

Abstract
Herein, we report a modular synthetic route to linear and branched homoallylic amines that operates through a sequential one-pot Lewis base/transition-metal catalyzed allylic alkylation/Hofmann rearrangement strategy. This protocol is operationally trivial, proceeds from simple and easily prepared substrates and catalysts, and enables all aspects of regio- and stereoselectivity to be controlled through a conserved experimental protocol. Overall, the high levels of enantio-, regio-, and diastereoselectivity obtained, in concert with the ability to access orthogonally protected or free amines, render this a straightforward and effective approach for the preparation of useful enantioenriched homoallylic amines. We have also demonstrated the utility of the products in the context of pharmaceutical synthesis.
Keywords: alkylation, amines, ammonium enolates, palladium, rearrangement
Introduction
Enantiomerically enriched carbinamines are among the most prevalent structural motifs in bioactive alkaloids and pharmaceuticals (Figure 1a).[1] They also constitute a diversity of essential chemical building blocks for applications in asymmetric synthesis and catalysis, and methods for their synthesis have been intensely pursued. Homoallylic amines, in particular, are widely employed for the synthesis of unnatural amino acids and bespoke heterocycles through the elaboration of the embedded amine and alkene functional groups.[2] Therefore, the development of operationally straightforward, direct, and modular synthetic routes to homoallylic amines that are capable of incorporating a diversity of functionality whilst simultaneously controlling reaction regio- and stereoselectivity is of continuing importance.
Figure 1.
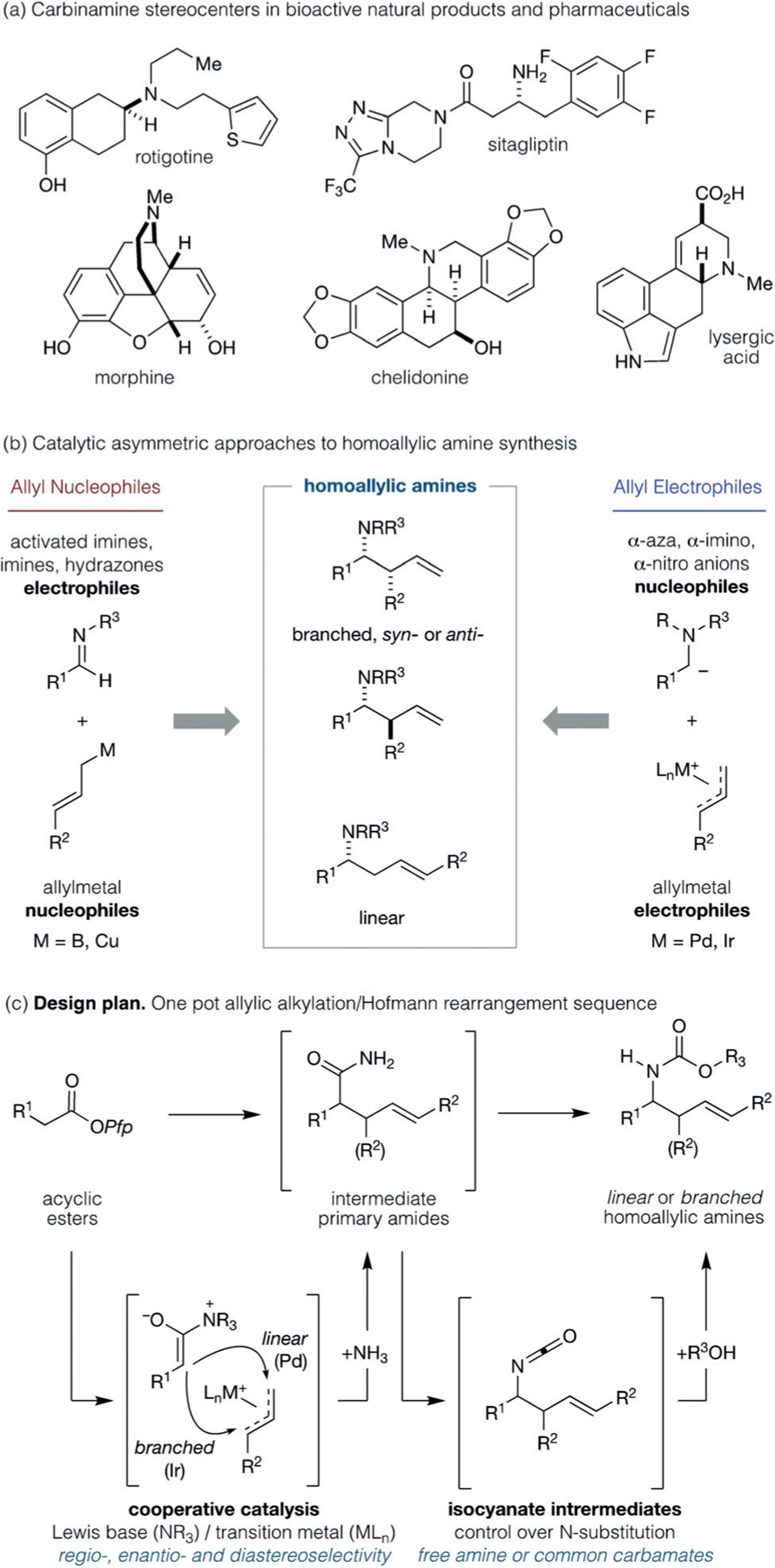
a) Examples of secondary-amine-containing bioactive molecules. b) Complementary polarity approaches to catalytic asymmetric homoallylic amine synthesis. c) This work: A modular, one-pot synthesis of homoallylic amines featuring a cooperative Lewis base/ transition-metal catalyzed regio- and stereodivergent allylic alkylation/ Hofmann rearrangement strategy.
