

Abstract
A convenient two-step method is reported for the ligation of alkoxyamine- or hydrazine- bearing cargo to proline N-termini. Using this approach, bifunctional proline N-terminal bionconjugates are constructed and proline N-terminal proteins are immobilized.
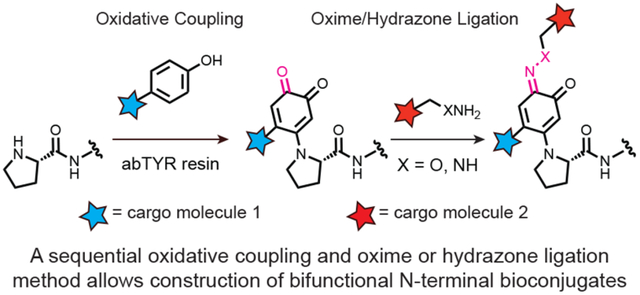
Graphical Abstract
Protein bioconjugates are an important class of biomaterials that harness the exquisite and diverse biochemical properties of proteins in practical contexts. These unique materials have wide-ranging applications, including the attachment of proteins to surfaces,1 the study and imaging of proteins in whole cells,2 and the delivery of cytotoxic payloads to malignant cell types.3 Common to all protein bioconjugates is the requirement to maintain proper protein folding, and thus function. In order to achieve this, chemistries that modify proteins must be performed at reduced temperatures (4 – 37 °C), in buffered aqueous solutions (pH 5 – 9), and with favourable kinetics so that large amounts of reagent are not required.4 The modification of nucleophilic groups in proteins relies on chemistries that meet these requirements. However, these reactions commonly suffer from poor site-selectivity, resulting in heterogenous products that are modified to different levels and in multiple sites. Notable and useful exceptions to this situation are modifications targeting cysteine residues and protein N-termini.5
Over the years, a variety of chemical strategies have been developed to target chemically distinct groups site-selectively. One of the earliest reported chemoselective reactions for protein modification involves the condensation of alkoxyamines and hydrazines with ketone and aldehyde moieties.6 Here, oxidative cleavage of N-terminal serine or threonine residues using NaIO4 selectively installs a ketone residue on the protein’s N-terminus (Fig 1a, top arrow). This ketone can then be modified covalently using an alkoxyamine- or hydrazine-containing reagent, producing an oxime or hydrazone linkage, respectively. In addition, our lab has focused on developing an array of pyridoxal phosphate-like reagents capable of transaminating a variety of N-terminal amino acids to afford the corresponding ketones for subsequent oxime or hydrazone ligation (Fig 1a, bottom arrow).7
Figure 1.
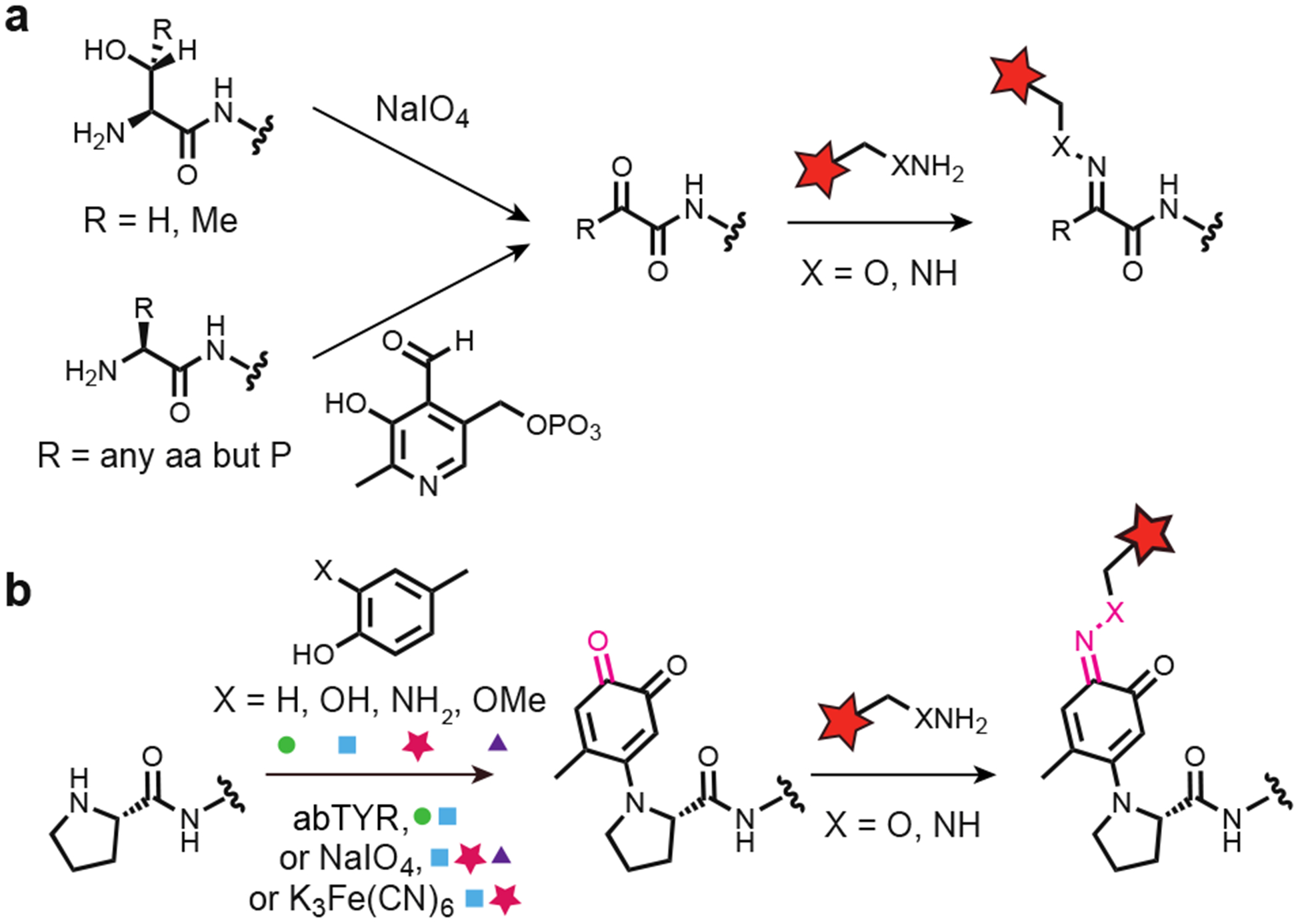
