

Abstract
Background
Antitumor therapies targeting programmed cell death-1 (PD-1) or its ligand-1 (PD-L1) are used in various cancers. However, in glioblastoma (GBM), the expression of PD-L1 varies between patients, and the relationship between this variation and the efficacy of anti-PD-1 antibody therapy remains unclear. High expression levels of PD-L1 affect the proliferation and invasiveness of GBM cells. As COX-2 modulates PD-L1 expression in cancer cells, we tested the hypothesis that the COX-2 inhibitor celecoxib potentiates anti-PD-1 antibody treatment via the downregulation of PD-L1.
Methods
Six-week-old male C57BL/6 mice injected with murine glioma stem cells (GSCs) were randomly divided into four groups treated with vehicle, celecoxib, anti-PD-1 antibody, or celecoxib plus anti-PD-1 antibody and the antitumor effects of these treatments were assessed. To verify the mechanisms underlying these effects, murine GSCs and human GBM cells were studied in vitro.
Results
Compared with that with each single treatment, the combination of celecoxib and anti-PD-1 antibody treatment significantly decreased tumor volume and prolonged survival. The high expression of PD-L1 was decreased by celecoxib in the glioma model injected with murine GSCs, cultured murine GSCs, and cultured human GBM cells. This reduction was associated with post-transcriptional regulation of the co-chaperone FK506-binding protein 5 (FKBP5).
Conclusions
Combination therapy with anti-PD-1 antibody plus celecoxib might be a promising therapeutic strategy to target PD-L1 in glioblastoma. The downregulation of highly-expressed PD-L1 via FKBP5, induced by celecoxib, could play a role in its antitumor effects.
Keywords: celecoxib, FKBP5, glioblastoma, PD-1, PD-L1
Importance of the Study.
Novel cancer therapies target programmed cell death-1 (PD-1) or its ligand (PD-L1). Although the expression of PD-L1 affects cell proliferation in human glioblastoma (GBM) cells, the effects of anti-PD-1 antibodies and the role of intrinsic PD-L1 remain unclear. We first document that the combination of an anti-PD-1 antibody plus the COX-2 inhibitor celecoxib synergistically exerts antitumor effects in a malignant glioma model of murine glioma stem cells (GSCs) with high PD-L1 expression. Furthermore, we established that celecoxib reduces PD-L1 protein expression via post-transcriptional regulation by FK506-binding protein 5 (FKBP5), a co-chaperone of isomerase activity, in murine GSCs, the GBM model, and in human GBM cells highly expressing PD-L1. The downregulation of PD-L1 via FKBP5 induced by celecoxib was also found to be associated with increased PD-1-positive cell accumulation and augmented antitumor effects in a GBM model receiving combination therapy. Such combination therapy might lead to new immunotherapies to treat GBM.
Key Points.
PD-L1 is downregulated by celecoxib via FKBP5 in GBM and murine glioma stem cells.
PD-L1 downregulation by celecoxib enhances anti-PD-1 antibody therapy.
Glioblastoma (GBM) is the most common primary malignant brain tumor. Despite surgery, chemotherapy, and radiation, its median survival is 14.6 months.1,2 Therefore, new therapeutic strategies are needed to improve prognosis for patients with GBM.
Programmed cell death-1 (PD-1) is an immune checkpoint receptor expressed on the surface of T- and B-cells. The activation of PD-1 after its interaction with the programmed cell death ligand-1 (PD-L1) results in the immediate dephosphorylation of key proteins downstream from the antigen receptors, endowing PD-1 with immunoregulatory functions.3 Anti-PD-1 antibody has shown antitumor efficacy in the treatment of multiple tumor types.4–7 The introduction of new and more effective therapies including treatments based on stimulating the immune response and targeting specific proteins has improved prognosis for patients with metastatic melanoma.7 However, CheckMate-143 (NCT 02017717), the first large randomized clinical trial with an anti-PD-1 antibody, showed that this treatment did not improve the overall survival of patients with recurrent GBM.8
The downregulation of PD-L1 decreases tumor proliferation, invasion, and cell cycle progression, whereas it enhances tumor-specific T-cell effector functions.9–12 COX-2 expression is positively correlated with PD-L1 expression in human melanoma13 and can modulate the expression of PD-L1 in human breast cancer cells.14 As COX-2 inhibitors are highly effective for the treatment of some cancers, an inhibitor based on the type of cancer might provide an effective adjuvant strategy.15 Fujita et al.16 demonstrated that COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. The selective COX-2 inhibitor celecoxib enhances cytotoxic T lymphocyte functions by blocking PD-L1 in chronic viral infections.17 Elsewhere, we demonstrated that celecoxib induces apoptosis and inhibits cell proliferation via the AKT/survivin- and Akt/ID3 pathway in low-grade glioma.18 Based on these findings, we hypothesized that adding celecoxib to anti-PD-1 antibody treatment would augment its antitumor effects against gliomas.
The COP9 signalosome 5 (CSN5) controls T-cell suppression via PD-L1 deubiquitination in breast cancer.19 FK506-binding protein 5 (FKBP5) plays a role in protein folding required for subsequent glycosylation.20 PD-L1 is stabilized by its glycosylation in the endoplasmic reticulum because the protein half-life of glycosylated PD-L1 is longer than that of the nonglycosylated form, which occurs by preventing degradation via the 26S proteasome.21 FKBP5 has been implicated in the regulation of PD-L1, which might be an immunotherapeutic target for patients with GBM.22 In addition, the expression of PD-L1 is regulated by the transcription factors HIF-1α, STAT3, and NFκB,23 which are associated with Akt inhibition. However, the significance of PD-L1 regulation has not been clarified in the glioma stem cells (GSCs).