The catalytic asymmetric synthesis of homoallylic amines continues to be the subject of considerable effort. The catalyzed addition of allylmetal nucleophiles to imine (and related) electrophiles has attracted significant attention (Figure 1b, left).[3] Transition-metal and Brønsted acid catalyzed addition of allylboron nucleophiles is especially effective and provides branched products with high levels of enantio- and diastereoselectivity.[4] More recently, allylcopper nucleophiles, which can be conveniently prepared in situ, were shown to undergo enantioselective reactions with aldimines and ketimines under the action of both metal and non-metal catalysts.[4i–k] The complementary polarity approach using π-allylmetal electrophiles (Figure 1b, right) has also been explored. Nitro-stabilized anions,[5–7] glycine-derived metal-loenolates,[8–10] and fluorenyl imine derived anions[11] have been most intensively studied and are effective nucleophiles for a host of enantioselective Pd- and Ir-catalyzed allylic alkylation reactions. While each of these methods is effective and efficient, they cannot be easily modified to rationally prepare linear or branched homoallylic amines. Thus a general and flexible process for their preparation remains a significant challenge.
As part of a long-standing program directed towards catalyzed and stereocontrolled carbon–carbon bond formation, we sought to provide a straightforward, operationally trivial, and modular-catalysis-based protocol for the regioand stereodivergent[12] synthesis of enantioenriched homoallylic amines.[13] Principal among our concerns was catalyst control over all aspects of selectivity during carbon-carbon bond formation, whilst also working within a useful N-protecting group regime. Herein, we describe such a protocol, which provides a straightforward and modular preparation of these valuable building blocks.
Reaction Design
We have invested considerable effort in the development of Lewis base/transition-metal cooperative catalysis to control enantioselective C(sp3)−C(sp3) bond formation between acyclic pentafluorophenyl esters (Pfp esters, hereafter) and allyl electrophiles.[14,15] This versatile platform proceeds via the union of C1-ammonium enolate nucleophiles[16,17] and π(allyl)Pd electrophiles,[18] and provides highly enantioenriched and functionalized α-branched esters that can be readily diversified without compromising optical purity.[19] Two crucial features of this orthogonal regime are relevant to our proposed synthesis of homoallylic amines (Figure 1c). First, all aspects of enantio-, regio-, and diastereodivergent C(sp3)−C(sp3) formation could be controlled by judicious choice of the Lewis base/transition-metal catalyst combination (see Figure 1c, cooperative catalysis). Second, rapid in situ conversion of product Pfp esters (not shown) into intermediate primary amides[14b,20] would permit subsequent stereospecific Hofmann-type rearrangement[21,22] to forge the necessary C(sp3)−N bond via isocyanates (see Figure 1c, isocyanate intermediates). These could be intercepted with an appropriate alcohol (R1-OH) to give the corresponding carbamate-protected or unprotected enantioenriched linear or branched homoallylic amines. We also expected that the projected efficiency of each constituent transformation in the sequence would provide ample opportunity for a sequential single-pot process.
Herein, we report the successful realization of this design, which provides a modular, operationally trivial, and convenient one-pot enantioselective process for the regio- and stereodivergent preparation of homoallylic amines.[23]
Results and Discussion
Oxidant Assessment and Optimization of a One-pot Process
Our initial efforts focused on identifying an appropriate oxidant to induce stereospecific Hofmann-type C→N rearrangement of enantioenriched amides.[24,25] In the presence ! of methanol, a variety of oxidants effectively mediated the Hofmann rearrangement of (R)-2-(4-methoxyphenyl)pent-4-enamide A, giving the corresponding methyl carbamate protected homoallylic amine (Table 1).[25a] We initially identified [bis(trifluoroacetoxy)iodo]benzene (PIFA, 1.5 equiv) in combination with potassium hydroxide (2.5 equiv) in methanol as an effective system, which gave the rearranged product in 90% yield upon isolation and with complete stereotransfer (96:4 er; entry 1). Further optimization (entries 6–9) resulted in a mixed solvent system of THF/MeOH in 2:1 ratio at 60°C from which the desired homoallylic methyl carbamate was isolated in quantitative yield (entry 9; for the full solvent screening, see the Supporting Information). These conditions obviated the need for potassium hydroxide, which not only simplified the protocol considerably but also raised the prospect of greater functional group tolerance. These conditions were then applied to a streamlined cooperative catalysis/rearrangement process. When Birman’s benzotetramisole (BTM)[26] was employed in combination with Buchwald’s Xantphos-ligated 3rd generation Pd precatalyst (XantphosPd G3),[27] allylation by enantioselective cooperative catalysis was followed by instantaneous formation of the corresponding primary amide by bubbling NH3 gas through the reaction mixture. Thereafter, treatment of the reaction mixture with PIFA and MeOH gave the methyl carbamate protected homoallylic amine in 67% yield (entry 10). Increasing the stoichiometry of PIFA to 2 equivalents gave the carbamate in an enhanced 73% isolated yield and with excellent enantioenrichment (97:3 er, Entry 11).
