

Abstract
N6-methyladenosine (m6A) regulates stability and translation of messenger RNA (mRNA) in various biological processes. In this work, we show that knockout of the m6A writer Mettl3 or the nuclear reader Ythdc1 in mouse embryonic stem cells increases chromatin accessibility and activates transcription in an m6A-dependent manner. We found that METTL3 deposits m6A modifications on chromosome-associated regulatory RNAs (carRNAs), including promoter-associated RNAs, enhancer RNAs, and repeat RNAs. YTHDC1 facilitates the decay of a subset of these m6A-modified RNAs, especially elements of the long interspersed element-1 family, through the nuclear exosome targeting–mediated nuclear degradation. Reducing m6A methylation by METTL3 depletion or site-specific m6A demethylation of selected carRNAs elevates the levels of carRNAs and promotes open chromatin state and downstream transcription. Collectively, our results reveal that m6A on carRNAs can globally tune chromatin state and transcription.
N6-methyladenosine (m6A) is an abundant modification on most eukaryote mRNAs (1, 2), regulated mainly by writer, eraser, and reader proteins (3). The mRNA m6A modification is installed by METTL3 (4) and can be removed by demethylases FTO and ALKBH5 (5, 6). Readers, including the YTH domain family and HNRNP proteins, directly or indirectly recognize the m6A-marked transcripts and affect mRNA metabolism (4, 7–9).
m6A plays critical roles in diverse biological processes, including the self-renewal and differentiation of embryonic and adult stem cells (3, 4). In mouse embryonic stem cells (mESCs), transcripts encoding pluripotency factors tend to be m6A-methylated and subjected to YTHDF2-mediated decay in cytoplasm, which affects their turnover during differentiation (10–12). However, m6A appears to also exhibit YTHDF2-independent regulations during early development, given that Ythdf2 knockout (KO) mice can survive to late embryonic developmental stages but Mettl3 KO results in early embryonic lethality (11, 13). Notably, mouse KO of the nuclear m6A reader Ythdc1 exhibits similar early mouse embryonic lethality to the Mettl3 KO (14). These observations imply that m6A could play additional roles in the nucleus that affect cell survival and differentiation. Previous studies have also suggested that m6A methylation on chromatin modifier transcripts or the chromosome binding of methyltransferase may affect transcription (15–17).
We investigated two independent Mettl3 KO mESC lines (Mettl3−/−−1 and Mettl3−/−−2) (10) and analyzed newly transcribed RNA levels. Mettl3 KO mESCs displayed marked increases in nascent transcripts synthesis compared with control wild-type (WT) mESCs (Fig. 1A). We generated stable rescue cell lines that express WT METTL3 or an inactive mutant METTL3 (fig. S1A). The increased transcription of nascent transcripts upon Mettl3 KO was reversed with WT but not mutant METTL3 (Fig. 1B).
