

Abstract
In recent years, an abundance of new molecular structures have been elucidated using cryo-electron microscopy (cryo-EM), largely due to advances in hardware technology and data processing techniques. Owing to these new exciting developments, cryo-EM was selected by Nature Methods as Method of the Year 2015, and the Nobel Prize in Chemistry 2017 was awarded to three pioneers in the field.
The main goal of this article is to introduce the challenging and exciting computational tasks involved in reconstructing 3-D molecular structures by cryo-EM. Determining molecular structures requires a wide range of computational tools in a variety of fields, including signal processing, estimation and detection theory, high-dimensional statistics, convex and non-convex optimization, spectral algorithms, dimensionality reduction, and machine learning. The tools from these fields must be adapted to work under exceptionally challenging conditions, including extreme noise levels, the presence of missing data, and massively large datasets as large as several Terabytes.
In addition, we present two statistical models: multi-reference alignment and multi-target detection, that abstract away much of the intricacies of cryo-EM, while retaining some of its essential features. Based on these abstractions, we discuss some recent intriguing results in the mathematical theory of cryo-EM, and delineate relations with group theory, invariant theory, and information theory.
I. Introduction
Structural biology studies the structure and dynamics of macromolecules in order to broaden our knowledge about the mechanisms of life and impact the drug discovery process. Owing to recent ground-breaking developments, chiefly in hardware technologies and data processing techniques, an abundance of new molecular structures have been elucidated to near-atomic resolutions using cryo-electron microscopy (cryo-EM) [44], [47], [8], [81], [52]. Cryo-EM was selected by Nature Methods as Method of the Year 2015, and the Nobel Prize in Chemistry 2017 was awarded to Jacques Dubochet, Joachim Frank and Richard Henderson “for developing cryoelectron microscopy for the high-resolution structure determination of biomolecules in solution1.”
In a cryo-EM experiment, biological macromolecules suspended in a liquid solution are rapidly frozen into a thin ice layer. The 3-D location and orientation of particles within the ice are random and unknown. An electron beam then passes through the sample and a 2-D tomographic projection, called micrograph, is recorded. The goal is to reconstruct a high-resolution estimate of the 3-D electrostatic potential of the molecule (in particular, its atomic structure) from a set of micrographs. The resolution measures the smallest detail that is distinguishable in a recovered 3-D structure; structures with better resolutions resolve finer features. For example, at resolutions of 9Å α-helices are resolved; at resolutions of 4.8Å individual β-strands are resolved; at resolutions of 3.5Å many amino acids side-chains are resolved [8]. Figure 1 shows a gallery of important biomedical structures solved by cryo-EM at increasingly higher resolutions. Figure 2 presents an example of a micrograph of the enzyme β-Galactosidase and the corresponding high-resolution 3-D reconstruction [15].
Fig. 1:

Gallery of important biomedical structures solved by single-particle cryo-EM at increasing resolutions. From left to right (lower to higher resolution): 4.5Å structure of the human rhodopsin receptor bound to an inhibitory G protein [43], a member of the family of G-protein coupled receptors which are the target of ~ 35% of FDA-approved drugs; 3.3Å map of a voltage-activated potassium channel, an integral membrane protein responsible for potassium ion transport [49];2.9Å cryo-EM structure of a CRISPR-Cas Surveillance Complex implicated in gene editing [36];2.4Å map of the T20s proteasome, a complex that degrades unnecessary or damaged proteins by proteolysis (data available from EMPIAR-10025) [1]; 2.3Å structure of human p97 AAA-ATPase, a key mediator of several protein homeostasis processes and a target for cancer [12]; 1.9Å structure of β-galactosidase enzyme in complex with a cell-permeant inhibitor (EMPIAR-10061) [15];1.8Å structure of the conformationally dynamic enzyme glutamate dehydrogenase [50]; and 1.6Å map of human apoferritin, a critical intracellular iron-storage protein (EMPIAR-10200).
Fig. 2:

High-resolution cryo-EM imaging of the β-galactosidase enzyme in complex with a cell-permeant inhibitor. A) Micrograph of β-galactosidase showing individual particle projections (indicated with white circle). B) Power spectra of image shown in panel A and estimated contrast transfer function (CTF) matching the characteristic Thon ring oscillations (see Section V-B). C) 1.9Å resolution map obtained from ~ 150, 000 individual particle projections extracted from the publicly available dataset EMPIAR-10061 [16], [15]. D) Close-up view of reconstruction shown in panel C highlighting high-resolution features of the map at the individual amino acid level.
The signal-to-noise ratio (SNR) of cryo-EM data is very low due to two compounding reasons. On the one hand, the micrograph’s contrast is low because of the absence of contrast enhancement agents, such as heavy-metal stains. On the other hand, the noise level is high because the electron doses must be kept low to prevent damage to the radiation sensitive biological molecules. The difficulty to estimate the 3-D structure in this low SNR regime, when the orientation and location of the particles are unknown, is the crux of the cryo-EM problem.
Forty years ago, Dubochet and colleagues devised a new technique to preserve biological samples within a thin layer of an amorphous solid form of water, called vitreous ice [27]. In contrast to “regular ice”, vitreous ice lacks crystalline molecular arrangement and therefore allows preservation of biological structures. As the vitreous ice is maintained at liquid nitrogen or liquid helium temperatures, the technique was named “cryo-EM”. In the next decades, the successful application of cryo-EM was limited to study large and highly symmetric structures, such as ribosomes and different types of viruses. Before 2013, only few structures were resolved at resolutions better than 7Å and the field was dubbed “blob-ology” due to the blobby appearance of structures at these resolutions. We refer the reader to [8], [52], [81], [30] for a more detailed historical account of the development of the technology.
Since 2013, single particle reconstruction using cryo-EM has been undergoing fast transformations, leading to an abundance of new high-resolution structures, reaching close to atomic resolution [15]. Figure 3 presents the historical growth in the number of high-resolution structures produced by cryo-EM. Several factors contributed to the “resolution revolution” [44]. First and foremost, a new generation of detectors was developed, called direct electron detectors (DED), that—in contrast to CCD cameras—do not convert electrons to photons. The DED dramatically improved image quality and SNR, and thus increased the attainable resolution of cryo-EM. These detectors have high output frame rates that allow to record multiple frames per micrograph (“movies”) rather than integrate individual exposures [58]. These movies can be used to compensate for motion induced by the electron beam to the sample; see Section V-A. In addition, recent hardware developments have enabled the acquisition and storage of huge amounts of data, which combined with readily access to CPUs and GPU resources, have helped propel the field forward.
Fig. 3:
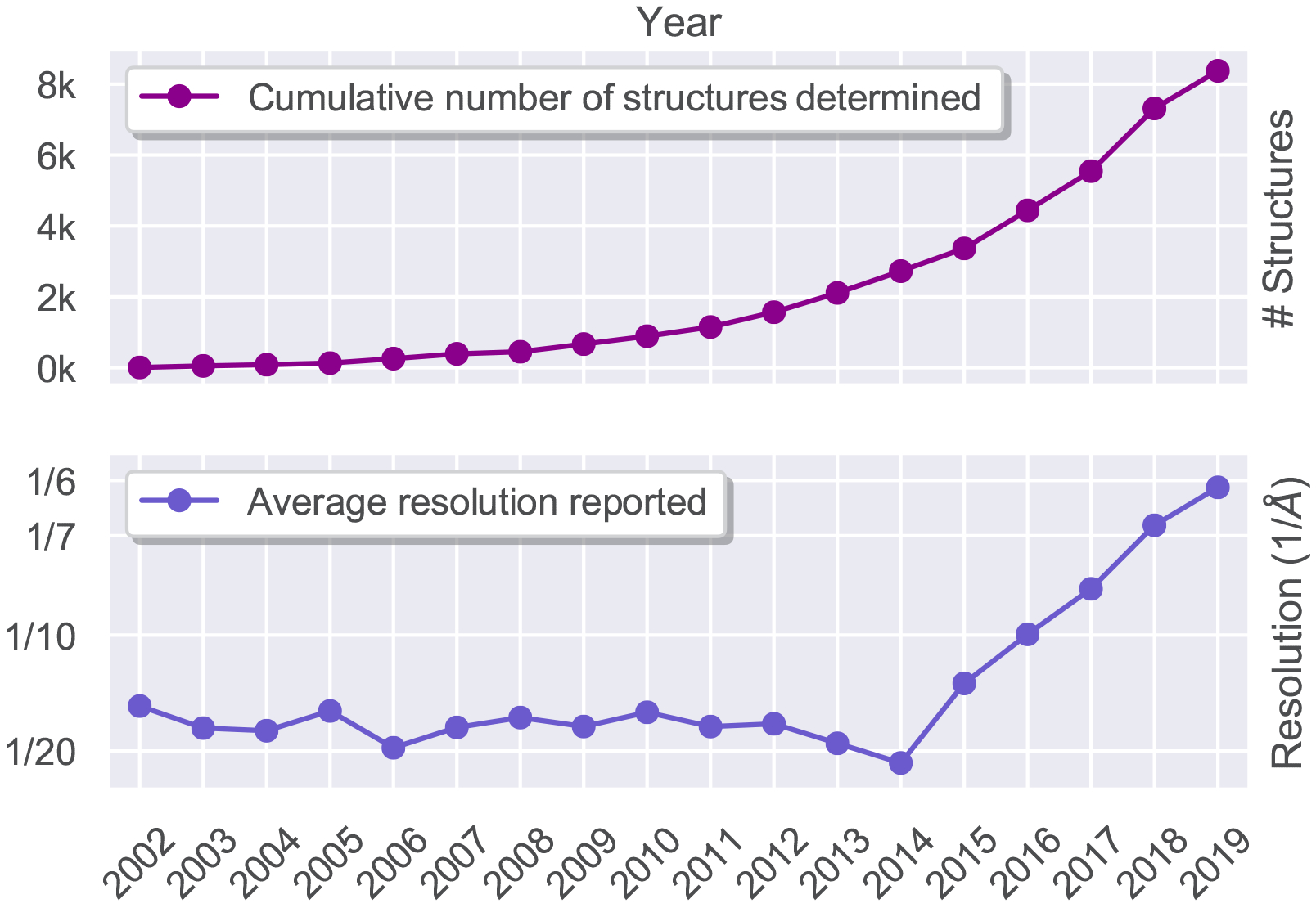
Recent growth in the number of high-resolution structures produced by single-particle cryo-EM. A) Cumulative number of structures solved by single-particle cryo-EM in the last 17 years as recorded in the Electron Microscopy Data Bank (EMDB) public repository [2]. B) Corresponding values for the average resolution of maps deposited in the database showing an inflection point after the year 2013, coinciding with the introduction of direct electron detectors technology in the cryo-EM field.
The prevalent technique in structural biology in the last half a century was X-ray crystallography. This technique suffers from three intrinsic weaknesses that can be mitigated by using cryo-EM imaging. First, many molecules, among them different types of membrane proteins, were notoriously difficult to crystallize. In contrast, the sample preparation procedure for a cryo-EM experiment is significantly simpler, does not require crystallization, and needs smaller amounts of sample. Second, crystal contacts may alter the structure of proteins making it difficult to recover their physiologically relevant conformation. Cryo-EM samples, instead, are rapidly frozen in vitreous ice which preserves the molecules in a near-physiological state. Third, the X-ray beam aggregates the information from all molecules simultaneously and thus hinders visualization of structural variability. In stark contrast, the projection of each particle in a cryo-EM experiment is recorded independently and thus multiple structures—associated with different functional states—can be estimated [74]. A third technique, nuclear magnetic resonance (NMR), can be used to elucidate molecular structures in physiological conditions (water solution at room temperature) but is restricted to small structures, up to ~50 kDa.
The main goal of this article is to introduce the unique and exciting algorithmic challenges and cutting-edge mathematical problems arising from cryo-EM research. Estimating molecular structures involves developing and adopting computational tools in signal processing, estimation and detection theory, high-dimensional statistics, convex and non-convex optimization, spectral algorithms, dimensionality reduction, and machine learning, as well as knowledge in group theory, invariant theory, and information theory. All tools from the aforementioned fields should be adapted to exceptional conditions: an extremely low SNR environment and the presence of missing data (for instance, 2-D location and 3-D orientation of samples in the micrograph). In addition, the devised algorithms should be efficient in order to run on massively large datasets in the order of several Terabytes. The article provides an account of the leading software packages in the field and discusses their underlying mathematical, statistical, and algorithmic principles [63],[76], [57], [33].