Table 1:
Optimization of the oxidative Hofmann rearrangement and application to the full one-pot allylation/Hofmann rearrangement sequence.
 | |||||
|---|---|---|---|---|---|
| Entry[a] | Oxidant | Base | Solvent | T [°C] | Yield [%][b] |
| 1 | PIFA | KOH | MeOH | r.t. | 90 |
| 2 | PIDA | KOH | MeOH | r.t. | 80 |
| 3 | PhI/oxone | – | MeOH | r.t. | - |
| 4 | Pb(OAc)4 | – | MeOH | r.t. | 89 |
| 5 | NBS | DBU | MeOH | r.t. | 76 |
| 6 | PIFA | KOH | THF/MeOH (25:1) | r.t. | 43 |
| 7 | PIFA | KOH | THF/MeOH (2:1) | r.t. | 66 |
| 8 | PIFA | – | THF/MeOH (2:1) | r.t. | 80 |
| 9 | PIFA | – | THF/MeOH (2:1) | 60 | 99 |
| 10[c] | PIFA | – | THF/MeOH (2:1) | 60 | 67 |
| 11[c,d] | PIFA | – | THF/MeOH (2:1) | 60 | 74 (73) |
Reactions run on 0.1 mmol scale from isolated amide.
1H NMR yield calculated using internal standard.
Full one-pot procedure starting from the Pfp ester (0.1 mmol scale).
With 2.0 equiv of PIFA.
Linear Homoallylic Amine Synthesis with Palladium
With these optimized conditions in hand, we examined the nucleophile scope (Scheme 1). A range of aryl and alkenyl acetic acid esters successfully afforded the corresponding carbamate-protected homoallylic amines in good isolated yields and with high levels of enantioenrichment. Noteworthy is the tolerance of aryl halides (5, 7, 8, and 10) electron-rich arenes (2–4, 6), and π-extended arenes (9 and 10) to this oxidative protocol. Finally, both conjugated and non-conjugated alkene-substituted esters were effective (see products 11–14); significant is the preparation of 14, which is chiral only by virtue of the alkene position. As yet, we have been unsuccessful in our attempts to engage either aliphatic esters or terminal alkyl-substituted electrophiles in this chemistry; both are the subject of ongoing study.
Scheme 1.

Preparation of enantioenriched linear homoallylic amines: Evaluation of nucleophile scope, N-substitution (via preparation of common carbamate-protected amines and free amine), and electrophile scope. A combination of these components in the synthesis of 37, a key intermediate in the synthesis of sertraline (38).
We then evaluated the scope of N-substitution (Scheme 1). In many approaches to homoallylic amine synthesis, nitrogen substitution needs to be tailored to enhance reactivity and/or provide a site for catalyst engagement. Here, simply changing the identity of the alcohol used to trap the putative isocyanates derived from oxidative Hofmann-type rearrangement provides access to the most commonly encountered and synthetically orthogonal carbamate protective groups;[28] tert-butoxycarbonyl- (Boc, 15), allyloxycarbonyl-(Alloc, 16), benzyloxycarbonyl- (Cbz, 17), and 2-(trimethylsilyl)ethoxycarbonyl-protected (Teoc, 18) amines were all obtained with high levels of enantioselectivity. Furthermore, employing water as the nucleophile provides the corresponding free amine 19 with similar enantioenrichment. Here, the lower isolated yield (42%) is due to more difficult chromatographic purification rather than compromised protocol efficiency, as evidenced by spectroscopic analysis of the crude material. Thereafter, we examined the scope of allyl sulfonate electrophiles (Scheme 1). Employing Pd[P(2-thienyl)3]3 (B) as the catalyst[29] enabled 2-substituted allyl electrophiles to react effectively, giving functionalized homoallylic amines 20–22,[14b] the latter containing a trimethylsilylacetylene substituent. Using the same catalyst, ester (23 and 24), Weinreb amide (25), and secondary amide (26) substituted allyl electrophiles were also effective.[14f] Recourse to P(2-furyl)3 (C) as a supporting ligand enabled the synthesis of silyl-substituted products 28 and 29.[14c] Functionalized electrophiles 23–29 are particularly noteworthy as their incorporation by allylmetal addition to imines would, beyond challenges associated with enantio- and regioselectivity, be precluded due to functional group compatibility issues and concerns over α- versus γ-nucleophile addition.[30] Finally, the preparation of an α,β-unsaturated ester substituted homobenzylic amine 27 was possible using a 2-naphthylic phenylphosphate electro-phile in combination with XantphosPd G3.[14d] Again, preparation of 27 by common imine addition reactions would be problematic. The major limitation of this process concerns the poor compatibility of the styrenyl motif within the electrophile scaffold, which leads to numerous unidentifiable products during the oxidative rearrangement. To address this limitation, benzyldimethylsilyl-substituted carbamate 29 was directly elaborated in diverse fashion by Pd-catalyzed Hiyama cross-couplings[31,32] with a range of (hetero)aryl and alkenyl iodides without erosion of enantioenrichment (Scheme 2; 30–35); ketone- (32), indole- (33), pyridine-(34), and dienoate-containing (35) products are all accessible from a common intermediate (29). We expect that late-stage diversification in this manner will prove convenient for the preparation of varied homoallylic amine structures of interest to therapeutic development. Finally, to demonstrate the utility of this method to pharmaceutically relevant molecules, we prepared Boc-protected homoallylic amine 36 using the protocol described and elaborated this compound by Hiyama cross-coupling to 37, which has been converted into selective serotonin reuptake inhibitor sertraline 38 (Scheme 1).[11a,33]
Scheme 2.
