

Abstract
Chemoradiation is an effective combined modality therapeutic approach that utilizes principles of spatial cooperation in order to combat the adaptability associated with cancer and to potentially expand the therapeutic window. Optimal therapeutic efficacy requires intelligent selection and refinement of radio-synergistic pharmaceutical agents, enhanced delivery methods, and temporal consideration. Here, a monodisperse sub-20 nm mixed poloxamer micelle (MPM) system was developed to deliver hydrophobic drugs intravenously, in tandem with ionizing radiation. This report demonstrates in vitro synergy and enhanced radiosensitivity when two molecularly targeted DNA repair inhibitors, talazoparib and buparlisib, are encapsulated and combined with radiation in a 4T1 murine breast cancer model. Evaluation of in vivo biodistribution and toxicity exhibited no reduction in particle accumulation upon radiation and a lack of both acute and chronic toxicity. In vivo efficacy studies suggested the promise of combining talazoparib, buparlisib, and radiation to enhance survival and control tumor growth. Tissue analysis suggests enhanced DNA damage leading to apoptosis, thus increasing efficacy. These findings highlight the challenges associated with utilizing clinically relevant inclusion criteria and treatment protocols as complete tumor regression and extended survival were masked by an aggressively metastasizing model. As with clinical treatment regimens, the findings here establish a need for further optimization of this multimodal platform.
Keywords: PARP, PI3K, Chemoradiotherapy, Combination Therapy, Synergy, Radiation
Graphical Abstract
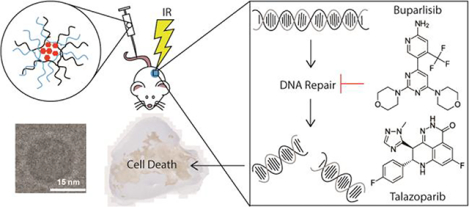
Environmental adaptability is a key paradigm cancer capitalizes on for survival and propagation. Oncologic diversity allows for adaptation and often places selective pressure on a therapeutically resistant sub-population of cells leading to subsequent metastatic spread due to an inadequate therapeutic response. Sub-optimal responses to individual therapeutics can be largely attributed to both inter- and intra-tumoral heterogeneity.1–4 As such, combined modality therapeutics have become a mainstay in oncology, owing to enhanced efficacy and the ability to reduce therapeutic resistance.5 In the interest of improving patient outcomes, there is continual interest in expanding the therapeutic window and enhancing the efficacy of combination therapies.6 One such approach involves combining radiation therapy (RT) with systemic chemotherapeutics or targeted therapy, to provide both local radiosensitization and disseminated disease control.6,7 However, to intelligently combine RT with other therapeutics, the biology and molecular mechanism of action for all agents must be considered. Ionizing radiation damages cellular DNA resulting in the manifestation of single (SSB) or double strand (DSB) breaks, some of which are simple breaks and others that may be considered complex breaks. In response to these strand breaks, the cell cycle arrests so the ensuing damage may be repaired. If cells sustained unrepairable damage due to complex break mechanisms, lethality follows due to the accumulation of chromosome aberrations.8 By utilizing DNA damage repair (DDR) inhibitors, it is possible to capitalize on these susceptibilities and negate cancer cell survival.
Two DDR proteins with crucial roles in this process are poly(ADP-ribose) polymerase (PARP) and breast cancer susceptibility proteins (BRCA). PARP is an important component in the SSB response, while BRCA is required for more intricate DSB repair.9 In patients that harbor a loss of function BRCA1 or BRCA2 mutation, PARP inhibition has been used to induce synthetic lethality. Unfortunately, its clinical utility is limited as these mutations are only observed in a minority (5–10%) of breast cancer patients.10 Efforts to broaden the scope of PARP therapy have included inhibition of the phosphoinositol-3-kinase (PI3K) family, whose members are implicated in the sensing and repair of genomic damage and chemotherapeutic resistance.11–13 In fact, the biological role of these proteins has inspired many clinical PI3K inhibitor-drug combination trials in order to overcome resistance and enhance chemotherapeutic efficacy.14 In particular, utilizing the PI3K inhibitor buparlisib (Bup) sensitized cells to PARP inhibition by reducing BRCA expression.12,15–17 In this way, administering a PI3K inhibitor to mimic BRCA mutated cancer revitalizes PARP therapy for a large subset of patients, previously unresponsive to PARP inhibition.
Despite the promise of combining PARP and PI3K inhibitors for enhanced chemoradiotherapy, these type of drugs commonly have poor in vivo stability limiting the dose and route of administration.18 An important hurdle to overcome when working with DDR inhibitors is their relative insolubility and high toxicity.13 As such, various drug carriers including micelles have been employed to solubilize these compounds and co-deliver them with traditional chemotherapeutics, each other, and/or radiation therapy.19–29 Nanoparticle (NP) formulation of therapeutics aims to address solubility and other drug delivery issues. NPs enhance the solubility of hydrophobic drugs, protect drugs from premature degradation, extend circulation times, reduce unintended off-target toxicity, enhance tumor uptake, and deliver drugs spatially to the desired location.30 Thus we hypothesized that development and implementation of a novel, mixed poloxamer micelle (MPM) formulation designed to deliver PARP inhibitor talazoparib (Tal) and PI3K inhibitor Bup in a single formulation during a course of radiotherapy, would increase the therapeutic ratio in a preclinical model. While there have been reports of dual PARP and PI3K inhibitor administration with and without radiation or as encapsulated agents, this report is the first to co-deliver Tal and Bup in a MPM during a course of RT.12,15–18,31–33 Herein, we demonstrate synergy between Tal and Bup in a BRCA proficient breast cancer model in vitro, as well as, the promise and challenges of translating this synergy to an aggressive, late-stage in vivo system. Altogether, this report demonstrates the critical importance of optimization, when administering multiple agents for enhancing the therapeutic window of combination therapies.
RESULTS AND DISCUSSION
Synthesis and Characterization of Micelles.
For the encapsulation of the two hydrophobic inhibitors, we employed a mixture of poloxamer polymers to enhance encapsulation while maintaining particle stability and solubility. Poloxamers consist of hydrophilic and hydrophobic units of various lengths that are characterized by different hydrophilic-lipophilic balance numbers. This amphiphilic nature gives these polymers surfactant properties rendering them ideal for the uptake and transfer of hydrophobic drugs under biological conditions.34 Here, formulations of MPMs with different payloads (empty, single drug, single fluorophore, combination drug) were prepared utilizing a nanoprecipitation method, by simply varying which drug(s) were added to the organic phase (Schematic 1).
Schematic 1.
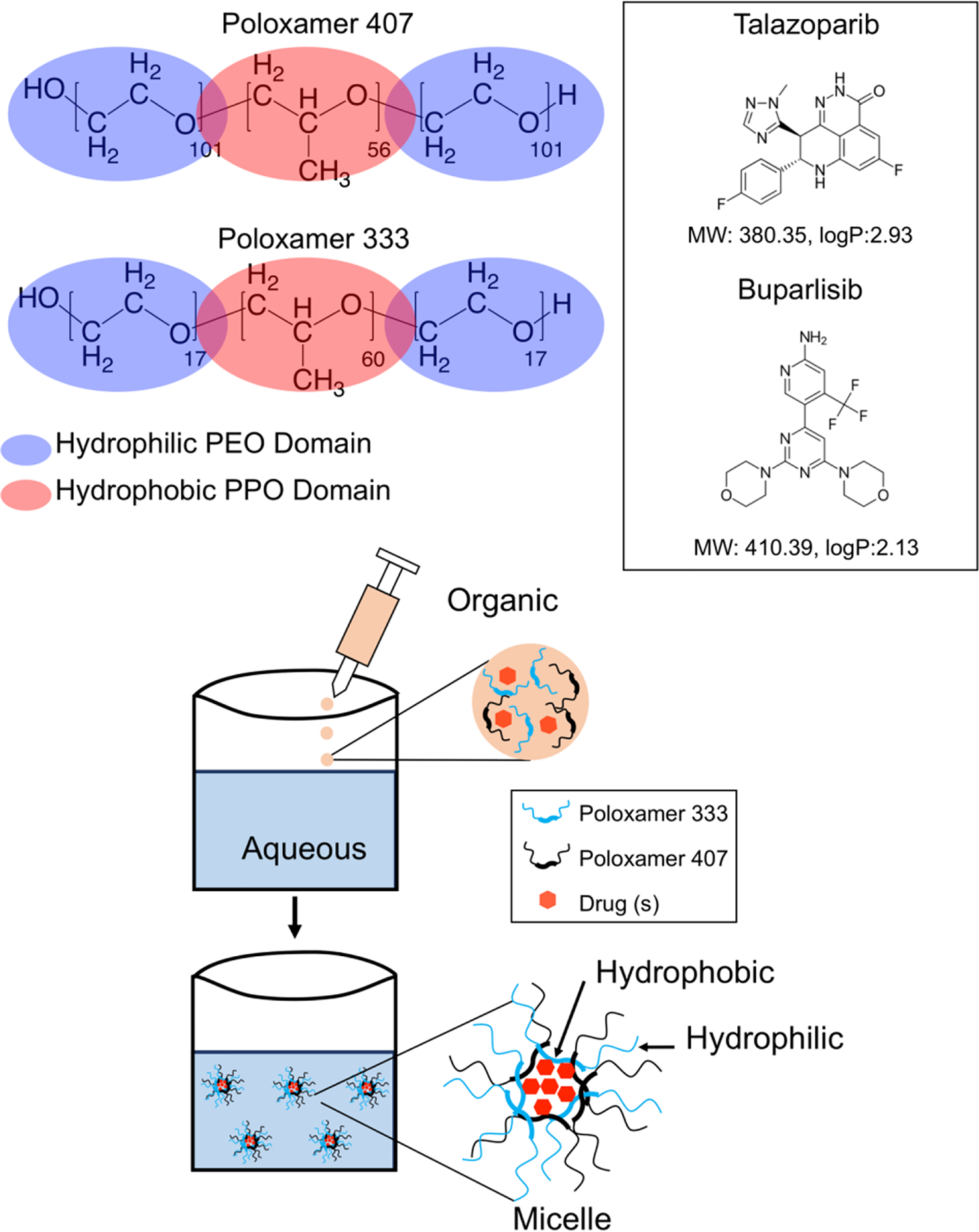
Structures of poloxamer 333 and 407 with poly-propylene oxide (PPO) and poly-ethylene oxide (PEO) chain lengths denoted and shown as hydrophilic or hydrophobic shown alongside structures of buparlisib and talazoparib with molecular weights and logP (partition coefficient) values highlighted.
The z-average and polydispersity index (PDI) of these formulations, as measured by dynamic light scattering (DLS), were found to vary slightly depending on the cargo with the average values of 17.4 ± 0.7 nm and 0.02 ± 0.01 respectively (Figure 1a–b, Figure S1a–d). Overall variation in the average values showed no statistical significance, indicating that encapsulation of the hydrophobic drug(s) does not drastically influencing the overall MPM size. Additionally, evaluation of MPMs in biologically relevant media (RPMI 1640 supplemented with 10% serum and 1% antibiotic) demonstrated a slight size increase from 17 nm to 23 nm (Figure S2b), likely due to adsorption of a protein corona.35 Importantly, significant stability of formulated MPMs was observed when stored at room temperature over the course of approximately two months (Figure S2a) demonstrating their off-the-shelf utility for therapeutic applications. This high MPM stability is promising for prolonging drug circulation times in vivo. Furthermore, the practicality of extending the MPMs shelf-life for long-term therapeutic regimens were demonstrated for both empty (E MPMs) and combination (TB MPMs) formulations, where upon freeze-dried and later reconstituted in PBS, the MPMs retained their size as visualized by DLS (data not shown). Transmission electron micrographs (TEM) demonstrate a dense hydrophobic core with a diameter of approximately 4 nm and a diffuse polymer shell (Figure 1c). However, cryoTEM images correlate well with DLS measurements confirming an overall diameter of 19 nm (Figure 1d).36 Collectively, these results demonstrate that the poloxamer polymers self-assemble to form highly uniform, stable MPMs with average sizes within the ideal range for biological uptake and PEGylation to avoid uptake by the reticuloendothelial system.30
Figure 1.