Chemoselective oxime and hydrazone formation for bioconjugation reactions using endogenous N-termini. (a) N-terminal ketones can be formed either via an oxidative cleavage of an N-terminal ser or thr using sodium periodate, or via a transamination reaction between the native N-terminus and PLP analogues. These reactive ketones readily undergo condensation reactions with either alkoxyamine- or hydrazine-bearing cargo, to form oxime and hydrazone linkages, respectively. (b) Reaction of a pro N-terminus via the oxidative coupling results in a product bearing a single ketone. This carbonyl could subsequently be reacted with an alkoxyamine or hydrazine, extending oxime and hydrazone formation to pro N-termini.
Over the last decade, our lab has also developed a series of oxidative coupling reactions that site-selectively modify proteins.8 In these reactions, a variety of phenolic compounds are oxidized to o-quinoid intermediates, which readily undergo reaction with proline N-termini to afford site-specific and covalent products (Fig 1b). A key feature of this chemistry is the variety of conditions that can be used to access the necessary o-quinoid intermediates, including the oxidation of aminophenols and catechols using K3Fe(CN)6 and the oxidation of these same phenolic compounds plus methoxyphenols with NaIO4.8 In addition, proline N-termini are easy to install in recombinant proteins expressed in bacterial and mammalian systems. Here, endogenous methionine aminopeptidases will completely remove the initial N-terminal methionine residue if it is followed by a small amino acid, like a proline, resulting in an easy to install reactive handle.9
More recently, we have shown that the enzyme tyrosinase from Agaricus bisporus (abTYR) can also convert catechols and phenols, which are far more synthetically tractable, to the same o-quinoid intermediates for N-terminal proline modification, as confirmed via protein sequencing.10 Regardless of the method used, all of the oxidative coupling conditions afford a proline covalently modified with an o-quinoid adduct.
Herein, we show that oxidative coupling at N-terminal proline sites installs a new ketone handle that can be used for subsequent condensation with alkoxyamines or hydrazines. We also show that this chemistry allows for the construction of bifunctional N-terminal bioconjugates by first installing primary cargo via the oxidative coupling and secondary cargo via oxime or hydrazone ligation, all the while remaining chemically orthogonal to cysteine residues. This opens the door for the synthesis of complex protein bioconjugates using endogenous amino acids as multiple reactive positions in a protein can be functionalized with cargo. Overall, this approach represents a useful two-step method for the site-selective installation of multiple cargo groups onto proteins using endogenous amino acids and further extends the applicability of multi-step protein bioconjugate synthesis.