We used a murine GSC-bearing glioma model to examine the antitumor effects of celecoxib, especially through the regulation of PD-L1. To elucidate the mechanisms underlying the regulation of PD-L1, we focused on these molecules in murine GSCs, a GSC-bearing glioma model, and human GBM cells. Here, we first document the antitumor effects of an anti-PD-1 antibody plus celecoxib in a malignant glioma model injected with murine GSCs and the mechanisms underlying the downregulation of PD-L1 by celecoxib in murine GSCs and human GBM primary culture cells.
Materials and Methods
Tissue Samples
This study was approved by our institutional Ethics Committee. Human tissue samples from GBM, anaplastic astrocytoma (AA), and diffuse astrocytoma (DA), n = 3 for each, were provided by the Department of Neurosurgery, Tokushima University hospital. All donors provided prior written informed consent to use their brain tissue material. All samples were classified by neuropathologists according to the World Health Organization (WHO) classification of brain tumors. A portion of each tissue sample was fixed in 4% formalin in phosphate-buffered saline (PBS) and processed for paraffin embedding. Sections from non-neoplastic regions (NNRs) were purchased from BioChain Institute (Newark, NJ).
Cell Lines
Murine GSCs were a gift from the Keio University laboratory of Prof. Saya.24,25 Human GBM cell lines U87 and U251 were purchased from the American Type Culture Collection (Manassas, VA) and the Health Science Research Resources Bank (Osaka, Japan), respectively. TGB-00 cells were primary GBM cells from a patient who granted prior informed consent for their use in this study. Murine GSCs were cultured (37°C; 5% CO2, 95% humidified air) in Dulbecco’s Modified Eagle’s medium/Ham’s F-12 nutrient mixture (Sigma-Aldrich, St. Louis, MO) supplemented with 20 ng/mL recombinant human epidermal growth factor (PeproTech, Rocky Hill, NJ), 20 ng/mL recombinant human basic fibroblast growth factor (PeproTech), B-27 supplement without vitamin A (Life Technologies, Carlsbad, CA), 200 ng/mL heparin sulfate, 100 U/mL penicillin, and 100 µg/mL streptomycin (Nacalai Tesque, Kyoto, Japan). U87-, U251-, and TGB-00 cells were cultured in RPMI-1640 with L-glutamine and phenol red (FUJI FILM Wako Pure Chemical Corp., Osaka, Japan) supplemented with 10% fetal bovine serum (Gibco-BRL, NY).
Animal Experiments
All animal experiments were approved by our institutional Ethics Committee. We used a malignant glioma model with murine GSCs as described previously.24,25 Six-week-old male C57BL/6 mice were anesthetized and a stereotactic apparatus was placed in the right brain. With a dental drill, a small hole was bored into the skull 2.0 mm lateral to the bregma. Murine GSCs (1 × 103) in 2 μL of Hank’s balanced salt solution (Sigma-Aldrich, St. Louis, MO) were injected into the right cerebral hemisphere 3 mm below the surface of the brain using a 10-µl Hamilton syringe with an unbeveled 30-gauge needle. The mice were randomized and treated with vehicle, celecoxib, anti-PD-1 antibody, or the combination of celecoxib plus anti-PD-1 antibody. Therapeutic anti-PD-1 antibody was administered intraperitoneally (i.p.) beginning on day 0 (20 mg/kg) after murine GSC implantation; repeat injections were delivered every 6 days (10 mg/kg) for 30 days. Celecoxib, prepared with dimethyl sulfoxide (DMSO) and hydroxypropyl-β-cyclodextrin (HBC), was injected i.p. starting on day 0 (10 mg/kg) after murine GSC implantation and again every day for 30 consecutive days.
The anti-PD-1 antibody was kindly gifted from ONO pharmaceutical Co. Ltd (Tokyo, Japan). Celecoxib was purchased from Sigma–Aldrich, Cat. # PHR1683 (Tokyo, Japan). Vehicle controls received equivalent doses of DMSO/HBC and normal saline on the same dosing schedule. On day 14, five mice from each group were euthanized and their brains were sliced on a brain slicer matrix at 1.0-mm intervals. Tumor volume, represented by the GFP-positive area, was calculated from microscopic measurements (Keyence BZ-X710). Survival rate was evaluated with the Kaplan–Meier method, which was repeated twice (12 mice in each group). To validate the effect of celecoxib on the expression of PD-L1, vehicle- and celecoxib-treated mice were euthanized, and their brains were analyzed by western blotting analysis on day 14.
Immunohistochemistry
Paraffinized human glioma tissue sections were dewaxed, rehydrated, and subjected to antigen retrieval. Murine tissue samples were fixed with 4% paraformaldehyde and 5-μm thick frozen sections were mounted on Matsunami adhesive saline (MAS)–coated glass slides (Matsunami Glass, Tokyo, Japan). The sections were blocked for 30 min with 1–3% hydrogen peroxide solution, covered overnight at 4°C with antibodies against PD-L1 (Cell Signaling Technology, Cat. # 13684), COX-2 (Abcam, Cat. # ab15191), FKBP5 (Proteintech, Cat. #14155-1-AP), CD16 (Santa Cruz Biotechnology, Cat. # SC-52376), and CD163 (Abcam, Cat. #ab182422), and then incubated with biotinylated secondary antibody (30 min, 30°C), visualized using DAB buffer tablets, and counterstained with hematoxylin. For immunofluorescence staining, the sections were immunoreacted with APC conjugated anti-PD-1 antibody (Becton, Dickinson and Company, Cat. #562671). All images were taken using a BZ-X710 microscope (Keyence, Osaka, Japan), and the positive area per 10000 μm2 was analyzed with BZ-X710 equipped image analysis software.
