

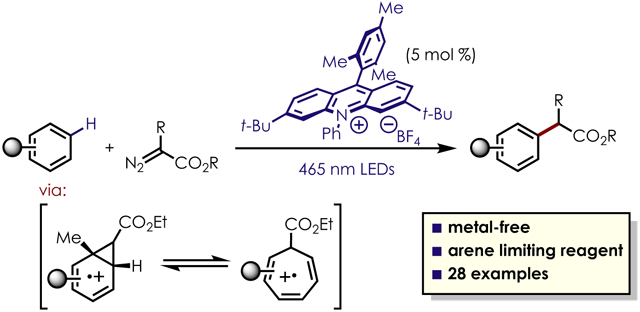
Abstract
Expanding the toolbox of C–H functionalization reactions applicable to the late stage modification of complex molecules is of interest in medicinal chemistry, wherein the preparation of structural variants of known pharmacophores is a key strategy for drug development. One manifold for the functionalization of aromatic molecules utilizes diazo-compounds and a transition metal catalyst to generate a metallocarbene species, which is capable of direct insertion into an aromatic C–H bond. However, these high energy intermediates can often requiring directing groups or large excess of substrate to achieve efficient and selective reactivity. Herein, we report that arene cation radicals generated via organic photoredox catalysis engage in formal C–H functionalization reactions with diazoacetate derivatives, furnishing sp2–sp3 coupled products with moderate to good regioselectivity. In contrast to previous methods utilizing metallocarbene intermediates, this transformation does not proceed via a carbene intermediate, nor requires the presence of a transition metal catalyst.
Keywords: Organic Chemistry, Methodology and Reactions, Alkylation, Arenes, Organic Photoredox Catalysis, Diazo Compounds
Graphical Abstract
INTRODUCTION
Metallocarbenes are reactive organometallic species which may be generated using a transition metal precursor and diazoacetate derivatives.[1–3] The resulting carbene intermediate may then undergo C-H insertion with reactive C-H bonds (often aryl or alkyl), affording the corresponding alkylated adducts. However, due to the high reactivity of metallocarbenes, aromatic substrates are often required to be present in excess for efficient reactivity.
Among methodologies based on metallocarbene C-H insertion that do not use several-fold excess of arene, directing groups are often required. Fox[4] and Wang[5] have reported ortho-selective C–H functionalization of heterocycles using diazoacetates. In addition, other directing groups such as purines,[6] azacycles,[7] disubstituted anilines,[8] and phenols,[9] have been utilized in metallocarbene C–H alkylation reactions. Collectively, these directing groups limit the synthetic applicability of metallocarbene C-H alkylation reactions.
Recently, Suero and coworkers reported the synthesis of aryl diazoacetate derivatives under photoredox conditions.[10] During the course of this work, it was noted that photolysis of aryldiazoacetates with blue LEDs yields free carbene species that undergo cyclopropanation reactions in the presence of styrene. Subsequently, Davies and coworkers investigated the reactivity of similar photogenerated free carbenes in combination with electron-rich aromatic substrates and alkenes.[11] In these cases, either cyclopropanation or formal C–H insertion was observed. However, the scope of this transformation is specific to aryl diazoacetates and a small number of arene coupling partners (which must be present in excess). This is largely due to the highly reactive nature of free carbenes, which readily undergo C-H insertion reactions with acidic C-H, O-H, and N-H bonds. The products of reaction with solvent are also typically observed in these cases. Due to the reactive nature of carbenes, methodologies that rely on their intermediacy can be incompatible with certain functional groups and low levels of regiocontrol are often observed.
In recent years, photoredox catalysis has served as the conceptual basis for a number of powerful aromatic C–H functionalization reactions.[12–14] Our group has previously reported arene C–H amination and cyanation methodologies which utilize acridinium salts as organic photoredox catalysts.[15,16] These methods rely on the catalytic generation of aromatic cation radicals via photoinduced electron transfer (PET) between the aromatic substrate and the excited acridinium catalyst. We sought to probe the reactivity of nucleophilic diazoacetates in combination with electrophilic aromatic cation radicals, as reactions of this type have not been investigated to date.