Our initial studies exploring the electrophilicity of the o-quinone-modified proline adduct showed modest conversion (~50%) when exposed to 10 mM benzylalkoxyamine.10 To increase conversion, we switched to methoxyamine, which more closely resembles the commonly employed alkyl alkoxyamines for oxime ligation. Incubation of 10 μM o-quinone modified pro-sfGFP with 5 mM CH3ONH2 in 50 mM pH 5.0 phosphate buffer overnight resulted in complete conversion to the expected oxime product (expected MW = 27724, Fig 2, middle trace). To assess the impact of the oxime modification protocol on protein yield, a BCA assay was used to monitor protein concentration at each step of the reaction. Compared to a negative control that was simply incubated in the reaction buffers at each step, no appreciable protein loss due to modification was observed (SI Fig S1).
Figure 2.
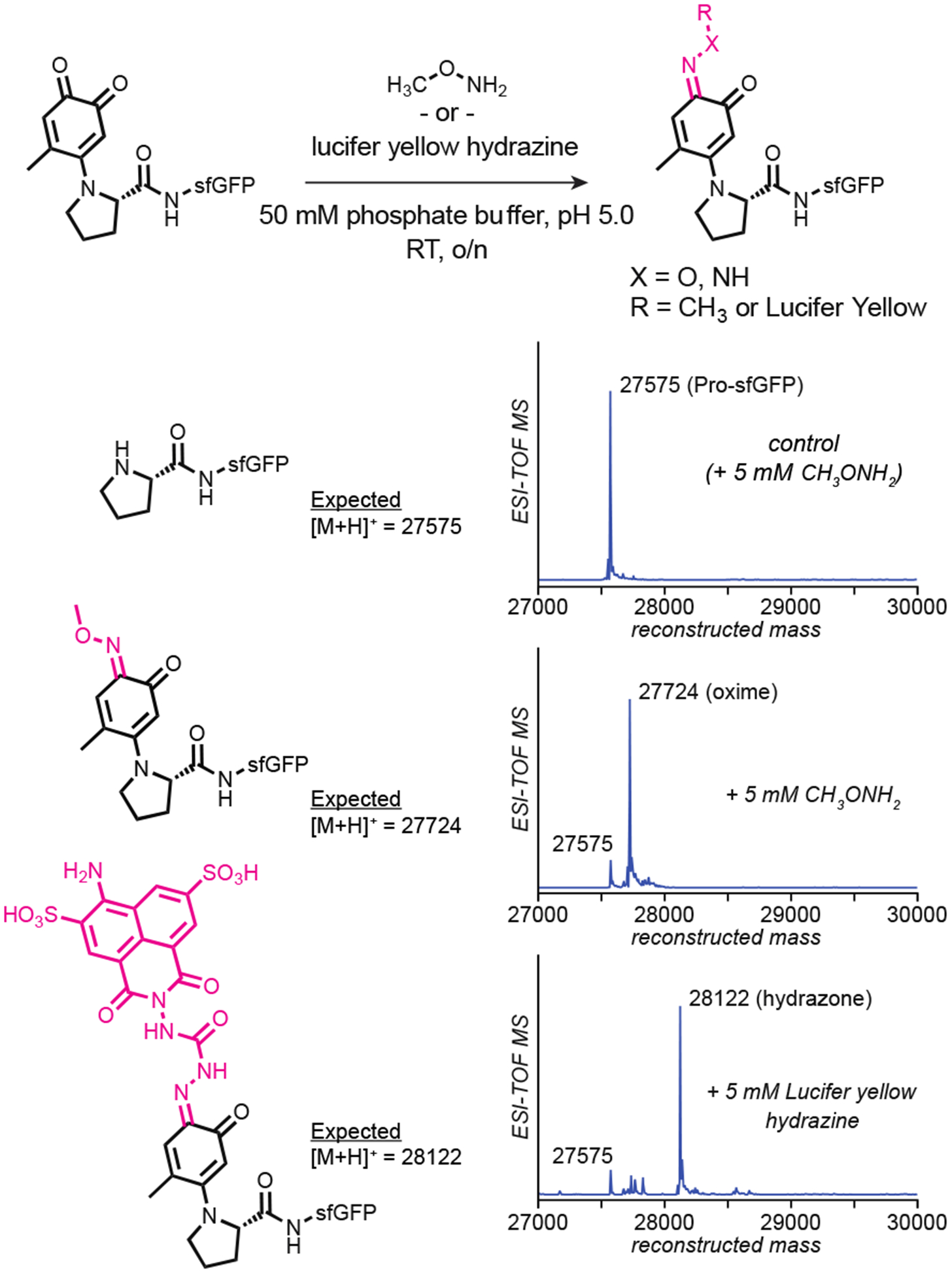
Chemoselective oxime and hydrazone formtion on oxidative coupling-modified proline N-termini. A variety of alkoxyamine and hydrazine containing compounds were reacted with a 4-methyl-o-quinone-pro N-terminus, acheiving complete conversion to the expected singly modified oxime or hydrazone product. Unmodified pro-sfGFP did not react as no ketone moiety was present on the protein.