The article is organized as follows. In Section II, we describe the generative (forward) model of cryo-EM images and formulate the inverse problem of estimating the 3-D structure of molecules. Section III stresses the main computational challenges in cryo-EM data processing: high noise level, missing data, and massive datasets. Section IV presents 3-D reconstruction methods. In particular, Section IV-A discusses high-resolution techniques and elaborates upon the maximum likelihood framework, which has been shown to be a highly effective tool. Then, Section IV-B introduces different methods for ab initio modeling, frequently used to constitute 3-D low-resolution estimates. Section V describes different building blocks in the algorithmic pipeline of cryo-EM, and relates them to detection, classification, denoising, and dimensionality reduction techniques. Section VI introduces recent results concerning two mathematical models: multi-reference alignment and multi-target detection. These models abstract the cryo-EM problem and provide a broader analysis framework. In Section VII we suggest future research directions and underscore important—computational and theoretical—open questions. Section VIII concludes the article.
Before moving on, we want to mention that topics which are of great importance to practitioners, such as the physics and optics of an electron microscope, sample preparation, and data acquisition are not discussed in this article. These topics are thoroughly covered by biological-oriented surveys, such as [81], [52], [30] and references therein.
II. The cryo-EM reconstruction problem
Modern electron microscopes produce multiple micrographs, each one of them composed of a series of frames (a “movie”). The first stage of any contemporary algorithmic pipeline is to align and average the frames in order to mitigate the effects of movement induced by the electron beam, and thus to improve the SNR. This process is called motion correction, or movie frame alignment. The next step, termed particle picking, consists in detecting and extracting the particles’ tomographic projections from the micrographs. Perfect detection requires finding the particles’ center of mass, which is difficult to estimate due to the characteristics of the Fourier transform of the microscope’s point spread function (PSF)—called the contrast transfer function (CTF); see Section V-B. The output of the particle picking stage is a series of images I1, …, IN from which we wish to estimate the 3-D structure; this section focuses on the latter problem of 3-D reconstruction from picked particles, which is the heart of the computational pipeline of single-particle reconstruction using cryo-EM. Motion correction and particle picking are discussed in more detail in Section V. Figure 4 shows the complete cryo-EM imaging pipeline.
Fig. 4:
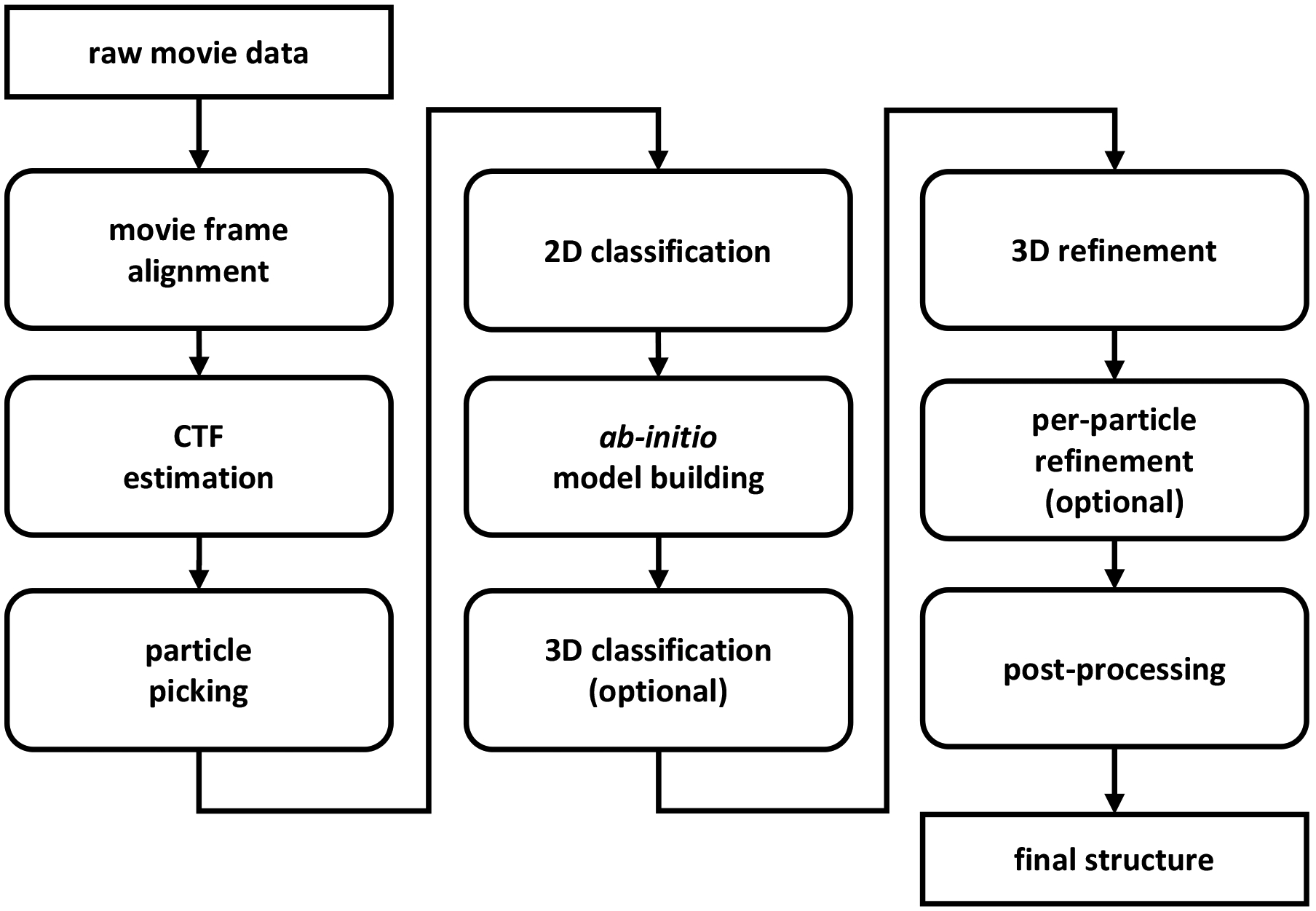
Flowchart diagram showing the computational pipeline required to convert raw movie data into high-resolution structures by single-particle cryo-EM. Raw data is first pre-processed at the movie frame alignment (Section V-A) and CTF estimation (Section V-B) steps followed by particle picking to detect and extract the individual projections from micrographs. Occasionally, particle picking is followed by a particle pruning stage to remove non-informative picked particles (Section V-C). The output of this stage is a set of 2-D images, each (ideally) contains a noisy tomographic projection taken from an unknown viewing direction. Particle images are then classified (Section V-D): the 2-D classes are used to construct ab initio models, as templates for particle picking, to provide a quick assessment of the particles, and for symmetry detection. Then, an initial, ab initio model is built using the 2-D classes or by alternative techniques (Section V-D). If the data contains structural variability or a mix of structures (see Sections II and VII-A), then a 3-D classification step is applied to cluster the projection images into the different structural conformations. Initial models are subjected to 3-D high-resolution refinement (Section IV-A) and an additional per-particle refinement may be applied. Finally, a post-processing stage is employed to facilitate interpretation of structures in terms of atomic models. Different software packages may use slightly different workflows and occasionally some of the steps are applied iteratively. For instance, one can use the 2-D classes to repeat particle picking with more reliable templates. This figure is adapted from [90].
Let represent the 3-D molecular structure to be estimated. Under the assumption that the particle picking is executed well (namely, each particle is detected up to a small translation; we assume for simplicity no false detection), each image I1, …, IN is formed by rotating ϕ by a 3-D rotation Rω, integrating along the z-axis (tomographic projection), shifting by a 2-D shift Tt, convolving with the PSF of the microscope hi, and adding noise:
| (II.1) |
where * denotes convolution. The model can be written more concisely as
| (II.2) |
where the tomographic projection is denoted by P, and (Rϕ)(r) = ϕ(RT r). The image is then sampled on a Cartesian grid. The integration along the z-axis is called the X-ray transform (not to be confused with the Radon transform in which the integration is over hyperplanes). The 3-D rotations describe the unknown 3-D orientation of the particles embedded in the ice, and can be thought of as random elements of the special orthogonal group ω ∈ SO(3). These rotations can be represented as 3 × 3 orthogonal matrices with determinant one or by quaternions. The 2-D shifts result from detection inaccuracies, which are usually small. The PSFs hi are assumed to be known and their Fourier transforms suffer from many zero-crossings, making deconvolution challenging; see Section V-B for further discussion.
The cryo-EM inverse problem of reconstruction consists in estimating the 3-D structure ϕ from the 2-D images I1, …, IN. Importantly, the 3-D rotations ω1, …,ωN and the 2-D shifts t1, …, tN are nuisance variables. Namely, while the rotations and the shifts are unknown a priori, their estimation is not an aim by itself. Figure 5 shows an example of the cryo-EM problem in its most simplified version, without noise, CTF, and shifts.
Fig. 5:
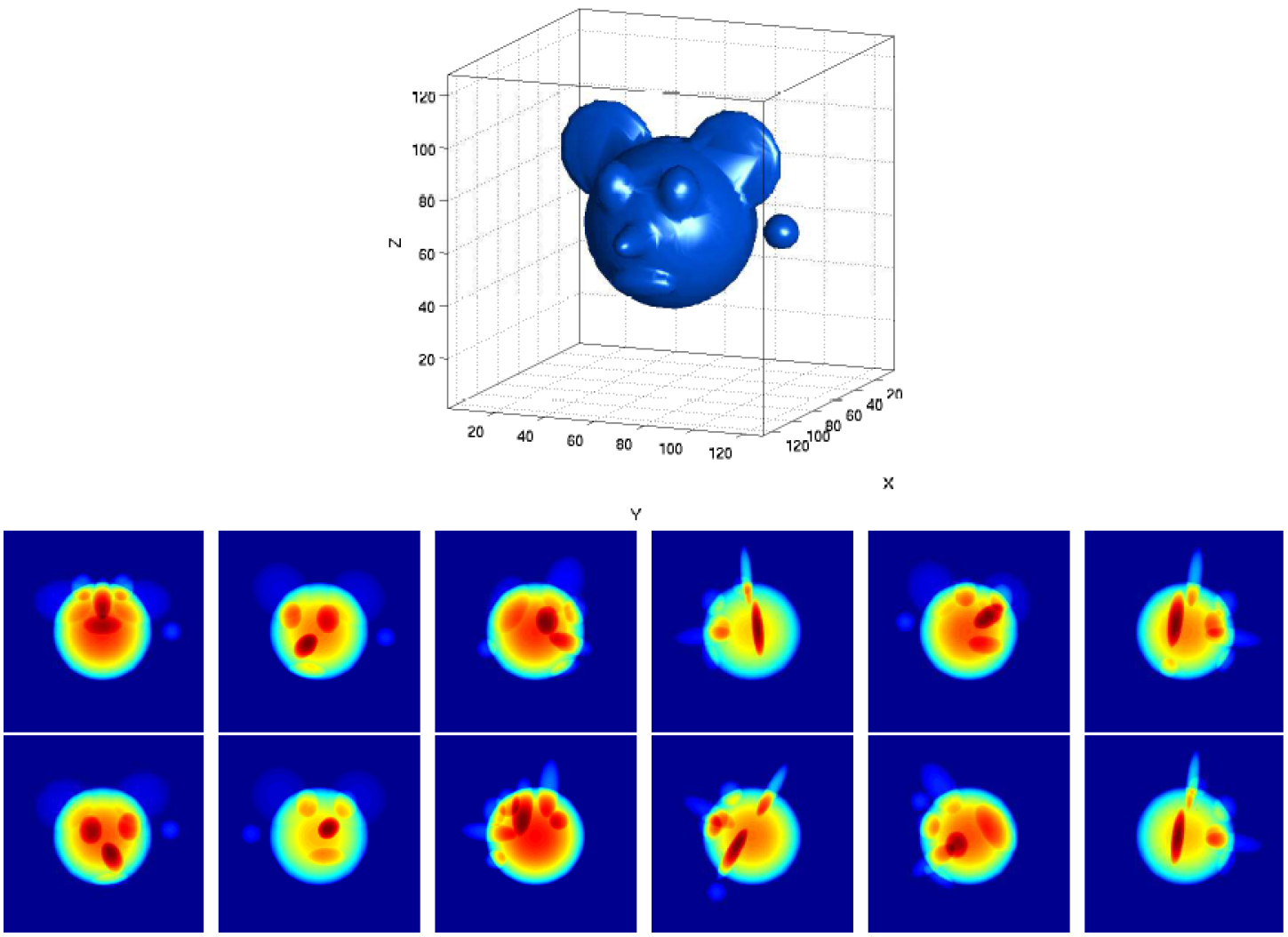
A simulated 3-D structure and a dozen of its noise-free tomographic projections from different viewing directions. The most simplified version of the cryo-EM problem is estimating the 3-D structure from the 2-D projection images when the viewing directions are unknown. In practice, the projections are highly noisy, slightly shifted, and convolved with the microscope’s PSF. Image credit: [71].