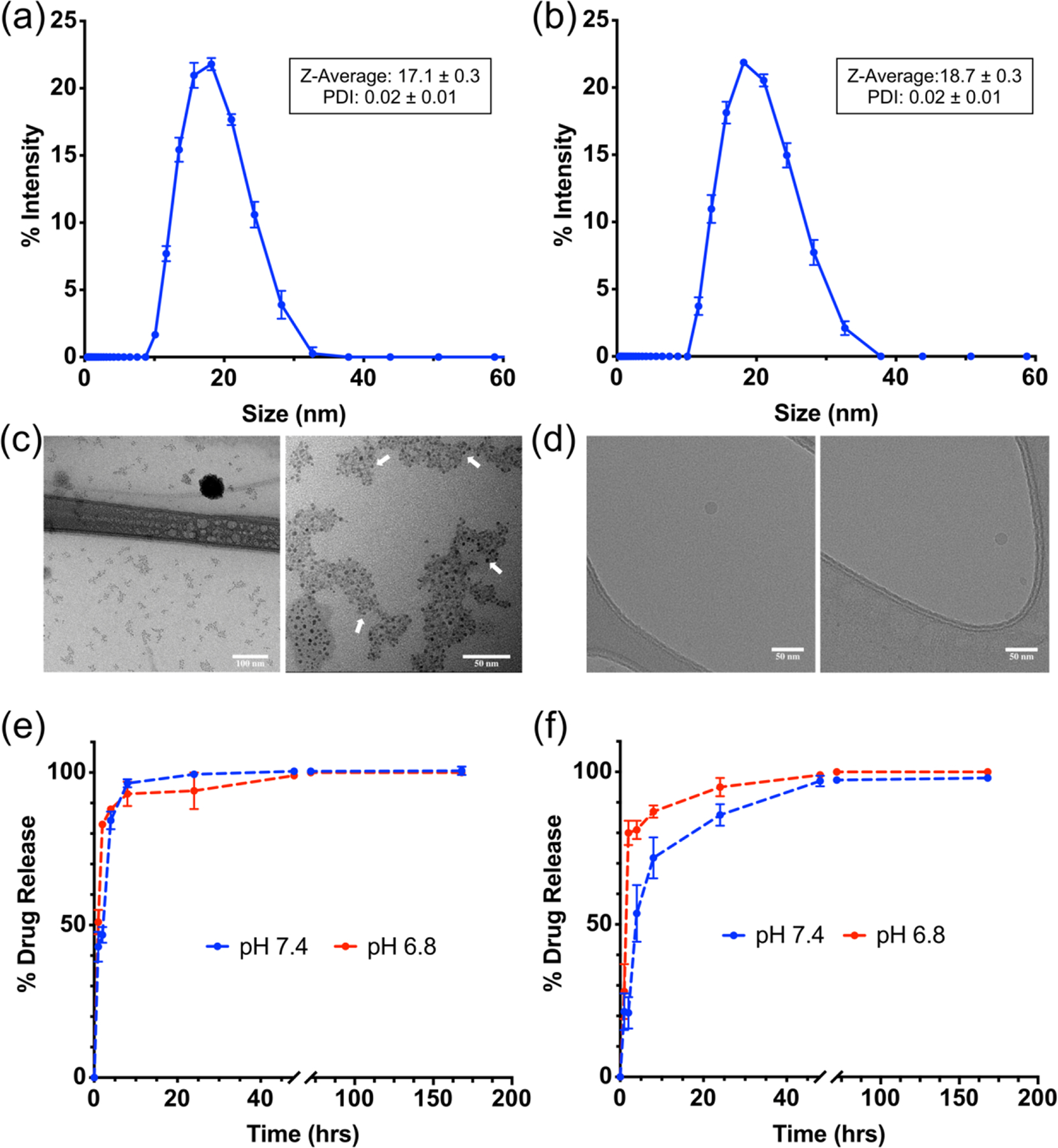
Intensity average hydrodynamic diameter measured by dynamic light scattering for (a) E MPMs (b) TB MPMs. (c) Transmission Electron Microscopy (TEM) shows a core size around 4 nm, individual cores shown by white arrows. Scale bar of left image is 100 nm and right image is 50 nm. (d) CryoTEM demonstrates an overall size closer to the hydrodynamic radius measured by DLS. Scale bar is 50 nm in both images. In vitro drug release profile of (e) talazoparib and (f) buparlisib from dual drug-loaded MPMs under sink conditions in PBS at pH 7.4 or 6.8 as determined by quantitative HPLC.
In order to optimize the MPM size and encapsulation efficiency in our formulation, varying ratios of poloxamers 333 and 407 (p333 and p407) were evaluated. At a constant concentration of p407, particle size decreased inversely with increasing p333 concentration while the opposite yielded no change in MPM size (Figure S3a). These findings suggest that the p333 concentration strongly influences MPM size and encapsulation while p407 acts as a stabilizing agent due to the increased hydrophilic block length.37
Initial studies aimed at optimizing MPM formulation evaluated micellar encapsulation of the fluorescent probe, Coumarin 153 (C153) chosen for its similarity to the target inhibitors. In this study, increases in the fluorescent intensity of encapsulated C153 were directly correlated to increases in the p333 concentration, indicating that p333 is critical for enhancing not only size, but also encapsulation (Figure S3b). Additionally, the near-infrared (NIR) fluorescent probe cyanine 7.5 carboxylic acid (Cy 7.5) was encapsulated within the MPMs for in vivo biodistribution studies. Encapsulation of Cy 7.5 fluorophore was confirmed by the presence of a hyperchromic shift in absorbance intensity as compared to the free Cy 7.5 in an aqueous environment (Figure S3c). Additionally, we observe a bathochromic shift, which is indicative of solvent influence on the fluorophore, when comparing encapsulated Cy 7.5 in an aqueous environment (max absorbance = 795 nm) to free dye in an organic environment (max absorbance = 785 nm). Evaluation of fluorescence intensity demonstrated a correlation between increased fluorophore loading and a corresponding decrease in fluorescence intensity (Figure S3d). This type of phenomena has been observed to occur at high fluorophore encapsulation values due to self-quenching, as a result of proximity constrains. As this quenching is unfavorable for evaluating biodistribution, we chose to utilize an encapsulation concentration that yielded strong signal with no quenching.
The final formulation was a balance between enhanced encapsulation at a higher p333 content and potential toxicity of increased p333 concentration. Optimization with C153 resulted in an MPM formulation boasting greater than 50% encapsulation efficiency for both drugs, as evaluated by high performance liquid chromatography (HPLC) (Table S1). Dual encapsulation did not significantly impair overall drug encapsulation efficiency. Interestingly, the drug with a slightly higher hydrophobicity (Tal logP = 2.93, Bup logP = 2.13) exhibited a lower encapsulation efficiency of 60 ± 6% Tal versus 75 ± 9% Bup. While the observed higher encapsulation of Bup could be due to its slightly increased size over Tal, it is more likely influenced by the hydrogen bonding ability of Bup.
Drug Release of Dual-Loaded MPMs in Biologically Relevant pH Environments.
MPMs encapsulating Tal and Bup were exposed to sink physiological conditions in order to simulate release in three biologically relevant pH environments over the course of a week. Investigated conditions included release in circulation, in the tumor microenvironment, and in the endosome. Under pH conditions mimicking circulation (pH 7.4, PBS, 25°C), approximately 100% of encapsulated Tal was released within the first 8 hours. Comparatively, Bup release was slower with full release achieved after 48 hours. Interestingly, pH conditions similar to the tumor microenvironment (pH 6.8, PBS, 25°C) had little influence on Tal release. In contrast, Bup demonstrated accelerated release under slightly acidic conditions with full release occurring within 24 hours. Under pH conditions meant to mimic the endosomal environment (pH 5.5, Citrate Buffer, 25°C) the MPMs underwent a significant degree of swelling resulting in the sudden and rapid release and precipitation of the drugs.
Overall, Tal release occurred rapidly (full release within 24 h) and with minimal dependence on pH changes. Whereas, the release of Bup was determined to be markedly slower with increased dependence on pH (Figure 1e–f). As described previously, these observed variations in MPMs drug release may be influenced by the overall drug properties (i.e., hydrogen bonding). Regardless of the mechanism, the pH-dependent release profile is promising for in vivo translation involving sequential release states of the MPMs. Ideally, as the MPMs circulate in the blood (pH 7.4), drug will be released at a steady rate. Upon distribution in the tumors cells (pH 6.8) the subsequent pH drop would result in accelerated release. Lastly, as the particles are endocytosed they would fall apart releasing the remainder of the drugs.
In vitro Toxicologic Evaluation and Synergistic Effects of Tal and Bup.
In order to confirm the efficacy of the DDR inhibitors, the cytotoxicity of Tal and Bup was evaluated individually and in combination for both the free and encapsulated agents in vitro. The cytotoxicity of Tal and Bup was evaluated in murine 4T1 breast cancer cells utilizing the CellTiter-Fluor assay. Figure 2a–b shows the viability curves for 4T1 cells treated with either Bup or Tal for 72 hours. Both drugs demonstrate moderate efficacy as individual agents with IC50 values of 4.7 and 1.4 μM respectively. Importantly, when combined at values near their IC50 values, we see viability values closer to 20% indicating a degree of synergy between the drugs (Figure 2c). In order to ascertain whether or not there was indeed synergy, the combination indexes (CI) were calculated at various fractions affected (fa) and plotted against each other.38 From this plot, values of CI>1 are antagonistic, CI=1 are additive, and CI<1 are synergistic. Figure S4A confirms that at fa values above 0.5 (or higher cell kill percentages) the two drugs indeed function synergistically at an approximately one-to-one weight ratio. Furthermore, Figure S4b demonstrates the enhanced toxicity of a combined micelle formulation of Tal and Bup.
Figure 2.
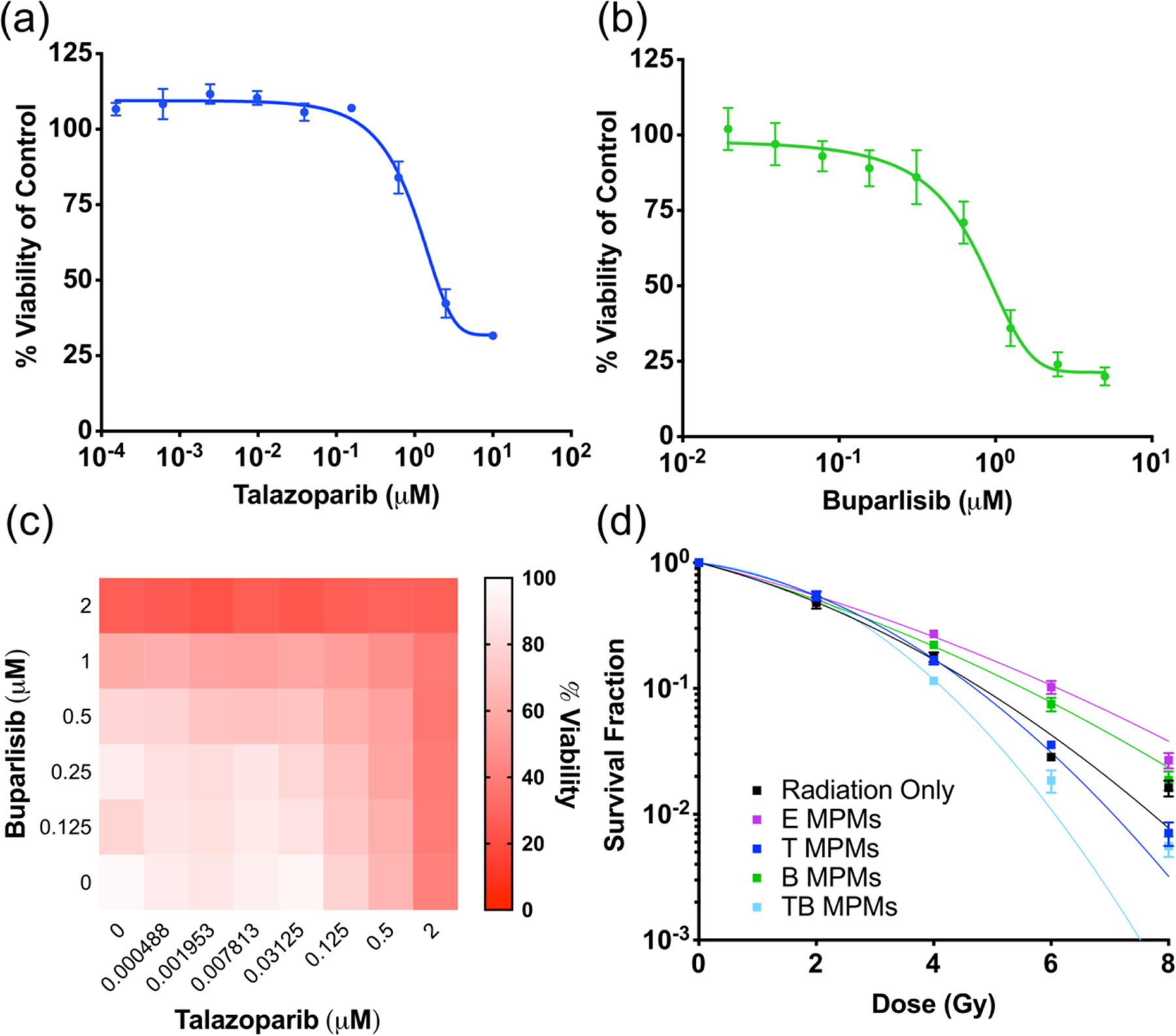
In vitro drug toxicity of free (a) talazoparib (IC50 1.412 μM) and (b) buparlisib (IC50 4.722 μM) in 4T1 breast cancer cells. (c) In vitro toxicity of free drugs in combination in 4T1 breast cancer cells. (d) In vitro evaluation of radiosensitization of various MPMs as evaluated by clonogenic assay. Curves are fit using the linear-quadratic equation.
Influence of Free and Encapsulated Drugs on Clonogenicity and DNA Damage.