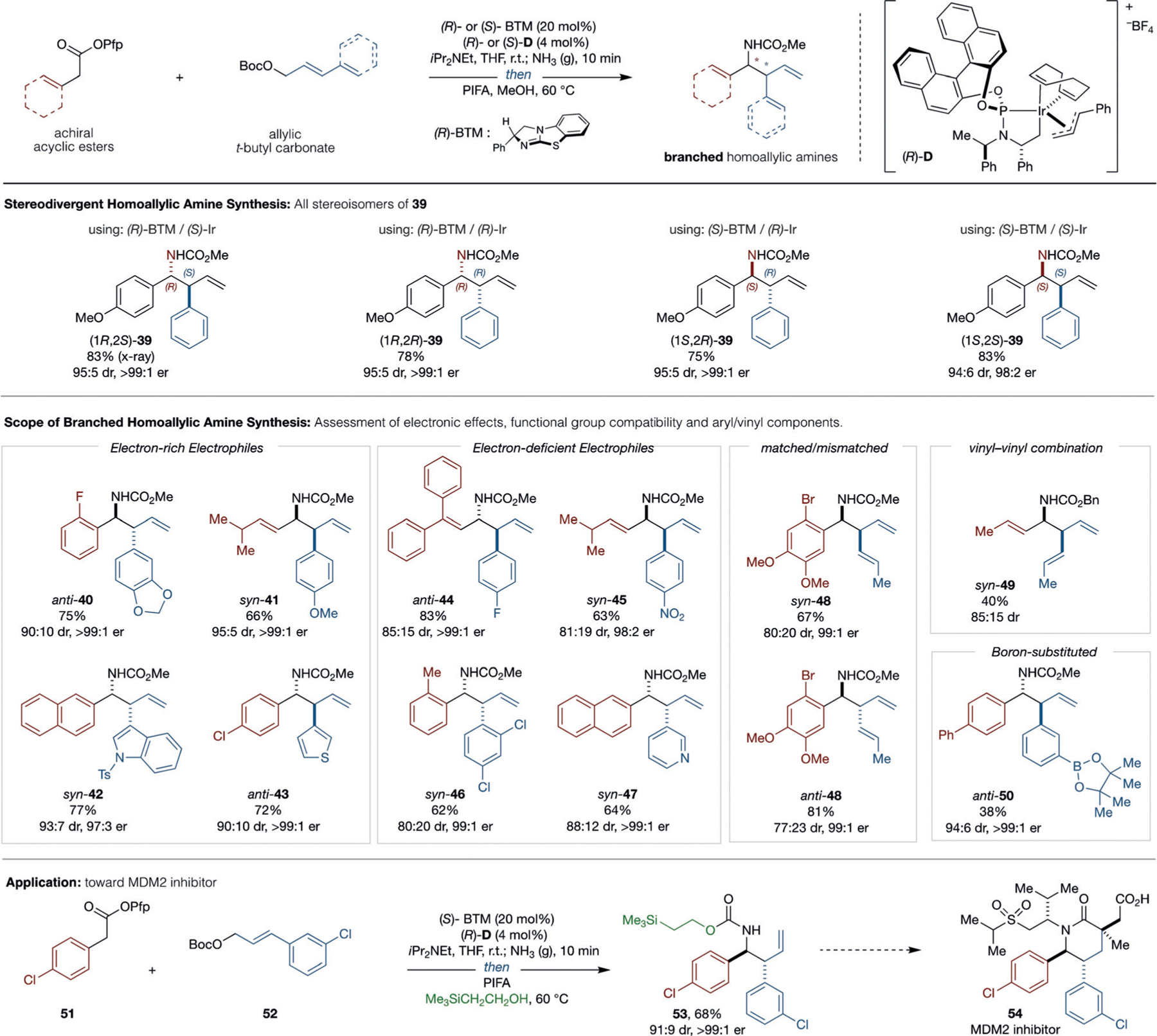
Preparation of enantioenriched branched homoallylic amines: Fully stereodivergent preparation of branched homoallylic amines 39 with different catalyst enantiomer combinations. Unbiased evaluation of scope by nucleophile/electrophile combinations and application to the synthesis of the MDM2 inhibitor core scaffold 53.