Such transformations would be reminiscent of known metallocarbenoid insertion reactions, while taking place in the absence of metals or superstoichiometric quantities of substrate. Furthermore, previous work from our group has shown moderate to excellent regioselectivity in nucleophilic additions to aromatic cation radicals. The observation of comparable levels of regioselectivity in the addition of diazoacetate derivatives would represent a major improvement over current aromatic C–H functionalization methodologies utilizing metallocarbene/carbene intermediates (Scheme 1). Such a reaction would also be mechanistically distinct from recent work published by Gryko which relies on the generation and alkylation of alpha-keto radicals from diazoacetate derivatives using photoredox catalysis.[17]
Scheme 1.
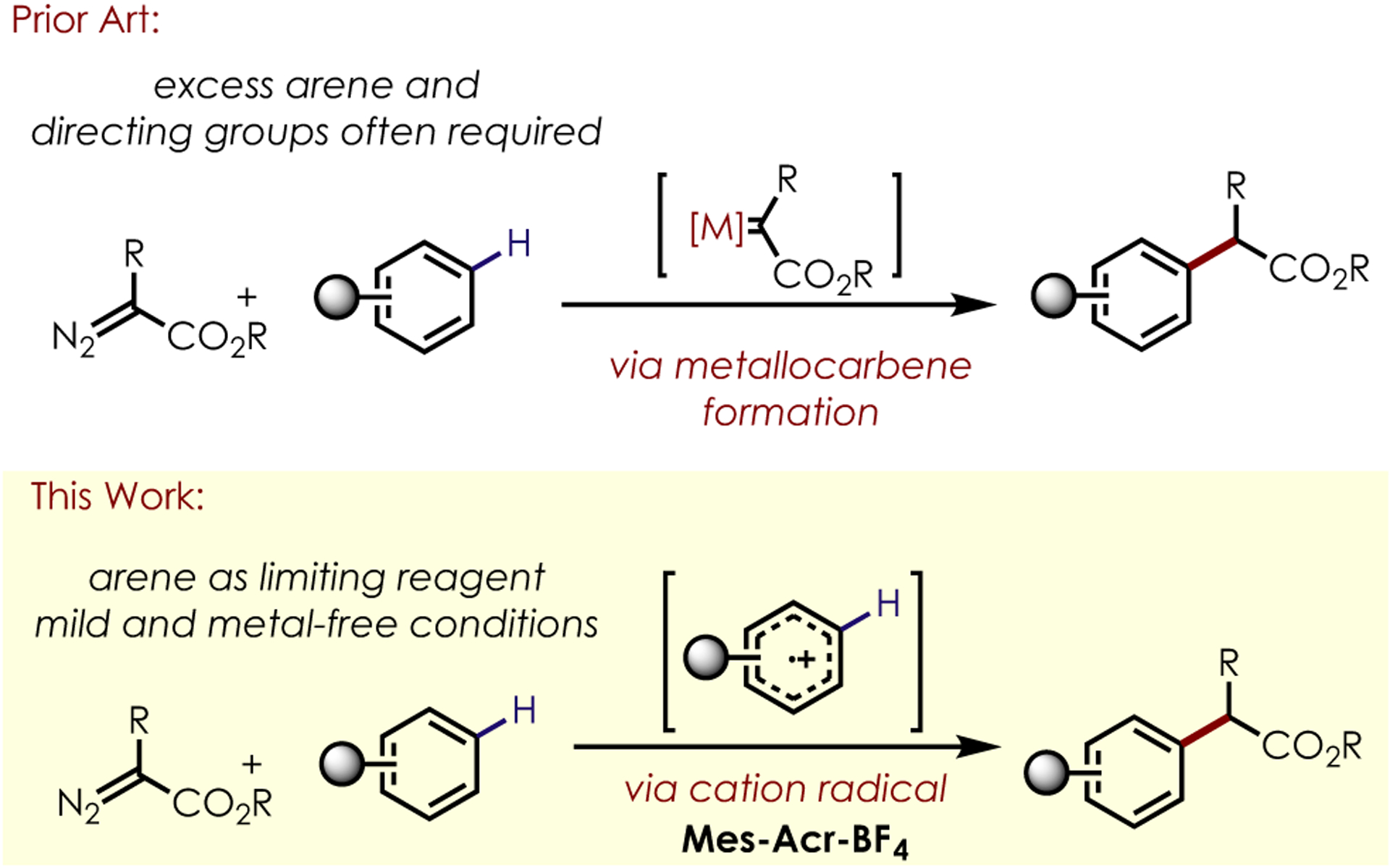
C-H alkylation via metallocarbenes or photoredox catalysis
Here we report a method for C–H functionalization reactions of arene cation radicals with diazoacetate derivatives generated via organic photoredox catalysis. Computational and experimental studies support a unique mechanism involving cation radical-meditated aromatic cyclopropanation followed by oxidative ring opening. These results demonstrate the utility of arene cation radicals as versatile intermediates for the construction of carbon-carbon bonds via organic photoredox catalysis.