Fig. 1. Mettl3 KO in mESCs leads to increased nascent RNA transcription and chromatin accessibility.
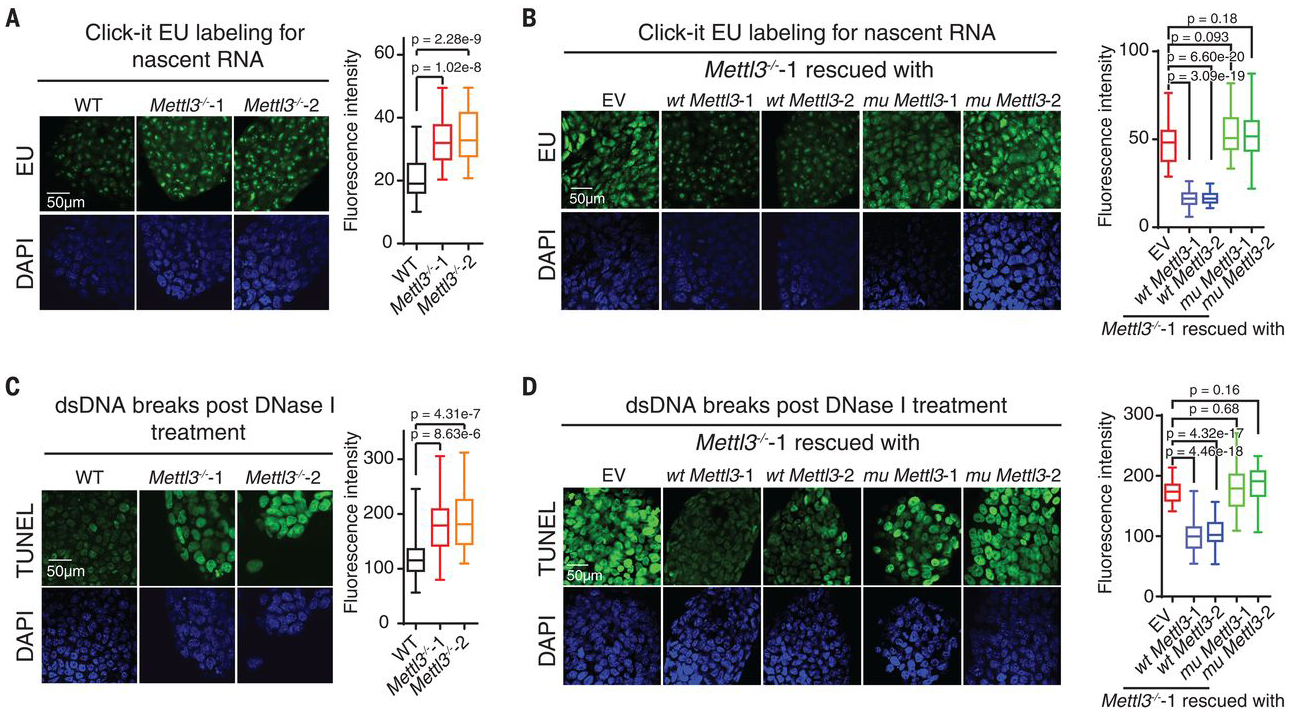
(A and B) Analysis of nascent RNA synthesis in WT or Mettl3−/− mESCs [(A), Mettl3−/−−1 and −2 are two independently generated KO lines], and Mettl3−/− mESCs rescued with WT or an inactive mutant Mettl3 (B). Nascent RNA synthesis was detected by using a click-it RNA Alexa fluor 488 imaging kit. EU, 5-ethynyl uridine; DAPI, 4′,6-diamidino-2-phenylindole. (C and D) Analysis of chromatin accessibility in WT or Mettl3−/− mESCs (C), and Mettl3−/− mESCs rescued with WT or mutant Mettl3 (D). DNase I–treated TUNEL assay was performed. For (A) to (D), the nucleus is counterstained by DAPI. EV (empty vector) refers to Mettl3−/− mESCs when transfected with empty vector plasmid. dsDNA, double-stranded DNA.
We next asked if the global chromatin state is affected by Mettl3 deletion. We performed deoxyribonuclease (DNase) I–treated terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay and observed a notable increase in chromatin accessibility in Mettl3 KO mESCs compared with wild type. Moreover, expression of WT but not mutant METTL3 reversed the increased chromatin accessibility, suggesting m6A dependence (Fig. 1, C and D).
We constructed conditional knockout (CKO) Ythdc1 (fig. S1B) and Ythdf2 (fig. S1C) mESCs. Ythdc1 CKO showed a similar increase in transcription and chromatin openness as Mettl3 KO (fig. S1, D and F), whereas Ythdf2 CKO showed minimal differences (fig. S1, E and G). The changes observed in Ythdc1 CKO mESCs were reversed by expressing WT but not mutant YTHDC1 (fig. S1, B, D, and F). Consistently, both H3K4me3 and H3K27ac, two histone marks associated with active transcription, were elevated upon Mettl3 and Ythdc1 depletion (fig. S1, H and I). Together, these data suggested a nuclear regulatory role for RNA m6A.
We next isolated nonribosomal RNAs from soluble nucleoplasmic and chromosome-associated fractions and quantified m6A/A by liquid chromatography–tandem mass spectrometry (LC-MS/MS) (fig. S2A). The m6A/A ratio in nonribosomal chromosome-associated RNAs (caRNAs) decreased the most (>50%) upon Mettl3 KO (Fig. 2A and fig. S2B), suggesting an effect of m6A on caRNAs. We immunoprecipitated ribosomal-RNA–depleted, m6A-containing caRNAs and performed high-throughput sequencing (methylated RNA immunoprecipitation sequencing, MeRIP-seq) in Mettl3 KO and WT mESCs. The m6A levels showed a global decrease after Mettl3 KO (fig. S2C), consistent with LC-MS/MS analysis (Fig. 2A). We identified ~40,000 peaks in each sample; both m6A levels (fig. S2D) and peaks (fig. S2E) were fully reproducible. Compared with wild type, Mettl3 KO samples showed more hypomethylated peaks (fig. S2F), with the largest reduction found at intergenic regions (fig. S2, G and H).
Fig. 2. Transcript turnover of carRNAs is regulated by m6A.