Branched Homoallylic Amine Synthesis with Iridium
Having established a general and modular protocol for the synthesis of carbamate-protected linear homoallylic amines, we sought to address the synthesis of branched regioisomers with control over diastereoselectivity (Scheme 2). We expected that this protocol would translate to the corresponding branched-selective cooperative BTM/iridium-catalyzed process, where control over both stereogenic centers should proceed under the independent direction of Lewis base and Ir catalysts, respectively, and provide stereodivergent access to any stereoisomer.[15b] Employing Hartwig’s cyclometalated iridum(I) phosphoramidite (S)-D [(S)-Ir] in combination with (R)-BTM within the same experimental protocol gave (1R,2S)-39 in 83% yield, 95:5 dr, and as a single enantiomer (for detailed optimization, see the Supporting Information). As previously described,[15b] judicious choice of catalyst permutations during allylic alkylation provides stereodivergent access to all possible stereoisomers of 39 with similar efficiency. Thus this protocol constitutes a formal catalytic stereodivergent cinnamylation of imines, and represents a completely catalyst-controlled evolution of Leighton’s branched-selective reagent-controlled synthesis of homoallylic amines.[34] As the stereodivergent nature of the allylic alkylation has previously been confirmed,[15b] we assessed the branched-selective synthesis of homoallylic amines with unbiased nucleophile, electrophile, and catalyst combinations. As expected, in this process, both aryl and alkenyl nucleophiles were effective in combination with a range of electro-philic partners (40–50). Furthermore, halide, nitroarene, indole, thiophene, pyridine, and pinacol borane ester moieties are all tolerated. More specifically, electron-rich aryl (anti-40, syn-41), indole (syn-42), and thiophene (anti-43) substituted electrophiles all provided the corresponding products with high levels of diastereo- and enantioselectivity, thus demonstrating excellent individual catalyst control. Interrogation of electron-deficient F-, NO2-, and Cl-substituted aryl electrophiles (anti-44, syn-45, syn-46) revealed an unexpected influence on diastereoselectivity (ca. 80:20). In each case, the major diastereoisomer was produced in high enantiopurity. This was also true when employing an electron-deficient pyridyl-substituted heteroaromatic electrophile (syn-47). Similarly, alkene-substituted electrophiles also exhibited more modest diastereoselectivity, which does not arise due to matched–mismatched interaction between catalysts (see syn-48 vs. anti-48). Again, the exceptional enantioselectivity of the major diastereoisomer is unaffected. Preparation of syn-49 demonstrated the tolerance of vinyl substituents on both reaction components. Boronic ester substituted electrophiles (anti-50) were also tolerated and provide another synthetic handle for further functionalization. Finally, this procedure was used to construct the core scaffold 53 of the MDM2 inhibitor 54[35] with excellent control over both diastereo- and enantioselectivity. Overall, when integrated with BTM/Ir-catalyzed allylic alkylation, this one-pot process provides robust stereocontrolled access to branched homo-allyl amines with incorporation of diverse functionality.[36]
Conclusion
By pairing cooperative Lewis base/transition-metal catalysis with stereospecific C–N bond formation, we have developed a unified and operationally simple experimental protocol for the catalytic enantioselective synthesis of homoallylic amines. This process exhibits broad functional group tolerance, and enantio-, regio-, and diastereoselectivity are addressed by judicious choice of the appropriate Lewis base/ transition-metal catalyst combination. Finally, in situ stereospecific C–N rearrangement enables nitrogen substitution to be augmented within the confines of a useful protecting group regime. Limitations to this process include the sensitivity of styrenyl motifs to the oxidation step; however, this can be circumvented by elaboration of a vinylsilane motif through Hiyama cross-coupling. Finally, neither aliphatic esters nor terminal-alkyl-substituted electrophiles are productive partners in this process; both aspects are the subject of current study within our laboratory. Nonetheless, we expect that this operationally straightforward and modular protocol will prove useful to those researchers requiring access to enantioenriched homoallylic amines. The starting Pfp esters are inexpensive to prepare in a single step from the precursor acids, and are of established utility as acyl donors for peptide coupling.[20] Similarly, the electrophiles used can be readily accessed using robust methods. Overall, this study demonstrates the potential of combining simultaneous catalysis events with subsequent value-added transformations in stereoselective chemical synthesis.
Supplementary Material
Acknowledgements
We gratefully acknowledge the National Institutes of Health (R01GM121573) for generous financial support. We also thank Dr. Maren Pink (IU) for X-ray crystallography.
Footnotes
Dedicated to Professor Alois Fürstner
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201905426.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Lawrence SA, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 2004. [Google Scholar]
