoids of the jejunum and ileum: an immunohistochemical and clinicopathologic study of 167 cases. Cancer 1997; 79(6):1086–93. [PubMed] [Google Scholar]
- 37.Boudreaux JP, Klimstra DS, Hassan MM, et al. The NANETS consensus guideline for the diagnosis and management of neuroendocrine tumors: well-differentiated neuroendocrine tumors of the jejunum, ileum, appendix, and cecum. Pancreas 2010;39(6):753–66. [DOI] [PubMed] [Google Scholar]
- 38.Pape UF, Niederle B, Costa F, et al. ENETS consensus guidelines for neuroendocrine neoplasms of the appendix (excluding goblet cell carcinomas). Neuroendocrinology 2016;103(2):144–52. [DOI] [PubMed] [Google Scholar]
- 39.Federspiel BH, Burke AP, Sobin LH, et al. Rectal and colonic carcinoids. A clinicopathologic study of 84 cases. Cancer 1990;65(1):135–40. [DOI] [PubMed] [Google Scholar]
- 40.Ramage JK, De Herder WW, Delle Fave G, et al. ENETS consensus guidelines update for colorectal neuroendocrine neoplasms. Neuroendocrinology 2016; 103(2):139–43. [DOI] [PubMed] [Google Scholar]
- 41.Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008;26(18):3063–72. [DOI] [PubMed] [Google Scholar]
- 42.Wang SC, Parekh JR, Zuraek MB, et al. Identification of unknown primary tumors in patients with neuroendocrine liver metastases. Arch Surg 2010;145(3):276–80. [DOI] [PubMed] [Google Scholar]
- 43.Keck KJ, Maxwell JE, Menda Y, et al. Identification of primary tumors in patients presenting with metastatic gastroenteropancreatic neuroendocrine tumors. Surgery 2017;161(1):272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lorenzo PI, Jimenez Moreno CM, Delgado I, et al. Immunohistochemical assessment of Pax8 expression during pancreatic islet development and in human neuroendocrine tumors. Histochem Cell Biol 2011;136(5):595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bellizzi AM. SATB2 in neuroendocrine neoplasms: strong expression is restricted to well-differentiated tumors of lower gastrointestinal tract origin and is more frequent in merkel cell carcinoma among poorly differentiated carcinomas. Histopathology 2019. 10.1111/his.13943. [DOI] [PMC free article] [PubMed]
- 46.Agaimy A, Erlenbach-Wunsch K, Konukiewitz B, et al. ISL1 expression is not restricted to pancreatic well-differentiated neuroendocrine neoplasms, but is also commonly found in well and poorly differentiated neuroendocrine neoplasms of extrapancreatic origin. Mod Pathol 2013;26(7):995–1003. [DOI] [PubMed] [Google Scholar]
- 47.Papaxoinis G, Lamarca A, Quinn AM, et al. Clinical and pathologic characteristics of pulmonary carcinoid tumors in central and peripheral locations. Endocr Pathol 2018;29(3):259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pelletier D, Sachs CR, Czeczok T, et al. Orthopedia homeobox (OTP) expression in a well-differentiated neuroendocrine tumor supports a bronchopulmonary origin. Lab Invest 2018;98(Supplement 1):236A. [Google Scholar]
- 49.Scarpa A, Chang DK, Nones K, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017;543(7643):65–71. [DOI] [PubMed] [Google Scholar]
- 50.Banck MS, Kanwar R, Kulkarni AA, et al. The genomic landscape of small intestine neuroendocrine tumors. J Clin Invest 2013;123(6):2502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simbolo M, Mafficini A, Sikora KO, et al. Lung neuroendocrine tumours: deep sequencing of the four World Health Organization histotypes reveals chromatinremodelling genes as major players and a prognostic role for TERT, RB1, MEN1 and KMT2D. J Pathol 2017;241(4):488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singhi AD, Liu TC, Roncaioli JL, et al. Alternative lengthening of telomeres and loss of DAXX/ATRX expression predicts metastatic disease and poor survival in patients with pancreatic neuroendocrine tumors. Clin Cancer Res 2017;23(2): 600–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim JY, Brosnan-Cashman JA, An S, et al. Alternative lengthening of telomeres in primary