The reconstruction of ϕ is possible up to three intrinsic ambiguities: a global 3-D rotation, the position of the center of the molecule (3-D location), and handedness. The latter symmetry, also called chirality, means that it is impossible to distinguish whether the molecule was reflected about a 2-D plane through the origin. The handedness of the structure cannot be determined from cryo-EM images alone because the original 3-D object and its reflection give rise to identical sets of projections related by the following conjugation , where J = diag(1, 1, −1).
In the presence of structural variability, ϕ may be thought of as a random signal with an unknown distribution defined over a space of possible structures (which might be unknown as well). In this case, the task is more ambitious and involves estimating the whole distribution of conformations. Usually, the distribution is assumed to be discrete (that is, in each measurement we observe one of few possible structures or conformational states) or to lie in a low-dimensional subspace or manifold. This subject is further discussed in Section VII-A and in a recent survey [74].
The chief noise source in cryo-EM at the frame level (before motion correction) is shot noise, which follows a Poisson distribution. After movie frames are averaged to produce micrographs, it is customary to assume that the noise is characterized by a Gaussian distribution. Indeed, all current algorithms build upon—implicitly or explicitly—the speculated Gaussianity of the noise. While the spectrum of the noise is not white—mainly due to inelastic scattering, variations in the thickness of ice, and the PSF of the microscope—it is assumed to be a 1-D radial function (namely, constant along the angular direction). The common practice is to estimate the parameters of the noise power spectrum during the 3-D reconstruction process, or from regions in the micrographs that, presumably, contain no signal.
III. Main computational challenges
The difficulty to determine high-resolution molecular structures using cryo-EM hinges upon three characteristic features of the cryo-EM data: high noise level, missing data, and massive datasets. This section elaborates on these unique features, while the next sections dive into the different tasks and algorithms involved in cryo-EM data processing.
a). High noise level:
In a cryo-EM experiment, the electron doses must be kept low to mitigate radiation damage due to the electron illumination. The low doses induce high noise levels on the acquired raw data frames. Together with the image’s low contrast, it results in SNR levels that are usually well below 0 dB and might be as low as −20 dB (namely, the power of the noise is 100 times greater than the power of the signal). Under such low SNR conditions, standard tasks, such as aligning, detecting, or clustering signals, become very challenging.
To comprehend the difficulty to perform estimation tasks in a low SNR environment, let us consider a simple rotation estimation problem. Let us denote by the samples of a periodic signal on the unit circle; x is assumed to be known. We wish to estimate a rotation θ ∈ [0, 2π) from a noisy measurement
| (III.1) |
where and Rθ denotes the rotation operator, that is, (Rθx)(t) = x(t − θ). To estimate θ, we correlate the signal x with rotated versions of y, and choose the inverse of the rotation that maximizes the correlation as the estimator—this technique is called template matching. In the absence of noise, template matching simply correlates the signal with its rotated versions; the maximum is attained when y is rotated by −θ. However, in the presence of noise, we get an additional stochastic term due to the correlation of the noise with the signal. Consequently, if σ2 is large, it is likely that the peak of the correlation will not be close to −θ. In particular, when σ → ∞ the location of the peak is distributed uniformly on the circle. This result can be derived formally using the Neyman-Pearson lemma and holds true for any estimation technique, not necessarily template matching [18]. Even if we collect N measurements (each with a different rotation), it is impossible to estimate the N rotations accurately. In [6], it was shown that the Cramér-Rao bound of this problem is proportional to σ2 and independent of N. Therefore, if the noise level is high, the variance of any estimator would be high as well.
The same conclusions that were derived for the simple rotation estimation problem (III.1) remain true for cryo-EM. For example, even if the 3-D structure is known, aligning a noisy raw image against multiple noiseless projection templates that correspond to rotations sampled from SO(3) will produce no salient peak in the correlation if the noise level is high. In particular, the higher the noise level, the flatter the distribution over SO(3).
b). Missing data—Unknown viewing directions and locations:
The viewing direction and location associated with each particle in a micrograph are unknown a priori. If they were known, estimating the structure ϕ would be a linear inverse problem—similarly to the reconstruction problem in computerized-tomography (CT). The recovery in this case is based on the Fourier slice theorem, stating that the 2-D Fourier transform of a tomographic projection is the restriction of the 3-D Fourier transform of ϕ to a 2-D plane. Mathematically, it can be written succinctly as
| (III.2) |
where F2, F3, P and S are, respectively, the 2-D Fourier, 3-D Fourier, tomographic projection and restriction operators; this theorem is a direct corollary of the fact that the Fourier operator F and the rotation operator R commute, i.e., RF = F R.
The Fourier slice theorem implies that acquiring tomographic projections from known viewing directions is equivalent to sampling the 3-D Fourier space. Hence, given enough projections, one can estimate ϕ to a certain resolution. This is the underlying principle behind many CT imaging algorithms, such as the classical filtered back projection (FBP). However, FBP is not optimal for cryo-EM (even if viewing directions are known) since not all viewing directions are necessarily represented in the data, and the sampling in Fourier domain is non-uniform; the coverage of viewing directions affects the quality of the solution. An alternative is the algebraic reconstruction technique (ART) which solves the linear inverse problem by iterative projections; this algorithm is called the Kaczmarz method in numerical linear algebra. However, ART is rarely used in cryo-EM because it is slow: it does not exploit the Fourier slice theorem and thus cannot be accelerated using FFTs. Modern approaches were developed to exploit the Fourier slice theorem and to account for non-uniform sampling; popular algorithms are based on efficient gridding methods (that compute a uniformly sampled version of a function from a nonuniformly sampled version by choosing proper weights) [55] and using non-uniform FFT packages to harness the structure of the linear system [35], [14].
While the viewing directions in cryo-EM are unknown, there is a rigorous technique to estimate them based on common-lines; see Section IV-B3 and [79], [72]. Unfortunately, any method to estimate the viewing directions is destined to fail when the SNR is low, for the same reasons that estimating θ in (III.1) would fail. Bearing in mind that the ultimate goal is to estimate the 3-D structure—not the viewing directions—it is essential to consider statistical methods that circumvent rotation estimation, such as maximum likelihood and the method of moments.
c). Massive datasets and high dimensionality:
A single session of data collection in a typical cryo-EM experiment produces a few thousand micrographs, each containing several hundred individual particle projections. Depending on the type of detector used during acquisition, micrographs can range in size from a few tens of Megapixels up to one hundred Megapixels for the newest detectors. Moreover, the new generation of detectors can record each micrograph as a rapid burst of frames, producing large movie files that result in datasets of several Terabytes in size. The sheer volume of data must be taken into account early on in the algorithmic design process, and in addition to storage considerations, steps must be taken to ensure the efficient use of computational resources in order to keep up with the ever-increasing throughput of data produced by modern cameras.
Another issue is the dimensionality of the reconstruction problem. The number of voxels of a typical 200×200×200 volume is 8 million. Estimating so many parameters poses a challenge—both from the computational complexity side (how to efficiently optimize; for example, how to find the maximum of the likelihood function of 8 million variables?) and also on the statistical estimation front (see Section V-E). The problem is even more severe when multiple structures, or even a continuum of structures, need to be estimated (see Section VII-A). Getting to high resolution is a major bottleneck because of a compounding computational burden effect: as more parameters need to be estimated, more data is required for their estimation, and the computation becomes severely more expensive and challenging.
IV. 3-D reconstruction from projections
A. High-resolution refinement
The reconstruction procedure of high-resolution 3-D structures is usually split into two stages: constructing an initial low-resolution model, which is later refined by applying an iterative algorithm (“refinement algorithm”); See Figure 4. This section is devoted to refinements techniques, while different approaches to constitute low-resolution estimates using ab initio modeling—initialization-free models—are discussed in the next section.
Refinement techniques for cryo-EM can be broadly classified into two categories: hard and soft angular assignment methods. The hard assignment approach is based on template matching. At each iteration, multiple projections are generated from the current estimate of the structure; the projections should, ideally, densely cover SO(3). Then, a single viewing direction is assigned to each experimental image based on the projection with which it correlates best. The angular assignment can be performed either in real or Fourier space. The advantage of working in Fourier space is that it is not necessary to rotate the molecule: projections can be computed fast using off-the-shelf non-uniform FFT packages (e.g., [35], [14]) as implied by the Fourier slice theorem. Moreover, using this representation, the CTF is simply a diagonal operator. Once the viewing directions of all experimental images are assigned, the 3-D structure is constructed using standard linear inversion techniques; see discussion in Section III. The algorithm iterates between hard angular assignment and structure construction until convergence. While the quality of hard angular assignments may be influenced by the high noise levels, there are several examples of packages that follow this type of approach, e.g., EMAN2 [76] and cisTEM [33], among others.
A second class of strategies are the soft assignment methods. Similarly to hard assignment methods, in each iteration the experimental images are correlated with multiple template projections. However, instead of choosing the best match, a similarity score is given to each pair of experimental image and projection. These scores, also called weights or responsibilities, are computed according to the generative statistical model of the experimental images and used for reconstruction as explained below.
The soft assignment methods are instances or variants of the well-known expectation-maximization (ML-EM) algorithm [25]. Notably, this relation classifies the 3-D reconstruction problem as a problem of maximum likelihood estimation or, more generally, a problem in Bayesian statistics, and thus provides a solid theoretical and algorithmic framework. In particular, it enables to incorporate priors that essentially act as regularizers in the reconstruction process. In the context of cryo-EM, ML-EM was first applied to 2-D cryo-EM images by Sigworth [68], and later implemented for 3-D reconstruction by the software packages RELION [63] and cryoSPARC [57].
In what follows, we describe the ML-EM algorithm for a more general statistical model, and then particularize it to the special case of cryo-EM. Suppose we collect N observations from the model:
| (IV.1) |
where Lθ is a linear transformation acting on the signal x, parameterized by a random variable θ, and . The goal is to estimate x, while θ1, …, θN are nuisance variables. A typical assumption is that x can be represented by a finite number of coefficients (for instance, its Fourier expansion is finite) and these coefficients were drawn from a Gaussian distribution with mean μ and covariance matrix Σ. The key for reliable estimation is to marginalize over the nuisance variables θ1, …, θN. Without marginalization (that is, when the goal is to jointly estimate x and θ1, …, θN), the number of parameters grows with the number of measurements indefinitely, and thus the maximum likelihood estimator may be inconsistent. This phenomenon is exemplified by the Neyman-Scott “paradox.”
In cryo-EM, the transformation Lθ is the operator described in (II.2). This operator rotates the volume, computes its 2-D tomographic projection, applies a 2-D translation and convolution with the PSF; θ is drawn from a distribution defined over the 5-D space of 3-D rotations and 2-D translations. The distribution of θ is generally unknown and should be estimated as part of the ML-EM algorithm.
Let us denote y = (y1, …, yN). The posterior distribution p(x|y) of (IV.1) is proportional to the product of the prior p(x) with the likelihood function
| (IV.2) |
where Θ is a discrete space on which θ is defined, and M is the length of each observation. If the entire posterior distribution could be computed, then one could obtain far more statistical information about x than just maximizing the likelihood. For instance, the best estimator in the minimum mean square error (MMSE) sense is obtained by marginilzing over the posterior:
| (IV.3) |
Unfortunately, the entire posterior can rarely be computed, especially in big data problems, such as cryo-EM. The ML-EM framework provides a simple, yet frequently very effective, iterative method that tries to compute the maximum a posteriori estimator (MAP), namely, the maximal value of p(x|y).
Each iteration of the ML-EM algorithm consists of two steps. The first step (E-step) computes the expected value of the log of the posterior with respect to the nuisance variables, conditioned on the current estimate of x, denoted here by xt, and the data:
| (IV.4) |
where
| (IV.5) |
If , then .
The second step, called the M-step, updates x by
| (IV.6) |
which is usually performed by setting the gradient of Q to zero. If x is assumed to be Gaussian, then the M-step reduces to solving a linear system of equations with respect to x. The E and M steps are applied iteratively. It is well-known that p(xt+1|y) ≥ p(xt|y); however, since the landscape of p(x|y) is usually non-convex, the iterations are not guaranteed to converge to the MAP estimator [25]. Usually, the ML-EM iterations halt when is smaller than some tolerance (but other criteria can be employed as well). The posterior distribution at each iteration can be evaluated according to (IV.2).