RESULTS AND DISCUSSION
Reaction development began using mesitylene as a substrate and ethyl diazoacetate as a nucleophile in the presence of 5 mol% Mes-Acr-BF4 under irradiation from blue LEDs. Following a screen of common solvents, a 1:1 mixture of trifluoroethanol (TFE) and acetonitrile was identified as a suitable medium for the reaction. With one equivalent of ethyl diazoacetate in a 1:1 TFE/acetonitrile solvent mixture, the desired C–H functionalized product 1a was isolated in 76% yield. In some cases, secondary alkylation of the desired product was observed as a minor byproduct at increased ethyl diazoacetate loadings (>2.0 eq.). Increasing diazoacetate loadings between 1.0 – 2.0 eq. did not result in a significant increase in yield, presumably due to competitive oxidation of the product of the reaction.
With optimized conditions in hand, the scope of the transformation with respect to both the arene and diazoacetate partners was explored. Simple aromatic substrates and electron-rich anisole derived substrates smoothly undergo the desired transformation in poor to good yields, tolerating synthetically useful cross-coupling functional handles such as aryl chlorides (6a, 8a), bromides (7a), and tosylates (14a). Pharmacologically-relevant heterocycles were competent substrates for the reaction, with 2,6-dimethoxypyridine and N-methylindazole furnishing the desired adducts, 16a and 17a with high regioselectivity, albeit in reduced yield. Tyrosine derivatives containing protected amines, acidic C–H bonds and benzylic hydrogens underwent the desired transformation selectively, yielding derivatives 19a and 21a in poor to moderate yield. This method was also applicable to the selective functionalization of drug-like motifs. Under the optimized conditions, flurbiprofen methyl ester and fenoprofen methyl ester furnished the desired products, 20a and 22a respectively with moderate to good regioselectivity, despite relatively low chemical yield. Functionalization ortho- to electron donating groups is observed in most cases, with para- selectivity favored in the presence of increased steric hinderance (8–14a). In biaryl systems, functionalization is favored on the more electron-rich ring system (4a, 8a). The mass balance for these reactions typically consists exclusively of returned starting material and C–H functionalized product.
Next, the scope of this transformation with respect to the diazo compound coupling partner was explored. This reaction proceeded efficiently with simple diazoesters possessing tert-butyl, benzyl and cyclopentyl diazoacetates, affording the desired adducts 1e, 1c, and 1d in good to moderate yields. Cyclic diazodihydrofuranone furnished product 1b in 61% yield. Less nucleophilic methyl diazomalonate formed product 1f, albeit with reduced yield. Following C-H alkylation, the resulting ester products can be converted into formal C-H methylation adducts via a simple hydrolysis/decarboxylation sequence, utilizing a photoredox hydrodecarboxylation method previously developed by our group.[18] The installation of tert-butyl, benzyl-, and malonate esters are also valuable due to the number of possible further transformations, which can be used to elaborate these products. For example, benzyl esters can be cleaved to the corresponding acid[19], tert-butyl esters can be converted to an acid chloride[20] and malonate derivatives can be used in the malonic ester synthesis to give substituted acids.[21]
To probe the mechanism of this transformation, we conducted a series of experimental and computational investigations. Based on previous studies on the reaction of diazoacetate derivatives with alkene cation radicals, we initially hypothesized that following nucleophilic addition to the aromatic cation radical, electron transfer from the reduced form of the catalyst to the generated cyclohexadienyl radical could trigger a 1,2-hydride shift and expulsion of N2, leading to the formation of the observed C–H functionalization product (IV, Figure 2). Such a mechanism would be reminiscent of that proposed for the cyclopropanation of alkene cation radicals by Ferreira and coworkers.[22] When utilizing d3-Mesitylene as a substrate, no deuterium incorporation was observed in the C–H alkylation product, suggesting that a 1,2-hydride shift is an unlikely step in the mechanism (II, Figure 2). When d1-TFE is used as the reaction solvent, a mixture of non-deuterated, mono-deuterated and di-deuterated products is observed with the mono-deuterated formed as the major product in 72% yield. The presence of non-deuterated and di-deuterated products in the presence of the singly labeled product was rationalized as the result of enolate equilibration. Treatment of the proteo-product with d1-TFE did not lead to any observable deuterium incorporation. This is consistent with a mechanism invoking the protonation of an enolate by the solvent as part of the mechanism.