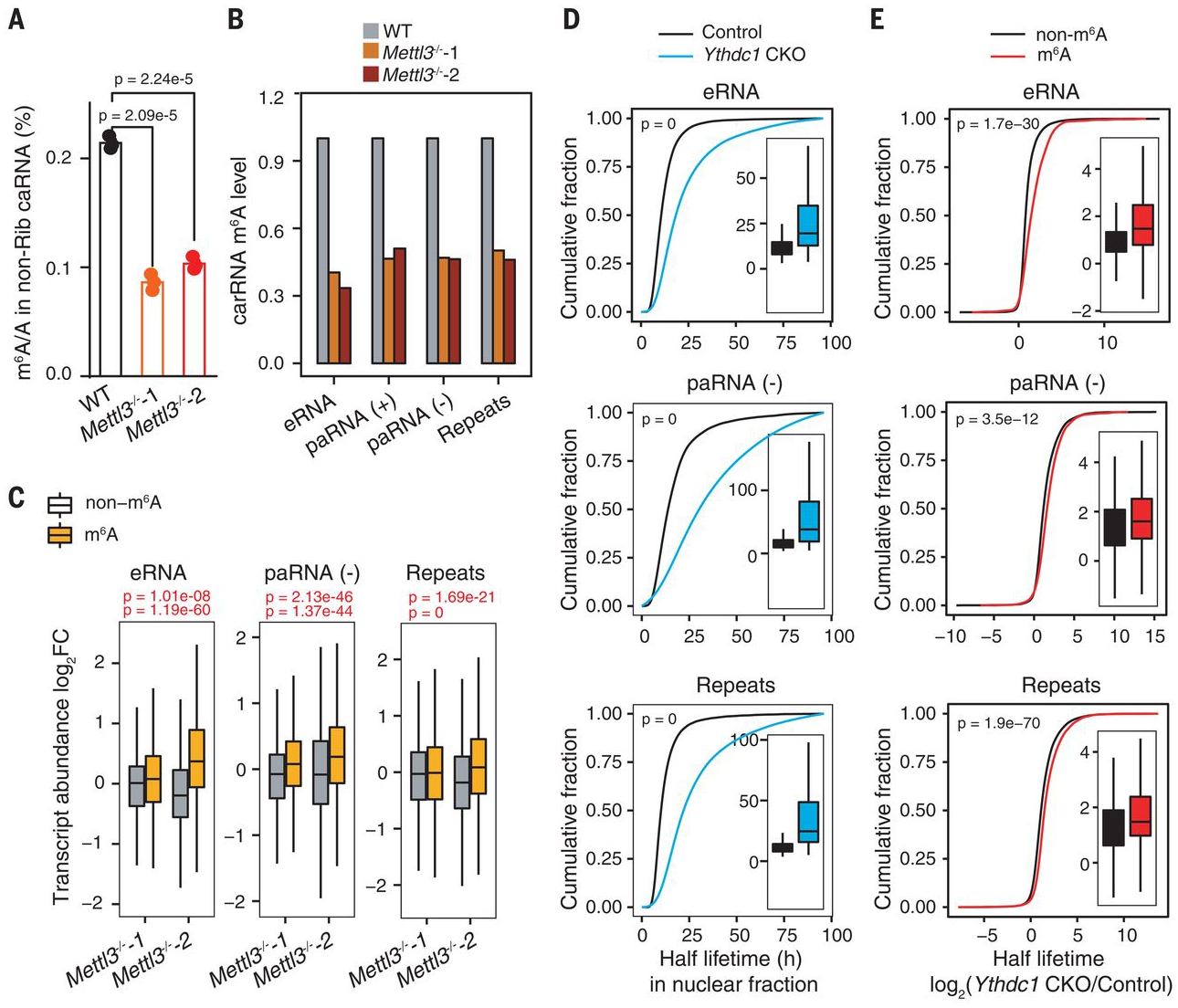
(A) LC-MS/MS quantification of the m6A/A ratio in nonribosomal (non-Rib) caRNAs (including pre-mRNA) extracted from WT or Mettl3−/− mESCs. n = 3 biological replicates; error bars indicate means ± SEM. (B) m6A level changes on carRNAs were quantified through normalizing m6A sequencing results with spike-in between WT and Mettl3 KO mESCs. n = 2 biological replicates. (C) carRNAs were divided into methylated (m6A) or nonmethylated (non-m6A) groups. The boxplot shows greater increases in transcript abundance fold changes of the m6A group versus the non-m6A group upon Mettl3 KO over WT mESCs. For (A) and (C), P values were determined by two-tailed t test. (D) Cumulative distribution and boxplots (inside) of nuclear carRNA half lifetime changes in CKO Ythdc1 and control mESCs. (E) Cumulative distributions and boxplots (inside) of the half lifetime changes of carRNAs upon Ythdc1 CKO. carRNAs were divided into methylated (m6A) or nonmethylated (non-m6A) groups. Depletion of YTHDC1 led to greater half lifetime increases of m6A-marked carRNAs than non-m6A–marked ones. For (D) and (E), P values were calculated by a nonparametric Wilcoxon-Mann-Whitney test. h, hours.
We analyzed three types of caRNAs with potential regulatory functions: promoter-associated RNA (paRNA), enhancer RNA (eRNA), andRNA transcribed from transposableelements (repeat RNA), which we termed chromosome-associated regulatory RNAs (carRNAs). The m6A levels of these carRNAs were markedly decreased in Mettl3 KO mESCs (Fig. 2B). Approximately 15 to 30% of all carRNAs contain m6A in mESCs, ~60% of which are regulated by METTL3 (fig. S3A). These m6A peaks contain GAC and AAC motifs, similar to those of the coding mRNAs (fig. S3, B and C). We categorized carRNAs into m6A-marked and non-m6A subgroups and found that the abundances of m6A-marked transcripts, but not non-m6A RNAs, were significantly elevated upon Mettl3 KO (Fig. 2C and fig. S3D). Additionally, changes in m6A levels negatively correlated with changes in expression levels for all three carRNA groups upon Mettl3 KO (fig. S3E). Together, these data suggest that m6A methylation destabilizes these carRNAs.