The implementation details of the ML-EM algorithm for cryo-EM vary across different software packages. The popular package RELION, for example, incorporates a prior of uniform distribution of rotations over SO(3) (although the distribution itself is usually non-uniform), and that each Fourier coefficient of the 3-D volume was drawn independently from a normal distribution [63]. The variance of the Fourier coefficients’ prior is updated at each iteration by averaging over concentric frequency shells of the current estimate (so it is a 1-D radial function). If the MLEM algorithm is initialized with a smooth volume (which is the common practice), the variance would be large only at low-frequencies, hence high frequencies would be severely regularized. As a result, the first ML-EM iterations would mostly update the low frequencies, but the high frequencies would also be slightly affected. As the algorithm proceeds, the structure would include more high frequency content, thus the variance of these frequency shells would increase, and the effective resolution of the 3-D map would gradually improve. This strategy of initializing with a low resolution structure and gradually increasing the resolution is called “frequency marching” and is shared by many packages in the field; see further discussion in Section IV-B5. Another popular package, cryoSPARC, also employs an ML-EM algorithm with the prior that each voxel in real space was drawn independently from an independent Poisson distribution with a constant parameter [57]. We mention that such statistical priors (e.g., each voxel or frequency is drawn from Poisson or normal distributions) are usually chosen out of mathematical and computational convenience and may bias the reconstruction process. It is an open challenge to replace those statistical priors by priors which are based on or inspired by biological knowledge, such as previously reconstructed structures.
The main drawback of the ML-EM approach, especially at high resolution, is the high computational load of the E-step in each iteration. Specifically, each ML-EM iteration requires correlating each experimental image (typically a few hundred thousands) with multiple synthetic projections of the current structure estimate (sampled densely over SO(3)) and their 2-D translations. To alleviate the computational burden, a variety of methods are employed to narrow the 5-D search space. Popular techniques are based on multi-scale approaches, where an initial search is done on a coarse grid followed by local searches on finer grids. A more sophisticated idea was suggested in [57] based on the “branch and bound” methodology that rules out regions of the search space that are not likely to contain the optimum of the objective function.
B. Ab initio modeling
In this section, we describe some of the intriguing ideas that were proposed to construct ab initio models, namely, models that do not require an initial guess. These methods usually result in low-resolution estimates that can be later refined as described in the previous section. To emphasize the importance of robust ab initio techniques, we begin by discussing model bias—a phenomenon of crucial importance in cryo-EM and statistics in general.
1). Model bias and validation:
The cryo-EM inverse problem is non-convex and thus the output of the 3-D reconstruction algorithms may depend on their initializations. This raises the validation problem: how can we verify that a given estimate is a faithful representation of the underlying data? Currently, a 3-D model is treated as valid if its structural features meet the common biological knowledge (primary structure, secondary structure, etc.) and if it passes some computational tests based on different heuristics. For instance, the reconstruction algorithm can be initialized from many different points. If the algorithm always converges to the same or similar structures, it strengthens the confidence in the attained solution. Moreover, since the data is usually uploaded to public repositories, different researchers can examine it and compare the results against each other [61]; see a more detailed discussion in Section VII-B.
Despite all precautions taken by researchers in the field, the verification methods are not immunized against systemic errors—this pitfall is dubbed model bias. One important example concerns the particle picking stage in which one aims to detect and extract particle projections from noisy micrographs. The majority of the detection algorithms in the field are based on template matching techniques, despite their intrinsic flaws. Specifically, choosing improper templates can lead to erroneous detection, which in turn biases the 3-D reconstruction algorithm toward the chosen templates; see [39] and further discussion on particle picking techniques in V-C.
The model bias phenomenon is exemplified by the “Einstein from noise” experiment [67]. In this experiment, N images of pure i.i.d. Gaussian noise are correlated with a reference image (in the original paper the authors chose an image of Einstein as the reference and thus the name). Then, each pure-noise image is shifted to best align with the reference image (based on the peak of their cross-correlation), and finally all images are averaged. Without bias, one would expect that averaging pure noise images will converge toward an image of zeros as N diverges. However, in practice the resulting image is similar to the reference image, that is, the algorithm is biased toward the reference image; see [67, Figure 2]. In the context of cryo-EM, the lesson is that without prudent algorithmic design, the reconstructed molecular structure may reflect the scientist’s presumptions rather than the structure that best explains the data.
We now turn our attention to some of the existing techniques for ab initio modeling.
2). Stochastic gradient descent over the likelihood function:
Stochastic gradient descent (SGD) gained popularity in recent years, especially due to its invaluable role in the field of deep learning [21]. The underlying idea is very simple. Suppose that we wish to minimize an objective function of the form
| (IV.7) |
A gradient descent algorithm is an optimization technique used to minimize the objective function by iteratively moving in the direction opposite to its gradient. Its tth iteration takes on the form
| (IV.8) |
where ∇ denotes a gradient, and ηt is called the step size, or learning rate. In SGD, we simply replace the full sum by a random element:
| (IV.9) |
where i is chosen uniformly at random from {1, …, N}. In expectation, the direction of each SGD step is the direction of the full gradient (IV.8). The main advantage of SGD is that each iteration is significantly cheaper to compute than the full gradient (IV.8). It is also easy to modify the algorithm to sum over few random elements at each iteration, rather than just one, to improve robustness at the cost of higher computational load per iteration. In addition, it is commonly believed that the inherent randomness of SGD helps to escape local minima in some non-convex problems.
This strategy was proven to be effective to construct ab initio models using cryo-EM, achieving resolutions of up to 10Å [57]. The objective function is the negative log of the posterior distribution. As the log posterior involves a sum over the experimental images (IV.2), the SGD scheme chooses at each iteration one random image (or a subset of images) to approximate the gradient direction.
3). Estimating viewing directions using common-lines:
If the orientations and locations of the particles in the ice are known (equivalently, the 3-D rotations and the 2-D translations in (II.2)), then recovering the 3-D structure is a linear problem that enjoys many solutions developed for CT imaging. In this section, we survey an analytical method to estimate the viewing directions using the common-lines property.
Due to the Fourier slice theorem, any pair of projection images has a pair of central lines (one in each image) on which their Fourier transforms agree. For generic molecular structures (with no symmetry), it is possible to uniquely identify this common-line, for example, by cross-correlating all possible central lines in one image with all possible central lines in the other image, and choosing the pair of lines with maximum cross-correlation.
The common-line pins down two out of the three Euler angles associated with the relative rotation between images Ii and Ij. A third image is required to determine the third angle: the three common-line pairs between three images uniquely determine their relative rotations. This technique is called angular reconstitution, and it was suggested, independently, by Vainshtein and Goncharov [78] and Van Heel [79]. The main drawback of this procedure is its sensitivity to noise; it requires the three pairs of common lines to be accurately identified. Moreover, estimating the rotations of additional images sequentially (using their common-lines with the previously rotationally assigned images) can quickly accumulate errors.
As a robust alternative, it was proposed to estimate the rotations R1, …, RN from the common-lines of all pairs Ii, Ij simultaneously; this framework is called synchronization. Since this strategy takes all pair-wise information into account, it has better tolerance to noise. The rotation assignment can be done using a spectral algorithm or semidefinite programming [72] and enjoys some theoretical guarantees; see for instance [70]. Nevertheless, as discussed in Section III, in the low SNR regime any method for estimating the rotations would fail. Therefore, the method requires as input high SNR images that can be obtained using a procedure called 2-D classification; see further discussion in Section V-D. 2-D classification, however, blurs the fine details of the images and therefore the attained resolution of the method is limited [34].
Three last comments are in order. First, if the structure possesses a nontrivial symmetry, there are multiple common-lines between pairs of images that should be considered [56]. Second, interestingly, this technique cannot work when the underlying object is a 2-D image (rather than 3-D as in cryo-EM). In this case, the Fourier transform of the tomographic projection (which is a 1-D function in that case) is a line that goes through the origin of the 2-D Fourier space of the underlying object. Therefore, the single common point of any two projections taken from different angles is the origin, and thus there is no way to find the relative angle between them. Finally, the common-lines method is effective for additional signal processing applications, such as the study of specimen populations [46].
4). The method of moments:
Suppose that a set of parameters x characterizes a distribution p(y|x) of a random variable y with L entries. We observe N i.i.d. samples of y and wish to estimate x. In the method of moments, the underlying idea is to relate the moments of the observed data with x. Those moments can be estimated from the data by averaging over the observations:
| (IV.10) |
where is a tensor with Lk entries and the entry indexed by is given by . By the law of large numbers, when N → ∞ the average converges almost surely to the expectation. The right-hand side underscores that the expectations are solely functions of x; the moment tensors M1, M2, …, Mk can be derived analytically. For instance, in cryo-EM, the expectation is taken against the random rotations, translations and the noise; the moments are functions of the 3-D volume, the 5-D distribution over the space of 3-D rotations and 2-D translations, and some parameters of the noise statistics (due to bias terms). The final step of the method of moments is to solve the system of equations (IV.10), which might be highly non-trivial. Importantly, since estimating the moments requires only one pass over the observations, it can be done potentially on the fly during data acquisition. This is especially important for cryo-EM in which hundreds of thousands of images need to be processed.
Computing the qth moment involves the product of q noisy terms and thus the variance of its estimation scales as σ2q/N. As a result, when invoking the method of moments it is crucial to determine the lowest-order moment that identifies the parameters x uniquely: this moment determines the estimation rate of the problem, that is, how many samples are required to accurately estimate the parameters. Remarkably, it was shown that under a general statistical model that is tightly-related to the cryo-EM problem (as will be described in Section VI-A), the first moment that distinguishes between different parameters determines the sample complexity of the problem (namely, how many observations are necessary to attain an accurate estimate) in the low SNR regime [4]. In addition, the number of equations in (IV.10) increases with the order of moments. Thus, using lower-order moments reduces the computational complexity.
The first to suggest the method of moments for single particle reconstruction, 40 years ago, was Zvi Kam [42]. Under the simplifying assumptions that the particles are centered, the rotations are uniformly distributed and by ignoring the CTF, he showed that the second moment of the experimental images determines the 3-D structure, up to a set of orthogonal matrices. To determine those matrices, he suggested to compute a subset of the third-order moment. Recently, it was shown that under the same assumptions the structure is indeed determined uniquely from the third moment [9]. Remarkably, if the distribution of rotations is non-uniform, then the second moment suffices [66]. Therefore, in light of [4], the sample complexity of the cryo-EM problem in the low SNR regime, under the specified conditions, scales as σ4 and σ6 for non-uniform and uniform distribution of rotations, respectively. It is still an open question how many images are required to solve the full cryo-EM problem; see further discussion in Section VII.
Interest in the method of moments has been recently revived, largely due to its potential application to X-ray free electron lasers (XFEL) [48], [82]. We believe that the method of moments has been largely overlooked by the cryo-EM community since Kam’s paper for three main reasons. First, Kam’s original formulation required uniform distribution of viewing directions, an assumption that typically does not hold in practice; a recent paper extends the method to any distribution over SO(3) [66]. Second, estimating the second and third order statistics accurately requires a large amount of data that was not available until recent years. Third, accurate estimation of high-order moments for high-dimensional problems requires modern statistical techniques, such as eigenvalue shrinkage in the spiked covariance model, that have only been introduced in the last decade [26].
5). Frequency marching:
Most of the cryo-EM reconstruction algorithms start with a low-resolution estimate of the structure, which is then gradually refined to higher resolutions. This process is dubbed frequency marching. Frequency marching can be done explicitly by constructing a low-resolution model using an ab initio technique that is later refined by ML-EM [57], or more implicitly by an iteration-dependent regularization [63].
In [13], a deterministic and mathematically rigorous method to gradually increase the resolution was proposed. The 3-D Fourier transform of the object is expanded by concentric shells. At each iteration, each experimental image is compared with many simulated projections of the current low-resolution estimate, and a viewing direction is assigned to each experimental image by template matching (hard angular assignment). Given the angular assignments, an updated structure with one more frequency shell is computed by solving a linear least squares problem, thus progressively increasing the resolution.
V. Building blocks in the computational pipeline
This section elaborates on some of the building blocks in the algorithmic pipeline of single particle reconstruction using cryo-EM. For each task, we introduce the problem and discuss the underlying principles behind some of the most commonly used solutions.
A. Motion correction
During data collection, the electron beam induces sample motion that mitigates high-resolution information. The modern detectors (direct electron detectors) acquire multiple frames per micrograph, allowing to partially correct the motion blur by aligning and averaging the frames. In essence, motion correction is an alignment problem (also referred to as registration or synchronization) that shares many similarities with classical tasks in signal processing and computer vision. The main challenge in alignment is the high noise levels that hamper precise estimation of relative shifts between frames.
Several solutions were proposed to the motion correction problem based on a variety of methods, such as pair-wise alignment of all frames and optical flow; see a survey on the subject in [58] and references therein. The first generation solutions aimed to estimate the movement of the entire micrograph, however, the drift is not homogeneous across the entire field of view, motivating the development of more accurate local techniques. One such strategy is implemented by the software MotionCor2 [88], which describes the motion as a block-based deformation that varies smoothly throughout the exposure. The micrograph is first divided into patches, and motions within each patch are estimated based on cross-correlation. Then, the local motions are fitted to a time varying 2-D polynomial; the polynomial is quadratic in the 2-D frame coordinates and cubic in the time axis. Finally, the frames are summed, with or without dose weighting which was determined according to a radiation damage analysis. More recently, strategies for per-particle motion correction have been proposed that yield significant improvements in resolution [15], [92]. Figure 6 demonstrates examples of different motion correction strategies.