As a control, a solution of 10 μM pro-sfGFP was exposed to the same conditions. This resulted in no modification (expected MW = 27575, Fig 2, top trace). The chromophore of GFP features a highly conjugated carbonyl that could be envisioned to react despite being inside the protein. This result indicates that it was the covalent installation of the o-quinoid adduct to the proline N-terminus that was necessary for oxime formation, and not the chromophore itself.
We also explored the potential of the previously reported p-phenylenediamine (pPDA) covalent catalyst to accelerate oxime formation on the o-quinone-modified proline N-terminus.11 We found that the addition of 10 mM pPDA can accelerate the attachment of CH3ONH2 (at 1 mM) to o-quinone-pro-sfGFP (at 10 μM) in 50 mM pH 5.0 phosphate buffer, leading to full conversion in under 4 h (SI Fig S2). Once again, a control reaction was run on unmodified pro-sfGFP using the same conditions. We observed no modification in this case. Further studies explored oxime and hydrazone ligation in the absence of catalyst, but note that it could be advantageous for applications that require it.
The oxidative coupling reaction works best for proline residues at the N-terminus; although we have observed modest conversion with other primary amine N-termini, we do not recommend their use in this transformation.8 It is also important to note that free cysteine residues need to be blocked using a disulphide exchange reagent, like Ellman’s reagent, to prevent them from participating in the reaction. The protected thiol can then be deprotected using a mild reductant such as TCEP, as shown previously.10
Next, we sought to install hydrazine-bearing cargo onto o-quinone-modified proline N-termini. Lucifer yellow hydrazine was chosen as a model substrate owing to its low price and use as a fluorescent probe. Employing the same conditions used with our oxime test reactions, we found that lucifer yellow hydrazine quantitatively converts o-quinone-pro-sfGFP to the expected hydrazone product (expected MW = 28122, Fig 2, bottom trace). This represents a convenient method for the site-selective installation of fluorophores onto proteins.
Oxime and hydrazone linkages are known to reverse under aqueous conditions with varying rates.12 We tested the stability of the oxime and hydrazone products formed from oxidatively-modified proline N-termini in 20 mM phosphate buffer at pH 7.2 for 24 h in various conditions. No apparent reversal was observed for the products in temperatures ranging from 4 to 37 °C over this time period (SI Fig S3). Oxidatively-modified proline N-termini had previously been shown to undergo addition with small molecule thiols, like reduced glutathione (GSH).10 Interestingly, incubation of 10 mM GSH with the oxime product showed no such addition, although some reversal to the o-quinone-pro-sfGFP starting material and addition into this electrophile were observed. The hydrazone product was completely stable toward 10 mM GSH and showed no reversal or addition (SI Fig S3).
Many applications will require removal of the tyrosinase enzyme from the coupling products. To facilitate this, a 2 mg/mL solution of abTYR was tethered through its surface exposed primary amines to NHS-activated sepharose.13 Initial tests showed that the resin-bound abTYR performed as well as it did in solution, with the added advantage that the bound enzyme could be removed readily from the oxidative coupling reactions using a 0.2 μm centrifugal filter (SI Fig S4 and S5). Additional tests showed that immobilizing the enzyme improved its storability, while simultaneously allowing recycling of the enzyme for multiple reactions (SI Fig S6). Immobilized tyrosinase was used for the remaining experiments described herein.
To explore the protein substrate scope, we installed o-quinoid adducts onto a variety of proline N-terminal proteins, including the chitin-binding domain of Pyrococcus furiosus (pro-CBD), the commercially available enzyme aldolase (pro-Ald), and a recombinantly engineered variant of the tobacco mosaic virus bearing an N-terminal proline (pro-TMV). The resin-bound abTYR successfully installed the o-quinone adduct onto the proline N-termini in all three cases. Subsequent incubation of these o-quinone-modified proline N-termini with 5 mM lucifer yellow hydrazine afforded the desired products (SI Figure S7). Ortho-quinone-modified pro-CBD and pro-Ald showed complete conversion to the hydrazone products. Ortho-quinone-pro-TMV showed some hydrazone formation, but protein degradation was observed through an unknown pathway. Overall, however, this approach shows promise in being a convenient method for N-terminal chromophore installation on a variety of proline N-termini.