Previous work has uncovered that YTHDC1 associates with components of the nuclear exosome targeting (NEXT) complex, which is responsible for degradation of certain non-coding nuclear RNAs (18). We confirmed that YTHDC1 interacts with the NEXT components RBM7 and ZCCHC8 (fig. S4A). Because YTHDC1 is a known m6A reader and Ythdc1 CKO induced transcription up-regulation (fig. S1D), we hypothesized that YTHDC1 recognizes a subset of m6A-marked carRNAs and triggers their decay through NEXT in mESCs. Consistently, depletion of Ythdc1 or Zcchc8, but not of Ythdf2, increased the m6A/A ratio of caRNAs (fig. S4B). We subsequently performed MeRIP-seq of nonribosomal caRNAs and observed consistently increased m6A levels after Ythdc1 depletion (fig. S4, C to E). We identified more hypermethylated peaks in Ythdc1 CKO mESCs compared with controls (fig. S4F). The distribution of m6A peaks on mRNA was not notably altered (fig. S4G); however, the proportion of m6A peaks at intergenic regions increased upon Ythdc1 CKO (fig. S4H). These observations suggest that YTHDC1, like METTL3, affects caRNAs transcribed mostly from intergenic regions.
We examined carRNAs and observed markedly increased m6A for repeat RNAs upon YTHDC1 depletion (fig. S5A). Specifically, ~20 to 30% of m6A-marked paRNAs and eRNAs and >60% of m6A-marked repeat RNAs are affected by YTHDC1 depletion, indicating a main role of YTHDC1 in affecting the stability of repeat RNAs in mESCs (fig. S5B). These m6A peaks in different regions share similar motifs to those we detected previously (fig. S5, C and D). Moreover, we correlated m6A fold changes on three carRNA groups separately and observed distinct negative correlations in all cases between Mettl3 and Ythdc1 depletion (fig. S5E), which further indicates that YTHDC1 promotes decay of a portion of these carRNAs. We next performed nuclear RNA decay assays and observed notably increased half lifetimes for all three groups of carRNAs upon Ythdc1 CKO (Fig. 2D and fig. S5F). Moreover, the m6A-marked RNAs from all three carRNA groups showed greater increases in half lifetime compared with those of non-m6A RNAs after Ythdc1 CKO (Fig. 2E and fig. S5G).
We then ranked repeats families according to their m6A peak enrichment fold changes in response to Mettl3 or Ythdc1 depletion and identified the long interspersed element-1 (LINE1) family as one of the most responsive in both cases (fig. S6, A and B). LINE1 elements are the most abundant class of mouse retrotransposon, transcribed in early embryos, and they play critical roles in development—particularly in remodeling chromatin structure and regulating transcription (19, 20). We observed that m6A levels of each subfamily of LINE1 negatively correlate with their divergence: younger LINE1 contains higher m6A levels (fig. S6, C and D) and shows more significant methylation fold changes (fig. S6, E and F) upon Mettl3 or Ythdc1 depletion. We next verified that the decay of L1Md_F, a representative young subfamily of LINE1, is regulated by YTHDC1 and METTL3 in an m6A-dependent manner (supplementary text and figs. S6G and S7).
paRNAs, eRNAs, and repeat RNAs, such as LINEs, can regulate transcription by affecting chromatin architecture at corresponding genomic loci. Because Mettl3 KO increases both transcription and chromatin accessibility (Fig. 1), we next examined whether these changes are regulated by methylation of these carRNAs. We performed time-course RNA sequencing of nascent transcripts as well as total nuclear RNAs and conducted mammalian native elongating transcript sequencing (mNET-seq) (21, 22) in Mettl3 KO and WT mESCs. Both the global expression level (Fig. 3, A and B) and transcription rate (Fig. 3, C and D, and fig. S8, A and B) increased upon Mettl3 KO. Genes that were up-regulated in Mettl3 KO mESCs tended to have upstream carRNAs marked with m6A more frequently than those that were down-regulated (Fig. 3, A and B). Moreover, genes with m6A-marked upstream carRNAs attained higher increases in transcription rate than those with non-m6A–marked upstream carRNAs. The same is true when sorting genes with their precursor mRNAs (pre-mRNAs) not subjected to m6A methylation (Fig. 3, E to F, and fig. S8, C to E), which indicates that the reduced m6A methylation of these carRNAs upon Mettl3 KO activates the transcription of downstream genes.
Fig. 3. The m6A level of carRNAs affects downstream gene expression and transcription rate.