Fig. 6:

Beam-induced motion correction. All movement trajectories in this figure are shown color coded, using cyan to represent the position of first frame and magenta to represent the position of the last frame in the sequence. A) Strategies for global motion correction compensate for movement of the specimen across the entire field of view containing multiple particles [32]. B) Semi-local strategies for motion correction align frames across sub-regions defined on a discrete grid [88]. C) Per-particle or local drift-correction allows accurate tracking of individual particles throughout the exposure to electrons [15].
B. CTF estimation and correction
Each cryo-EM image is affected by the PSF of the microscope through convolution with a kernel hi; see (II.2). The Fourier transform of the PSF—the CTF—suffers from multiple zero-crossings, making its inversion (i.e., deconvolution) challenging. To compensate for the missing frequencies, the standard routine is to apply different PSFs to different micrographs. Thus, spectral information that was suppressed in one micrograph may appear in another.
The CTF cannot be specified precisely by the user. Instead, the number and location of the zero-crossings is given by the specific experimental parameters of the microscope, which are hard to control. Thus, a preceding step entails estimating the CTF from the acquired data. Specifically, the CTF is modeled as
| (V.1) |
where k is the frequency index, α is a small phase shift term, λ is the electron wavelength, Δf is the objective defocus, Cs is the spherical aberration, and E(|k|) is an exponentially decaying envelope function—specified by a parameter called B-factor—due to the beam energy spread, the beam coherence, and the sample drift [59]. The CTF mitigates the very low frequencies, and therefore centering projection images (that is, finding their center of mass) is challenging. In fact, the center of mass depends on the CTF, and thus two projection images with the same viewing angles but different CTFs (e.g., different defocus values) will have different centers in real space. The CTF is approximately constant along concentric rings, although in practice those rings might be slightly deformed; this deformation can be modeled by an additional parameter called astigmatism). The defocus value of the microscope has a pivotal role: high defocus values enhance low-resolution features and improve the contrast of the image (increase the SNR), whereas low defocus values enhance high-resolution features (fine details) at the cost of lower contrast (lower SNR).
CTF estimation begins by computing the power spectrum of the whole micrograph. The power spectrum exhibits “rings”, called Thon rings, that correspond to the oscillations of the sine function (V.1); those rings are used to fit the CTF’s parameters (see Figure 2B). Two popular software packages CTFFIND4 [59] and Gctf [85] estimate those parameters by generating multiple templates according to the postulated model (V.1). The template that best fits the measured Thon rings is used as the estimated CTF.
Once the CTF was estimated, the goal it to invert its action. The challenge stems from the structure of the CTF (V.1) because it has many small values and thus direct inversion is impossible. The most popular technique for CTF correction, “phase flipping”, is embarrassingly simple: it disregards the information about amplitude changes and corrects the data only for the sign of the CTF, and thus obtains the correct phases in Fourier space. Namely, if a micrograph is represented in Fourier domain by
| (V.2) |
where I(k) is the micrograph before the CTF action, then the corrected image would be
| (V.3) |
Another approach is to apply Wiener filtering that attempts to correct both the amplitudes and the phases [29], [69]. However, this approach requires the knowledge of the SNR (which is not trivial to estimate in a low SNR environment) and cannot correct for missing frequencies.
Instead of inverting the CTF explicitly, an attractive alternative is to incorporate the CTF into the forward model of the reconstruction algorithm. This can be done naturally in the ML-EM approach, the method of moments, and the SGD framework. Because different CTFs are applied to different micrographs, in principle, full CTF correction is feasible.
C. Particle picking
A micrograph consists of regions which contain only noise, regions with noisy 2-D projections, and regions with other contaminants, such as carbon film. In particle picking, the goal is to detect and extract the 2-D projections (particles) from the noisy micrographs. High-resolution reconstruction typically requires hundreds of thousands of particles, and thus manual picking is time consuming and tedious. In addition, it may introduce subjective bias into the procedure.
Many solutions were proposed in the literature for the particle picking problem, based on standard edge detection techniques, machine learning [84], [91], [83], and template matching. For the latter, a popular strategy is the one implemented in RELION. In this framework, the user manually selects a few hundreds of particles. These particle images are then 2-D classified (see next section), and the 2-D classes are used as templates for an automatic particle picking based on template matching [64].
As discussed in Section IV-B1, a major concern of any particle picking algorithm is a systematic bias. To alleviate the risk for model bias, a fully automated technique was proposed in [37]. Rather than templates, the algorithm selects automatically a set of reference windows that include both particle and noise windows. The selection is based on the local mean and variance: regions with particles typically have lower mean and higher variance than regions without particles. Then, regions in the micrograph are correlated with the reference windows. The regions which are most likely to contain a particle (high correlation) and those which are least likely (low correlation with all reference windows) are used to train a support vector machine classifier. The output of this linear classifier is used to pick the particles.
In practice, a considerable number of picked particles are usually non-informative, containing adjacent particles that are too close to each other, only part of a particle, or just pure noise. Consequently, it is common to try and prune out these outliers. In [62], it was proposed to employ several particle pickers simultaneously and compute a consensus between them using a deep neural network. In [90], a simple pruning is devised by viewing the output of the particle picking algorithm as a mixture of Gaussians.
D. 2-D classification
As experimental images corresponding to similar viewing directions tend to be very much alike (due to the smoothness of the molecular structure), it is common to divide the images into several classes (i.e., clustering) and average them to increase the SNR. This process is called 2-D classification and the averaged images are referred to as “class averages.” Since the global in-plane rotation of each micrograph is arbitrary, the clustering should be invariant under in-plane rotations: two images from a similar viewing direction but with a different in-plane rotation are supposed to be grouped together after appropriate alignment. The class averages are used for a variety of tasks: to construct ab initio models, as templates for particle picking, to provide a quick assessment of the particles, to remove picked particles which are associated with non-informative classes, and for symmetry detection.
There are three main computational aspects that make the 2-D classification task quite challenging. First, as already mentioned, low SNR impedes accurate clustering and alignment. Second, the high computational complexity of finding the optimal clustering among hundreds of thousands of images may be prohibitive unless designed carefully. Third, it is not clear what is the appropriate similarity metric to accurately compare images.
Many different strategies for 2-D classification were proposed; see for instance [80], [73]. A popular 2-D classification algorithm, implemented in RELION [65], is based on the ML-EM scheme. In this algorithm, each observed image is modeled as a sample from the statistical model
| (V.4) |
where k is distributed uniformly over {1, …, K} (these are the K class averages to be estimated), θ is distributed uniformly over θ ∈ [0, 2π), t is drawn from an isotropic 2-D Gaussian, hi is the estimated PSF, and . Given this model, it is straight-forward to implement an ML-EM algorithm to estimate x1, …, xK following the guidelines of Section IV. This algorithm, albeit popular, suffers from three imperative weaknesses. First, the computational burden of running ML-EM is a major hurdle as the algorithm needs to go through all experimental images at each iteration. Second, it assumes that each experimental image is originated from only K possible class averages. However, this is an inaccurate model as the orientation of particles in the ice layer are distributed continuously over SO(3) (in addition to structural variability that may be present). Finally, ML-EM typically suffers from the “winner takes all” phenomenon: most experimental images would correlate well with, and thus be assigned to the class averages that enjoy higher SNR. As a result, ML-EM tends to output only a few, low-resolution classes. This phenomenon was already recognized by [73] and is also present for ML-EM based 3-D classification and refinement.
An alternative solution was proposed in [87]. In this framework, each image is averaged with its few nearest neighbors, after a proper in-plane alignment. The nearest neighbor search is executed efficiently over the bispectra of the images. The bispectrum is a rotationally-invariant feature of an image— that is, it remains unchanged under an in-plane rotation. To reduce the computational complexity and denoise the data, each image is first compressed using a dimensionality reduction technique—called steerable PCA—which is the main topic of the next section.
E. Denoising and dimensionally reduction techniques
Principal component analysis (PCA) is a widely used tool for linear dimensionality reduction and denoising, dating back to Pearson [54], [41]. PCA computes the best (in the least squares sense) low-dimensional affine space that approximates the data by projecting the images into the space spanned by the leading eigenvectors of their covariance matrix. In cryo-EM, covariance estimation was introduced by Kam [42] and used for dimensionality reduction and image classification for the first time by van Heel and Frank [80]. PCA is often used to denoise the experimental images and as part of the 2-D classification stage.
Cryo-EM images are equally likely to appear in any in-plane rotation. Consequently, when performing PCA, it makes sense to take all in-plane rotations of each image into account. Luckily, there is a simple way to account for all in-plane rotations without rotating the images explicitly. This can be done by expanding the images in a steerable basis: a basis formed as an outer product of radial functions with an angular Fourier basis; examples are Fourier Bessel, 2-D prolate spheroidal wave functions, and Zernike polynomials. When integrating over all possible in-plane rotations, the covariance matrix of the expansion coefficients enjoys a block-diagonal structure, reducing the computational load significantly: while the covariance matrix of images of size L × L has L4 entries, the block diagonal structure guarantees that merely O(L3) entries are non-zero. This in turn reduces the computational complexity of steerable PCA from O(NL4+L6) to O(NL3 + L4): the first term is the cost of computing the sample covariance over N images, and the second term is the cost of the eigendecomposition over all blocks [86].
Classical covariance estimation techniques usually assume that the number of samples is considerably larger than the signal’s dimension. However, this is not the typical case in cryo-EM in which the dimensionality of the molecule is of the same order as the number of measurements. In this regime, one can take advantage of recent developments in high-dimensional statistics under the “spiked covariance model” [26]. These techniques, based on eigenvalue shrinkage, were recently applied successfully to denoise cryo-EM images [20].
VI. Mathematical frameworks for cryo-em data analysis
Inspired by the cryo-EM problem, a couple of abstract mathematical models were studied in recent years. These models provide a general framework for analyzing cryo-EM from theoretical and statistical perspectives, while removing some of its complications. In what follows, we introduce the models and succinctly describe some intriguing results.
A. Multi-reference alignment
The multi-reference alignment (MRA) model reads [10]:
| (VI.1) |
where G and Ti are, respectively, a known compact group and linear operators, and . The signal is assumed to lie in a known space (say, the space of signals with a finite spectral expansion) that random elements of G act upon. The task is to estimate the signal x from the observations y1, …, yN, while the group elements g1, …, gN are unknown. Since the statistics of y are invariant under the action of a constant g on x, the recovery is possible up to left multiplication by some group element g ∈ G—namely, we wish to estimate the orbit of x under the group action. Figure 7 shows an example of three discrete 1-D MRA observations under the group of cyclic shifts at different noise levels. If we assume perfect particle picking, then the cryo-EM model (II.2) is a special case of (VI.1) when G is the group of 3-D rotations SO(3) and T is the linear operator that takes the rotated structure, integrates along the z-axis (tomographic projection), convolves with the PSF, and samples it on a Cartesian grid. The MRA model formulates many additional applications, including structure from motion in computer vision [5], localization and mapping in robotics [60], study of specimen populations [46], optical and acoustical trapping [28], and denoising of permuted data [53].
Fig. 7:
An example of multi-reference alignment observations at different noise levels σ. Each column consists of three different cyclic shifts (the group actions) of a 1-D periodic discrete signal (the linear operator T is the identity operator). Clearly, if the noise level is low, estimating the signal is easy: one can align the observations (i.e., undo the group action) and average out the noise. The challenge is to estimate the signal when high noise levels hinder alignment (right column).
While surveying all recent results about the MRA model is beyond the scope of this article, we wish to present a remarkable information-theoretic result about the sample complexity of the problem (and therefore also for cryo-EM under the stated assumptions). In many cases, among them when T = I and the cryo-EM setup (without shifts so that G = SO(3)), one can estimate the group elements gi from the measurements yi in the high SNR regime using one of many synchronization techniques (based on spectral methods, semidefinite programming, or non-convex programming) [70], [72]. Once the group elements were identified, one can estimate the signal by aligning all observations (undo the group actions) and averaging. The variance of averaging over i.i.d. Gaussian variables is proportional to 1/(N · SNR)—namely, the number of measurements should be proportional to 1/SNR to achieve an accurate estimate. In this case, we say that the sample complexity of the problem in the high SNR regime is 1/SNR: this is a common scenario in many data processing tasks. In the low SNR regime the situation is radically different. Specifically, it was shown that when σ → ∞, the sample complexity of the MRA problem is determined by the first moment that distinguishes between different signals. More precisely, in the asymptotic regime where N and σ diverge, the estimation error of any method is bounded away from zero if is bounded from above. We then say that the sample complexity in the low SNR regime is proportional to .