pancreatic neuroendocrine tumors is associated with aggressive clinical behavior and poor survival. Clin Cancer Res 2017;23(6):1598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fishbein L, Khare S, Wubbenhorst B, et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun 2015;6:6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Czeczok TW, Stashek KM, Maxwell JE, et al. Clusterin in neuroendocrine epithelial neoplasms: absence of expression in a well-differentiated tumor suggests a jejunoileal origin. Appl Immunohistochem Mol Morphol 2018;26(2):94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roquiz W, Maxwell JE, Pelletier D, et al. Comparison of serotonin to the midgut marker CDX2 to assign site of origin in a well-differentiated neuroendocrine tumor. Lab Invest 2018;98(Supplement 1):237A. [Google Scholar]
- 57.Viale G, Doglioni C, Gambacorta M, et al. Progesterone receptor immunoreactivity in pancreatic endocrine tumors. An immunocytochemical study of 156 neuroendocrine tumors of the pancreas, gastrointestinal and respiratory tracts, and skin. Cancer 1992;70(9):2268–77. [DOI] [PubMed] [Google Scholar]
- 58.Stashek KM, Czeczok TW, Bellizzi AM. Extensive evaluation of immunohistochemistry to assign site of origin in well-differentiated neuroendocrine tumors: a study of 10 markers in 265 tumors. Mod Pathol 2014;27(Supplement 2):160A. [Google Scholar]
- 59.Ralston J, Chiriboga L, Nonaka D. MASH1: a useful marker in differentiating pulmonary small cell carcinoma from Merkel cell carcinoma. Mod Pathol 2008; 21(11):1357–62. [DOI] [PubMed] [Google Scholar]
- 60.Shuda M, Arora R, Kwun HJ, et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int J Cancer 2009;125(6):1243–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Busam KJ, Jungbluth AA, Rekthman N, et al. Merkel cell polyomavirus expression in merkel cell carcinomas and its absence in combined tumors and pulmonary neuroendocrine carcinomas. Am J Surg Pathol 2009;33(9):1378–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Czeczok TW, Gailey MP, Hornick JL, et al. High-grade neuroendocrine carcinomas are characterized by marked transcription factor lineage infidelity: an evaluation of 36 diagnostic markers in 83 tumors. Mod Pathol 2014; 27(Supplement 2):152A. [Google Scholar]
- 63.George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524(7563):47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rekhtman N, Pietanza MC, Hellmann MD, et al. Next-generation sequencing of pulmonary large cell neuroendocrine carcinoma reveals small cell carcinoma like and non-small cell carcinoma-like subsets. Clin Cancer Res 2016;22(14): 3618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yachida S, Vakiani E, White CM, et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol 2012;36(2):173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alkapalan D, Maxwell JE, O’Dorisio TM, et al. Prospective experience with routine SSTR2A immunohistochemistry in neuroendocrine epithelial neoplasms. Mod Pathol 2016;29(Suppl 2):145A.26820331 [Google Scholar]
- 67.Bellizzi A, Czeczok T, McMullen E. Somatostatin receptor subtype 2a is frequently expressed by poorly differentiated neuroendocrine carcinomas: a potential novel therapeutic target. Mod Pathol 2015;28(Supplement 2):132A. [Google Scholar]
- 68.Pelletier D, Mott SL, O’Dorisio MS, et al. CXCR4 is highly expressed by poorly differentiated neuroendocrine carcinoma: a novel diagnostic, prognostic, and potential therapeutic target. Lab Invest 2018;98(Supplement 1):236A. [Google Scholar]
- 69.College of American Pathologists Cancer Protocols. Available at: https://www.cap.org/protocols-and-guidelines/cancer-reporting-tools/cancer-protocol-templates. Accessed July 29, 2019.
- 70.Bergsland EK, Woltering EA, Rindi G, et al. Neuroendocrine tumors of the pancreas In: Amin MB, editor. AJCC cancer staging manual. 8th edition. New York: Springer; 2017. p. 407–19. [Google Scholar]