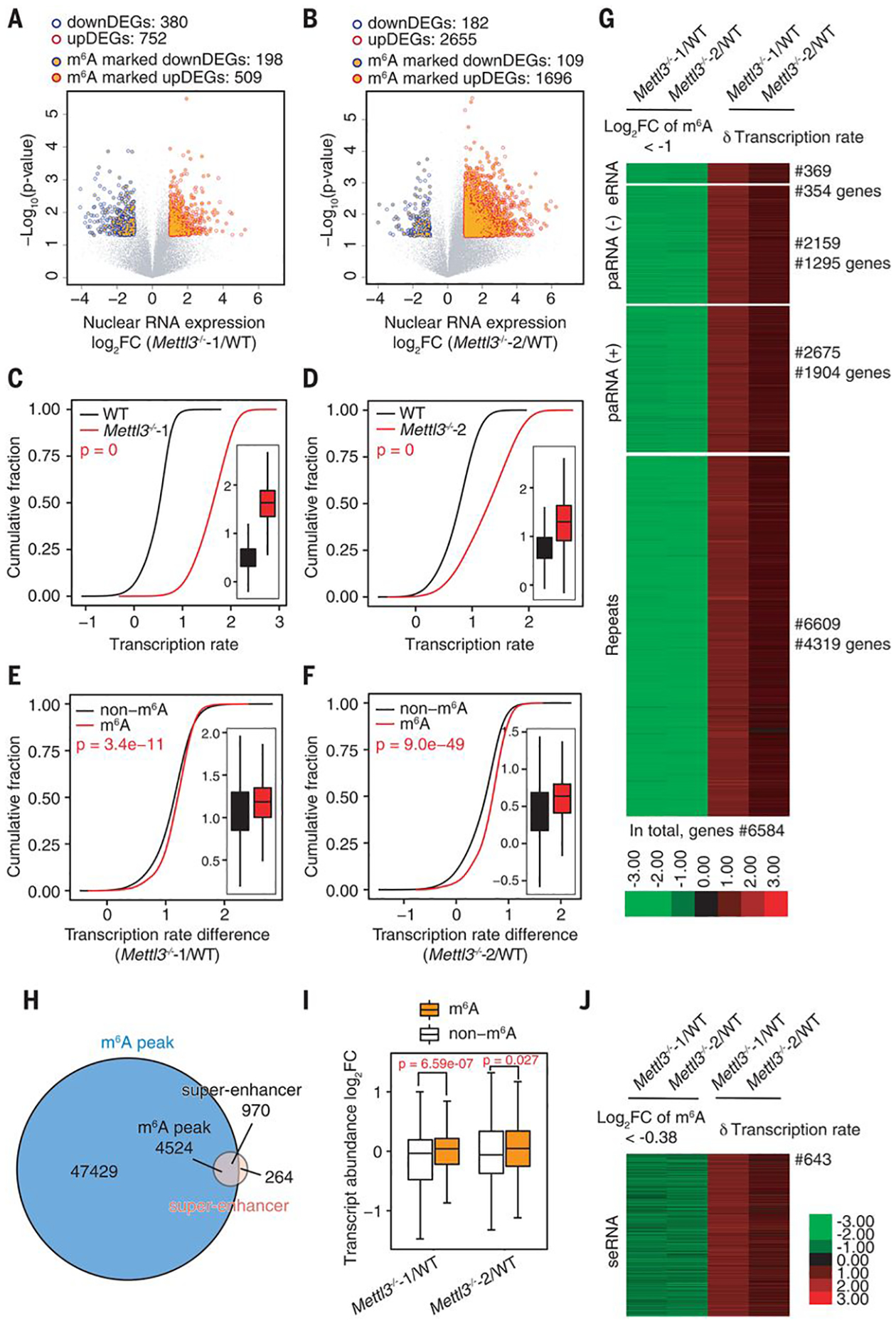
(A and B) Volcano plot of genes with differential expression levels in Mettl3−/−−1 (A) or Mettl3−/−−2 (B) versus WT mESCs [P < 0.05 and |log2FC| > 1 (FC, fold change)]. Genes with upstream, m6A-marked carRNAs are shown with orange circles. Gene expression level was normalized to ERCC spike-in with linear regression method. DEG, differentially expressed gene. (C and D) Cumulative distribution and boxplot (inside) of gene transcription rate in Mettl3−/−−1 (C) or Mettl3−/−−2 (D) versus WT mESCs. (E and F) Cumulative distribution and boxplot (inside) of transcription rate difference between Mettl3−/−−1 (E) or Mettl3−/−−2 (F) versus WT mESCs. Genes were categorized into two subgroups according to whether their upstream carRNAs contain m6A (m6A) or not (non-m6A). For (C) to (F), P values were calculated by a nonparametric Wilcoxon-Mann-Whitney test. (G) Heatmap showing the m6A level fold changes (log2FC < −1) on carRNAs and downstream gene transcription rate difference between Mettl3 KO and WT mESCs. (H) Venn diagram showing the overlap between the m6A peaks and super-enhancer peaks in mESCs. (I) Boxplot showing fold changes of the abundance of m6A-marked and non-m6A–marked seRNAs between Mettl3−/− and WT mESCs. For (A), (B), and (I), P values were determined by two-tailed t test. (J) Heatmap showing fold change (log2FC < −0.38) of m6A level of seRNAs and transcription rate difference of their downstream genes between Mettl3 KO and WT mESCs.
Notably, we found that all m6A-dependent genes that showed reduced upstream carRNA methylation upon Mettl3 depletion (~6584 genes) exhibited increased transcription rates (Fig. 3G). We identified a subset of these genes that demonstrated transcription rate differences >1 upon Mettl3 KO (fig. S9A) and found that these genes are mainly involved in transcription regulation, chromatin modification, and stem cell population maintenance (fig. S9B). Hence, the reduced m6A methylation of carRNAs not only promotes downstream transcription but may activate genes involved in chromatin opening, initiating a positive feedback loop. We further analyzed Prdm9, Kmt2d (encoding two H3K4me3 methyltransferases), Esrrb, and Ranbp17 (related with differentiation), all of which possess upstream carRNAs with reduced m6A level upon Mettl3 KO (fig. S10). Consistently, the half lifetime of these carRNAs and the transcription rate of their downstream genes both increased upon Mettl3 KO, and these changes could be rescued by WT but not mutant METTL3 (fig. S11).