For the cryo-EM setup, assuming perfect particle picking and no CTF, it was shown that the sample complexity depends on the distribution of rotations: for uniform distribution the sample complexity scales as 1/SNR3 (that is, the structure is determined by the third moment), while for generic non-uniform distribution the second moment suffices, and thus the sample complexity scales as 1/SNR2 [9], [66]. Similar results were derived for simpler setups, for instance, when a discrete signal is acted upon by cyclic shifts (as shown in Figure 7) [19], [3], [9].
The pivotal role of moments in sample complexity analysis led naturally to the design of algorithms based on the method of moments. Some of these algorithms hinge on the tightly-related notion of invariant polynomials. A polynomial p is called invariant under the action of a group G if for any signal x in the specified space and any g ∈ G, it satisfies p(g ∘ x) = p(x). One particular important invariant is the third-order polynomial, the bispectrum—originally proposed by John Tukey [77]—that was used for 2-D classification [87] and ab initio modeling [42]. We refer the readers to [71] and references therein for a more detailed account on the MRA problem.
B. Multi-target detection
Multi-target detection (MTD) is the problem of estimating a signal that occurs multiple times at unknown locations in a noisy measurement. In its simplest form, the MTD problem is an instance of the 1-D blind deconvolution problem and can be written as
| (VI.2) |
where is the target signal, s ∈ {0, 1}N−L+1 is a binary signal whose 1’s indicate the location of the signal copies in the measurement , and ; see Figure 8. Detecting the signal occurrences in the data (that is, estimating the signal s) is the analog of particle picking in cryo-EM. Clearly, if the noise level is low, one can estimate s (analogously, detect the particle projections in the micrograph), extract the signal occurrences, and average them to suppress the noise. However, as aforementioned, low SNR precludes detection (and particle picking) and therefore one must estimate x directly, without explicit estimation of s. In [18], it was shown that under certain generative models of s, the signal x can be estimated provably, at any noise level, from the bispectrum (third-order statistics). More ambitiously, numerical experiments suggest that the bispectrum suffices to estimate multiple signals x1, …, xK from a mix of blind deconvolution problems
| (VI.3) |
without explicit estimation of the binary signals s1, …, sK that indicate the location of the corresponding signals.
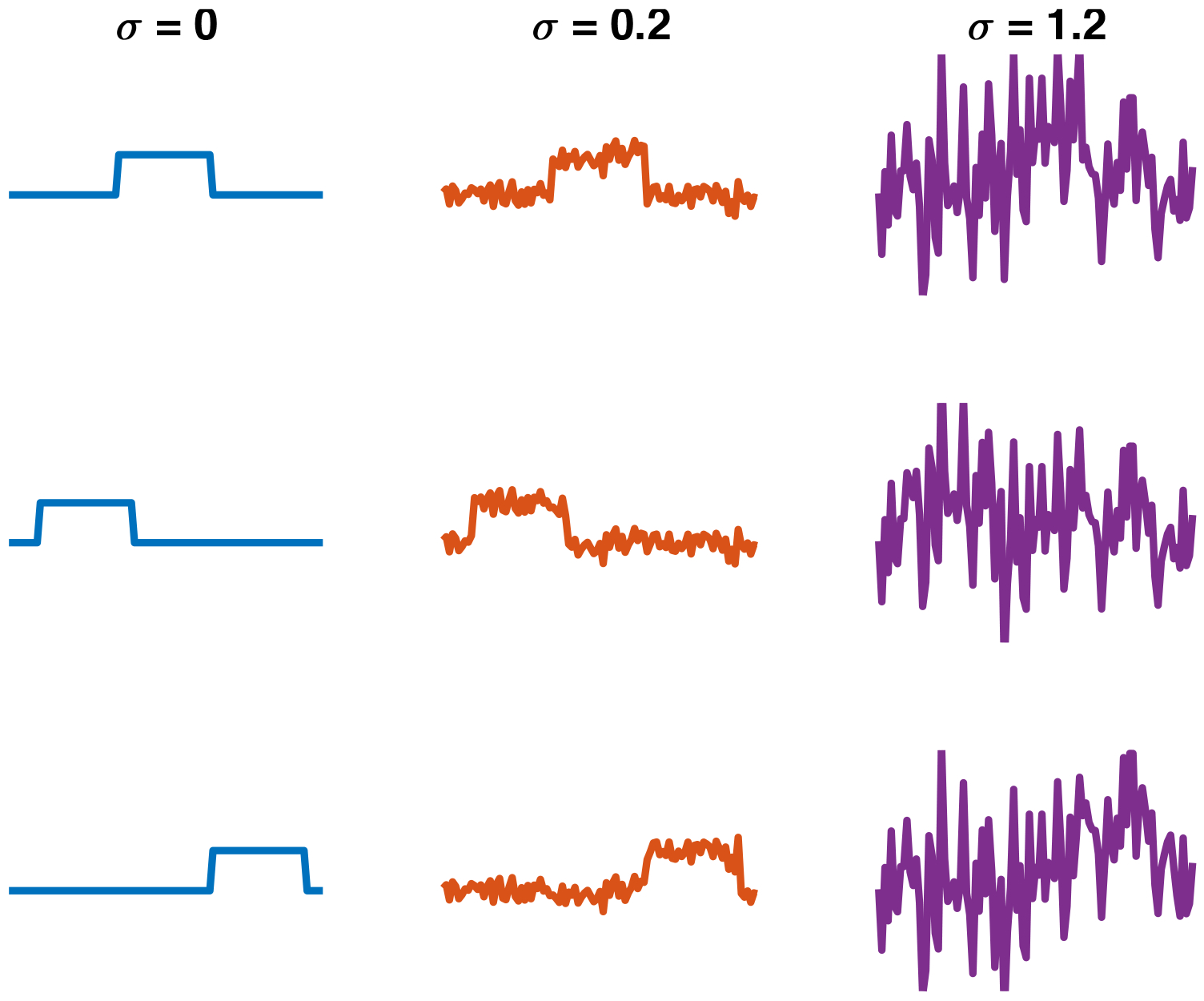
Fig. 8:
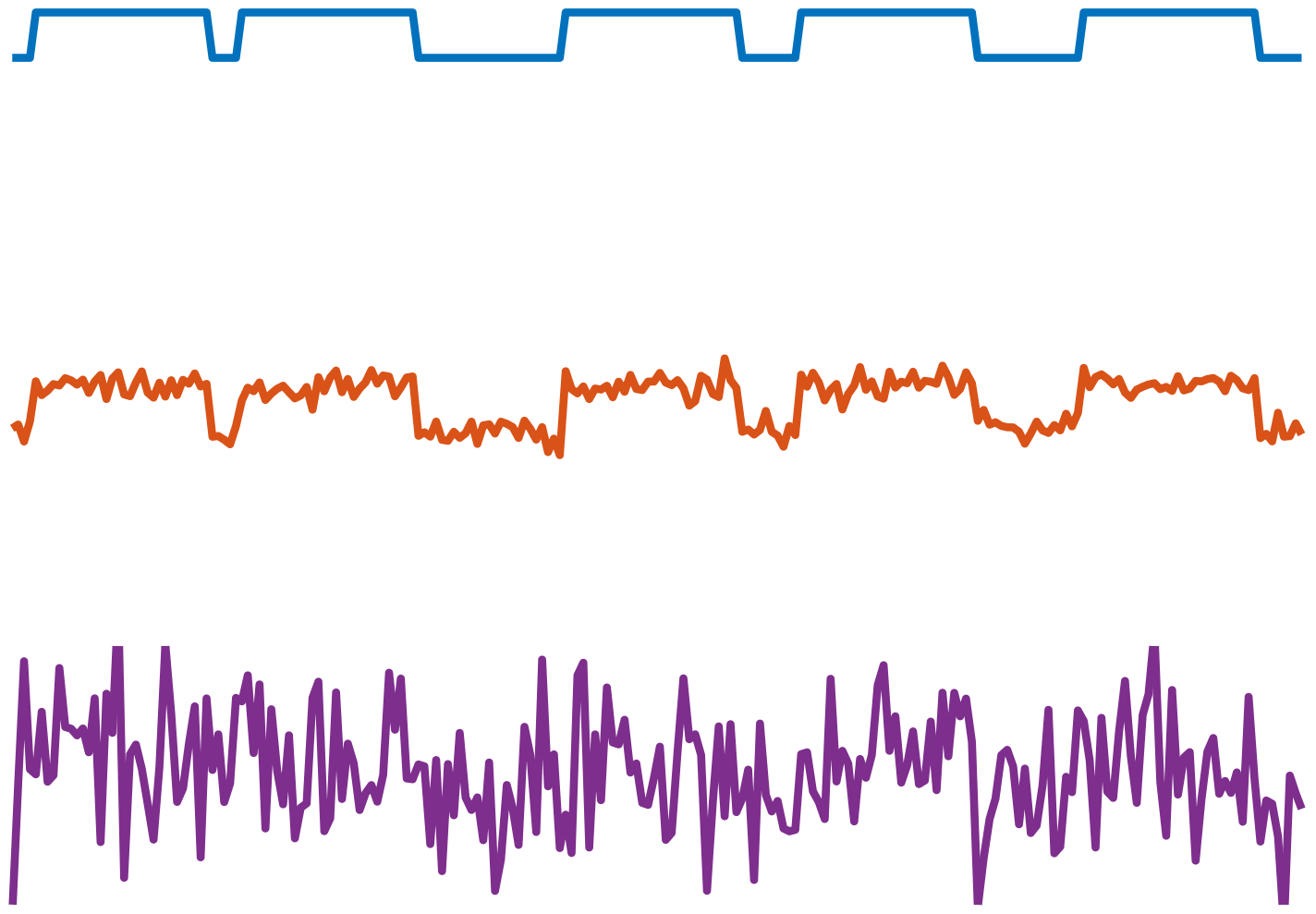
An example of a multi-target detection observation with five signal occurrences at different noise levels (VI.2). From top to bottom: σ = 0, σ = 0.2, and σ = 1.2. When the noise level is low, it is easy to detect the signal occurrences and estimate the underlying signal by averaging out the noise. However, when the noise level is high (bottom), reliable detection is rendered challenging. The task is then to estimate the underlying signal directly, without intermediate detection of its occurrences.
The MTD model can be extended to formulate a generative model of a micrograph: the key is to treat x as a random signal that represents the tomographic projections of the molecule ϕ, rather than a deterministic signal as in (VI.2) and (VI.3). Specifically, locations are chosen in the 2-D plane: these are the positions of the embedded samples in the micrograph. For each location, a signal is drawn from a probability distribution described by the model
| (VI.4) |
where the 3-D rotation Rω is applied to the volume ϕ according to a (possibly unknown) distribution of ω over SO(3), P is a tomographic projection, and h is the microscope’s PSF; the goal is to estimate ϕ. Therefore, the MTD model paves the way to fully model the cryo-EM problem, including most of its important features [18]. In particular, MTD provides a mathematical and computational framework for reconstructing ϕ directly from micrographs, without intermediate particle picking [17]. A full analysis of this model is still lacking.
VII. Remaining computational and theoretical challenges
To conclude the article, we would like to discuss some of the interesting and important computational and theoretical challenges ahead for single particle reconstruction using cryo-EM.
A. Conformational heterogeneity: modeling and recovery
One of the important opportunities offered by cryo-EM is its ability to analyze different functional and conformational states of macromolecules. Mathematically, it entails estimating multiple 3-D structures simultaneously; we refer to this problem as the heterogeneity problem. There is no consensus about the proper way to model the heterogeneity problem and the computational tools are not well-established. We therefore believe that it offers an opportunity for researchers with strong mathematical and computational background to make a profound impact on the field of structural biology.
We now briefly survey different approaches to handle the heterogeneity problem. To this end, we extend the basic cryo-EM model (II.2) to account for conformational variability:
| (VII.1) |
where the goal is to estimate the distribution from which the structures ϕi, …, ϕN are sampled from. Using this formulation, the problem is ill-posed: there is not enough information in N 2-D images to recover N 3-D structures. Hence, additional assumptions on the structures must be made. The different techniques in the field can be broadly classified into two categories: discrete and continuous models. The discrete heterogeneous model assumes that the measurements stem from a few independent volumes (that is, a discrete distribution). Under this model, each projection image can be written as
| (VII.2) |
where ϕk, k = 1, …, K, represent the K different volumes. The advantage of this approach is evident: it is easy to extend the ML-EM framework to incorporate several volumes simultaneously. Alternatively, some software packages apply a preliminary 3-D classification stage, in which the 2-D experimental images are being classified into different structures. CryoSPARC runs multiple trials of an SGD algorithm from different initializations. This procedure occasionally leads to multiple low-resolution estimates, and each of those is refined by an ML-EM algorithm [57]. While the simplicity of the discrete model is a big advantage, its drawbacks are apparent: it does not scale well for large K and ignores the correlation between different functional states of the molecule and thus overlooks important information.