Cell Viability Assay
Murine GSCs were plated in 96-well microtiter plates at 1 × 104 per well in 100 µL of medium and treated with celecoxib (30 and 60 µM) or DMSO as a vehicle control. Viability was assessed using WST-8 reagent (Dojindo, Kumamoto, Japan) and a microplate reader (TECAN, InfiniteR 200 PRO, Kanagawa, Japan). Cell viabilities were calculated relative to those with vehicle controls.
Quantitative Real-Time Polymerase Chain Reaction
Murine GSCs were seeded at 2 × 105 per 100-mm cell culture dish and treated for 24 h with celecoxib (60 µM) or vehicle. Human TGB-00 cells were seeded at 3 × 105 per 100-mm cell culture dish and treated three times for 24 h with celecoxib (20 µM) or vehicle. Total RNA isolated from cultured cells was purified with the MagNA pure compact RNA isolation kit (Roche Diagnostics) according to the manufacturer’s protocol and reverse-transcribed with Transcriptor Universal cDNA Master (Roche). Quantitative real-time polymerase chain reaction (qRT–PCR) was performed under the conditions recommended by the manufacturer on a Light Cycler rapid thermal cycler (Roche). The primers reported in Supplementary Table 1 were purchased from Nihon Gene Research Laboratories, Inc. (Miyagi, Japan). Quantitative gene expression data were normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA content.
Western Blotting
Murine GSCs and human TGB-00 cells were grown and treated as described. Cells or tissue samples were homogenized in RIPA buffer containing a protease/phosphatase inhibitor cocktail (Cell Signaling Technology, Cat. #5872). Protein concentration was assayed using a BCA kit (Thermo Fisher Scientific, USA). Protein (50 µg) was separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes (immune-blot PVDF membrane, BIO-RAD) by electroblotting. The membranes were immersed for 1 hour in blocking buffer (5% skim milk or 2% BSA in TBS) and incubated with primary antibodies against PD-L1 (Cell Signaling Technology, Cat. #13684), FKBP5 (Proteintech, Cat. #14155-1-AP), CSN5 (Cell Signaling Technology, Cat. #6895), HIF-1α (Cell Signaling Technology, Cat. #3716), STAT3 (Santa Cruz Biotechnology, Cat. #sc-7179), p-STAT3 (Cell Signaling Technology, Cat. #9131) p-Akt (Cell Signaling Technology, Cat. #4060), p-NF-κB p65 (Cell Signaling Technology, Cat. #3033), and β-actin (Sigma–Aldrich, Cat. #A5441). After washing and a 1-hour incubation with horseradish peroxidase-conjugated secondary antibody, protein–antibody complexes were detected with Amersham ECL prime western blotting detection reagents (GE Healthcare, UK) using a Lumino image analyzer (Image Quant LAS4000 mini, GE Healthcare, UK) and NIH ImageJ 1.52 software. All experiments were performed in triplicate.
Statistical Analyses
All analyses were performed with Prism software (GraphPad V6.0). Data were analyzed by a Student’s t-test for two-group comparisons. Survival estimates and median survivals were determined based on Kaplan–Meier survival curves. A log-rank (Mantel-Cox) test was performed to calculate P values for the Kaplan–Meier survival curves. One-way ANOVA followed by Tukey and Dunnett tests was used to determine the statistical significance for multiple comparisons. Error bars indicate standard deviations (SDs). Differences with P-values less than .05 were considered statistically significant.
Results
PD-L1 and COX-2 Are Expressed at High Levels in GBM and the Murine Malignant Glioma Model
In human malignant brain tumors, the expression of PD-L1 correlated with WHO grading (Figure 1A). This finding agrees with the report that the high expression of PD-L1 is correlated with grade IV glioma (GBM).26 The expression of COX-2 was higher in GBM than in the NNR (Figure 1B).
Figure 1.

PD-L1 and COX-2 are expressed at high levels in glioblastoma (GBM) and a murine malignant glioma model. (A) Human: Representative immunohistochemical staining for PD-L1 in normal brain and glioma samples (n = 6). The expression of PD-L1 increased with the WHO grade. Scale bar = 200 µm. (B) Human: Representative immunohistochemical staining for COX-2 in normal brain and GBM tissue (n = 6). Scale bar = 200 µm. (C) Mouse: Representative immunohistochemical staining for PD-L1 and COX-2 in brain samples from malignant glioma model (n = 6). The expression of PD-L1 and COX-2 was increased in brain tumor tissues. Scale bar = 100 µm. (D) Mouse: Representative images of western blot analysis for PD-L1 and COX-2 in brain tissue from malignant glioma model. Densitometry shows the relative protein expression normalized by β-actin. Each experiment was repeated three times (mean ± SD). **P < .01 vs. normal by Student’s t-test.
In the murine malignant glioma model, the expression of PD-L1 and COX-2 was significantly higher in tumors than in normal areas (Figure 1C). This observation agreed with the results from western blotting (Figure 1D). Thus, the high expression of PD-L1 and COX-2 in the murine malignant glioma model is similar to expression levels in GBM patients.