Figure 2:
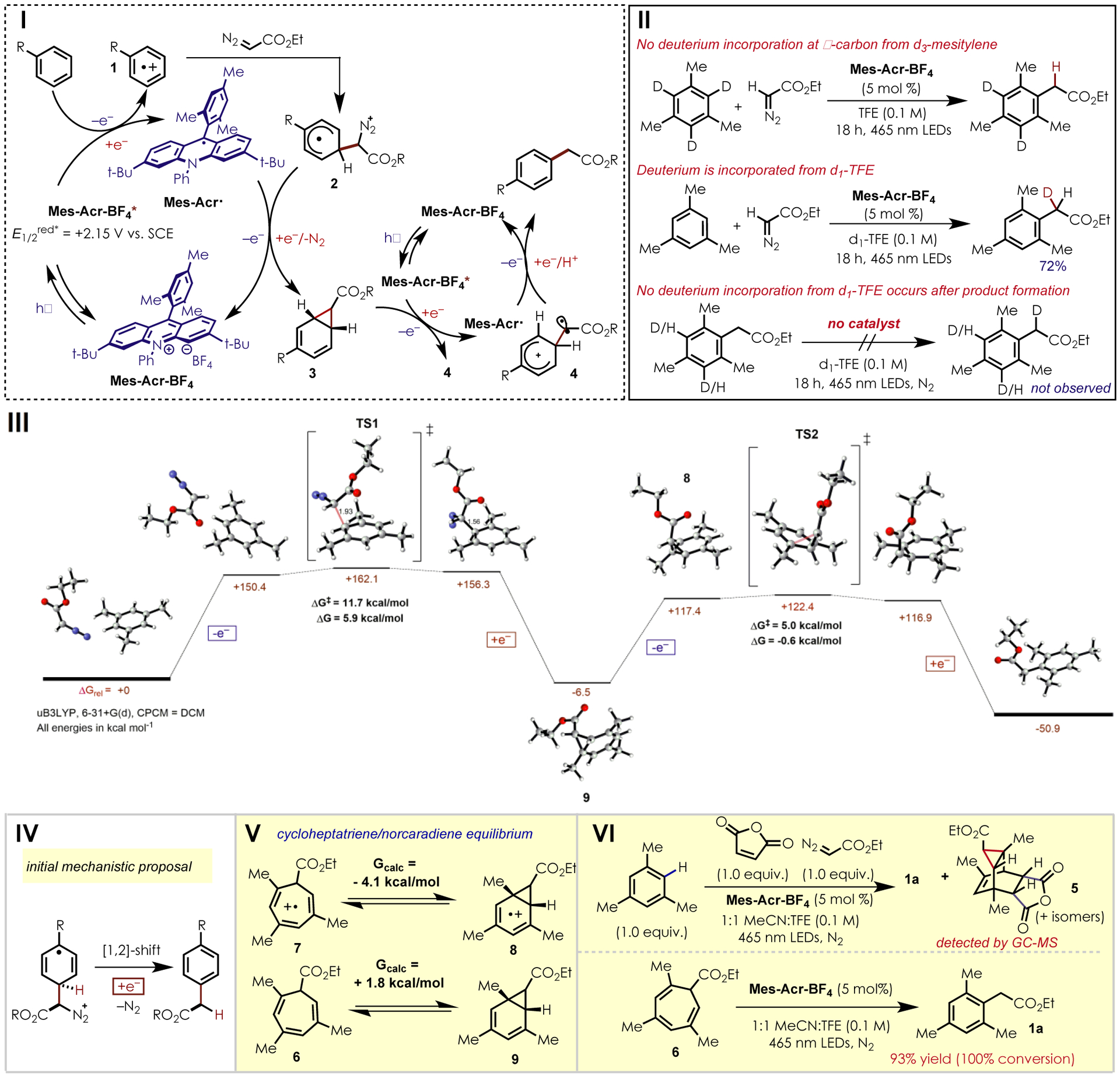
Investigation of the mechanism of an organic photoredox C-H alkylation. I: Current mechanistic proposal. II: Deuterium labeling studies with d3-mesitylene and d1-TFE. III: Reaction energy diagram for an arene cation radical cyclopropanation. IV: Initial mechanistic proposal involving a 1,2-hydride shift. V: Thermodynamics of the norcaradiene and cycloheptatriene equilibrium. VI: Evidence for a norcaradiene intermediate.