To demonstrate enhanced radiosensitivity after dual administration of Tal and Bup, we investigated the clonogenicity of 4T1 cells treated with increasing doses of radiation and drugs. Upon fitting the linear-quadratic (LQ) equation to our experimental data, the curves shown in Figure 2d are able to be compared. These curves are defined by the LQ model:
where S is the survival fraction, α is the linear portion of the curve, β is the quadratic portion of the curve, and D is the dose.8,39 From this equation, we can compare the α/β ratio values for each treatment group. Lower ratios α/β indicate tissues that would benefit from fractionated dosing as they are sensitive to a lower dose, which could be utilized to reduce toxicity to normal tissues that normally have high α/β ratios.40 The evaluation of generated survival curves in Figure 2d demonstrates that dual-encapsulated TB MPMs (0.625/0.375 μM Tal/Bup) create a more radiosensitive environment over other micelle treatments delivered at higher doses of individual drugs (0.945 μM Tal or 0.505 μM Bup). This further validates the synergy between Tal and Bup. Additionally, it is observed that E MPMs and buparlisib (B MPMs) offer no radiosensitization whereas talazoparib (T MPMs) provide radiosensitization to a lesser extent than TB MPMs (Table S2). These results are similar to the free drug combination studies report by Jang et al where it was observed that in comparison to monotherapy, co-administration of PI3K (PI-103) and PARP (Olaparib) inhibitors led to synergy at doses of radiation from 2 to 8 Gy.18 In our case, the lack of an observed clonogenic synergistic index (SI) may be a product of the lower drug concentrations utilized in the combinatorial therapeutic strategy (0.625/0.375 μM Tal/Bup) than the individual treatments (0.945 μM Tal or 0.505 μM Bup).
In further evaluation of DNA damage, quantification of γH2AX was utilized to determine the efficacy of therapy when combined with radiation. Utilizing a modified speckles pipeline from CellProfiler, γH2AX puncta were identified giving both an average number of foci per nucleus and percentage of nuclei positive for at least one focus (Figure 3a and S5).41 In terms of foci, TB MPMs were observed to induce the largest amount per cell both with (~7.5 foci/cell) and without irradiation (~6 foci/cell), demonstrating the utility of this combination therapy. Furthermore, TB MPMs combined with 5 Gy irradiation demonstrate a statistically significant increase in γH2AX foci per cell over all other treatment groups. Comparatively, TB MPMs without irradiation demonstrate a statistically significant increase in γH2AX foci over all treatment groups except TB MPMs + IR and B MPMs + IR. When evaluating the percentage of nuclei expressing at least one focus, TB MPMs boasted the largest number (86%) positive nuclei in unirradiated samples. However, Figure 3b demonstrates that when irradiation was added, TB MPMs express the second highest number (86%) of positive nuclei losing to B MPMs (89%). Surprisingly, the addition of radiation to TB MPM therapy does not increase the percent of cells expressing foci but does increase the number of foci per cell. The reduced amount of Bup utilized in the TB MPM formulation may explain the absence of an increase in foci positive cells. Although it would be ideal to detect increases in both values, visualizing an increase in the amount foci per cell corresponds to an increased amount of DNA damage and therefore a greater chance of cell death. In addition, through qualitative analysis we observe that the overall intensity of the γH2AX channel appears to increase from unirradiated to irradiated samples (Figure 3c).
Figure 3.
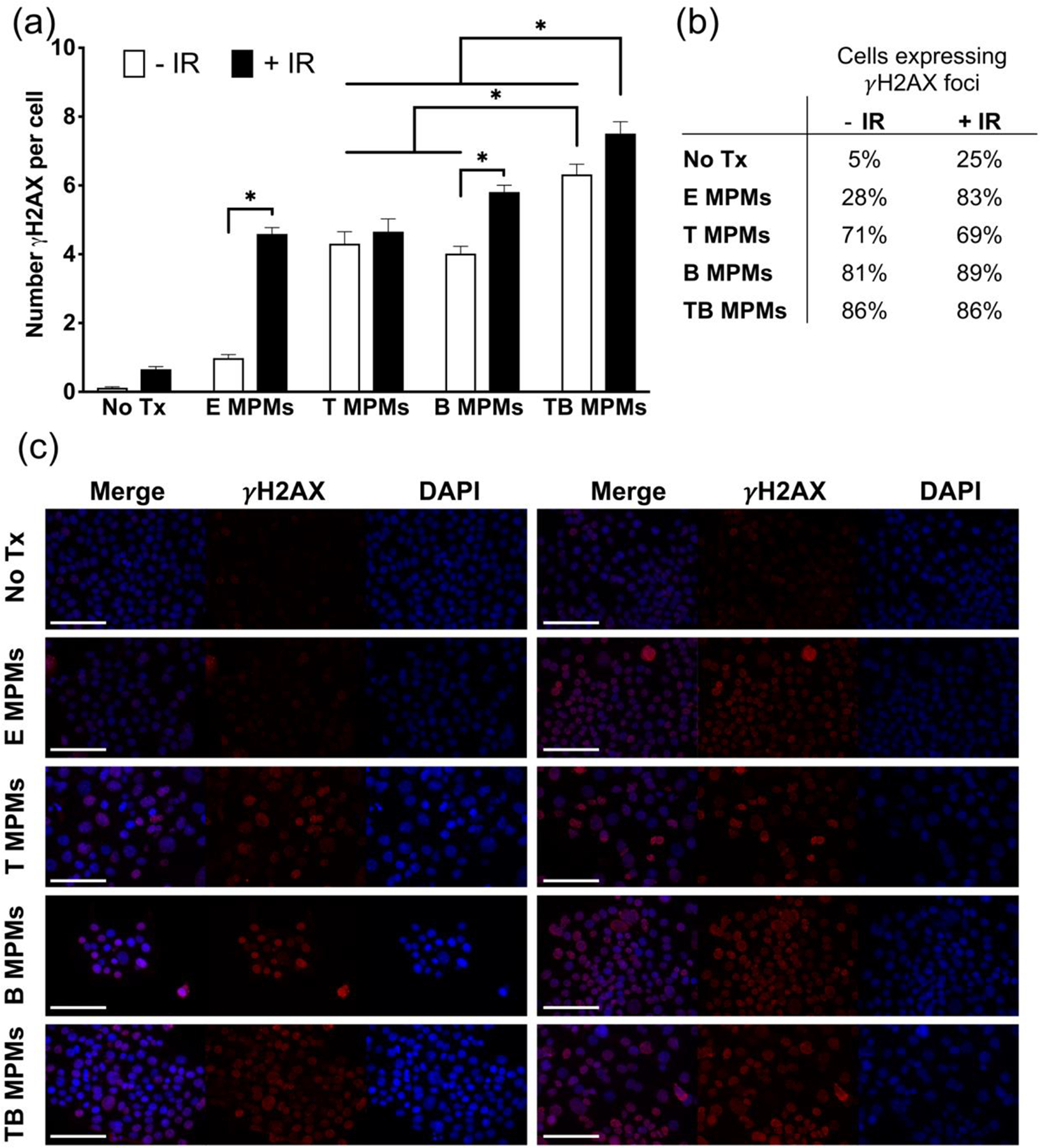
(a) Average number of γH2AX foci per cell and (b) Percentage of cells positive for γH2AX foci for various treatment conditions. (c) Representative images of γH2AX foci in cells treated with no drug, E MPMs, T MPMs, B MPMs, or TB MPMs and unirradiated (left) or irradiated (right). Nuclei stained for DAPI (blue) and γH2AX (red). Scale bar represents 100 microns. * denotes statistically significant difference (p < 0.05).
Interestingly, E MPMs demonstrate increased levels of DNA damage compared to no treatment both with and without radiation. This data correlates well with a report that co-administration of Pluronic™ L10 and 3 Gy results in an increased amount of γH2AX foci attributed to a decrease in the expression of a stress response proteins Hsp90 and Hsp70, which are implicated as DDR chaperones.42 Furthermore, several other poloxamer-based formulations have been shown to influence the expression of Hsp70 over a period of 6 hours after hyperthermia treatment, suggesting the impact of stress-induced cellular responses to these block copolymers.43
In Vitro Cellular Uptake and In Vivo Biodistribution of Cy 7.5-Loaded MPMs.
During the nanoparticle formulation process, two highly sought-after traits include the enhancement of cellular uptake and preferential organ biodistribution. In order to elucidate the uptake and biodistribution of these MPMs, a hydrophobic NIR dye, Cy 7.5, was encapsulated to enable imaging at wavelengths with greater depth penetration than typical fluorophores. The Cy 7.5-loaded MPMs demonstrated quick, robust intracellular uptake in the 4T1 cancer cell line at 2 hours (Figure 4a). Interestingly, there is a unique time-dependent uptake of 125 ng/mL Cy 7.5-loaded MPMs. From zero to eight hours, there is a gradual increase in fluorescent signal (Figure 4b). At 24 hours, the fluorescent signal dropped below the signal observed at 8 hours. These observations could indicate micelle swelling and subsequent release of the fluorophore from the hydrophobic core. This hypothesis is supported by the drug release data, where it became evident that MPMs in an endosomal environment (pH 5.5) undergo substantial swelling resulting in the entire payload being released. As such, this observed particle instability may account for the decrease in signal observed after 24 hours of uptake. Alternatively, a maximum amount of uptake may have occurred between 8 and 24 hours, leading to MPM self-quenching within the cell, similar to what was observed when MPM loading concentrations were high (Figure S3d). These MPMs also exhibit a concentration-dependent uptake between 1.95 and 500 ng/mL of loaded Cy 7.5 (Figure 4c).
Figure 4.
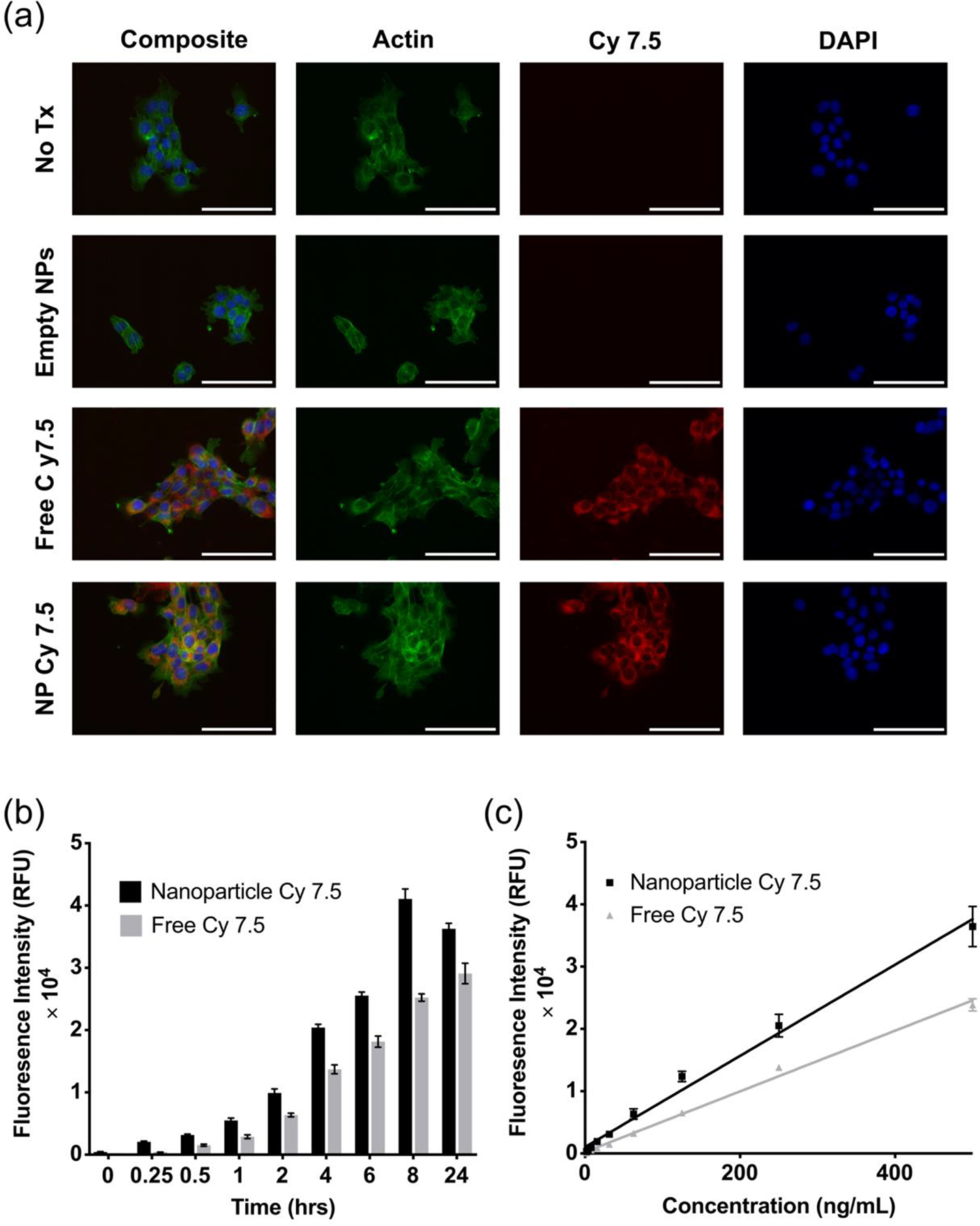
(a) Uptake of 1000 ng/mL Cy 7.5-loaded MPMs in 4T1 at 2 hours. Scale bar represents 100 microns. (b) Time-dependent uptake of 125 ng/mL Cy 7.5-loaded MPMs. (c) Concentration-dependent uptake of Cy 7.5-loaded MPMs at 2 hours.