Celecoxib Plus the Anti-PD-1 Antibody Synergistically Improves Survival Rates in a Murine Malignant Glioma Model
We assessed the effects of therapy with the combination of celecoxib and the anti-PD-1 antibody in a murine malignant glioma model (Figure 2A). Compared with those with monotherapies with celecoxib or anti-PD-1 antibody, the combination decreased tumor volumes significantly (Figure 2B) and markedly prolonged survival (Figure 2C). To elucidate the mechanisms underlying the success of this treatment, we focused on the regulation of PD-L1 expression in the mouse brain tumors for two reasons: COX-2 affects the expression of PD-L1 in melanoma and silencing PD-L1 affects cell proliferation.12,13 We first confirmed that in our murine malignant glioma model, monotherapy with the COX-2 inhibitor celecoxib decreased the expression of PD-L1 protein in tumors (P < .05; Figure 2D).
Figure 2.

The combination of celecoxib plus anti-PD-1 antibody synergistically improves survival rates in a murine malignant glioma model. (A) Experimental protocol; an anti-PD-1 antibody was intraperitoneally (i.p.) injected at 20 mg/kg on days 0 and 10 mg/kg every 6 days until day 30 following the injection of murine glioma stem cells (GSCs). Celecoxib was i.p. injected at 10 mg/kg, every day for 30 days. (B) Tumor volumes in mice sacrificed on day 14 (mean ± SD, n = 5). *P < .05, **P <.01 vs. vehicle control (VC), ANOVA followed by the Dunnett test. (C) Kaplan–Meier survival curves (each experiment was repeated two times, 12 mice/group). **P < .01 in relation to the other groups based on a Mantel-Cox test. (D) Expression of PD-L1 protein as measured by western blotting. Each experiment was repeated three times (mean ± SD). *P < .05 vs. VC based on a Student’s t-test.
Celecoxib Decreases PD-L1 Protein Expression, But Not PD-L1 mRNA Expression
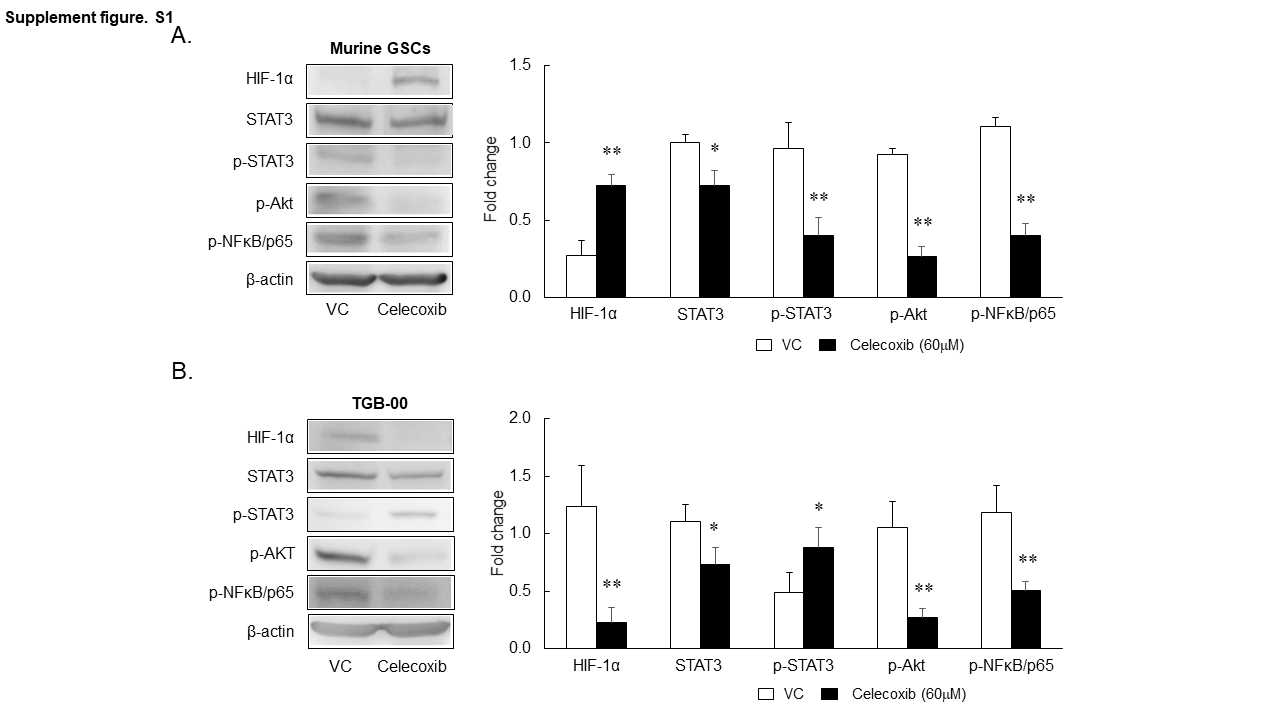
To examine the mechanisms underlying the reduction of PD-L1 by celecoxib in the murine model, we used cultured murine GSCs exposed to 30 or 60 µM celecoxib (Figure 3A) with the dosage based on our preliminary study. The viability of the GSCs was significantly reduced by treatment with celecoxib in a dose-dependent manner (Figure 3B). PD-L1 protein expression in murine GSCs was also significantly inhibited by treatment with celecoxib in a dose-dependent manner (Figure 3C), whereas its mRNA levels increased (Figure 3D). The reduction of PD-L1 protein might not be associated with transcriptional regulation, although the transcriptional regulation of PD-L1 via HIF-1α, STAT3, or NFκB has been reported.23 We further confirmed that HIF-1α protein expression was increased by celecoxib treatment in murine GSCs (Supplementary Figure 1A), whereas the protein expression of phosphorylated STAT3, Akt, and NF-kB was decreased. Increased HIF-1α might compensate for the reduced expression of PD-L1 protein by celecoxib in murine GSCs.