The interactions between super-enhancers and their target genes are known to be affected by transcription of exosome-regulated transcripts in mESCs (23). We examined super-enhancers and found that ~80% of super-enhancer RNAs (seRNAs) contain m6A peaks (Fig. 3H and fig. S12A). The m6A methylation level of seRNAs decreased (fig. S12, B and C) and the m6A-marked seRNAs showed a greater increase in abundances compared with non-m6A–marked ones upon Mettl3 KO (Fig. 3I). seRNAs that showed reduced m6A level upon Mettl3 KO were associated with increased transcription rates at downstream genes (Fig. 3J); genes with transcription rate differences larger than one were mainly involved in transcription regulation, chromatin modification, and stem cell maintenance (fig. S12, D and E), consistent with results obtained from other carRNAs (fig. S9). Moreover, we found that genes regulated by m6A-marked upstream seRNAs tended to exhibit greater increases in transcription rate than those regulated by m6A-marked upstream typical eRNAs upon Mettl3 KO (fig. S12F).
We next investigated chromatin state changes affected by altered carRNA methylation. We performed chromatin immunoprecipitation sequencing (ChIP-seq) and observed global increases of these two active marks, H3K4me3 and H3K27ac, upon Mettl3 KO (fig. S13, A and B), consistent with the Western blot results (fig. S1H). Moreover, genes with m6A-marked upstream carRNAs showed greater increases in H3K4me3 and H3K27ac than genes with non-m6A upstream carRNAs in Mettl3 KO mESCs (Fig. 4A). Likewise, Mettl3 KO mESCs showed obvious increases in both marks at regions that flank METTL3-enriched DNA loci (fig. S13, C and D). These data suggested that stabilizing the upstream carRNAs in Mettl3 KO mESCs could increase the deposition of active histone marks.
Fig. 4. The m6A level of carRNAs affects local chromatin state and downstream transcription.
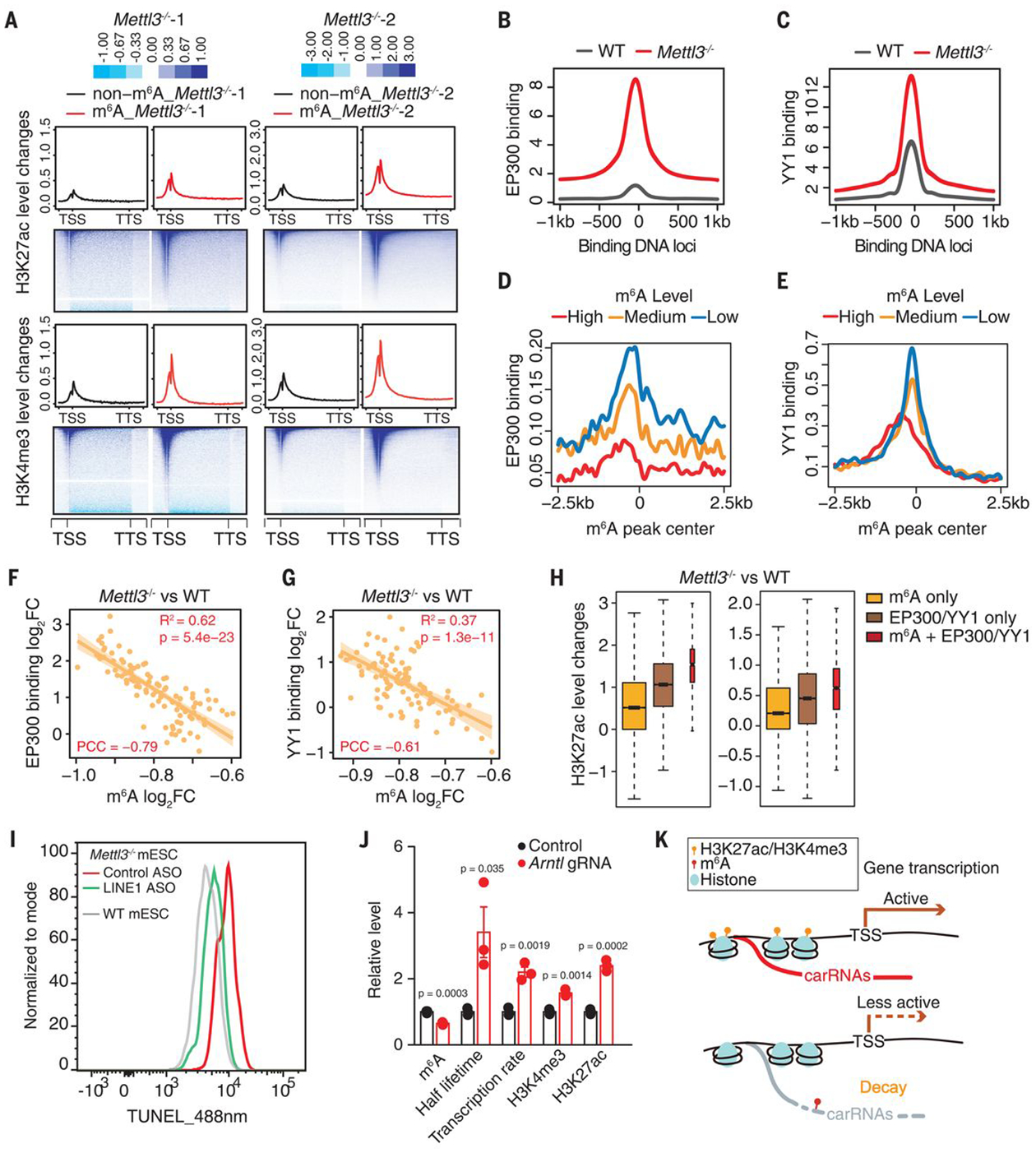
(A) Profiles of H3K27ac (top) and H3K4me3 (bottom) level changes on gene body together with 2.5 kb upstream of the TSS (transcription start site) and 2.5 kb downstream of the TTS (transcription termination site) in WT and Mettl3 KO mESCs. Genes were categorized into two groups according to whether they harbor upstream m6A-marked carRNAs (m6A) or not (non-m6A). (B and C) Profiles of EP300 (B) or YY1 (C) DNA binding at their peak center and flanking 2.5 kb regions in WT and Mettl3 KO mESCs. (D and E) Profiles of EP300 (D) and YY1 (E) DNA binding at the center of m6A peaks overlapped with carRNAs and its flanking 2.5 kb regions in WT mESCs. m6A peaks were categorized into highly (high), moderately (medium), or lowly (low) methylated groups, according to their m6A levels in WT mESCs. (F and G) The correlation between changes in m6A level of the carRNAs and changes