Based on this evidence, we propose an alternative mechanism as follows: following excitation by blue light, Mes-Acr-BF4* engages in photoinduced electron transfer (PET) with an arene, producing cation radical 1 and the reduced form of the catalyst (Mes-Acr·). Polar nucleophilic addition of ethyl diazoacetate forms distonic cation radical 2, which is reduced by acridine radical (Mes-Acr·) to form the intermediate norcaradiene 3 and regenerate ground state Mes-Acr-BF4. Following a second PET event, intermediate 3 undergoes ring-opening to form distonic benzylic radical cation 4. Reduction of this intermediate by Mes-Acr·, deprotonation (presumably by conjugate base of the solvent) to rearomitize the product, and protonation of the resulting enolate by the solvent closes the second catalytic cycle.
To investigate the energetic feasibility of the proposed mechanism, the reaction between mesitylene and ethyl diazoacetate was modeled using DFT (B3LYP, 6–31+g(d)) calculations (see SI for complete computational details). Following oxidation of mesitylene, ethyl diazoacetate undergoes nucleophilic addition to the arene cation radical with a transition state energy of +11.7 kcal/mol and an overall free energy change of +5.9 kcal/mol. The calculated endergonicity of this process is in agreement with the known equilibrium between arene cation radical/nucleophile pairs and the corresponding σ-adducts. [23,24] Following single electron transfer from the reduced form of the catalyst to the σ-adduct, a barrierless loss of N2 and cyclization forms norcaradiene 9 with an overall free energy change of -6.5 kcal/mol relative to the starting materials. A second single electron transfer event generates cation radical intermediate 8 which then undergoes ring opening to form a distonic cation radical. This process has a calculated transition state energy of +5.0 kcal/mol and an overall free energy change of -0.6 kcal/mol. Following electron transfer from the reduced catalyst and proton transfer, the desired C–H alkylation product is formed. The free energy change for this overall process was calculated to be -50.9 kcal/mol.
Norcaradienes are known to exist as equilibrium mixtures with the corresponding cycloheptatrienes.[25,26] To further explore the impact of this equilibrium in our system, the free energy change for the interconversion of these species was modeled for intermediates 6 and 7 (V, Figure 2). In the neutral species, the cycloheptatriene form (6) is favored by 1.8 kcal/mol. However, upon oxidation of this species, the equilibrium was calculated to favor norcaradiene cation radical 8 by 4.1 kcal/mol. This suggests that, under the reaction conditions, the equilibrium between norcaradiene and cycloheptriene may be driven towards products via electron transfer.
To support this proposal, we subjected cycloheptatriene 6 to our optimized conditions (VI, Figure 2). Following irradiation for 18 h, full conversion of 6 to the desired product 1a was observed. Control experiments indicated that the presence of Mes-Acr-BF4 is required for conversion. A mass corresponding to the desired [4+2] adduct (5) was observed by GC-MS upon the inclusion of maleic anhydride (1 equiv.) as a Diels-Alder trap under otherwise optimized conditions, supporting the formation of norcaradiene 9 during the reaction (VI, Figure 2). Diazoacetates bearing aryl substituents, which are known to form free carbenes upon irradiation with blue light, gave solely the product resulting from O-H insertion of trifluoroethanol into the corresponding carbene. Ethyl diazoacetate, which absorbs at 371 nm with no overlap at 465 nm (SI 35), shows no formation of these O-H insertion byproducts. This suggests that under these reaction conditions a carbene is not formed and instead ethyl diazoacetate behaves as a polar nucleophile. Control experiments also indicated that the blue LEDs used for this reaction are unable to trigger photolysis of ethyl diazoacetate in a similar manner to aryl diazoacetates. This is in accordance with other experimental and computational data in support of the proposed mechanism given above.