Figure 3.

Celecoxib decreases PD-L1 protein expression, but not PD-L1 mRNA expression. (A) Timing of cell-viability, western blotting, and qRT–PCR analyses. (B, C) Cell viability (B, n = 8) and PD-L1 expression (C, n = 3) in murine glioma stem cells (GSCs) treated with 30 or 60 µM celecoxib for 24 hours (mean ± SD). **P < .01, in relation to the other groups, ANOVA followed by the Dunnett test. Scale bar = 50 µm. (D) PD-L1 mRNA levels as assessed by qRT-PCR (mean ± SD, n = 6). **P < .01 vs. the vehicle control (VC) based on a Student’s t-test. (E) Schematic of regulation of PD-L1 expression by CSN5. Ub; ubiquitin. (F, G) CSN5 expression (F, n = 3) and CSN5 mRNA (G, n = 6) levels in murine GSCs treated with 60 µM celecoxib. Each column of data indicates mean ± SD, ns; not significant, Student’s t-test.
Lim et al.19 found that CSN5 enzyme activity controls T-cell suppression via PD-L1 deubiquitination in breast cancer cell lines. Based on their findings, we further examined the regulation of CSN5 by celecoxib in murine GSCs to elucidate the mechanisms underlying the celecoxib-induced reduction of PD-L1 expression (Figure 3E). Celecoxib affected neither the mRNA nor the protein levels of CSN5 (Figure 3F and 3G), indicating that it does not function by increasing enzyme levels.
Celecoxib Induces the Downregulation of PD-L1 by Inhibiting FKBP5
We then focused on FKBP5, which has been shown to increase the expression of PD-L1 by catalyzing the protein-folding required for subsequent glycosylation in gliomas.20 The celecoxib-induced decrease in PD-L1 expression was accompanied by the reduction of FKBP5 at both the protein and mRNA levels (Figure 4A and 4B). To examine whether FKBP5 also modulates the expression of PD-L1 in murine GSCs, we used the FKBP5 inhibitors FK506 and rapamycin.27 In murine GSCs, FK506 (25 µM) or rapamycin (10 nM) at IC50 doses was applied according to the protocol shown in Figure 4C. Western blotting revealed that in murine GSCs exposed to FK506 or rapamycin, the expression of PD-L1 and FKBP5 was significantly decreased (Figure 4D and 4E). These findings are consistent with the FKBP5-mediated celecoxib-induced downregulation of PD-L1 in murine GSCs.
Figure 4.

Celecoxib induces the downregulation of PD-L1 by inhibiting FKBP5. (A, B) Expression of FKBP5 and PD-L1 by western blot (WB) analysis repeated three times and mRNA levels of FKBP5 (n = 6) in murine glioma stem cells (GSCs) exposed to 60 µM celecoxib for 24 hours (mean ± SD). *P < .05, **P < .01 vs. vehicle control (VC) based on a Student’s t-test. (C–E) WB analysis of murine GSCs exposed to 25 µM FK506 or 10 nM rapamycin for 24 hours (C). Representative images of murine GSCs and the expression of FKBP5 and PD-L1, which was decreased by FK506 (D) or rapamycin (E). Each data point indicates mean ± SD (n = 3). *P < .05, **P < .01 vs. VC based on a Student’s t-test. Scale bar = 50 µm.
PD-L1 Downregulation by Celecoxib Is Associated With the Downregulation of FKBP5 in Primary GBM Cells
To test whether treatment with celecoxib induces the downregulation of PD-L1 in the human TGB-00 cell lines as it did in the murine malignant glioma model and murine GSCs, we confirmed the high expression of PD-L1 and FKBP5 protein in the human GBM cell lines U87 and U251, as well as in the primary human GBM cell line TGB-00 established in our laboratory (Figure 5A). The optimal conditions for reducing PD-L1 expression by celecoxib were one treatment with 20 µM of celecoxib daily for 3 days (Figure 5B). The viability of TGB-00 cells was reduced by celecoxib (Figure 5C). We found that celecoxib significantly decreased the expression of both PD-L1 and FKBP5 at the protein level without changing the mRNA levels of PD-L1 (Figure 5D and 5E). Thus, we observed that in human GBM cells, as well as in murine GSCs, celecoxib induces a reduction in PD-L1 via FKBP5 (Figure 5F). We also confirmed that celecoxib reduced the protein expression of HIF-1α, STAT3, phosphorylated Akt, and NF-kB (Supplementary Figure 1B), whereas it increased the protein expression of phosphorylated STAT3. Although the compensatory pathway might be different between murine GSCs and human GBM, the upregulation of these molecules might be reciprocal for the downregulation of PD-L1 protein (Supplementary Figure 1A and 1B).
Figure 5.