- [2].Weiner B, Szymański W, Janssen DB, Minnaard AJ, Feringa BL, Chem. Soc. Rev 2010, 39, 1656–1691. [DOI] [PubMed] [Google Scholar]
- [3]. For reviews, see:; a) Yus M, Gonzàlez-Gómez JC, Foubelo F, Chem. Rev 2013, 113, 5595–5698; [DOI] [PubMed] [Google Scholar]; b) Yus M, Gonzàlez-Gómez JC, Foubelo F, Chem. Rev 2011, 111, 7774–7854; [DOI] [PubMed] [Google Scholar]; c) Kobayashi S, Mori Y, Fossey JS, Salter MM, Chem. Rev 2011, 111, 2626–2704. [DOI] [PubMed] [Google Scholar]
- [4]. For reviews, see:; a) Diner C, Szabo KJ, J. Am. Chem. Soc 2017, 139, 2–14; [DOI] [PubMed] [Google Scholar]; b) Ramadhar TR, Batey RA, Synthesis 2011, 1321–1346; [Google Scholar]; c) Huo H-X, Duvall JR, Huang M-Y, Hong R, Org. Chem. Front 2014, 1, 303–320; for selected examples, see: [Google Scholar]; d) Fujita M, Nagano T, Schneider U, Hamada T, Ogawa C, Kobayashi S, J. Am. Chem. Soc 2008, 130, 2914–2915; [DOI] [PubMed] [Google Scholar]; e) Jonker SJ, Diner C, Schulz G, Iwamoto H, Eriksson L, Szabó K, Chem. Commun 2018, 54, 12852–12855; [DOI] [PubMed] [Google Scholar]; f) Morrison RJ, Hoveyda AH, Angew. Chem. Int. Ed 2018, 57, 11654–11661; Angew. Chem. 2018, 130, 11828 – 11835; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Jang H, Romiti F, Torker S, Hoveyda AH, Nat. Chem 2017, 9, 1269–1275; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) van der Mei FW, Miyamoto H, Silverio DL, Hoveyda AH, Angew. Chem. Int. Ed 2016, 55, 4701–4706; Angew. Chem. 2016, 128, 4779 – 4784; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Silverio DL, Torker S, Pilyugina T, Vieira EM, Snapper ML, Haeffner F, Hoveyda AH, Nature 2013, 494, 216–221; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Vieira EM, Snapper ML, Hoveyda AH, J. Am. Chem. Soc 2011, 133, 3332–3335; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Lou S, Moquist PN, Schaus SE, J. Am. Chem. Soc 2007, 129, 15398–15404; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Yeung K, Ruscoe RE, Rae J, Pulis AP, Procter DJ, Angew. Chem. Int. Ed 2016, 55, 11912–11916; Angew. Chem. 2016, 128, 12091 – 12095; [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Alam B, Diner C, Jonker S, Eriksson L, Szabo KJ, Angew. Chem. Int. Ed 2016, 55, 14417–14421; Angew. Chem. 2016, 128, 14629 – 14633; [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Zhang P, Roundtree IA, Morken JP, Org. Lett 2012, 14, 1416–1419; [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Dutheuil G, Selander N, Szabo KJ, Aggarwal VK, Synthesis 2008, 14, 2293–2297; [Google Scholar]; p) Murata M, Watanabe S, Masuda Y, Tetrahedron Lett. 2000, 41, 5877–5880; [Google Scholar]; q) Ishiyama T, Ahiko T-A, Miyaura N, Tetrahedron Lett. 1996, 37, 6889–6892. [Google Scholar]
- [5].a) Trost BM, Surivet J-P, Angew. Chem. Int. Ed 2000, 39, 3122–3124; Angew. Chem. 2000, 112, 3252 – 3254; [DOI] [PubMed] [Google Scholar]; b) Trost BM, Surivet J-P, J. Am. Chem. Soc 2000, 122, 6291–6292. [Google Scholar]
- [6].Ohmatsu K, Ito M, Kunieda T, Ooi T, Nat. Chem 2012, 4, 473–477. [DOI] [PubMed] [Google Scholar]
- [7].Yang X-F, Yu W-H, Ding C-H, Ding Q-P, Wan S-L, Hou X-L, Dai L-X, Wang P-J, J. Org. Chem 2013, 78, 6503–6509. [DOI] [PubMed] [Google Scholar]
- [8].Nakoji M, Kanayama T, Okino T, Takemoto Y, J. Org. Chem 2002, 67, 7418–7423. [DOI] [PubMed] [Google Scholar]
- [9].Kanayama T, Yoshida K, Miyabe H, Takemoto Y, Angew. Chem. Int. Ed 2003, 42, 2054–2056; Angew. Chem. 2003, 115, 2100 – 2102. [DOI] [PubMed] [Google Scholar]
- [10].a) Huo X, Zhang J, Fu J, He R, Zhang W, J. Am. Chem. Soc 2018, 140, 2080–2084; [DOI] [PubMed] [Google Scholar]; b) Huo X, He R, Fu J, Zhang J, Yang G, Zhang W, J. Am. Chem. Soc 2017, 139, 9819–9822. [DOI] [PubMed] [Google Scholar]
- [11].a) Liu J, Cao C-G, Sun H-B, Zhang X, Niu D, J. Am. Chem. Soc 2016, 138, 13103–13106; [DOI] [PubMed] [Google Scholar]; b) Wan L, Tian L, Liu J, Niu D, Synlett 2017, 28, 2051–2056. [Google Scholar]
- [12]. For a recent review on catalyst-controlled stereodivergence, see:; a) Krautwald S, Carreira EM, J. Am. Chem. Soc 2017, 139, 5627–5639; for selected additional examples, see: [DOI] [PubMed] [Google Scholar]; b) Cruz FA, Dong VM, J. Am. Chem. Soc 2017, 139, 1029–1032; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kassem S, Lee AT, Leigh DA, Marcos V, Palmer LI, Pisano S, Nature 2017, 549, 374–378; [DOI] [PubMed] [Google Scholar]; d) Sandmeier T, Krautwald S, Zipfel HF, Carreira EM, Angew. Chem. Int. Ed 2015, 54, 14363–14367; Angew. Chem. 2015, 127, 14571 – 14575; [DOI] [PubMed] [Google Scholar]; e) Krautwald S, Schafroth MA, Sarlah D, Carreira EM, J. Am. Chem. Soc 2014, 136, 3020–3023; [DOI] [PubMed] [Google Scholar]; f) Krautwald S, Sarlah D, Schafroth MA, Carreira EM, Science 2013, 340, 1065–1068. [DOI] [PubMed] [Google Scholar]
- [13]. For an excellent modular stereodivergent approach to enantioenriched allylic amines bearing vicinal stereogenic centers via three sequential catalyst-controlled reactions, see:; Tosatti P, Campbell AJ, House D, Nelson A, Marsden SP, J. Org. Chem 2011, 76, 5495–5510. [DOI] [PubMed] [Google Scholar]