CONCLUSION
In conclusion, we have developed an operationally simple, metal-free site-selective aromatic C–H alkylation which utilizes equimolar quantities of coupling partners, unlike known metallocarbene chemistry which requires excess arene substrate to achieve efficient reactivity. This transformation is compatible with a range of aromatic substrates, including those containing known pharmaceutical motifs and cross-coupling handles. Experimental and computational studies suggest a unique mechanism involving cyclopropanation of arene cation radicals and subsequent oxidative ring opening. This work represents both a fundamental insight into the reactivity of aromatic cation radicals and a valuable approach for the formation of sp3–sp2 bonds through a C–H functionalization manifold in the absence of transition metal catalysts.
Supplementary Material
Figure 1:
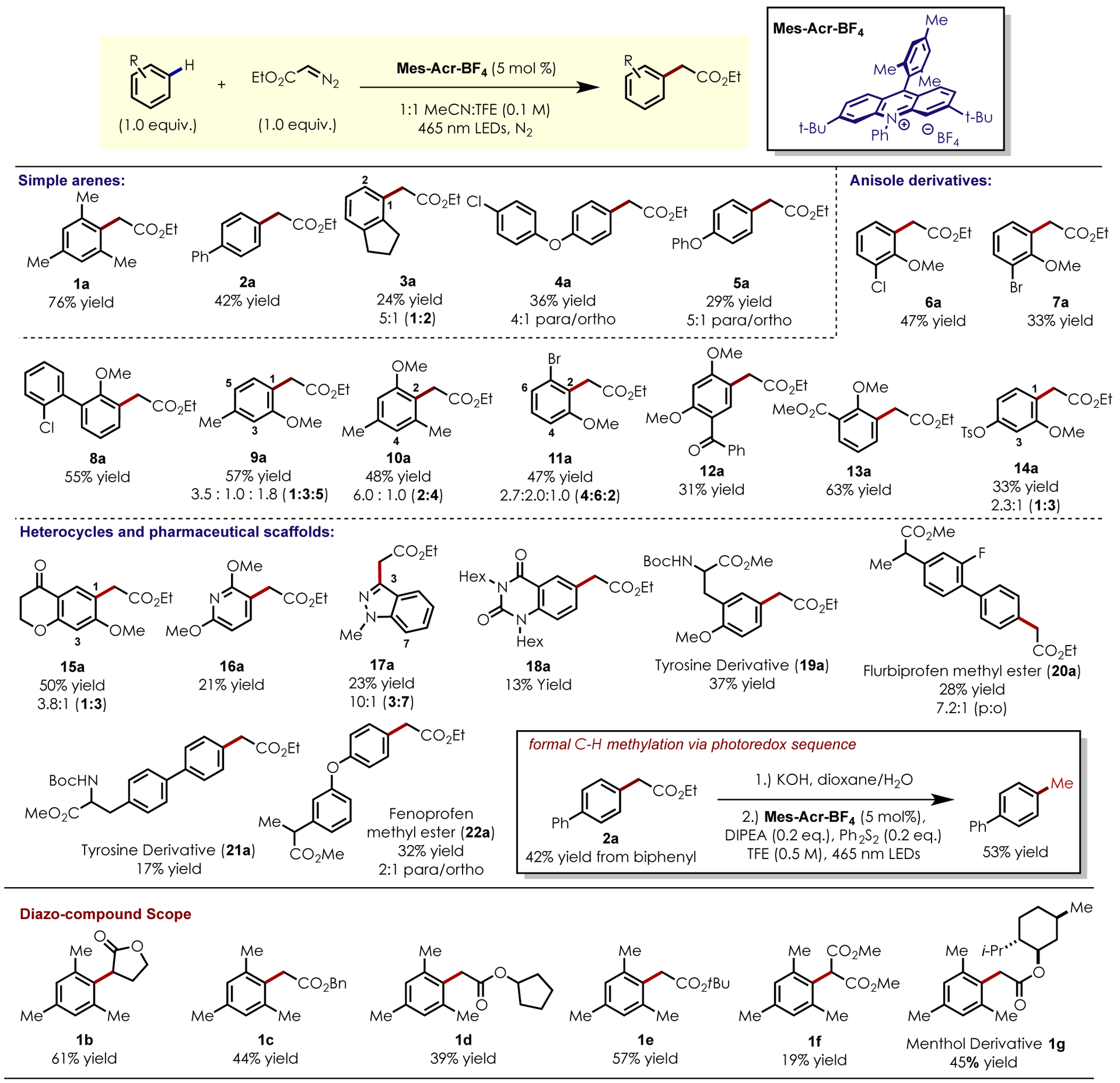
Scope of photoredox-catalyzed C-H alkylation
Acknowledgements
This work was supported by the National Institutes of Health (NIGMS) Award No. R01 GM120186. Photophysical measurements were performed in the AMPED EFRC Instrumentation Facility established by the Alliance for Molecular PhotoElectrode Design for Solar Fuels (AMPED), an Energy Frontier Research Center (EFRC) funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0001011. The authors would additionally like to thank the Johnson laboratory at UNC-Chapel Hill for their generous donation of several diazo-compounds.