The second approach, referred to as continuous heterogeneity, assumes that ϕ1, …, ϕN can be embedded in a low-dimensional space. For instance, one approach is to assume that the set of conformations lies in a linear subspace (that can be learned using PCA), or in more intricate low-dimensional manifolds (that could be learned by other spectral methods, such as diffusion maps). An alternative suggestion was to model the structure as a set of rigid domains that can move one with respect to the other. We refer the reader to a thorough survey on the subject in [74].
B. Verification
Given a 3-D reconstruction, how do we verify that it is a reliable and faithful representation of the underlying molecule? This is a question of crucial importance for any scientific field. Nevertheless, there are no agreed upon computational verification methods for cryo-EM. Several validation techniques were proposed in the cryo-EM literature. For example, it is possible to check the consistency of the 3-D map by recording pairs of images of the same particles at different tilt angles and comparing the relative angle between orientations assigned to each projection: ideally it should agree with the relative rotation angle of the microscope’s specimen holder used during the experiment. This approach is called the tilt-pair validation technique and is useful only for intermediate resolution structures [40], [61]. In practice, structure validation is based on a set of heuristics and the experts’ knowledge and experience. For instance, it is common to initialize 3-D reconstruction algorithms from multiple random points; if all instances attain similar structures, it serves as a validation fidelity. If the same molecule was reconstituted by other technologies, such as X-ray crystallography and NMR, then structures can be compared. In addition, experimental data is usually uploaded to public repositories and thus other researchers can process the same data, experiment with different computational techniques, and compare the results [61].
Verification is also related to the question of determining the resolution of a recovered structure. The current convention is to reconstruct two structures independently, each from one half of the data. The two subsets are chosen at random. The highest frequency for which the two structures agree (up to some tolerance) determines the resolution [75]. This process can be understood as an indication for the confidence we have in the structure at a given resolution. However, this method is susceptible to systematic flaws; if the same refinement procedure is applied to both halves of the data, it can induce correlated blunders (although the data is independent) and the resolution determination would be unsound. In [24], a simple computational procedure was proposed to validate a structure by assessing the amount of overfitting that is present in the 3-D map. Establishing computational tools that provide confidence intervals to estimated structures and are immunized against systematic errors is one of the remaining challenges in the field.
C. Theoretical foundations of cryo-EM
Many mathematical and statistical properties of the cryo-EM problem are still unexplained (and even unexplored). A prominent example is the sample complexity of the full cryo-EM problem: given a fixed SNR level (that might be very low) and a fixed setup, how many particles are required to achieve a desired resolution? An initial analysis was conducted in [9], [66]; see Section VI-A.
Another interesting question concerns computational complexity and information-computational gaps [11]. The fact that there is enough information to solve a problem (sample complexity) does not immediately imply that there is an efficient (polynomial time) algorithm to solve it. For example, it might be that for non-uniform distribution of rotations the second moment would suffice to estimate a 3-D structure from an information-theoretic perspective [66], but at the same time it could be computationally hard in that regime. In that case, we might need to consider the third-order moment in order to design a computationally efficient algorithm. Information-computational gaps have been observed empirically in variants of the MRA and MTD models [22], [18]. Related questions—that might be even more challenging—deal with the properties of specific algorithms, such as ML-EM, the method of moments, and SGD that are not well-understood.
Another interesting research thread regards the size limit of molecular structures that can be elucidated by cryo-EM. The common belief in the community is that very small molecules cannot be visualized using cryo-EM. The logic is simple: small molecules induce low contrast, and thus low SNR on the micrograph, which in turn hinders detection (particle picking) [38]. A recent paper contends this belief and suggests that it is possible, at least in principle, to reconstruct structures directly from the micrograph, without particle picking and at any SNR level, given enough data [17].
D. Machine learning
The groundbreaking advances in machine learning in the last decade reshaped dramatically many computational fields and penetrated into some scientific applications. Naturally, learning techniques based on deep neural networks have been applied to cryo-EM as well. Examples are particle picking [84], [91], [83] (as discussed in Section V-C), validation [7], 3-D reconstruction [89], and particle pruning [62]. In addition, manifold learning techniques were designed for 3-D heterogeneity analysis [31], [51] and deniosing [45]. Modern learning techniques were also implemented to other cryo-EM applications that do not involve single particle reconstruction; see for instance an application to feature extraction in cellular electron cryotomography [23].
Deep learning gained its popularity in applications with low noise levels. The performance of these techniques in more challenging environments, such as highly contaminated data, is not clear yet. In addition, supervised learning techniques are susceptible to model bias—the reconstruction will depend heavily on the training data, rather than on the experimental images. This explains why the impact of deep learning on the cryo-EM field—especially on the more involved tasks, such as 3-D reconstruction and the heterogeneity problem—is limited at the moment. We expect that more efforts in this direction will be made in the coming years. In particular, it is still to be clarified whether this set of computational tools can outperform current tools in the field, which are based on more classical statistics.
VIII. Perspective
Single particle reconstruction using cryo-EM is an alluring research area for researchers interested in developing modern computational tools and sophisticated mathematical models to an emerging scientific field. In this article, we have introduced the problem of constituting 3-D molecular structures using cryo-EM and described its unique computational characteristics and challenges. We have delineated relations between the cryo-EM inverse problem and a variety of disciplines at the core of signal processing, information theory, statistics, machine learning, and group theory. We believe that contributions from these areas have the potential to drive the field forward. New ideas and solutions can, and should, be tested on experimental cryo-EM datasets publicly available online [1], [2]. We have also reviewed two abstract frameworks to conveniently study the cryo-EM inverse problem from computational, statistical and theoretical perspectives.
We believe that the challenges arising in cryo-EM research provide an ample of opportunities to investigate and test novel algorithms and advanced mathematical techniques in order to impact a task of paramount importance: to broaden our understanding of fundamental mechanisms of life.
Acknowledgment
We thank the anonymous reviewers and the editor for their valuable comments and suggestions. AS was supported in part by Award Number R01GM090200 from the NIGMS, FA9550-17-1-0291 from AFOSR, Simons Foundation Math+X Investigator Award, the Moore Foundation Data-Driven Discovery Investigator Award, and NSF BIGDATA Award IIS-1837992.
Footnotes
References
- [1]. https://www.ebi.ac.uk/pdbe/emdb/empiar/.
- [2]. http://www.ebi.ac.uk/pdbe/emdb/.
- [3].Abbe E, Bendory T, Leeb W, Pereira JM, Sharon N, and Singer A, “Multireference alignment is easier with an aperiodic translation distribution,” IEEE Transactions on Information Theory, vol. 65, no. 6, pp. 3565–3584, 2018. [Google Scholar]
- [4].Abbe E, Pereira JM, and Singer A, “Estimation in the group action channel,” in 2018 IEEE International Symposium on Information Theory (ISIT). IEEE, 2018, pp. 561–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Agarwal S, Furukawa Y, Snavely N, Simon I, Curless B, Seitz SM, and Szeliski R, “Building Rome in a day,” Communications of the ACM, vol. 54, no. 10, pp. 105–112, 2011. [Google Scholar]
- [6].Aguerrebere C, Delbracio M, Bartesaghi A, and Sapiro G, “Fundamental limits in multi-image alignment,” IEEE Transactions on Signal Processing, vol. 64, no. 21, pp. 5707–5722, 2016. [Google Scholar]
- [7].Avramov TK, Vyenielo D, Gomez-Blanco J, Adinarayanan S, Vargas J, and Si D, “Deep learning for validating and estimating resolution of cryo-electron microscopy density maps,” Molecules, vol. 24, no. 6, p. 1181, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bai X-C, McMullan G, and Scheres SH, “How cryo-EM is revolutionizing structural biology,” Trends in biochemical sciences, vol. 40, no. 1, pp. 49–57, 2015. [DOI] [PubMed] [Google Scholar]
- [9].Bandeira AS, Blum-Smith B, Kileel J, Perry A, Weed J, and Wein AS, “Estimation under group actions: recovering orbits from invariants,” arXiv preprint arXiv:1712.10163, 2017. [Google Scholar]
- [10].Bandeira AS, Chen Y, and Singer A, “Non-unique games over compact groups and orientation estimation in cryo-EM,” arXiv preprint arXiv:1505.03840, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bandeira AS, Perry A, and Wein AS, “Notes on computational-to-statistical gaps: predictions using statistical physics,” Portugaliae Mathematica, vol. 75, no. 2, pp. 159–186, 2018. [Google Scholar]
- [12].Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P et al. , “2.3Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition,” Science, vol. 351, no. 6275, pp. 871–875, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Barnett A, Greengard L, Pataki A, and Spivak M, “Rapid solution of the cryo-EM reconstruction problem by frequency marching,” SIAM Journal on Imaging Sciences, vol. 10, no. 3, pp. 1170–1195, 2017. [Google Scholar]
- [14].Barnett AH, Magland J, and af Klinteberg L, “A parallel nonuniform fast Fourier transform library based on an “exponential of semicircle” kernel,” SIAM Journal on Scientific Computing, vol. 41, no. 5, pp. C479–C504, 2019. [Google Scholar]
- [15].Bartesaghi A, Aguerrebere C, Falconieri V, Banerjee S, Earl LA, Zhu X, Grigorieff N, Milne JL, Sapiro G,Wu X, and Subramaniam S, “Atomic resolution cryo-EM structure of β-galactosidase,” Structure, vol. 26, no. 6, pp. 848–856, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bartesaghi A, Merk A, Banerjee S, Matthies D, Wu X, Milne JL, and Subramaniam S, “2.2Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor,” Science, vol. 348, no. 6239, pp. 1147–1151, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bendory T, Boumal N, Leeb W, Levin E, and Singer A, “Toward single particle reconstruction without particle picking: Breaking the detection limit,” arXiv preprint arXiv:1810.00226, 2018. [Google Scholar]
- [18].——, “Multi-target detection with application to cryo-electron microscopy,” Inverse Problems, 2019. [Google Scholar]
- [19].Bendory T, Boumal N, Ma C, Zhao Z, and Singer A, “Bispectrum inversion with application to multireference alignment,” IEEE Transactions on Signal Processing, vol. 66, no. 4, pp. 1037–1050, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bhamre T, Zhang T, and Singer A, “Denoising and covariance estimation of single particle cryo-EM images,” Journal of structural biology, vol. 195, no. 1, pp. 72–81, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bottou L, Curtis FE, and Nocedal J, “Optimization methods for large-scale machine learning,” Siam Review, vol. 60, no. 2, pp. 223–311, 2018. [Google Scholar]
- [22].Boumal N, Bendory T, Lederman RR, and Singer A, “Heterogeneous multireference alignment: A single pass approach,” in 2018 52nd Annual Conference on Information Sciences and Systems (CISS) IEEE, 2018, pp. 1–6. [Google Scholar]
- [23].Chen M, Dai W, Sun SY, Jonasch D, He CY, Schmid MF, Chiu W, and Ludtke SJ, “Convolutional neural networks for automated annotation of cellular cryo-electron tomograms,” Nature methods, vol. 14, no. 10, p. 983, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SH, and Henderson R, “High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy,” Ultramicroscopy, vol. 135, pp. 24–35, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dempster AP, Laird NM, and Rubin DB, “Maximum likelihood from incomplete data via the EM algorithm,” Journal of the Royal Statistical Society: Series B (Methodological), vol. 39, no. 1, pp. 1–22, 1977. [Google Scholar]
- [26].Donoho DL, Gavish M, and Johnstone IM, “Optimal shrinkage of eigenvalues in the spiked covariance model,” Annals of statistics, vol. 46, no. 4, p. 1742, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dubochet J, Lepault J, Freeman R, Berriman J, and Homo J-C, “Electron microscopy of frozen water and aqueous solutions,” Journal of Microscopy, vol. 128, no. 3, pp. 219–237, 1982. [Google Scholar]
- [28].Elbau P, Ritsch-Marte M, Scherzer O, and Schmutz D, “Inverse problems of trapped objects,” arXiv preprint arXiv:1907.01387, 2019. [Google Scholar]