- [14].a) Schwarz KJ, Amos JL, Klein JC, Do DT, Snaddon TN, J. Am. Chem. Soc 2016, 138, 5214–5217; [DOI] [PubMed] [Google Scholar]; b) Schwarz KJ, Pearson CM, Cintron-Rosado GA, Liu P, Snaddon TN, Angew. Chem. Int. Ed 2018, 57, 7800–7803; Angew. Chem. 2018, 130, 7926 – 7929; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fyfe JWB, Kabia OM, Pearson CM, Snaddon TN, Tetrahedron 2018, 74, 5383–5391; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Schwarz KJ, Yang C, Fyfe JWB, Snaddon TN, Angew. Chem. Int. Ed 2018, 57, 12102–12105; Angew. Chem. 2018, 130, 12278 – 12281; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Scaggs WR, Snaddon TN, Chem. Eur. J 2018, 24, 14378–14381; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Hutchings-Goetz L, Yang C, Snaddon TN, ACS Catal. 2018, 8, 10537–10544; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Rush Scaggs W, Scaggs TD, Snaddon TN, Org. Biomol. Chem 2019, 17, 1787–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. For other examples of C1-ammonium enolates being employed in conjunction with transition-metal catalysis, see:; a) Spoehrle SS, West TH, Taylor JE, Slawin AMZ, Smith AD, J. Am. Chem. Soc 2017, 139, 11895–11902; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jiang Z, Beiger JJ, Hartwig JF, J. Am. Chem. Soc 2017, 139, 87–90; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lu X, Ge L, Cheng C, Chen J, Cao W, Wu X, Chem. Eur. J 2017, 23, 7689–7693; [DOI] [PubMed] [Google Scholar]; d) Song J, Zhang Z-J, Gong L-Z, Angew. Chem. Int. Ed 2017, 56, 5212–5216; Angew. Chem. 2017, 129, 5296 – 5300; [DOI] [PubMed] [Google Scholar]; e) Song J, Zhang Z-J, Chen S-S, Fan T, Gong L-Z, J. Am. Chem. Soc 2018, 140, 3177–3180. [DOI] [PubMed] [Google Scholar]
- [16]. For reviews on C1-ammonium enolate generation and reactivity, see:; a) Hartley WC, O’Riordan TJC, Smith AD, Synthesis 2017, 49, 3303–3310; [Google Scholar]; b) Morrill LC, Smith AD, Chem. Soc. Rev 2014, 43, 6214–6226; [DOI] [PubMed] [Google Scholar]; c) Gaunt MJ, Johansson CC, Chem. Rev 2007, 107, 5596–5605; [DOI] [PubMed] [Google Scholar]; d) Paull DH, Weatherwax A, Lectka T, Tetrahedron 2009, 65, 6771–6803; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) France S, Guerin DJ, Miller SJ, Lectka T, Chem. Rev 2003, 103, 2985–3012. [DOI] [PubMed] [Google Scholar]
- [17]. For computation supporting the structure of benzotetramisole-derived C1-ammonium enolates, including a discussion of stereocontrol, see:; a) West TH, Walden DM, Taylor JE, Brueckner AC, Johnstone RC, Cheong PH-Y, Lloyd-Jones GC, Smith AD, J. Am. Chem. Soc 2017, 139, 4366–4375; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Robinson ERT, Waldon DM, Fallan C, Green-halgh MD, Cheong PH-Y, Smith AD, Chem. Sci 2016, 7, 6919–6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. For reviews concerning catalyzed enantioselective reactions proceeding via p(allyl)Pd electrophiles, see:; a) Tsuji J, Tetrahedron 2015, 71, 6330–6348; [Google Scholar]; b) Trost BM, Tetrahedron 2015, 71, 5708–5733; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Weaver JD, Recio III A, Grenning AJ, Tunge JA, Chem. Rev 2011, 111, 1846–1913; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Trost BM, Machacek MR, Aponick AP, Acc. Chem. Res 2006, 39, 747–760; [DOI] [PubMed] [Google Scholar]; e) Trost BM, Crawley ML, Chem. Rev 2003, 103, 2921–2944; [DOI] [PubMed] [Google Scholar]; f) Trost BM, Van Vranken DL, Chem. Rev 1996, 96, 395–422. [DOI] [PubMed] [Google Scholar]
- [19]. For computation concerning benzotetramisole-containing C1-ammonium enolates reacting with cationic π(allyl)PdII electrophiles through an outer-sphere mechanism, see Ref. [14b].
- [20]. Pentafluorophenyl esters are established acyl donors in peptide bond formation; for examples, see:; a) Gayo LM, Suto MJ, Tetrahedron Lett. 1996, 37, 4915–4918; [Google Scholar]; b) Green M, Berman J, Tetrahedron Lett. 1990, 31, 5851–5852; [Google Scholar]; c) Atherton E, Cameron LR, Sheppard RC, Tetrahedron 1988, 44, 843–857; [Google Scholar]; d) Kisfaludy L, Roberts J, Johnson R, Mayers GL, Kovacs J, J. Org. Chem 1970, 35, 3563–3565. [DOI] [PubMed] [Google Scholar]
- [21].Kurti L, Czakó B, Strategic Applications of Named Reactions in Organic Synthesis, Elsevier, Amsterdam, 2005. [Google Scholar]
- [22]. There are isolated reports of the Hofmann rearrangement being used to prepare homoallylic amines; however, none of these constitutes a general method for the regio- and stereodivergent synthesis of enantioenriched homoallylic amines; see:; a) Kou KGM, Kulyk S, Marth CJ, Lee JC, Doering NA, Li BX, Galego GM, Lebold TP, Sarpong R, J. Am. Chem. Soc 2017, 139, 13882–13896; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Marth CJ, Galego GM, Lee JC, Lebold TP, Kulyk S, Kou KGM, Qin J, Lilien R, Sarpong R, Nature 2015, 528, 493–498; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ito M, Kondon Y, Nambu H, Anada M, Takeda K, Hashimoto S, Tetrahedron Lett. 2015, 56, 1397–1400; [Google Scholar]; d) Umezaki S, Yokoshima S, Fukuyama T, Org. Lett 2013, 15, 4230–4233; [DOI] [PubMed] [Google Scholar]; e) Sosale C, Rao MV, Beilstein J Org. Chem 2012, 8, 1393–1399; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Ye J-H, Huang Y, Chen R-Y, Org. Prep. Proced. Int 2003, 35, 429–432; [Google Scholar]; g) Newlander KA, Callahan JF, Moore ML, Tomaszek TA Jr., Huffmann WF, J. Med. Chem 1993, 36, 2321–2331. [DOI] [PubMed] [Google Scholar]
- [23]. For an excellent recent example of stereodivergence via the addition of stereodefined E- or Z-configured allylboronic acid nucleophiles to indoles and dihydroisoquinolines, see Ref. [4m].