PD-L1 downregulation by celecoxib is associated with the downregulation of FKBP5 in primary glioblastoma (GBM) cells. (A) Representative baseline expression of FKBP5 and PD-L1 in the human GBM cell lines U87 and U251 and in primary cultures of human GBM cells, TGB-00. Western blot (WB) analysis was repeated three times. (B–E) WB and qRT–PCR analysis of TGB-00 cells treated three times with 20 µM celecoxib or vehicle for 24 hours (B). Representative images of TGB-00 cells (C), protein expression (D, n = 3), and the mRNA levels (E, n = 6) of FKBP5 and PD-L1 in TGB-00 cells. Each data point indicates mean ± SD, *P < .05, **P < .01 vs. vehicle (VC) based on a Student’s t-test. ns; not significant. Scale bar = 50 µm. (F) Schematic of the regulation of PD-L1 expression via the inhibition of FKBP5 mediated by celecoxib.
Downregulation of PD-L1 and FKBP5 induced by celecoxib is associated with PD-1-positive cell accumulation in a murine malignant glioma model
Lastly, we examined the expression of PD-L1 and FKBP5 mediated by celecoxib in the murine malignant glioma model. Western blot analysis and immunohistochemical findings showed that monotherapy with celecoxib significantly decreased the expression of COX-2, FKBP5, and PD-L1 protein in the tumors of the murine model (P < 0.01; Figure 6A and 6B). Meanwhile celecoxib increased CD16-positive cells (pro-inflammatory M1-type macrophages) and decreased CD163-positive cells (anti-inflammatory M2-type macrophages; Figure 6C). Importantly, PD-1-positive cells with the combination therapy were significantly increased (Figure 6D). Taken together, anti-PD-1 antibody and celecoxib combination therapy might augment the antitumor effects by downregulating PD-L1 via FKBP5, increasing PD-1-positive cells, and regulating peripheral macrophage recruitment in the tumor and its microenvironment.
Figure 6.

Celecoxib-mediated downregulation of PD-L1 via FKBP5 in a murine malignant glioma model. (A) Expression of COX-2, FKBP5, and PD-L1 protein, as measured by western blot analysis in a murine malignant glioma model (vehicle control [VC] and monotherapy with celecoxib group). Brain tissue samples were analyzed on day 14 as in Figure 2A. Each experiment was repeated three times (mean ± SD). **P < .01 vs. VC based on a Student’s t-test. (B) Immunohistochemistry for COX-2, FKBP5, and PD-L1; positive cells were analyzed by KEYENCE BZ-X710 equipped image analysis (mean ± SD, n = 5). Scale bar = 50 µm. *P < .05, **P < .01 vs. VC based on a Student’s t-test. (C) Expression of CD16 and CD163 in brain tumor tissues from murine malignant glioma model (mean ± SD, n = 5). Scale bar = 50 µm. *P < .05, **P < .01 vs. VC based on a Student’s t-test. (D) Immunohistochemical staining for PD-1 in brain tumor tissues from murine malignant glioma model. Scale bar = 100 µm. **P < .01 relative to the other groups, based on ANOVA followed by a Tukey test.
Discussion
We first documented that the addition of celecoxib enhances the antitumor effects of an anti-PD-1 antibody in a malignant glioma animal model. Second, we demonstrated that the downregulation of PD-L1 via FKBP5, mediated by celecoxib, contributes at least partly to the mechanisms underlying the antitumor effects augmented by celecoxib. Although monotherapy with anti-PD-1 antibody or celecoxib was insufficient to improve the survival rate in our mouse model, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) blockade is most commonly combined with anti-PD-1 antibody to enhance antitumor efficacy.28 Combination therapy with anti-PD-1 antibody plus CTLA-4 blockade was curative for 75% of mice in the GBM model, even in the presence of advanced-stage tumors.29 Triple-targeting immunosuppression with IDO, CTLA-4, and PD-L1 was found to be highly effective for mice with brain tumors.30 In combination, anti-PD-1 antibodies plus stereotactic radiation also improve survival rates in a murine glioma model31 and the combination of an anti-PD-1 antibody, anti-T-cell immunoglobulin, and mucin domain-3 (TIM-3) antibody, plus focal radiation was found to result in the regression of murine glioma.32 In tumor-bearing mice, interferon-α plus an anti-PD-1 antibody effectively augments antitumor immunity.33 Murine glioma models using the cell line GL261 further revealed the efficacy of combination therapy with anti-PD-1 antibodies.29–32,34 Together, these findings suggest that anti-PD-1 immunotherapy should be combined with some other therapy to enhance antitumor effects for glioma treatment.
A few studies have investigated the efficacy of anti-PD-1 antibodies combined with low-molecular-weight compounds. In an orthotopic mouse glioma model, anti-PD-1 antibody plus temozolomide significantly improved survival time.34 Using a murine model of melanoma and metastatic lung cancer, celecoxib plus an anti-PD-1 antibody, delivered in alginate hydrogel, elicited potent antitumor effects by remodeling the immune response and improved the angiogenic tumor microenvironment.35 Elsewhere, we reported the antitumor effects exerted by celecoxib via the AKT/survivin- and AKT/ID3 pathway in low-grade glioma cells.18 We also demonstrated that Akt2 and Akt3 might hold promise as candidate targets for therapy in patients with malignant glioma.36 Here, we first confirmed the high expression of COX-2 and PD-L1 in the murine malignant glioma model. Furthermore, we documented the augmented antitumor effects of combination therapy comprising anti-PD-1 plus celecoxib in a malignant glioma model. Thus, this combination might provide a potential approach for the treatment of glioma.