Additionally, we verified that o-quinone-modified pro-Ald maintained activity after modification with methylalkoxyamine. NADH production was measured via its absorbance at 340 nm when either unmodified or oxime-modified pro-Ald was incubated with fructose-bisphosphate and NAD+. No differences in product formation were observed between the modified pro-Ald and unmodified control, indicating that the dual-modification at the proline N-terminus does not hinder enzymatic activity (SI Fig S8).
This two-step approach to oxime or hydrazone formation also represents a new way to construct bifunctional N-terminal proline bioconjugates. In this case, an oxidative coupling reaction can first be used to install a useful cargo group, while simultaneously introducing a ketone handle for further functionalization via oxime or hydrazone ligation (Fig 3a). This strategy is particularly amenable to modification at protein N-termini, as these are often solvent accessible in a majority of proteins and therefore minimize reductions to the rate of bioconjugation.14 If necessary, this accessibility can be improved via the addition of linker groups that minimize both protein perturbations and steric impediments to the reaction.
Figure 3.
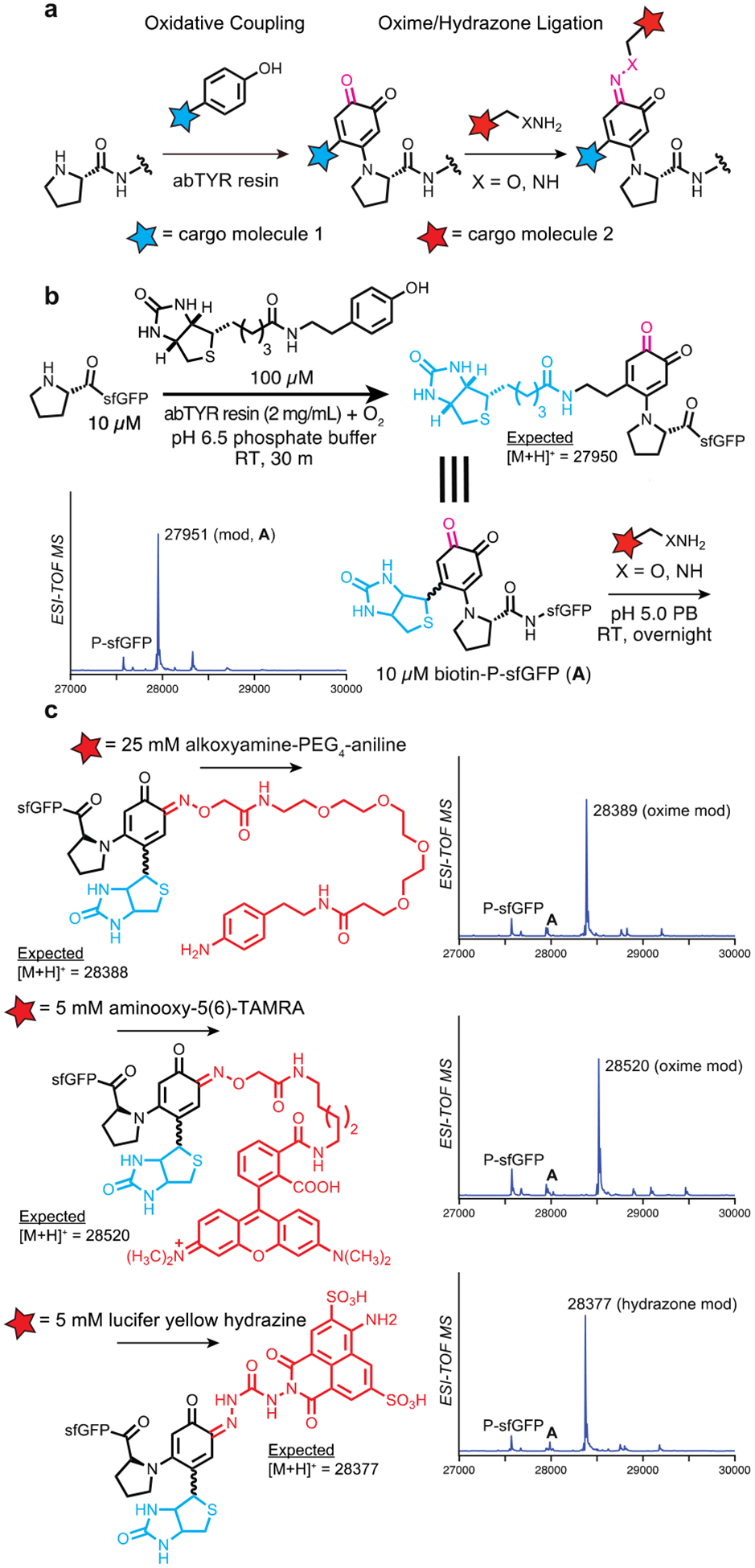
Construction of bifunctional proline N-terminal bioconjugates using a sequentional oxidative coupling and oxime/hydrazone ligation, (a) Using an initial oxidative coupling reaction, primary cargo can be installed at the proline N-terminus. Subsequent oxime or hydrazone ligation can then be used to install secondary cargo at the N-terminus using the oxidative coupling product, (b) Synthesis of N-terminally biotinylated pro-sfGFP A was carried out using abTYR resin and a biotin-phenol, installing both ketone and biotin moieties at the proline N-terminus. (c) Secondary cargo installation was performed on product A using an alkoxyamine-aniline, alkoxyamine-TAMRA, and a lucifer yellow hydrazine. All reagents afforded near-complete conversion to the expected products when incubated with A overnight.