in EP300 (F) or YY1 (G) DNA binding at genomic regions that show m6A differences with Mettl3 KO. The genomic regions were categorized into 100 bins on the basis of fold change rank of m6A level upon Mettl3 KO. (H) Barplots showing H3K27ac level changes at genomic regions that are m6A methylated (m6A only, without EP300 and YY1 binding), bound by EP300 or YY1 (EP300/YY1 only, without m6A carRNA), and m6A methylated with EP300 and YY1 binding (m6A + EP300/YY1). The last group showed the highest increase upon Mettl3 KO. (I) Analysis of chromatin accessibility in Mettl3 KO mESCs treated with control or LINE1 antisense oligos (ASOs). DNase I–treated TUNEL assay was performed. (J) A dCas13b-FTO (WT or inactive mutant) construct with gRNA targeting the seRNA of Arntl was used to reduce the m6A level of Arntl seRNA. After treatment, increased half lifetime of the target seRNA, elevated local H3K27ac and H3K4me3 levels, and increased Arntl transcription rate were observed, accompanied by the decreased seRNA m6A level. (K) A schematic model showing how m6A affects transcription by regulating the decay of upstream carRNAs stability and chromatin state.
We suspect that carRNAs could recruit proteins, such as CBP/EP300 and YY1, to promote open chromatin and activate transcription (24, 25). In fact, YY1 was identified as one of the top enriched transcription factors (TFs) at genomic regions that harbor m6A-marked carRNAs (fig. S14A). Our ChIP-seq experiments revealed global increases of EP300 and YY1 binding in Mettl3 KO mESCs (Fig. 4, B and C), which correlated well with higher nearby eRNAs or paRNAs abundances (fig. S14, B and C). Moreover, both EP300 and YY1 binding negatively correlate with the m6A level of nearby carRNAs (Fig. 4, D and E), and Mettl3 KO leads to elevated EP300 and YY1 binding at regions that lose m6A (Fig. 4, F and G). The genomic regions with both m6A-marked carRNAs and binding of EP300 or YY1 showed the greatest increase in H3K27ac compared with regions with just m6A or just EP300/YY1 binding upon Mettl3 KO (Fig. 4H). We also performed ChIP-seq of JARID2, a component of the polycomb repressive complex 2 (PRC2), because RNA transcripts could repel PRC2 binding to maintain chromatin openness (26). We observed a globally decreased JARID2 binding, correlating well with the abundance increases of eRNAs and repeats transcripts upon Mettl3 KO (fig. S14, D and E). Furthermore, JARID2 tends to bind to regions with high m6A methylation of carRNAs (fig. S14F), with a positive correlation between the m6A level on carRNA and local JARID2 binding changes upon Mettl3 KO (fig. S14G). Therefore, these m6A-regulated carRNAs may stabilize the open chromatin state by not only recruiting active TFs but also repelling repressive factors, such as PRC2, upon loss of methylation.