Botti et al.13 documented that celecoxib downregulates both overall protein levels and the surface expression of PD-L1 in the human malignant melanoma cell lines A375 and SK-MEL-2. Markosyan et al.14 found that COX-2 knockdown reduced the expression of PD-L1 via a pathway other than PGE2 in mammary tumor cells. Therefore, to elucidate the mechanisms underlying the regulation of PD-L1 expression, we focused on the regulatory effects of celecoxib. Unexpectedly, celecoxib increased the mRNA level of PD-L1, whereas its protein expression decreased, suggesting that celecoxib might play a role predominantly at the protein level and not via transcriptional regulation. The elevated mRNA level of PD-L1 induced by celecoxib might be associated with compensatory activity mediated by different pathways in murine GSCs and human TGB-00 (Supplementary Figure 1A and 1B)
D’Arrigo et al.20,37 demonstrated that targeting of FKBP5 with si-RNA or an inhibitor lowered PD-L1 expression and induced antitumor effects in GBM cells based on in vitro and in vivo studies. Similarly, we found that celecoxib reduced the expression of both PD-L1 and FKBP5 in murine GSCs and in human GBM cells, with the reduction in PD-L1 expression associated with reduced viability. In patients with GBM, high expression of PD-L1 was correlated with poor outcomes.38,39 An analysis of PD-L1 DNA abundance in circulating extracellular vesicles from GBM patients revealed a significant correlation with tumor volume.40 PD-L1 promotes epithelial–mesenchymal transition (EMT) via Ras-MEK/Erk-EMT signaling and confers GBM cell malignancy and aggressiveness.9 In contrast, the downregulation of PD-L1 decreases tumor proliferation, invasion, and cell cycle progression; furthermore, it enhances tumor-specific T-cell effector functions.9–12 Consistent with these studies, we found that the downregulation of PD-L1 by celecoxib was associated with not only the enhanced antitumor effects but also an increase in PD-1-positive cell accumulation in tumors of the glioma model after combination therapy with celecoxib and the anti-PD-1 antibody.
Annovazzi et al. reported that in high-grade GBM, the anti-inflammatory CD163-positive cells associated with cell growth and survival were increased but that there was no effect on pro-inflammatory CD16-positive cells associated with cell death, resulting in an imbalance in CD16/163 positive cells.41 We found that celecoxib decreased CD163-positive cells in PD-L1-positive brain tumor tissue, whereas it increased CD16-positive cells. The celecoxib-induced downregulation of PD-L1 via FKBP5 inhibition might account for some or all of the antitumor effects of anti-PD-1 antibody treatment.
Our study has some limitations. Although we confirmed the high expression of COX-2 and PD-L1 in GBM and the murine malignant glioma model and found that the expression of FKBP5 was related to PD-L1 downregulation by celecoxib, we did not assess the expression of PD-L1 in GSCs from human glioma tissue. We showed that celecoxib decreased the expression of PD-L1 in murine GSCs and human GBM cells and induced the downregulation of COX-2, FKBP5, PD-L1, and CD163-positive cells in a glioma model; however, we could not evaluate the number of viable cells expressing PD-L1, COX-2, and FKBP5 based on gated live cells by flow cytometry with murine GSCs and human GBM cells. Furthermore, we cannot rule out the role of CSN5 in regulating PD-L1 because we did not assay its enzymatic activity. As PD-L1 regulation might involve multiple signaling pathways, we exclude the possibility that other mechanisms underlie the regulation of PD-L1.
In conclusion, combination therapy comprising anti-PD-1 antibody drugs plus celecoxib synergistically enhances the antitumor effects of either agent in a malignant glioma model. The celecoxib-induced downregulation of PD-L1 protein via FKBP5 might contribute to the antitumor effects with murine GSCs. The current findings warrant further studies to determine the antitumor effects of combined therapy with anti-PD-1 antibody drugs and celecoxib in the clinical setting.
Funding
Funding for this work was provided by a Grant-in-Aid for Scientific Research (No. 18K16586 [to K.S.]) and (No. 25462264 [to Y.M.]) from Japan Society for the Promotion of Science.
Conflict of Interest:
All authors declare no potential conflicts of interest.
Authorship statement.
Conceived and designed the project: I.Y. and K.K. Performed experiments: I.Y. and K.S. Analyzed data: I.Y., K.N., K.S., Y.M., T.F., E.S., T.Y., K.K., and Y.T. Provided mice glioma stem cells and contributed to experiments: O.S. and H.S. Wrote the manuscript: I.Y.
Supplementary Material
{kind=link}
Acknowledgments
We thank Emiko Nishikawa and Akiko Sumi for excellent technical support.