REFERENCES
- [1].Yu Z, Li Y, Zhang P, Liu L, Zhang J, Chem. Sci 2019, 10, 6553–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Conde A, Sabenya G, Rodríguez M, Postils V, Luis JM, Díaz-Requejo MM, Costas M, Pérez PJ, Angew. Chem. Int. Ed 2016, 55, 6530–6534. [DOI] [PubMed] [Google Scholar]
- [3].Doyle MP, Duffy R, Ratnikov M, Zhou L, Chem. Rev 2010, 110, 704–724. [DOI] [PubMed] [Google Scholar]
- [4].DeAngelis A, Shurtleff VW, Dmitrenko O, Fox JM, J. Am. Chem. Soc 2011, 133, 1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hu F, Xia Y, Ma C, Zhang Y, Wang J, Chem. Commun 2015, 51, 7986–7995. [DOI] [PubMed] [Google Scholar]
- [6].Vorobyeva DV, Vinogradov MM, Nelyubina YV, Loginov DA, Peregudov AS, Osipov SN, Org. Biomol. Chem 2018, 16, 2966–2974. [DOI] [PubMed] [Google Scholar]
- [7].Yu X, Yu S, Xiao J, Wan B, Li X, J. Org. Chem 2013, 78, 5444–5452. [DOI] [PubMed] [Google Scholar]
- [8].Tayama E, Yanaki T, Iwamoto H, Hasegawa E, Eur. J. Org. Chem 2010, 2010, 6719–6721. [Google Scholar]
- [9].Yu Z, Ma B, Chen M, Wu H-H, Liu L, Zhang J, J. Am. Chem. Soc 2014, 136, 6904–6907. [DOI] [PubMed] [Google Scholar]
- [10].Wang Z, Herraiz AG, del Hoyo AM, Suero MG, Nature 2018, 554, 86–91. [DOI] [PubMed] [Google Scholar]
- [11].Jurberg ID, Davies HML, Chem. Sci 2018, 9, 5112–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brachet E, Ghosh T, Ghosh I, König B, Chem. Sci 2015, 6, 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hari DP, Schroll P, König B, J. Am. Chem. Soc 2012, 134, 2958–2961. [DOI] [PubMed] [Google Scholar]
- [14].Margrey KA, Levens A, Nicewicz DA, Angew. Chem. Int. Ed Engl 2017, 56, 15644–15648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Romero NA, Margrey KA, Tay NE, Nicewicz DA, Science 2015, 349, 1326–1330. [DOI] [PubMed] [Google Scholar]
- [16].McManus JB, Nicewicz DA, J. Am. Chem. Soc 2017, 139, 2880–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ciszewski ŁW, Durka J, Gryko D, Org. Lett 2019, 21, 7028–7032. [DOI] [PubMed] [Google Scholar]
- [18].Griffin JD, Zeller MA, Nicewicz DA, J. Am. Chem. Soc 2015, DOI 10.1021/jacs.5b07770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mandal PK, McMurray JS, J. Org. Chem 2007, 72, 6599–6601. [DOI] [PubMed] [Google Scholar]
- [20].Greenberg JA, Sammakia T, J. Org. Chem 2017, 82, 3245–3251. [DOI] [PubMed] [Google Scholar]
- [21].Cope AC, Holmes HL, House O, in Org. React, 1957, pp. 107–331. [Google Scholar]
- [22].Sarabia FJ, Ferreira EM, Org. Lett 2017, 19, 2865–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hammerich O, Parker VD, in Adv. Phys. Org. Chem, Elsevier, 1984, pp. 55–189. [Google Scholar]
- [24].Parker VD, Acc. Chem. Res 1984, 17, 243–250. [Google Scholar]
- [25].Mackay WD, Johnson JS, Org. Lett 2016, 18, 536–539. [DOI] [PubMed] [Google Scholar]
- [26].Maier G, Angew. Chem. Int. Ed. Engl 1967, 6, 402–413. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.