Lastly, elevated LINE1 may also modulate global chromatin accessibility (19). We blocked LINE1 RNA in Mettl3−/− mESCs and observed an overall reduction in chromatin accessibility (Fig. 4I) (20). We employed a fused CRISPR-dCas13b system (27) with either WT or inactive mutant FTO (fig. S15A). Only when targeting LINE1 by guide RNA (gRNA) with dCas13b-wt FTO, but not dCas13b-mu FTO, did we observe decreased m6A level and increased half lifetime for LINE1, together with a globally increased chromatin accessibility (fig. S15, B to E). We then applied the dCas13b-FTO system to genes that harbor m6A-marked upstream carRNAs (fig. S16). When targeting carRNAs using gRNAs and dCas13b-wt FTO, we observed site-specific methylation reduction, with little methylation change observed using dCas13b-mu FTO or negative control gRNAs (Fig. 4J and fig. S17). We also observed increased half lifetime of these carRNAs, up-regulation of downstream gene transcription, and elevated local H3K4me3 and H3K27ac levels (Fig. 4J and fig. S18). Together, these results strongly support our discovery that m6A methylation of carRNAs controls carRNA stability and downstream gene transcription.
To explore functional relevance of this m6A-mediated regulation, we modulated the LINE1 RNA level in WT and Mettl3 KO mESCs. LINE1 abundance was elevated in Mettl3 KO mESCs (fig. S19A) (11). Blocking LINE1 elevated differentiation capacity and decreased cell renewal in Mettl3 KO mESCs (fig. S19, B to D). In contrast, targeting LINE1 using gRNA with dCas13b-wt FTO resulted in decreased differentiation capacity and increased cell renewal in WT mESCs but not with Cas13b-mu FTO (fig. S19, B to D). We also confirmed regulatory functions of m6A-marked carRNAs in endometrial cancer progression, in which down-regulation of METTL3 increases cell proliferation, migration, and tumor growth (supplementary text and figs. S20 to S23) (28).
In this work, we report that carRNAs can be m6A methylated by METTL3. A subset of these m6A-marked carRNAs (mainly LINE1 repeats) is destabilized by YTHDC1 via the NEXT complex. We show that m6A serves as a switch to affect abundances of these carRNAs, thus tuning nearby chromatin state and downstream transcription (Fig. 4K). Effects of m6A methylation on carRNAs could vary; m6A may stabilize modified carRNAs in different cell types. In mESCs, the transcription activation induced by m6A depletion is coupled with the increased chromatin accessibility, enrichment of certain TFs, and elevated histone marks, revealing a direct cross-talk between carRNA m6A methylation and chromatin state. Our findings demonstrate an additional layer of regulatory effect of carRNA m6A on transcription.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Chang for Mettl3 KO mESCs and J. Tauler and A. Andersen from Life Science Editor for editing.
Funding: This study was supported by the National Institute of Health HG008935 to C.H.; Strategic Priority Research Program XDA16010506 to D.H.; National Key R&D Program of China 2018YFA0109700 to D.H.; National Institute of Health ES030546 to C.H.; National Key R&D Program of China 2016YFA0100400 to Y.G. and Chuan C.; the China Scholarship Council (CSC) for the visit of Y.G. to the University of Chicago; National Science Fund for Excellent Young Scholars 31922017 to D.H.; and the CAS Hundred Talent Program to D.H. The Mass Spectrometry Facility of the University of Chicago is funded by the National Science Foundation (CHE-1048528). C.H. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: C.H. is a scientific founder and a member of the scientific advisory board of Accent Therapeutics, Inc.
Data and materials availability: All sequencing data have been deposited in Gene Expression Omnibus (GSE133600 and GSE140561). All other data are available in the manuscript or the supplementary materials.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/367/6477/580/suppl/DC1
Materials and Methods
Supplementary Text
Figs. S1 to S23
Tables S1 and S2
References (29–44)
REFERENCES AND NOTES
- 1.Dominissini D et al. , Nature 485, 201–206 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Meyer KD et al. , Cell 149, 1635–1646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frye M, Harada BT, Behm M, He C, Science 361, 1346–1349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roundtree IA, Evans ME, Pan T, He C, Cell 169, 1187–1200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jia G et al. , Nat. Chem. Biol 7, 885–887 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng G et al. , Mol. Cell 49, 18–29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X et al. , Nature 505, 117–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X et al. , Cell 161, 1388–1399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao W et al. , Mol. Cell 61, 507–519 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Batista PJ et al. , Cell Stem Cell 15, 707–719 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geula S et al. , Science 347, 1002–1006 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Wang Y et al. , Nat. Cell Biol 16, 191–198 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivanova I et al. , Mol. Cell 67, 1059–1067.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasowitz SD et al. , PLOS Genet. 14, e1007412 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y et al. , Nat. Neurosci 21, 195–206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbieri I et al. , Nature 552, 126–131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slobodin B et al. , Cell 169, 326–337.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lubas M et al. , Mol. Cell 43, 624–637 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Jachowicz JW et al. , Nat. Genet 49, 1502–1510 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Percharde M et al. , Cell 174, 391–405.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayer A et al. , Cell 161, 541–554 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nojima T et al. , Cell 161, 526–540 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pefanis E et al. , Cell 161, 774–789 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bose DA et al. , Cell 168, 135–149.e22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sigova AA et al. , Science 350, 978–981 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X et al. , Nat. Struct. Mol. Biol 24, 1028–1038 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rauch S, He C, Dickinson BC, J. Am. Chem. Soc 140, 11974–11981 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J et al. , Nat. Cell Biol 20, 1074–1083 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.