References
- 1. Stupp R, Hegi ME, Mason WP, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. [DOI] [PubMed] [Google Scholar]
- 2. Stupp R, Mason WP, van den Bent MJ, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 3. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14(12):1212–1218. [DOI] [PubMed] [Google Scholar]
- 4. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferris RL, Blumenschein G Jr, Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Motzer RJ, Escudier B, McDermott DF, et al. ; CheckMate 025 Investigators Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. [DOI] [PubMed] [Google Scholar]
- 8. Filley AC, Henriquez M, Dey M. Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget. 2017;8(53):91779–91794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qiu XY, Hu DX, Chen WQ, et al. PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochim Biophys Acta Mol Basis Dis. 2018;1864(5 Pt A):1754–1769. [DOI] [PubMed] [Google Scholar]
- 10. Audrito V, Serra S, Stingi A, et al. PD-L1 up-regulation in melanoma increases disease aggressiveness and is mediated through miR-17-5p. Oncotarget. 2017;8(9):15894–15911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iwamura K, Kato T, Miyahara Y, et al. siRNA-mediated silencing of PD-1 ligands enhances tumor-specific human T-cell effector functions. Gene Ther. 2012;19(10):959–966. [DOI] [PubMed] [Google Scholar]
- 12. Li Y, Wang J, Li C, Ke XY. Contribution of PD-L1 to oncogenesis of lymphoma and its RNAi-based targeting therapy. Leuk Lymphoma. 2012;53(10):2015–2023. [DOI] [PubMed] [Google Scholar]
- 13. Botti G, Fratangelo F, Cerrone M, et al. COX-2 expression positively correlates with PD-L1 expression in human melanoma cells. J Transl Med. 2017;15(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Markosyan N, Chen EP, Evans RA, Ndong V, Vonderheide RH, Smyth EM. Mammary carcinoma cell derived cyclooxygenase 2 suppresses tumor immune surveillance by enhancing intratumoral immune checkpoint activity. Breast Cancer Res. 2013;15(5):R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hashemi Goradel N, Najafi M, Salehi E, Farhood B, Mortezaee K. Cyclooxygenase-2 in cancer: a review. J Cell Physiol. 2019;234(5):5683–5699. [DOI] [PubMed] [Google Scholar]
- 16. Fujita M, Kohanbash G, Fellows-Mayle W, et al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011;71(7):2664–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen JH, Perry CJ, Tsui YC, et al. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med. 2015;21(4):327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sato A, Mizobuchi Y, Nakajima K, et al. Blocking COX-2 induces apoptosis and inhibits cell proliferation via the Akt/survivin- and Akt/ID3 pathway in low-grade-glioma. J Neurooncol. 2017;132(2):231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lim SO, Li CW, Xia W, et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30(6):925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. D’Arrigo P, Russo M, Rea A, et al. A regulatory role for the co-chaperone FKBP51s in PD-L1 expression in glioma. Oncotarget. 2017;8(40):68291–68304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li CW, Lim SO, Xia W, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang W, Cazacu S, Xiang C, et al. FK506 binding protein mediates glioma cell growth and sensitivity to rapamycin treatment by regulating NF-kappaB signaling pathway. Neoplasia. 2008;10(3):235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016;27(3):409–416. [DOI] [PubMed] [Google Scholar]
- 24. Sampetrean O, Saya H. Characteristics of glioma stem cells. Brain Tumor Pathol. 2013;30(4):209–214. [DOI] [PubMed] [Google Scholar]
- 25. Sampetrean O, Saga I, Nakanishi M, et al. Invasion precedes tumor mass formation in a malignant brain tumor model of genetically modified neural stem cells. Neoplasia. 2011;13(9):784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garber ST, Hashimoto Y, Weathers SP, et al. Immune checkpoint blockade as a potential therapeutic target: surveying CNS malignancies. Neuro Oncol. 2016;18(10):1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gaali S, Kirschner A, Cuboni S, et al. Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat Chem Biol. 2015;11(1):33–37. [DOI] [PubMed] [Google Scholar]
- 28. Johnson CB, Win SY. Combination therapy with PD-1/PD-L1 blockade: an overview of ongoing clinical trials. Oncoimmunology. 2018;7(4):e1408744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reardon DA, Gokhale PC, Klein SR, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016;4(2):124–135. [DOI] [PubMed] [Google Scholar]
- 30. Wainwright DA, Chang AL, Dey M, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20(20):5290–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zeng J, See AP, Phallen J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim JE, Patel MA, Mangraviti A, et al. Combination therapy with Anti-PD-1, Anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23(1):124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Terawaki S, Chikuma S, Shibayama S, et al. IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186(5):2772–2779. [DOI] [PubMed] [Google Scholar]
- 34. Dai B, Qi N, Li J, Zhang G. Temozolomide combined with PD-1 Antibody therapy for mouse orthotopic glioma model. Biochem Biophys Res Commun. 2018;501(4):871–876. [DOI] [PubMed] [Google Scholar]
- 35. Li Y, Fang M, Zhang J, et al. Hydrogel dual delivered celecoxib and anti-PD-1 synergistically improve antitumor immunity. Oncoimmunology. 2016;5(2):e1074374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mure H, Matsuzaki K, Kitazato KT, et al. Akt2 and Akt3 play a pivotal role in malignant gliomas. Neuro Oncol. 2010;12(3):221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. D’Arrigo P, Digregorio M, Romano S, et al. The splicing FK506-binding protein-51 isoform plays a role in glioblastoma resistance through programmed cell death ligand-1 expression regulation. Cell Death Discov. 2019;5:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nduom EK, Wei J, Yaghi NK, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016;18(2):195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pratt D, Dominah G, Lobel G, et al. Programmed Death Ligand 1 Is a Negative Prognostic Marker in Recurrent Isocitrate Dehydrogenase-Wildtype Glioblastoma. Neurosurgery. 2019;85(2):280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ricklefs FL, Alayo Q, Krenzlin H, et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci Adv 2018;4(3):eaar2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Annovazzi L, Mellai M, Bovio E, Mazzetti S, Pollo B, Schiffer D. Microglia immunophenotyping in gliomas. Oncol Lett. 2018;15(1):998–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.