- [29].Frank J and Penczek P, “On the correction of the contrast transfer function in biological electron microscopy,” Optik, vol. 98, no. 3, pp. 125–129, 1995. [Google Scholar]
- [30].Frank J, “Advances in the field of single-particle cryo-electron microscopy over the last decade,” Nature protocols, vol. 12, no. 2, p. 209, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Frank J and Ourmazd A, “Continuous changes in structure mapped by manifold embedding of single-particle data in cryo-EM,” Methods, vol. 100, pp. 61–67, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Grant T and Grigorieff N, “Measuring the optimal exposure for single particle cryo-EM using a 2.6Å reconstruction of rotavirus VP6,” Elife, vol. 4, p. e06980, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Grant T, Rohou A, and Grigorieff N, “cisTEM, user-friendly software for single-particle image processing,” Elife, vol. 7, p. e35383, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Greenberg I and Shkolnisky Y, “Common lines modeling for reference free ab-initio reconstruction in cryo-EM,” Journal of structural biology, vol. 200, no. 2, pp. 106–117, 2017. [DOI] [PubMed] [Google Scholar]
- [35].Greengard L and Lee J-Y, “Accelerating the nonuniform fast Fourier transform,” SIAM review, vol. 46, no. 3, pp. 443–454, 2004. [Google Scholar]
- [36].Guo TW, Bartesaghi A, Yang H, Falconieri V, Rao P, Merk A, Eng ET, Raczkowski AM, Fox T,Earl LA et al. , “Cryo-EM structures reveal mechanism and inhibition of DNA targeting by a CRISPR-Cas surveillance complex,” Cell, vol. 171, no. 2, pp. 414–426, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Heimowitz A, Andén J, and Singer A, “APPLE picker: Automatic particle picking, a low-effort cryo-EM framework,” Journal of structural biology, vol. 204, no. 2, pp. 215–227, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Henderson R, “The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules,” Quarterly reviews of biophysics, vol. 28, no. 2, pp. 171–193, 1995. [DOI] [PubMed] [Google Scholar]
- [39].——, “Avoiding the pitfalls of single particle cryo-electron microscopy: Einstein from noise,” Proceedings of the National Academy of Sciences, vol. 110, no. 45, pp. 18 037–18 041, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Henderson R, Chen S, Chen JZ, Grigorieff N, Passmore LA, Ciccarelli L, Rubinstein JL, Crowther RA, Stewart PL, and Rosenthal PB, “Tilt-pair analysis of images from a range of different specimens in single-particle electron cryomicroscopy,” Journal of molecular biology, vol. 413, no. 5, pp. 1028–1046, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jolliffe I, Principal component analysis. Springer, 2011. [Google Scholar]
- [42].Kam Z, “The reconstruction of structure from electron micrographs of randomly oriented particles,” Journal of Theoretical Biology, vol. 82, no. 1, pp. 15–39, 1980. [DOI] [PubMed] [Google Scholar]
- [43].Kang Y, Kuybeda O, de Waal PW, Mukherjee S, Van Eps N, Dutka P, Zhou XE, Bartesaghi A, Erramilli S, Morizumi T et al. , “Cryo-EM structure of human rhodopsin bound to an inhibitory G protein,” Nature, vol. 558, no. 7711, p. 553, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kühlbrandt W, “The resolution revolution,” Science, vol. 343, no. 6178, pp. 1443–1444, 2014. [DOI] [PubMed] [Google Scholar]
- [45].Landa B and Shkolnisky Y, “The steerable graph Laplacian and its application to filtering image datasets,” SIAM Journal on Imaging Sciences, vol. 11, no. 4, pp. 2254–2304, 2018. [Google Scholar]
- [46].Levis A, Schechner YY, and Talmon R, “Statistical tomography of microscopic life,” in Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, 2018, pp. 6411–6420. [Google Scholar]
- [47].Liao M, Cao E, Julius D, and Cheng Y, “Structure of the TRPV1 ion channel determined by electron cryomicroscopy,” Nature, vol. 504, no. 7478, p. 107, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liu H, Poon BK, Saldin DK, Spence JC, and Zwart PH, “Three-dimensional single-particle imaging using angular correlations from X-ray laser data,” Acta Crystallographica Section A: Foundations of Crystallography, vol. 69, no. 4, pp. 365–373, 2013. [DOI] [PubMed] [Google Scholar]
- [49].Matthies D, Bae C, Toombes GE, Fox T, Bartesaghi A, Subramaniam S, and Swartz KJ, “Single-particle cryo-EM structure of a voltage-activated potassium channel in lipid nanodiscs,” Elife, vol. 7, p. e37558, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA,Milne JL et al. , “Breaking cryo-EM resolution barriers to facilitate drug discovery,” Cell, vol. 165, no. 7, pp. 1698–1707, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Moscovich A, Halevi A, Andén J, and Singer A, “Cryo-em reconstruction of continuous heterogeneity by laplacian spectral volumes,” Inverse Problems, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Nogales E and Scheres SH, “Cryo-EM: a unique tool for the visualization of macromolecular complexity,” Molecular cell, vol. 58, no. 4, pp. 677–689, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pananjady A, Wainwright MJ, and Courtade TA, “Denoising linear models with permuted data,” in 2017 IEEE International Symposium on Information Theory (ISIT) IEEE, 2017, pp. 446–450. [Google Scholar]
- [54].Pearson K, “On lines and planes of closest fit to systems of points in space,” The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science, vol. 2, no. 11, pp. 559–572, 1901. [Google Scholar]
- [55].Penczek PA, Renka R, and Schomberg H, “Gridding-based direct Fourier inversion of the three-dimensional ray transform,” JOSA A, vol. 21, no. 4, pp. 499–509, 2004. [DOI] [PubMed] [Google Scholar]
- [56].Pragier G and Shkolnisky Y, “A common lines approach for ab-initio modeling of cyclically-symmetric molecules,” arXiv preprint arXiv:1901.10888, 2019. [Google Scholar]
- [57].Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA, “cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination,” Nature methods, vol. 14, no. 3, p. 290, 2017. [DOI] [PubMed] [Google Scholar]
- [58].Ripstein Z and Rubinstein J, “Processing of cryo-EM movie data,” in Methods in enzymology. Elsevier, 2016, vol. 579, pp. 103–124. [DOI] [PubMed] [Google Scholar]
- [59].Rohou A and Grigorieff N, “CTFFIND4: Fast and accurate defocus estimation from electron micrographs,” Journal of structural biology, vol. 192, no. 2, pp. 216–221, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rosen DM, Carlone L, Bandeira AS, and Leonard JJ, “SE-sync: A certifiably correct algorithm for synchronization over the special Euclidean group,” The International Journal of Robotics Research, vol. 38, no. 2–3, pp. 95–125, 2019. [Google Scholar]
- [61].Rosenthal PB and Rubinstein JL, “Validating maps from single particle electron cryomicroscopy,” Current opinion in structural biology, vol. 34, pp. 135–144, 2015. [DOI] [PubMed] [Google Scholar]
- [62].Sanchez-Garcia R, Segura J, Maluenda D, Carazo JM, and Sorzano COS, “Deep consensus, a deep learning-based approach for particle pruning in cryo-electron microscopy,” IUCrJ, vol. 5, no. 6, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Scheres SH, “RELION: implementation of a Bayesian approach to cryo-EM structure determination,” Journal of structural biology, vol. 180, no. 3, pp. 519–530, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].——, “Semi-automated selection of cryo-EM particles in RELION-1.3,” Journal of structural biology, vol. 189, no. 2, pp. 114–122, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Scheres SH, Valle M, Nuñez R, Sorzano CO, Marabini R, Herman GT, and Carazo J-M, “Maximum-likelihood multi-reference refinement for electron microscopy images,” Journal of molecular biology, vol. 348, no. 1, pp. 139–149, 2005. [DOI] [PubMed] [Google Scholar]
- [66].Sharon N, Kileel J, Khoo Y, Landa B, and Singer A, “Method of moments for 3-D single particle ab initio modeling with non-uniform distribution of viewing angles,” arXiv preprint arXiv:1907.05377, 2019. [Google Scholar]
- [67].Shatsky M, Hall RJ, Brenner SE, and Glaeser RM, “A method for the alignment of heterogeneous macromolecules from electron microscopy,” Journal of structural biology, vol. 166, no. 1, pp. 67–78, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sigworth F, “A maximum-likelihood approach to single-particle image refinement,” Journal of structural biology, vol. 122, no. 3, pp. 328–339, 1998. [DOI] [PubMed] [Google Scholar]
- [69].Sindelar CV and Grigorieff N, “An adaptation of the Wiener filter suitable for analyzing images of isolated single particles,” Journal of structural biology, vol. 176, no. 1, pp. 60–74, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Singer A, “Angular synchronization by eigenvectors and semidefinite programming,” Applied and computational harmonic analysis, vol. 30, no. 1, pp. 20–36, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].——, “Mathematics for cryo-electron microscopy,” Proceedings of the International Congress of Mathematicians, 2018. [Google Scholar]
- [72].Singer A and Shkolnisky Y, “Three-dimensional structure determination from common lines in cryo-EM by eigenvectors and semidefinite programming,” SIAM journal on imaging sciences, vol. 4, no. 2, pp. 543–572, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Sorzano C, Bilbao-Castro J, Shkolnisky Y, Alcorlo M, Melero R, Caffarena-Fernández G, Li M, Xu G,Marabini R, and Carazo J, “A clustering approach to multireference alignment of single-particle projections in electron microscopy,” Journal of structural biology, vol. 171, no. 2, pp. 197–206, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sorzano C, Jiménez A, Mota J, Vilas J, Maluenda D, Martínez M, Ramírez-Aportela E, Majtner T, Segura J, Sánchez-García R et al. , “Survey of the analysis of continuous conformational variability of biological macromolecules by electron microscopy,” Acta Crystallographica Section F: Structural Biology Communications, vol. 75, no. 1, pp. 19–32, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sorzano C, Vargas J, Otón J, Abrishami V, de la Rosa-Trevín J, Gómez-Blanco J, Vilas J, Marabini R, and Carazo J, “A review of resolution measures and related aspects in 3D electron microscopy,” Progress in biophysics and molecular biology, vol. 124, pp. 1–30, 2017. [DOI] [PubMed] [Google Scholar]
- [76].Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ, “EMAN2: an extensible image processing suite for electron microscopy,” Journal of structural biology, vol. 157, no. 1, pp. 38–46, 2007. [DOI] [PubMed] [Google Scholar]
- [77].Tukey J, “The spectral representation and transformation properties of the higher moments of stationary time series,” Reprinted in The Collected Works of John W. Tukey, vol. 1, pp. 165–184, 1953. [Google Scholar]
- [78].Vainshtein B and Goncharov A, “Determination of the spatial orientation of arbitrarily arranged identical particles of unknown structure from their projections,” in Soviet Physics Doklady, vol. 31, 1986, p. 278. [Google Scholar]
- [79].Van Heel M, “Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction,” Ultramicroscopy, vol. 21, no. 2, pp. 111–123, 1987. [DOI] [PubMed] [Google Scholar]
- [80].Van Heel M and Frank J, “Use of multivariates statistics in analysing the images of biological macromolecules,” Ultramicroscopy, vol. 6, no. 1, pp. 187–194, 1981. [DOI] [PubMed] [Google Scholar]
- [81].Vinothkumar KR and Henderson R, “Single particle electron cryomicroscopy: trends, issues and future perspective,” Quarterly reviews of biophysics, vol. 49, 2016. [DOI] [PubMed] [Google Scholar]
- [82].von Ardenne B, Mechelke M, and Grübmuller H, “Structure determination from single molecule X-ray scattering with three photons per image,” Nature communications, vol. 9, no. 1, p. 2375, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wagner T, Merino F, Stabrin M, Moriya T, Antoni C, Apelbaum A, Hagel P, Sitsel O, Raisch T, Prumbaum D et al. , “SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM,” Communications Biology, vol. 2, no. 1, p. 218, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wang F, Gong H, Liu G, Li M, Yan C, Xia T, Li X, and Zeng J, “Deeppicker: a deep learning approach for fully automated particle picking in cryo-EM,” Journal of structural biology, vol. 195, no. 3, pp. 325–336, 2016. [DOI] [PubMed] [Google Scholar]
- [85].Zhang K, “Gctf: Real-time CTF determination and correction,” Journal of structural biology, vol. 193, no. 1, pp. 1–12, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zhao Z, Shkolnisky Y, and Singer A, “Fast steerable principal component analysis,” IEEE Transactions on Computational Imaging, vol. 2, no. 1, pp. 1–12, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Zhao Z and Singer A, “Rotationally invariant image representation for viewing direction classification in cryo-EM,” Journal of structural biology, vol. 186, no. 1, pp. 153–166, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA, “MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy,” Nature methods, vol. 14, no. 4, p. 331, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zhong ED, Bepler T, Davis JH, and Berger B, “Reconstructing continuously heterogeneous structures from single particle cryo-EM with deep generative models,” arXiv preprint arXiv:1909.05215, 2019. [Google Scholar]
- [90].Zhou Y, Moscovich A, Bendory T, and Bartesaghi A, “Unsupervised particle sorting for high-resolution single-particle cryo-EM,” arXiv preprint arXiv:1910.10051, 2019. [Google Scholar]
- [91].Zhu Y, Ouyang Q, and Mao Y, “A deep convolutional neural network approach to single-particle recognition in cryo-electron microscopy,” BMC bioinformatics, vol. 18, no. 1, p. 348, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zivanov J, Nakane T, and Scheres SH, “A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis,” IUCrJ, vol. 6, no. 1, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]