This two-step labelling approach was demonstrated using a biotin-bearing o-quinone on the proline N-terminus of sfGFP. Site-selective protein biotinylation is a common approach for the immobilization of proteins onto streptavidin modified surfaces. When combined with site-selective chromophore-labelling, typically on engineered thiol residues, surface-bound biotinylated-proteins are a convenient means to study protein folding and enzyme function via FRET.15 However, controlling both the stoichiometry and placement of both functional groups for such applications can be challenging.
Using the abTYR resin and a previously reported biotin-phenol, we successfully biotinylated the N-terminus of pro-sfGFP (Fig 3b). The o-quinoid adduct on pro-sfGFP then underwent a secondary modification via oxime ligation. Using an alkoxyamine-PEG4-aniline, we were able to install an aniline moiety onto the biotinylated-proline N-terminus, converting biotinylated A to the expected product with near quantitative yield (MW = 28389, Fig 3c). In addition, biotinylated A readily coupled with common dyes using 5 mM of either aminooxy-5(6)-TAMRA or lucifer yellow hydrazine and the conditions reported above (MW = 28520 and 28377, respectively, Fig 3c). Together, these results demonstrate the ease and applicability of this two-step method for the multistep synthesis of protein bioconjugates bearing multiple functional cargo molecules.
This sequential coupling approach can also be applied for the immobilization of o-quinone-modified proline N-termini. To demonstrate this, we first converted primary amine PEGA resin to an alkoxyamine resin (Fig 4a). We then verified that the o-quinoid-proline N-terminus was capable of coupling to the resulting polymer support. Briefly, 50 μM of o-quinone-pro-sfGFP (OQP-sfGFP) was incubated with ~10 μL of alkoxyamine resin in 50 mM pH 5.0 phosphate buffer. As controls, 50 μM pro-sfGFP was incubated with the alkoxyamine resin and 50 μM OQP-sfGFP was incubated with the initial primary amine PEGA resin. After incubation at room temperature overnight, the resins were washed with 20 mM pH 7.2 phosphate buffer and imaged with a fluorescence microscope. Covalent labelling of the alkoxyamine-PEGA beads was only seen when pro-sfGFP was previously functionalized with an o-quinone (Fig 4b). Together, these results represent a convenient, site-selective immobilization strategy for proteins.
Figure 4.