Moving forward in vivo, the Cy 7.5 MPMs were used to understand the biodistribution of the MPMs and the potential influence of radiation on organ accumulation. Here we employed a syngeneic breast cancer mouse model. BALB/c mice were implanted with 4T1 tumors in the mammary fat pad, randomized to radiation or no radiation, and injected with Cy 7.5-loaded MPMs either 24 or 72 hours after radiation. Whole-body fluorescent images were obtained 4 or 24-hours after injection (Figure 5a and b) to qualitatively evaluate the influence of time on clearance and tumor accumulation. These images demonstrated that particles were able to continue circulating over the course of 24-hours and were not immediately cleared by the RES. 24hours after MPM injection, organs were collected and imaged ex vivo. Regions of interest (ROI) were determined, individual organ flux was quantified, all organ flux was summed, and relative organ accumulation was quantified (Figure 5c–d and Figure S6). As expected, due to the nature of clearance organs, the highest signal was observed in the liver, followed by the kidneys and spleen. These results coincide well with reports by Stapleton et. al and Yang et. al both of which demonstrated that radiation was not able to enhance the tumor accumulation of nanotherapeutics in a 4T1 model.19,44 Interestingly, Yang et. al saw that despite a lack of increasing accumulation after IR, the tumor cell uptake was improved leading to synergistic therapeutic outcomes. Several other reports have also demonstrated with macromolecules and nanoparticles that radiation delivered in a single dose or in multiple fractions can lead to a decrease in nanoparticle accumulation as compared to an unirradiated control.45–47 Importantly, here we observed that the 3 by 5 Gy fractionation schedule did not markedly influence the biodistribution of MPMs, aside from a statistically significant decrease in spleen accumulation when MPMs were injected 24 hours after irradiation (Figure 5c).
Figure 5.
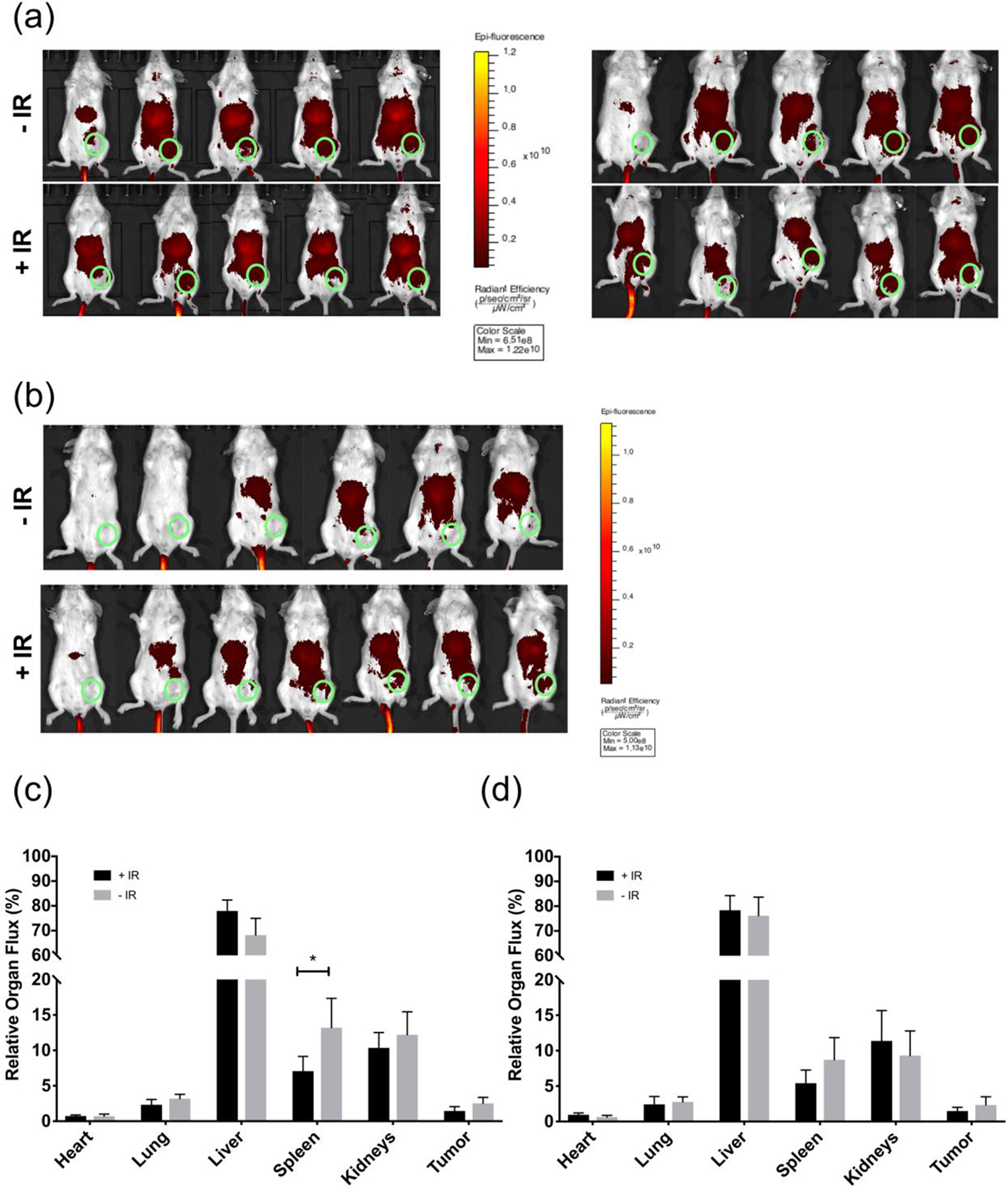
Whole-body fluorescent images of mice radiated (5 Gy) 3 consecutive days (a) injected with nanoparticles 24 hours after irradiation, imaged at 4 (left) and 24 (right) hours or (b) injected with nanoparticles 72 hours after irradiation and imaged 24 hours later. Green circle represents tumor location. Quantification of signal from organs (c) (Fig. 5a) and (d) (Fig. 5b) harvested at 24 hours. * denotes statistically significant difference compared to No IR control (p < 0.05).
In Vivo Tumor Growth Inhibition and Efficacy of Combination MPM + IR Therapeutic Regimen.
To investigate the potential of encapsulated DDR inhibitor delivery in tandem with radiation for enhancing tumor control and survival, BALB/c mice bearing subcutaneous 4T1 tumors were treated and evaluated for therapeutic response (Schematic S1). As the goal of this study was to evaluate the efficacy of this therapeutic regimen in an aggressive, late-stage cancer model, treatment was initiated on palpable rather than unmeasurable or newly inoculated tumors. Furthermore, in order to more closely mimic current practice, mice were randomized into four treatment groups (n=4 for E MPMs, n=5 for all others) when tumors reached a size between 60–175 mm3, approximately 11–25 days after inoculation. Initiation of treatment at a later timepoint when tumors are more mature, theoretically creates a scenario more representative of current issues encountered in oncology, allowing for a more realistic understanding of true patient outcomes. All four groups received intravenous (i.v.) injections three times weekly until an endpoint (described in Schematic S1) was reached, starting on the day of randomization (Figure 6a). In groups that received IR, the doses were administered directly following the first dose of MPMs, at 24 hours post-injection, and at 48 hours post-injection immediately before a second MPM injection (5 Gy per treatment). As demonstrated in Figure 6b and c, there was no enhancement of tumor control when utilizing TB MPMs or E MPMs in the absence of radiation. In the radiation groups, IR elicited statistically significant tumor control by day 7 as compared to both non-IR groups. In comparison to E MPMs, treatment with TB MPMs increased survival from 10.5 to 14 days. IR lead to statistically significant increases in survival times over vehicle control from 10.5 to 23 and 24 days respectively for E MPMs + IR and TB MPMs + IR.
Figure 6.
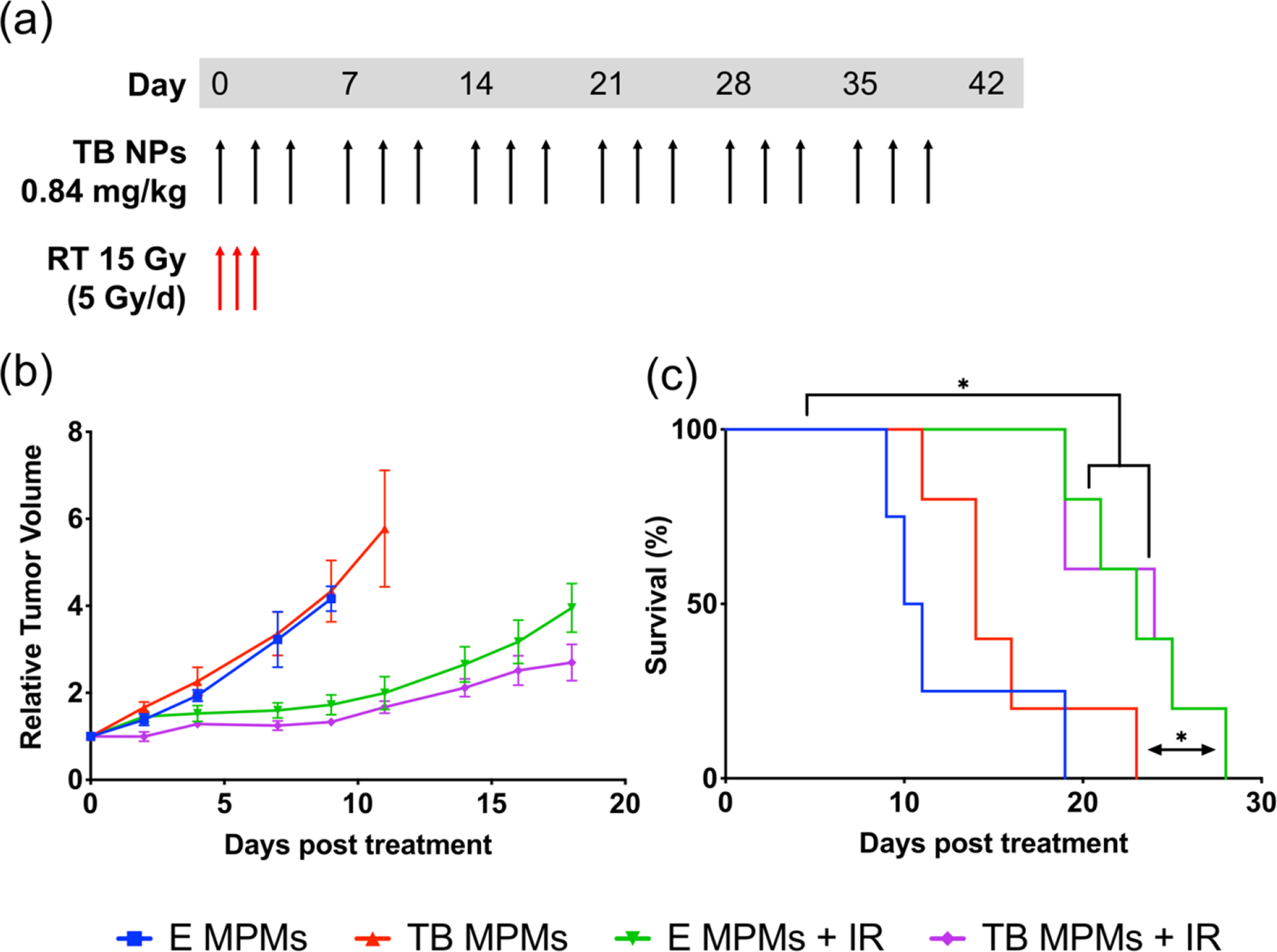
(a) Therapeutic dosing regimen. (b) Average Relative Tumor Volume. (c) Kaplan-Meier survival curve where differences in survival were calculated according to the logrank (Mantel-Cox) test. * denotes statistically significant difference (p < 0.05).
A main component of combination therapy is to elicit additivity or synergy between multiple agents, ultimately enhancing their therapeutic index. In previous reports of PARPi administration to BRCA proficient cancers, efficacy was observed when the inhibitor was administered alongside a systemic DNA damaging agent such as temozolomide or RT but demonstrated minimal efficacy as single agents.48–51 Despite efficacy, direct combination of PARP inhibitors with systemic DNA damaging chemotherapeutics, such as cisplatin or temozolomide, frequently results in undesirable side effects and dose-limiting, systemic toxicity.15 Such toxicity may be attributed to the ability of PARP inhibitors to trap PARP on any site of DNA damage creating cytotoxic complexes throughout the body.49 Therefore, employing local RT rather than systemic chemotherapeutics for induction of DNA damage may provide a more targeted approach, isolating PARP trapping and synthetic lethality to a smaller, tumoral region.15
Combination PARPi strategies typically utilize aggressive once to twice daily administration, in order to maintain concentrations within the therapeutic window throughout the course of study. For this study, MPMs were only administered three times weekly. In particular, this treatment regimen was chosen to enhance practicality (fewer injections would enhance patient compliance and clinical translatability) and because micelles are known to exhibit increased circulation times over free drug counterparts. Furthermore, Tal was intentionally encapsulated at a concentration that would reach IC50 levels and remain below the MTD over a 21-day period, even if only 1% of the administered MPMs reached the tumor site. For Bup, delivered concentrations were significantly reduced compared to single agent concentrations in order to avoid dose-limiting toxicities associated with combination pan-PI3K therapy.53–57
In this study, the control radiation dose was too effective, or the concentration of drugs utilized was too low to see a statistically significant difference between the two IR groups. However, despite no statistical significance, mice treated with TB MPMS and radiation demonstrated controlled tumor growth more consistently than radiation alone (Figure S7a–b). Theoretically, increasing the dose of PI3K inhibitor administered and/or decreasing the amount of radiation delivered should enhance the separation between radiation only and radiation in combination with DDR inhibitors. Interestingly, three of the mice treated with TB MPMs and RT did not reach the tumor size or ulceration endpoint, but rather rapidly lost weight reaching 20% requiring sacrifice (Figure S7c). Visual organ evaluation at sacrifice showed that the weight loss was not due to therapeutic toxicity, but rather metastases that had developed in the lungs and chest cavity. Histologic evaluation, however, demonstrated lung metastases in other treatment groups that did not succumb to the metastases (Figure S8). The observed gross metastases provide some explanation as to the similar survival rates between the two IR groups. Comparatively, the E MPM + IR mice lacked tumor control and reached primary tumor endpoints, whereas the TB MPM + IR mice demonstrated primary tumor control but reached endpoints due to metastasis. A recent report noted that PARP inhibitor therapy may induce cancer cell PD-L1 expression leading to immune suppression, possibly allowing for metastatic spread.58 However, investigation into the mechanism of metastatic escape during therapy and mitigation of metastases by administration of higher doses of Bup or co-administration of an anti-PD-L1 checkpoint inhibitor is ongoing.59,60 If the metastases are due to an upregulation of PD-L1 on tumor cells, incorporation of a PD-L1 or PD-1 checkpoint inhibitor would possibly aid in extending the survival times of the TB MPM + IR. Future iterations of this therapeutic strategy will focus on optimizing the temporal aspect of combining DDR inhibitors with radiation therapy, solidifying doses of each agent that are maximally minimized yet efficacious, and investigating the immune influence on metastatic escape.