- [24]. For a recent review, see:; Yoshimura A, Zhdankin VV, Chem. Rev 2016, 116, 3328–3435. [DOI] [PubMed] [Google Scholar]
- [25].a) Zagulyaeva AA, Banek CT, Yusubov MS, Zhdankin VV, Org. Lett 2010, 12, 4644–4647; [DOI] [PubMed] [Google Scholar]; b) Liu P, Wang Z, Hu X, Eur. J. Org. Chem 2012, 1994–2000; [Google Scholar]; c) Yoshimura A, Middleton KR, Luedtke MW, Zhu C, Zhdankin VV, J. Org. Chem 2012, 77, 11399–11404; [DOI] [PubMed] [Google Scholar]; d) Yoshimura A, Luedtke MW, Zhdankin VV, J. Org. Chem 2012, 77, 2087–2091. [DOI] [PubMed] [Google Scholar]
- [26].a) Liu P, Yang X, Birman VB, Houk KN, Org. Lett 2012, 14, 3288–3291; [DOI] [PubMed] [Google Scholar]; b) Bumbu VD, Birman VB, J. Am. Chem. Soc 2011, 133, 13902–13905; [DOI] [PubMed] [Google Scholar]; c) Yang X, Lu G, Birman VB, Org. Lett 2010, 12, 892–895; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Birman VB, Li X, Org. Lett 2006, 8, 1351–1354; for a discussion of isothioureas as enantioselective Lewis base catalysts, see: [DOI] [PubMed] [Google Scholar]; e) Merad J, Pons J-M, Chuzel O, Bressy C, Eur. J. Org. Chem 2016, 5589–5560. [Google Scholar]
- [27].Bruno NC, Tudge MT, Buchwald SL, Chem. Sci 2013, 4, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kocienski PJ, Protecting Groups, 3rd ed., Thieme, Stuttgart, 2005. [Google Scholar]
- [29].Li W, Han Y, Li B, Liu C, Bo Z, J. Polym. Sci. Part A 2008, 46, 4556–4563. [Google Scholar]
- [30]. For access to related products by vinylogous Mannich reactions, see:; Sickert M, Schneider C, Angew. Chem. Int. Ed 2008, 47, 3631–3634; Angew. Chem. 2008, 120, 3687 – 3690. [DOI] [PubMed] [Google Scholar]
- [31]. For reviews on Pd-catalyzed cross-couplings of organosilicon compounds, see:; a) Nakao Y, Hiyama T, Chem. Soc. Rev 2011, 40, 4893–4901; [DOI] [PubMed] [Google Scholar]; b) Denmark SE, Liu J, Angew. Chem. Int. Ed 2010, 49, 2978–2986; Angew. Chem. 2010, 122, 3040 – 3049; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sore HF, Galloway WR, Spring DR, Chem. Soc. Rev 2012, 41, 1845–1866. [DOI] [PubMed] [Google Scholar]
- [32].a) Trost BM, Machacek MR, Ball ZT, Org. Lett 2003, 5, 1895–1898; [DOI] [PubMed] [Google Scholar]; b) McLaughlin MG, McAdam CA, Cook MJ, Org. Lett 2015, 17, 10–13. [DOI] [PubMed] [Google Scholar]
- [33].Kalshetti R, Venkataramasubramanian V, Kamble S, Sudalai A, Tetrahedron Lett. 2016, 57, 1053–1055. [Google Scholar]
- [34].a) Huber JD, Leighton JL, J. Am. Chem. Soc 2007, 129, 14552–14553; [DOI] [PubMed] [Google Scholar]; b) Huber JD, Perl NR, Leighton JL, Angew. Chem. Int. Ed 2008, 47, 3037–3039; Angew. Chem. 2008, 120, 3079 – 3081. [DOI] [PubMed] [Google Scholar]
- [35].Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, Canon J, Chen A, Chen X, Chow D, Deignan J, Duquette J, Eksterowicz J, Fisher B, Fox BM, Fu J, Gonzalez AZ, Gonzalex-Lopez De Turiso F, Houze JB, Huang X, Jiang M, Jin L, Kayser F, Liu J, Lo M-C, Long AM, Lucas B, McGee LR, McIntosh J, Mihalic J, Oliner JD, Osgood T, Peterson ML, Roveto P, Saiki AY, Shaffer P, Toteva M, Wang Y, Wang YC, Wortman S, Yakowec P, Yan X, Ye Q, Yu D, Yu M, Zhao X, Zhou J, Zhu J, Olsen SH, Medina JC, J. Med. Chem 2014, 57, 1454–1472. [DOI] [PubMed] [Google Scholar]
- [36]. Our efforts to incorporate analogous Curtius and Lossen rearrangements (via acyl azides and hydroxamic acids, respectively) into a similar general one-pot procedure have been unsuccessful.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.