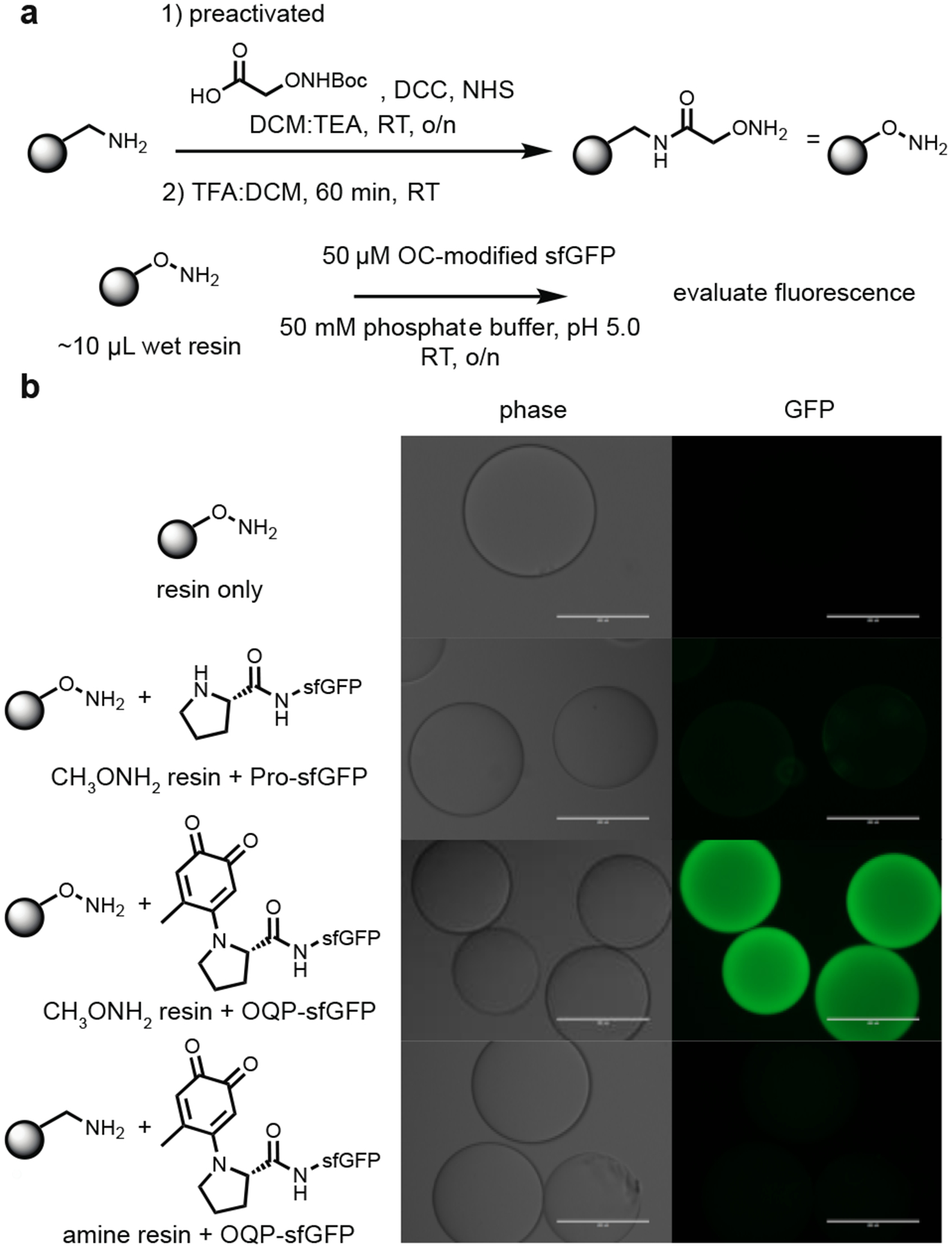
Immobilization of oxidative coupling-modified pro-sfGFP. (a) Alkoxyamine-PEGA resin was synthesized using an NHS-activated Boc-aminooxyacetic acid. Subsequent deprotection using 50% TFA:DCM afforded the desired alkoxyamine functionalized resin. (b) Alkoxyamine-PEGA resin was incubated with pro-sfGFP and o-quinone-pro-sfGFP (OQP-sfGFP). Successful conjugation of the sfGFP to the alkoxyamine resin only occurred when the sfGFP had previously been modified through oxidative coupling. As an additional control, unmodified primary amine PEGA resin was incubated with OQP-sfGFP. No conjugation was observed. Bars denote 200 μm.
In this report, we have demonstrated the utility of ketone groups introduced onto proline N-termini in a mild manner via enzymatic oxidative coupling reactions. Subsequent modification of these groups with alkoxyamine or hydrazine-containing compounds can be used to generate highly functionalized protein bioconjugates site-specifically. Overall, this method extends the scope of oxime and hydrazone ligation to include proline N-termini and furthers the examples of multistep synthesis for the construction of complex protein bioconjugates.