Tumor Histology.
To evaluate therapeutic efficacy further, tumor tissue was extracted and stained for hematoxylin and eosin (H&E), cleaved caspase-3 (CC3), an apoptotic marker, and γH2AX. Tumor H&E (Figure 7a and S9a) and CC3 staining (Figure 7b and S9b) demonstrate differences in tumor size and tissue damage. In E MPM and TB MPM treated tumor tissue CC3 staining appears to surround necrotic tissue while tumors treated with IR tend to have CC3 staining in necrotic and non-necrotic regions, particularly the TB MPM + IR tissue where large sections of apoptotic and necrotic tissue can be seen. In the E MPM + IR treatment group, we see that the CC3 expression appears to occur sporadically near outer edges of the tumor while in the TB MPM + IR treatment group there is extensive CC3 expression throughout the innermost region of the tumor. The low energy of radiation delivered in this study may explain the minimal amount of apoptosis in the E MPM treatment group as lower energy radiation is less penetrating and may not have reached the entire mass uniformly. However, TB MPM delivery to the tumor via circulation likely played a role in the increased expression of apoptosis. γH2AX evaluation in tumor tissue follows a similar trend to that observed in vitro, where the largest amount of signal is observed in groups treated with TB MPMs (Figure 7c). Signal was specific as TB MPM + IR treated tumor tissue stained with only secondary antibody did not demonstrate any expression. Largely, the combination of TB MPMs and radiation appears to induce the more extensive apoptosis which partially results from persistent DNA damage.
Figure 7.
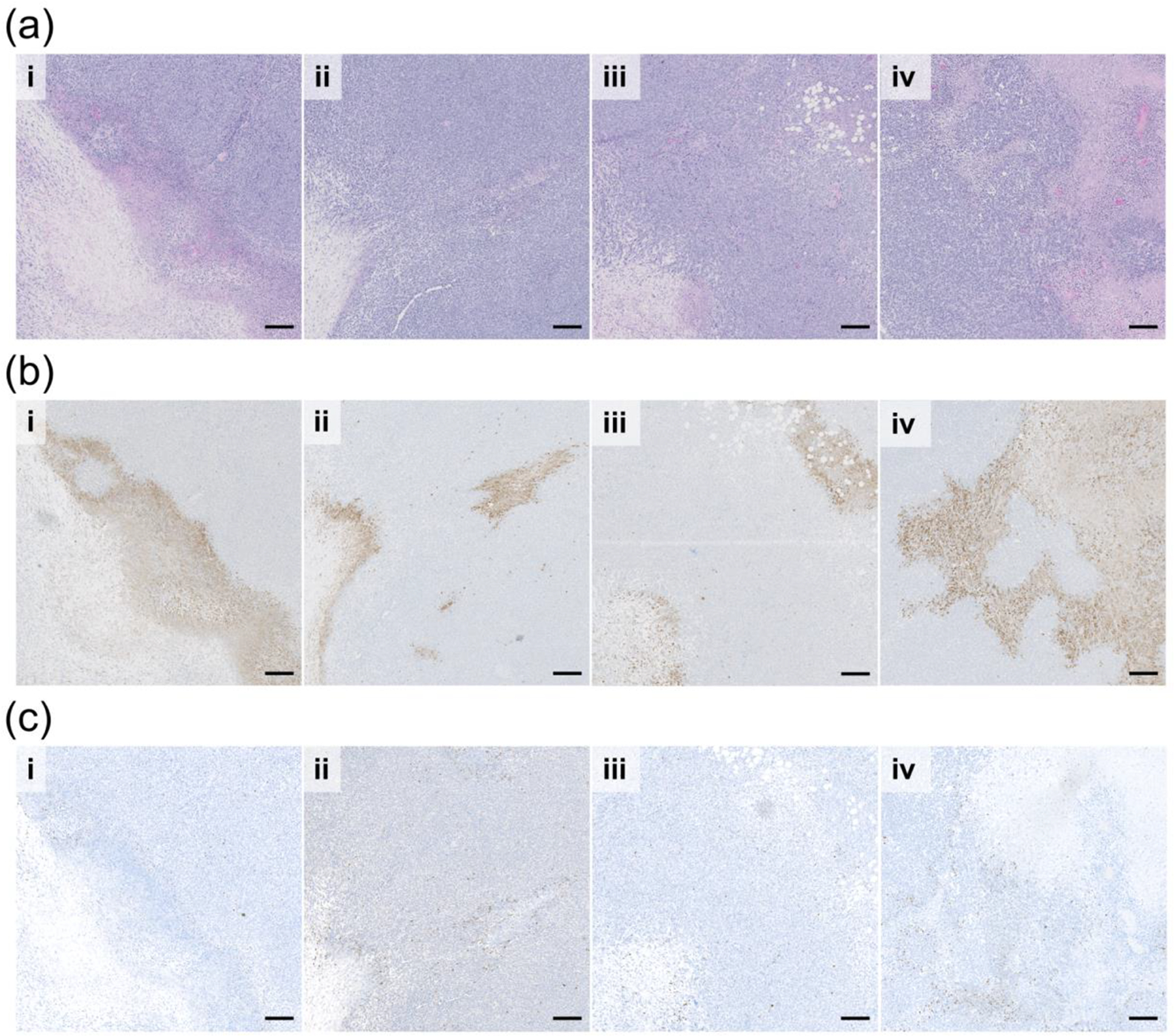
Sequential tumor sections of (a) H&E (b) apoptosis (Cleaved Caspase-3 stained in brown), and (c) DNA Damage (γH2AX stained in brown) evaluation of (i) E MPM (ii) TB MPM (iii) E MPM + IR and (iv) TB MPM+ IR therapeutic regimen efficacy. Scale bar represents 200 microns.
Safety Profile of Empty and Dual-Loaded Micelles.
Therapeutics that are efficacious but demonstrate a poor safety profile can hinder bench to bedside translation.61 In order to prevent administration of therapeutics with unacceptable side effects, pre-clinical in vivo studies must be conducted evaluating not only efficacy but also, more importantly, safety. Towards this end, both the acute and chronic toxicities associated with our MPMs were investigated. Acute toxicity was evaluated 4 days after a single injection of empty or dual drug-loaded MPMs to healthy, tumor-free mice. Blood was collected terminally via cardiac puncture and sent for clinical chemistry evaluation of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), Blood Urea Nitrogen (BUN), albumin/globulin (A/G), and total protein (TP) as well as complete blood counts (CBC). As shown in Figure 8a, administration of TB MCMs did not significantly alter markers associated with liver (ALT, ALP, AST), kidney (BUN), and liver/kidney (albumin/globulin A/G, total protein TP) function compared to the control group (PBS). Additionally, TP, BUN, and AST all fell within the reference ranges for BALB/c mice as described by Charles River.62 ALT and ALP were slightly lower than the published range, but low levels of these parameters are not concerning as higher levels are what indicate liver disease or dysfunction. As common side effects associated with Tal administration include increased ALT, AST and ALP levels, these findings are promising when assessing MPM encapsulation and dose reduction due to combination therapy for expanding the therapeutic index.63
Figure 8.
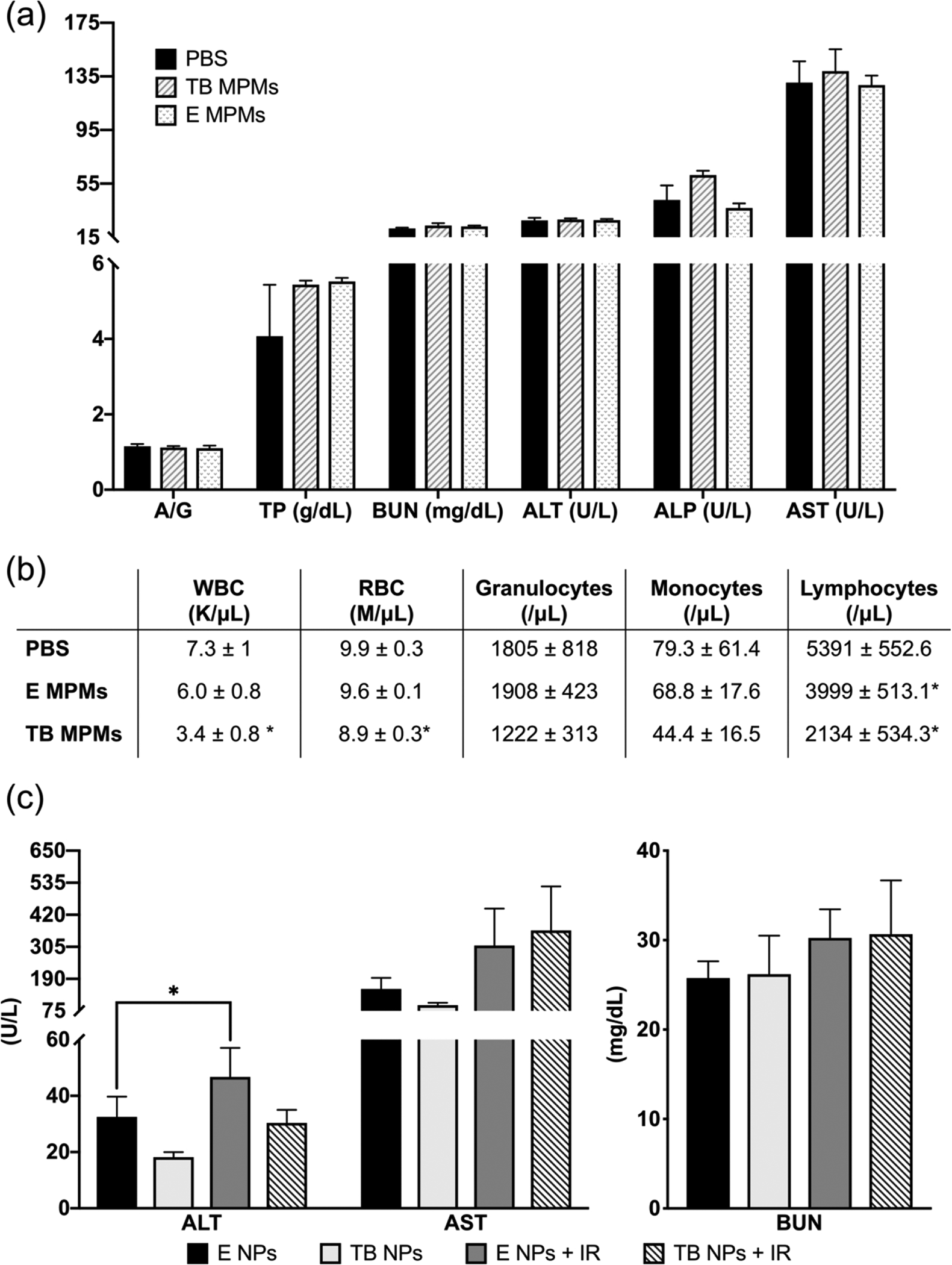
In vivo evaluation of acute toxicity of encapsulated drugs and carrier. (a) Plasma clinical chemistry markers of liver and kidney function and (b) Hematology toxicity parameters from healthy, tumor-free mice 96 hours after a single dose of PBS (control), E MPMs (310 mg/kg poloxamers), or TB MPMs (1.3 mg/kg tal, 1.23 mg/kg bup). (c) Plasma clinical chemistry markers of chronic toxicity of encapsulated drugs and carrier monitoring liver (ALT and AST) and kidney (BUN function.) * denotes statistically significant difference compared to PBS control (p < 0.05).
In addition to hepatotoxicity and renal toxicity, CBCs, were performed in order to assess overall hematological toxicity. As shown in Figure 8b, E MPM treatment had no impact on white blood cell (WBC), red blood cell (RBC), granulocyte, or monocyte counts as compared to the PBS control. Interestingly, E MPMs did significantly decrease lymphocyte counts from PBS controls, but they remained in the normal range for BALB/c mice. In comparison, administration of TB MPMs resulted in statistically significant decreases in WBC, RBC, and lymphocyte counts. This is likely due to the effect of Tal which is known to cause anemia (low RBC count) and low lymphocyte counts (and subsequently low WBC count).63 Overall, despite some evidence of hematological toxicities characteristic of Tal therapy, TB MPMs demonstrated no significant acute renal or hepatic toxicities. A nanoparticle mediated increase in circulation time and a decrease in amount distributed to the liver could account for the presence of hematologic toxicity and absence hepatic toxicity.
Oftentimes, therapeutic regimens consist of many administrations over a longer period of time. This makes chronic toxicologic evaluation an essential investigation. Our proposed therapeutic regimen is not only the administration of dual drug-loaded MPMs, but also the incorporation of radiation to the dosing schedule. Thus, chronic toxicity from E MPMs and TB MPMs alone, as well as in conjunction with radiation was evaluated. In the efficacy study, once a terminal endpoint was reached, blood was collected for clinical chemistry evaluation of liver and kidney toxicity and organs were collected for H&E staining to evaluate whole-body therapeutic toxicity. As shown in Figure 8c and Figure S10, markers of liver and kidney disease/dysfunction are not significantly different from each other in all groups, apart from an increase in ALT levels when E MPMs + IR are administered in comparison to E MPMs alone. The increase in ALT could be attributed to the crude shielding that occurs during radiotherapy as radiotherapy has been correlated with increases in ALT levels.64 In addition, body weight was evaluated throughout the course of study to monitor toxicity (Figure S11). A loss of greater than 20% would indicate a toxic therapeutic regimen or alternatively, cancer associated disease progression.
To further evaluate long-term MPM toxicity in vital organs; heart, lung, liver, spleen, and kidneys were harvested at efficacy study endpoint and subjected to H&E staining and histologic evaluation. As demonstrated in Figure 9, there is no obvious treatment related toxicity to the vital tissue of mice treated with MPMs or MPMs in combination with IR as compared to tumor-free, untreated mice. There is, however, evidence of metastatic infiltration in the liver of mice treated with E MPMs, TB MPMs, and E MPMs + IR (Figure 9c). In tumored mice treated with E MPMs alone, in addition to metastasis, large amounts of hepatocyte vacuolation can be seen indicating some level of liver damage. The addition of radiation to E MPMs slightly reduces vacuolation while administration of Tal and Bup seems to combat vacuolation. Interestingly, PARP inhibitors have previously been demonstrated to help reduce hepatocellular vacuolation associated with acetaminophen overdose.65
Figure 9.
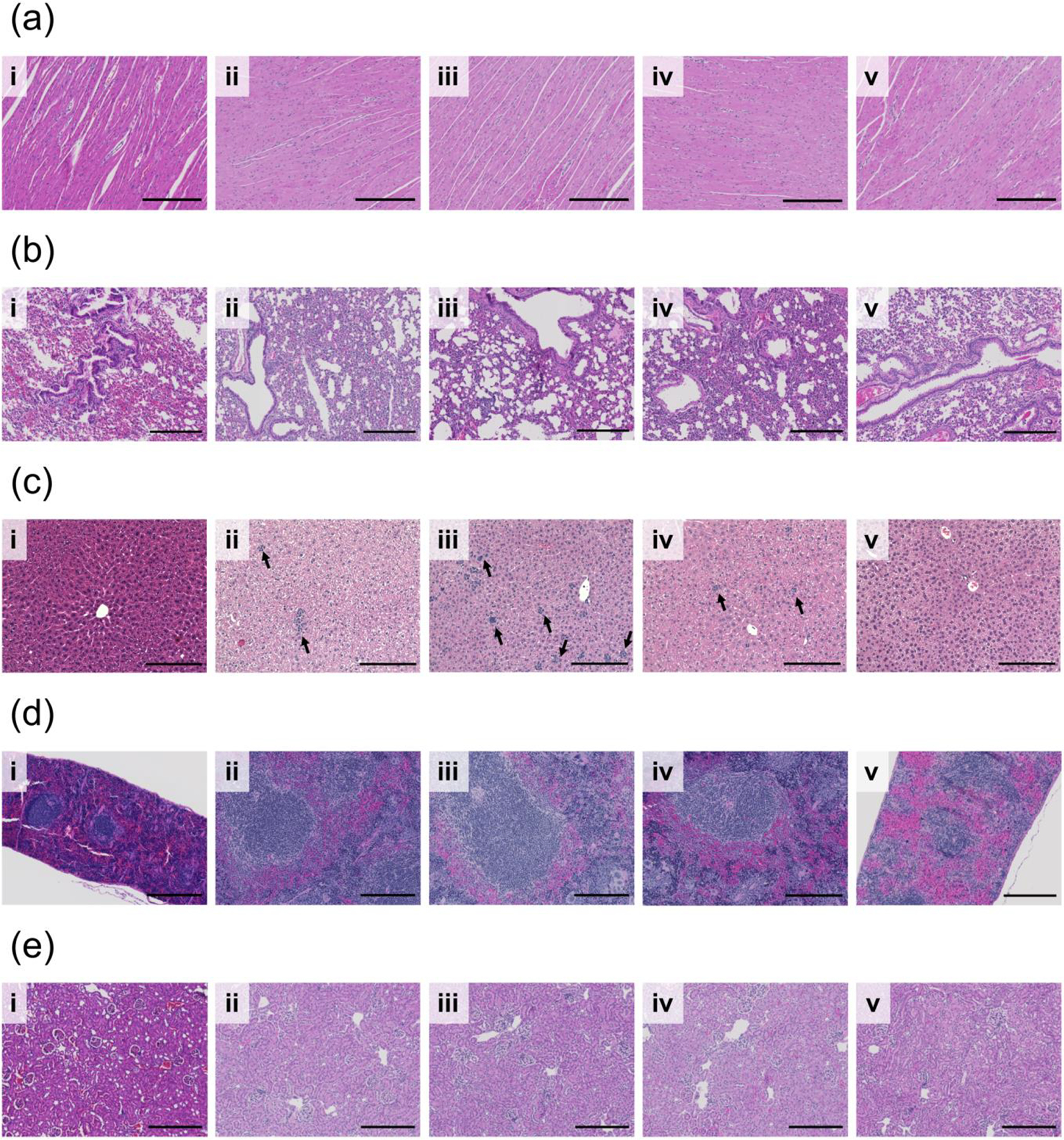
H&E staining evaluating safety of various treatments (i) naïve (ii) E MPM (iii) TB MPM (iv) E MPM + IR and (v) TB MPM+ IR on vital organs (a) heart (b) lung (c) liver (d) spleen (e) kidney during the course of study. Scale bar represents 200 microns in all images. Arrows identify regions of micrometastasis. Darkness of staining is a function of staining time, rather than histologic differences.
Another observation worth noting, is the difference in spleen size between treatment groups. Mice treated with E MPMs, TB MPMs, and E MPMS + IR demonstrate significantly enlarged spleens. While this observation alone is not noteworthy, as the 4T1 model is known to induce splenomegaly, it is worth noting that mice treated with TB MPMs + IR which developed lung metastases presented normal spleen sizes (Figure 9d). This could be a function of the combination of a PI3K inhibitor and IR, both of which alone have shown some efficacy in reducing splenomegaly.66,67
These data present evidence that this formulation of MPMs does not exhibit unacceptable or unusual toxicological effects when delivered at the doses prescribed and in combination with radiotherapy, despite evidence of cancer related toxicity.
CONCLUSION
In this report, a novel mixed poloxamer micelle (MPM) formulation was used to deliver both Tal and Bup in a single formulation during a course of radiotherapy. These near monodisperse sub-20 nm particles exhibited excellent colloidal stability (2 months) and versatility in addition to high encapsulation rates of the two hydrophobic DDR inhibitors. Particle stability and release were determined to be pH-dependent with both drugs exhibiting faster release in acidic environments, particularly those mimicking an endosomal environment. Metabolic assays confirmed that utilization of both Tal and Bup synergistically enhanced cytotoxicity in vitro. The ability of the therapeutics to enhance radiosensitivity was demonstrated in vitro using both a colony forming assay and DNA damage assay. Cell uptake experiments demonstrated that the MPMs were rapidly taken up over a 24-hour time period both in vitro and in vivo, with in vivo biodistribution demonstrating a tumor accumulation around one to three percent of the injected dose. Importantly, both short- and long-term administration of MPMs did not lead to any appreciable toxicity as evaluated through blood chemistry, histology, and weight change. In vivo experiments for evaluating therapeutic efficacy in an environment comparable to the current clinical context demonstrated that concurrent administration of TB MPMs during a course of fractionated radiotherapy exhibited promise through enhanced tumor control, as well as, the induction of γH2AX and subsequent apoptosis, despite absence of complete tumor regression. The unforeseen, sudden progression of gross metastases in TB MPM + IR treated mice may have obscured tumor control resulting in an absence of extended survival as compared to E MPM + IR treated mice.
Moving forward, additional adjustments to the treatment protocol such as alteration of the dosing schedule, the per injection dose, total radiation dose and dose per fraction, will likely lead to enhanced outcomes. The continued development and advancement of combination therapies is a critical component in cancer-based research when aiming to enhance the outcomes of patients with late-stage and metastatic disease. In order for this to be achieved, steps must to be taken towards the ongoing development and evaluation of potential drug combinations and the biological response. As such, this work provides valuable insight into factors that must be considered when utilizing DDR inhibitors for primary tumor control and the potential challenges associated with their use. Taken together, the results shown herein demonstrate the exciting potential of these novel nanomaterials for expanding the oncological therapeutic window, thus meriting further investigation.
MATERIALS AND METHODS
Materials.
The triblock copolymers Pluronic® F127 (Poloxamer P407 - MW12,600) and P103 (Poloxamer P333 - MW 4650) were supplied by BASF (Ludwigshafen, Germany). Coumarin 153, crystal violet dye, acetonitrile (HPLC grade), and methanol (HPLC grade) were purchased from Fisher Scientific (Hampton, NH, USA). Cyanine 7.5 Carboxylic Acid was purchased from Lumiprobe (Hunt Valley, MD, USA), Tal and Bup were purchased from MedChemExpress (Monmouth Junction, NJ, USA). Phosphate Buffered Saline (PBS) 1X and RPMI 1640 medium were purchased from Corning Inc. (Corning, NY, USA). Amicon Ultra centrifugal filters and 10% (v/v) neutral buffered formalin solution were obtained from Millipore Sigma (Burlington, MA, USA). DAPI (4’,6-Diamidino-2-Phenylindole, Dilactate) and Alexa Fluor™ 488 Phalloidin were purchased from Thermo Fisher Scientific (Carlsbad, CA, USA). 4T1 cells were kindly donated by the Dr. Lei Xing (Stanford University, CA, USA) and maintained in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin.
Preparation and Characterization of Drug Encapsulated Micelles.
Mixed poloxamer micelles (MPMs) were prepared via a modified nanoprecipitation method.68 In brief, all components are dissolved separately in acetonitrile at respective concentrations of 1000 mg/ml and 200 mg /ml for poloxamers 333 and 407. Following dissolution, stock solutions of p333, p407, and the corresponding compounds (Cy 7.5, C153, Tal, Bup) were added to acetonitrile for a total volume of 1 mL. To achieve MPM formation, the resulting organic solution was added to the aqueous phase, PBS, at a 1:9 volume ratio and briefly vortexed. The resulting MPM solution was diluted 1:1 into PBS and filtered at 4696 × g (max setting for centrifuge) for 15 minutes using a 10 kDa MW cutoff Amicon Ultra centrifugal filter to remove solvent and free drug/polymer. Following centrifugation, the MPMs were resuspended to the starting volume using PBS. Drug free MPMs were prepared in the same manner without the addition of drug.
Purified MPMs were characterized by Dynamic Light Scattering (DLS) and Transmission electron microscopy (TEM). DLS was performed using a Malvern Nano ZSP (Malvern Panalytical, Malvern, UK) to determine the size, zeta potential, and polydispersity. TEM images were obtained with a Tecnai iCorr while cryoTEM images were obtained with a Titan Krios Cryo-TEM (FEI, Hillsboro, OR, USA). Drug loading was quantified using a Shimadzu SPD-20A high performance liquid chromatography (HPLC) instrument (Torrance, CA, USA) equipped with UV/Vis detector and an Agilent Zorbax Rapid Resolution SBC-18 column (4.6 × 100 mm 3.5 μm; Santa Clara, CA, USA). Samples were diluted in methanol (1:9 dilution) and sonicated in order to break apart MPMs prior to HPLC analysis. Tal and Bup MPMs were run with a 65% to 70% methanol in water gradient over 2 minutes with a flow rate of 1 mL/min and a 311 nm detection wavelength. Tal eluted first (retention time = 1.3 min) with Bup following (retention time = 2.3 min).
In vitro drug release for the two inhibitors were determined by subjecting MPMs to dialysis using Slide-A-Lyzer MINI dialysis microtubes with a molecular weight cutoff of 10 kDa. MPM samples were loaded into dialysis microtubes and dialyzed against a > 400-fold excess of buffer (pH 7.4 or 6.8 PBS) with gentle stirring at ambient conditions. 0.1 mL samples were collected at designated time points (0, 1, 2, 4, 24, 48, 72 hours, or 7 days), diluted in methanol (1:9), and concentrations determined by HPLC as previously described above. Drug release profiles were plotted as a cumulative percentage of total drug release vs. time. All drug release measurements were performed in triplicate.
In Vitro Cellular Uptake Study.
4T1 cells were seeded into a 96-well plate (10,000 cells/well) or an 8-well chamber slide (80,000 cells/well) and allowed to settle for 24 hours. When cells had settled, for the 96-well plate, cells were treated with either an increasing concentration (1.95 ng/mL to 500 ng/mL) of Cy 7.5-loaded MPMs for 2 hours or a constant concentration (125 ng/mL) of Cy-7.5-loaded MPMs in fresh media for desired times between 0 and 24 hours. A maximum concentration of 500 ng/mL was chosen in order to avoid quantification interference due to intramolecular quenching. At study endpoint, the cells were washed with PBS, formalin fixed, and analyzed using a Tecan Infinite M200 Pro plate reader at an excitation and emission of 750 and 830 nm. When cells had settled in the 8-well chamber slides, cells were treated with empty MPMs, Cy 7.5-loaded MPMs (1000 ng/mL), or left untreated for 2 hours, washed with PBS, formalin fixed and stained with DAPI and Phalloidin 488 for nuclear and actin labeling. Microscopy was performed on an EVOS FL Auto microscope at 40 × magnification.