Supplementary Material
Acknowledgements
This work was supported by the National Science Foundation (CHE 1808189), as well as the Chemical Biology Graduate Program at UC Berkeley (NIH T32-GM066698). J.C.M. was supported by a UC Berkeley Fellowship for Graduate Studies and the NSFGRFP. Entry vectors used in this paper were a gift from the Danielle Tullman-Ercek lab at Northwestern University. Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA is acknowledged for additional financial support and helpful discussions.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Hutsell SQ, Kimple RJ, Siderovski DP, Willard FS and Kimple AJ, Methods Mol Biology Clifton N J, 2010, 627, 75–90; [DOI] [PMC free article] [PubMed] [Google Scholar]; Malmqvist M and Karlsson R, Curr Opin Chem Biol, 1997, 1, 378–383. [DOI] [PubMed] [Google Scholar]
- 2.O’Hare HM, Johnsson K and Gautier A, Curr Opin Struc Biol, 2007, 17, 488–494; [DOI] [PubMed] [Google Scholar]; Krall N, da Cruz FP, Boutureira O and Bernardes GJ, Nature Chemistry, 2016, 8, 103–113. [DOI] [PubMed] [Google Scholar]
- 3.Alley SC, Okeley NM and Senter PD, Curr Opin Chem Biol, 2010, 14, 529–537; [DOI] [PubMed] [Google Scholar]; Chudasama V, Maruani A and Caddick S, Nat Chem, 2016, 8, nchem.2415. [DOI] [PubMed] [Google Scholar]
- 4.Sletten EM and Bertozzi CR, Angewandte Chemie Int Ed, 2009, 48, 6974–6998; [DOI] [PMC free article] [PubMed] [Google Scholar]; Rosen CB and Francis MB, Nature Chemical Biology, 2017, 13, 697–705. [DOI] [PubMed] [Google Scholar]
- 5.Baslé E, Joubert N and Pucheault M, Chem Biol, 2010, 17, 213–227. [DOI] [PubMed] [Google Scholar]
- 6.Geoghegan KF and Stroh JG, Bioconjugate Chem, 1992, 3, 138–146. [DOI] [PubMed] [Google Scholar]
- 7.Witus LS, Netirojjanakul C, Palla KS, Muehl EM, Weng C-H, Iavarone AT and Francis MB, J Am Chem Soc, 2013, 135, 17223–17229; [DOI] [PMC free article] [PubMed] [Google Scholar]; Palla KS, Witus LS, Mackenzie KJ, Netirojjanakul C and Francis MB, J Am Chem Soc, 137, 1123–1129. [DOI] [PubMed] [Google Scholar]
- 8.Obermeyer AC, Jarman JB and Francis MB, Journal of the American Chemical Society, 2014, 136, 9572–9; [DOI] [PMC free article] [PubMed] [Google Scholar]; ElSohly AM, MacDonald J, Hentzen NB, Aanei I, Muslemany KM and Francis MB, J Am Chem Soc, 2017, 139, 3767–3773. [DOI] [PubMed] [Google Scholar]
- 9.Hirel, Schmitter M, Dessen P, Fayat G and Blanquet S, Proceedings of the National Academy of Sciences, 1989, 86, 8247–8251; [DOI] [PMC free article] [PubMed] [Google Scholar]; Xiao Q, Zhang F, Nacev BA, Liu JO and Pei D, Biochemistry-us, 2010, 49, 5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maza J, Bader DL, Xiao L, Marmelstein AM, Brauer DD, ElSohly AM, Smith MJ, Krska SW, Parish CA and Francis MB, J Am Chem Soc, 2019, 141, 3885–3892. [DOI] [PubMed] [Google Scholar]
- 11.Wendeler M, Grinberg L, Wang X, Dawson PE and Baca M, Bioconjugate Chem, 2014, 25, 93–101; [DOI] [PubMed] [Google Scholar]; Dirksen A, Hackeng TM and Dawson PE, Angewandte Chemie Int Ed, 2006, 45, 7581–7584. [DOI] [PubMed] [Google Scholar]
- 12.Kalia J and Raines RT, Angewandte Chemie (International ed. in English), 2008, 47, 7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Oliveira K, Mischiatti K, Fontana J and de Oliveira B, J Chromatogr B, 2014, 945, 10–16; [DOI] [PubMed] [Google Scholar]; Lasmi K, Derder H, Kermad A, Sam S, Boukhalfa-Abib H, Belhousse S, Tighilt F, Hamdani K and Gabouze N, Appl Surf Sci, 2018, 446, 3–9. [Google Scholar]
- 14.Jacob E and Unger R, Bioinformatics, 2007, 23, e225–e230. [DOI] [PubMed] [Google Scholar]
- 15.Wilson DS and Nock S, Curr Opin Chem Biol, 2002, 6, 81–85; [DOI] [PubMed] [Google Scholar]; Groll J, Amirgoulova EV, Ameringer T, Heyes CD, Röcker C, Nienhaus UG and Möller M, J Am Chem Soc, 2004, 126, 4234–4239; [DOI] [PubMed] [Google Scholar]; Chung H, Louis JM and Eaton WA, Biophys J, 2010, 98, 696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.