In Vitro Cytotoxicity.
To determine the efficacy of Tal or Bup as single or combination agents, 4T1 cells were seeded at 2 × 103 cells per well in a 96-well plate and allowed to settle overnight. Cells were then treated with free drugs (alone or in combination) at various concentrations (Figure 2a–c) and incubated at 37°C. After 72 hours, cell viability was determined by CellTiter-Fluor assay (Promega, Madison, WI). Sample fluorescence at 380 and 505 nm was evaluated using an Infinite M200 Pro plate reader (Tecan US Inc, Morrisville, NC, USA). Cell viability was normalized to untreated cells and IC50 values were computed by fitting the data to a four-parameter dose response, variable slope model using GraphPad Prism 7. Combination indexes (CI) were determined utilizing CompuSyn (ComboSyn Inc, Paramus, NJ, USA). All cell viability measurements were performed in triplicate.
Clonogenic Assay.
The effect of dual drug-loaded MPMs and radiation was assayed by seeding 4T1 cells into T75 flasks, which were allowed to settle overnight and subsequently treated for 72 hours. After therapeutic incubation, cells were trypsinized, seeded into 12-well plates, and irradiated (CellRad X-ray Cabinet Irradiator, Faxitron, 130 kV, 5 mA, 0.5 mm aluminum filter, ~1.2 Gy/min). The irradiated cells were then allowed to proliferate the equivalent of 6 doubling times. The number of cells seeded varied based on the dose of radiation (0, 2, 4, 6, 8 Gy) and drug treatment conditions (0.945 μM T MPMs, 0.505 μM B MPMs, 0.625/0.375 μM TB MPMs, 1.8 mg/mL E and all other MPMs) ranging from 200 cells/well for no treatment to 9600 cells/well for 8 Gy plus encapsulated Tal/Bup. Colonies were washed with PBS, fixed with 4% v/v formalin, and stained with 0.1% w/v crystal violet dye. After staining cells were gently submerged in a bath of deionized water, dried, and counted. The survival fraction (SF) was determined by calculating a plating efficiency (PE) for each treatment and dividing that value by the PE of the untreated control, where PE is the number of colonies formed divided by the number of cells seeded.
Immunofluorescent γH2AX DNA Damage Assay.
40,000 4T1 cells were plated in a gelatin-coated 8-well chamber slide and allowed to settle for 24 hours. At 24 hours, media was replaced with media containing drug or MPM treatment. Cells were incubated with various drug or MPM treatments (Figure 3) for 72 hours and then irradiated with 5 Gy (CellRad). Three hours after irradiation, cells were washed 3× with PBS and formalin fixed. Formalin was removed by washing with PBS and then blocked and permeabilized using 5% Goat Serum and 0.3% Triton X-100. Following blocking and permeabilization, cells were incubated with a rabbit anti-Phospho-Histone H2AX (Ser139) Antibody (#2577, Cell Signaling Technologies) at a 1:800 dilution for 1 hour at room temperature in antibody dilution buffer (1% BSA and 0.3% Triton X-100 in PBS). Next, cells were washed with PBS and incubated with a goat anti-rabbit IgG H&L (Cy 5) antibody (6564, Abcam) at a 1:1600 dilution in antibody dilution buffer in the dark for 1 hour at room temperature. Lastly, cells were once more washed with PBS and imaged using an EVOS FL Auto microscope at 40 × magnification. Using 40× EVOS images (> 150 cells/ treatment group in various images), DNA damage quantification was automated by cell profiler overlaying the DAPI stained nucleus with the Cy 5 stained γH2AX foci for a foci/cell count. To be counted, a focus had to be greater than 7 pixels.
In Vivo Studies.
All animal studies were approved by and conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Oregon Health and Sciences University. For all imaging studies and when noted, mice were anesthetized using 2–3% isoflurane (Piramal Enterprises Limited, Telangana, India).
Evaluation of acute MPM effects in mice.
Toxicity of the MPM formulations were evaluated in healthy, tumor-free female BALB/c mice aged 12-weeks. Treatments were as follows: PBS, E MPMs (310 mg/kg poloxamer), or TB MPMs (1.30 mg/kg Tal, 1.23 mg/kg Bup). 96 hours after treatment, mice were anesthetized, and blood was collected into 1 mL lithium heparin coated tubes (Grenier Bio-One, Kremsmünster, Austria) via cardiac puncture and stored at 4°C prior to analysis by IDEXX Laboratories for overall toxicity.
Establishment of tumor xenografts in mice.
Subcutaneous 4T1 tumor xenografts were established by injecting cell suspensions (5 × 104 cells) into the left inguinal mammary fat pad (biodistribution) or right flanks (efficacy) of BALB/c mice (Charles River Laboratories, Wilmington, MA).
Evaluation of biodistribution in mice.
For biodistribution studies, BALB/c mice bearing inguinal mammary fat pad tumors were irradiated with 3 fractions of 5 Gy delivered on 3 consecutive days. At a given timepoint after irradiation (24 or 72 hours), mice were injected intravenously with 100 μL 0.1 mg/mL Cy 7.5-loaded MPMs. For MPMs injected 24 hours after irradiation, whole-body fluorescent images were obtained using an IVIS Lumina XRMS (Perkin-Elmer, Waltham, MA) at 4 and 24 hours post-i.v. injection using excitation and emission filters of 780 and 845 respectively. For mice injected 72 hours after irradiation, whole-body fluorescent images were obtained using an IVIS Lumina XRMS at only 24 hours post-i.v. injection using the same wavelengths. At 24 hours post-injection, mice were euthanized by isoflurane overdose and organs were collected for ex vivo imaging using the IVIS settings described above. Organ biodistribution was determined utilizing Living Image Software (Perkin-Elmer, Waltham, MA) by summing flux for all organs, based on organ traced region of interests, and dividing the value for each organ of interest by the total value.
Evaluation of antitumor efficacy and long-term effects in mice.
When tumors reached 60–175 mm3, mice were randomized to one of four treatment groups (n = 4–5 per group). Treatment groups were as follows: (1) Empty micelles (E MPMs) control, (2) Tal and Bup micelles (TB MPMs) (0.84 mg/kg tal, 0.78 mg/kg Bup), (3) E MPMs + IR, or (4) TB MPMs (0.84 mg/kg tal, 0.78 mg/kg Bup) + IR. Mice were treated via tail vein injection 3× weekly starting on day 0 and continuing throughout the course of the study. Mice in groups 3 and 4 were irradiated with 5 Gy on days 0, 1, and 2 for a total of 15 Gy. Radiation was delivered (CellRad, Faxitron, 130 kV, 5 mA, 0.5 mm aluminum filter, ~1.2 Gy/min) selectively to tumors by covering mice with half-moon cutout lead shields (Precision X-Ray, North Branford, CT). Mouse body weight and average tumor diameter (1/2 × length × width2) were recorded thrice per week prior to MPM injection. Mice were euthanized per IACUC guidelines once tumors began to develop cavitated ulcerations, weight loss reached 20% of starting weight, or any diameter reached 2 cm. Cardiac puncture was utilized to collect terminal blood samples for clinical chemistry. Organs from at least one mouse per treatment group were harvested for H&E staining to evaluate therapeutic toxicity. Additionally, tumors from these mice were stained for γH2AX and Cleaved Caspase-3 to evaluate prevalence of DNA DSBs or apoptosis respectively. A Kaplan-Meier curve was developed for each treatment group to examine differences in survival.
Tumor Histology.
Tumors from mice in the antitumor efficacy treatment protocol were collected and fixed in 10% (v/v) neutral buffered formalin solution (4% formaldehyde) at 4°C overnight before being transferred to 70% ethanol and submitted to the OHSU Histopathology Shared Resource Core for tissue clearing, slicing (5 μm), and subsequent staining. Stains performed were hematoxylin and eosin (H&E), Caspase-3 (Promega), and γH2AX (Cell Signaling Technologies). Antibodies were diluted according to supplier recommendations before staining. Imaging was performed on a Zeiss Axio Scan.Z1 Slide Scanner by the OHSU Advanced Light Microscopy Core at 20X.
Statistical analysis.
All data are expressed as mean ± SD. Statistical differences and significance were evaluated using one-way ANOVA or logrank (Mantel-Cox) test for survival curves in the Graph Pad Prism 7 software pack. p < 0.05 was considered statistically significant and represented by *.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the Histology Core, Multiscale Microscopy Core, and Advanced Light Microscopy Core at Oregon Health & Sciences University for their assistance with various procedures presented in this study. This work was supported by the NIH NIGMS as a Maximizing Investigators’ Research Award, 1R35GM119839-01 (C.S.), NIH NIBIB 1R15EB021581-01 (G.S.) and Oregon State University College of Pharmacy Startup Funds.
Footnotes
SUPPORTING INFORMATION
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
Additional experimental details and data: DLS graphs of individually loaded MPMs; graph detailing long-term stability of empty and TB MPMs; formulation details and data for MPMs utilizing fluorescent probe C153 and Cy 7.5; Combination Index plot of Tal and Bup in 4T1 cells; γH2AX images and quantification for free Tal, Bup, and the combination; IVIS images of organs quantified for biodistribution study; tumor volume change, tumor volume, and weight change of individual mice in efficacy study; histology H&E staining of lung tissue demonstrating metastases; whole tumor images of H&E and cleaved caspase-3 staining; kidney and liver clinical chemistry markers from long-term toxicity study; average weight change of mice treated with MPMs demonstrating long-term particle safety; in vivo experimental design flow chart; table detailing nanoparticle characteristics; table detailing clonogenic linear-quadratic line fit parameters
REFERENCES
- (1).Holohan C; Van Schaeybroeck S; Longley DB; Johnston PG Cancer Drug Resistance: An Evolving Paradigm. Nature Reviews Cancer 2013, 13, 714–726. [DOI] [PubMed] [Google Scholar]
- (2).Dagogo-Jack I; Shaw AT Tumour Heterogeneity and Resistance to Cancer Therapies. Nature Reviews Clinical Oncology 2018, 15, 81–94. [DOI] [PubMed] [Google Scholar]
- (3).Mroz EA; Rocco JW The Challenges of Tumor Genetic Diversity: Tumor Diversity. Cancer 2017, 123, 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bozic I; Reiter JG; Allen B; Antal T; Chatterjee K; Shah P; Moon YS; Yaqubie A; Kelly N; Le DT; Lipson EJ; Chapman PB; Diaz LA Jr; Vogelstein B; Nowak MA Evolutionary Dynamics of Cancer in Response to Targeted Combination Therapy. eLife 2013, 2, e00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bayat Mokhtari R; Homayouni TS; Baluch N; Morgatskaya E; Kumar S; Das B; Yeger H Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022–38043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gordon Steel G; Peckham MJ Exploitable Mechanisms in Combined Radiotherapy-Chemotherapy: The Concept of Additivity. International Journal of Radiation Oncology*Biology*Physics 1979, 5, 85–91. [DOI] [PubMed] [Google Scholar]
- (7).Seiwert TY; Salama JK; Vokes EE The Concurrent Chemoradiation Paradigm—General Principles. Nature Clinical Practice Oncology 2007, 4, 86–100. [DOI] [PubMed] [Google Scholar]
- (8).Hall EJ; Giaccia AJ Cell Survival Curves In Radiobiology for the Radiologist; Lippincott Williams & Wilkins: Philadelphia, PA, 2012; pp 30–46. [Google Scholar]
- (9).Aly A; Ganesan S BRCA1, PARP, and 53BP1: Conditional Synthetic Lethality and Synthetic Viability. J Mol Cell Biol 2011, 3, 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chan SL; Mok T PARP Inhibition in BRCA-Mutated Breast and Ovarian Cancers. The Lancet 2010, 376, 211–213. [DOI] [PubMed] [Google Scholar]
- (11).Kumar A; Fernandez-Capetillo O; Carrera AC Nuclear Phosphoinositide 3-Kinase β Controls Double-Strand Break DNA Repair. PNAS 2010, 107, 7491–7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Philip C-A; Laskov I; Beauchamp M-C; Marques M; Amin O; Bitharas J; Kessous R; Kogan L; Baloch T; Gotlieb WH; Yasmeen A Inhibition of PI3K-AKT-MTOR Pathway Sensitizes Endometrial Cancer Cell Lines to PARP Inhibitors. BMC Cancer 2017, 17, 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).West KA; Sianna Castillo S; Dennis PA Activation of the PI3K/Akt Pathway and Chemotherapeutic Resistance. Drug Resistance Updates 2002, 5, 234–248. [DOI] [PubMed] [Google Scholar]
- (14).Criscitiello C; Viale G; Curigliano G; Goldhirsch A Profile of Buparlisib and Its Potential in the Treatment of Breast Cancer: Evidence to Date. Breast Cancer (Dove Med Press) 2018, 10, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Dréan A; Lord CJ; Ashworth A PARP Inhibitor Combination Therapy. Critical Reviews in Oncology/Hematology 2016, 108, 73–85. [DOI] [PubMed] [Google Scholar]
- (16).Juvekar A; Burga LN; Hu H; Lunsford EP; Ibrahim YH; Balmañà J; Rajendran A; Papa A; Spencer K; Lyssiotis CA; Nardella C; Pandolfi PP; Baselga J; Scully R; Asara JM; Cantley LC; Wulf GM Combining a PI3K Inhibitor with a PARP Inhibitor Provides an Effective Therapy for BRCA1-Related Breast Cancer. Cancer discovery 2012, 2, 1048–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ibrahim YH; García-García C; Serra V; He L; Torres-Lockhart K; Prat A; Anton P; Cozar P; Guzmán M; Grueso J; Rodríguez O; Calvo MT; Aura C; Díez O; Rubio IT; Pérez J; Rodón J; Cortés J; Ellisen LW; Scaltriti M; Baselga J PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov 2012, 2, 1036–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jang NY; Kim DH; Cho BJ; Choi EJ; Lee J-S; Wu H-G; Chie EK; Kim IA Radiosensitization with Combined Use of Olaparib and PI-103 in Triple-Negative Breast Cancer. BMC Cancer 2015, 15, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yi X; Chen L; Chen J; Maiti D; Chai Z; Liu Z; Yang K Biomimetic Copper Sulfide for Chemo-Radiotherapy: Enhanced Uptake and Reduced Efflux of Nanoparticles for Tumor Cells under Ionizing Radiation. Advanced Functional Materials 2018, 28, 1705161. [Google Scholar]
- (20).Mizrachi A; Shamay Y; Shah J; Brook S; Soong J; Rajasekhar VK; Humm JL; Healey JH; Powell SN; Baselga J; Heller DA; Haimovitz-Friedman A; Scaltriti M Tumour-Specific PI3K Inhibition via Nanoparticle-Targeted Delivery in Head and Neck Squamous Cell Carcinoma. Nature Communications 2017, 8, 14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Au KM; Min Y; Tian X; Zhang L; Perello V; Caster JM; Wang AZ Improving Cancer Chemoradiotherapy Treatment by Dual Controlled Release of Wortmannin and Docetaxel in Polymeric Nanoparticles. ACS Nano 2015, 9, 8976–8996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Caster JM; Sethi M; Kowalczyk S; Wang E; Tian X; Hyder SN; Wagner KT; Zhang Y-A; Kapadia C; Au KM; Wang AZ Nanoparticle Delivery of Chemosensitizers Improve Chemotherapy Efficacy without Incurring Additional Toxicity. Nanoscale 2015, 7, 2805–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pandey A; Kulkarni A; Roy B; Goldman A; Sarangi S; Sengupta P; Phipps C; Kopparam J; Oh M; Basu S; Kohandel M; Sengupta S Sequential Application of a Cytotoxic Nanoparticle and a PI3K Inhibitor Enhances Antitumor Efficacy. Cancer Res 2014, 74, 675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang M; Hagan CT; Min Y; Foley H; Tian X; Yang F; Mi Y; Au KM; Medik Y; Roche K; Wagner K; Rodgers Z; Wang AZ Nanoparticle Co-Delivery of Wortmannin and Cisplatin Synergistically Enhances Chemoradiotherapy and Reverses Platinum Resistance in Ovarian Cancer Models. Biomaterials 2018, 169, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Maiti D; Chao Y; Dong Z; Yi X; He J; Liu Z; Yang K Development of a Thermosensitive Protein Conjugated Nanogel for Enhanced Radio-Chemotherapy of Cancer. Nanoscale 2018, 10, 13976–13985. [DOI] [PubMed] [Google Scholar]
- (26).Zhang Z; Yang J; Min Q; Ling C; Maiti D; Xu J; Qin L; Yang K Holo-Lactoferrin Modified Liposome for Relieving Tumor Hypoxia and Enhancing Radiochemotherapy of Cancer. Small 2019, 15, 1803703. [DOI] [PubMed] [Google Scholar]
- (27).Song G; Liang C; Gong H; Li M; Zheng X; Cheng L; Yang K; Jiang X; Liu Z Core–Shell MnSe@Bi2Se3 Fabricated via a Cation Exchange Method as Novel Nanotheranostics for Multimodal Imaging and Synergistic Thermoradiotherapy. Advanced Materials 2015, 27, 6110–6117. [DOI] [PubMed] [Google Scholar]
- (28).Song G; Chao Y; Chen Y; Liang C; Yi X; Yang G; Yang K; Cheng L; Zhang Q; Liu Z All-in-One Theranostic Nanoplatform Based on Hollow TaOx for Chelator-Free Labeling Imaging, Drug Delivery, and Synergistically Enhanced Radiotherapy. Advanced Functional Materials 2016, 26, 8243–8254. [Google Scholar]
- (29).Song G; Cheng L; Chao Y; Yang K; Liu Z Emerging Nanotechnology and Advanced Materials for Cancer Radiation Therapy. Advanced Materials 2017, 29, 1700996. [DOI] [PubMed] [Google Scholar]
- (30).Blanco E; Shen H; Ferrari M Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nature Biotechnology 2015, 33, nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yi YW; Park J-S; Kwak S-J; Seong Y-S Co-Treatment with BEZ235 Enhances Sensitivity of BRCA1-Negative Breast Cancer Cells to Olaparib. Anticancer Res 2015, 35, 3829–3838. [PubMed] [Google Scholar]
- (32).Wang D; Li C; Zhang Y; Wang M; Jiang N; Xiang L; Li T; Roberts TM; Zhao JJ; Cheng H; Liu P Combined Inhibition of PI3K and PARP Is Effective in the Treatment of Ovarian Cancer Cells with Wild-Type PIK3CA Genes. Gynecologic Oncology 2016, 142, 548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Patel P; Misra S; Rodriguez NS; Vulugundam G; Oh AL; Senyuk V; Mahmud N; Rondelli D; Pan D Combined Nanoparticle Delivery of PARP and DNA-PK Inhibition for Multiple Myeloma. Blood 2017, 130, 1809–1809.28807982 [Google Scholar]
- (34).Batrakova EV; Kabanov AV Pluronic Block Copolymers: Evolution of Drug Delivery Concept from Inert Nanocarriers to Biological Response Modifiers. Journal of Controlled Release 2008, 130, 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wolfram J; Yang Y; Shen J; Moten A; Chen C; Shen H; Ferrari M; Zhao Y The Nano-Plasma Interface: Implications of the Protein Corona. Colloids and Surfaces B: Biointerfaces 2014, 124, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Stewart PL Cryo-Electron Microscopy and Cryo-Electron Tomography of Nanoparticles. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology 2017, 9, e1417. [DOI] [PubMed] [Google Scholar]
- (37).Batrakova EV; Bronich TK; Vetro JA; Kabanov AV Polymer Micelles as Drug Carriers In Nanoparticulates as Drug Carriers; Imperial College Press: Covent Garden, London, 2006; pp 57–93. [Google Scholar]
- (38).Chou T-C Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res 2010, 70, 440–446. [DOI] [PubMed] [Google Scholar]
- (39).Unkel S; Belka C; Lauber K On the Analysis of Clonogenic Survival Data: Statistical Alternatives to the Linear-Quadratic Model. Radiat Oncol 2016, 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Williams MV; Denekamp J; Fowler JF A Review of Aβ Ratios for Experimental Tumors: Implications for Clinical Studies of Altered Fractionation. International Journal of Radiation Oncology*Biology*Physics 1985, 11, 87–96. [DOI] [PubMed] [Google Scholar]
- (41).Jones TR; Kang IH; Wheeler DB; Lindquist RA; Papallo A; Sabatini DM; Golland P; Carpenter AE CellProfiler Analyst: Data Exploration and Analysis Software for Complex Image-Based Screens. BMC Bioinformatics 2008, 9, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Perera RH; Patel R; Wu H; Gangolli M; Traughber B; Oleinick N; Exner AA Preclinical Evaluation of Radiosensitizing Activity of Pluronic Block Copolymers. Int J Radiat Biol 2013, 89, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Perera RH; Krupka TM; Wu H; Traughber B; Dremann D; Broome AM; Exner AA Role of Pluronic Block Copolymers in Modulation of Heat Shock Protein 70 Expression. International Journal of Hyperthermia 2011, 27, 672–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Stapleton S; Dunne M; Milosevic M; Tran CW; Gold MJ; Vedadi A; Mckee TD; Ohashi PS; Allen C; Jaffray DA Radiation and Heat Improve the Delivery and Efficacy of Nanotherapeutics by Modulating Intratumoral Fluid Dynamics. ACS Nano 2018, 12, 7583–7600. [DOI] [PubMed] [Google Scholar]
- (45).Harrington KJ; Rowlinson-Busza G; Syrigos KN; Uster PS; Vile RG; Peters AM; Stewart JSW The Effect of Irradiation on the Biodistribution of Radiolabeled Pegylated Liposomes. International Journal of Radiation Oncology*Biology*Physics 2001, 50, 809–820. [DOI] [PubMed] [Google Scholar]
- (46).Single-Fraction Irradiation Has No Effect on Uptake of Radiolabeled Pegylated Liposomes in a Tumor Xenograft Model. International Journal of Radiation Oncology*Biology*Physics 2001, 49, 1141–1148. [DOI] [PubMed] [Google Scholar]
- (47).Kobayashi H; Reijnders K; English S; Yordanov AT; Milenic DE; Sowers AL; Citrin D; Krishna MC; Waldmann TA; Mitchell JB; Brechbiel MW Application of a Macromolecular Contrast Agent for Detection of Alterations of Tumor Vessel Permeability Induced by Radiation. Clin Cancer Res 2004, 10, 7712–7720. [DOI] [PubMed] [Google Scholar]
- (48).Smith MA; Reynolds CP; Kang MH; Kolb EA; Gorlick R; Carol H; Lock RB; Keir ST; Maris JM; Billups CA; Lyalin D; Kurmasheva RT; Houghton PJ Synergistic Activity of PARP Inhibition by Talazoparib (BMN 673) with Temozolomide in Pediatric Cancer Models in the Pediatric Preclinical Testing Program. Clin Cancer Res 2015, 21, 819–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hopkins TA; Shi Y; Rodriguez LE; Solomon LR; Donawho CK; DiGiammarino EL; Panchal SC; Wilsbacher JL; Gao W; Olson AM; Stolarik DF; Osterling DJ; Johnson EF; Maag D Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Molecular Cancer Research 2015, 13, 1465–1477. [DOI] [PubMed] [Google Scholar]
- (50).Laird JH; Lok BH; Ma J; Bell A; de Stanchina E; Poirier JT; Rudin CM Talazoparib Is a Potent Radiosensitizer in Small Cell Lung Cancer Cell Lines and Xenografts. Clin Cancer Res 2018, 24, 5143–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Gadducci A; Guerrieri ME PARP Inhibitors Alone and in Combination with Other Biological Agents in Homologous Recombination Deficient Epithelial Ovarian Cancer: From the Basic Research to the Clinic. Critical Reviews in Oncology/Hematology 2017, 114, 153–165. [DOI] [PubMed] [Google Scholar]
- (52).Wilhelm S; Tavares AJ; Dai Q; Ohta S; Audet J; Dvorak HF; Chan WCW Analysis of Nanoparticle Delivery to Tumours. Nature Reviews Materials 2016, 1, 16014. [Google Scholar]
- (53).de Gooijer MC; Zhang P; Buil LCM; Çitirikkaya CH; Thota N; Beijnen JH; van Tellingen O Buparlisib Is a Brain Penetrable Pan-PI3K Inhibitor. Scientific Reports 2018, 8, 10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Netland IA; Førde HE; Sleire L; Leiss L; Rahman MA; Skeie BS; Miletic H; Enger PØ; Goplen D Treatment with the PI3K Inhibitor Buparlisib (NVP-BKM120) Suppresses the Growth of Established Patient-Derived GBM Xenografts and Prolongs Survival in Nude Rats. J Neurooncol 2016, 129, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Brachmann SM; Kleylein-Sohn J; Gaulis S; Kauffmann A; Blommers MJJ; Kazic-Legueux M; Laborde L; Hattenberger M; Stauffer F; Vaxelaire J; Romanet V; Henry C; Murakami M; Guthy DA; Sterker D; Bergling S; Wilson C; Brummendorf T; Fritsch C; Garcia-Echeverria C; Sellers WR; Hofmann F; Maira S-M Characterization of the Mechanism of Action of the Pan Class I PI3K Inhibitor NVP-BKM120 across a Broad Range of Concentrations. Molecular Cancer Therapeutics 2012, 11, 1747–1757. [DOI] [PubMed] [Google Scholar]
- (56).Niessner H; Schmitz J; Tabatabai G; Schmid AM; Calaminus C; Sinnberg T; Weide B; Eigentler TK; Garbe C; Schittek B; Quintanilla-Fend L; Bender B; Mai M; Praetorius C; Beissert S; Schackert G; Muders MH; Meinhardt M; Baretton GB; Dummer R; Flaherty K; Pichler BJ; Kulms D; Westphal D; Meier F PI3K Pathway Inhibition Achieves Potent Antitumor Activity in Melanoma Brain Metastases In Vitro and In Vivo. Clinical Cancer Research 2016, 22, 5818–5828. [DOI] [PubMed] [Google Scholar]
- (57).Pons-Tostivint E; Thibault B; Guillermet-Guibert J Targeting PI3K Signaling in Combination Cancer Therapy. Trends in Cancer 2017, 3, 454–469. [DOI] [PubMed] [Google Scholar]
- (58).Jiao S; Xia W; Yamaguchi H; Wei Y; Chen M-K; Hsu J-M; Hsu JL; Yu W-H; Du Y; Lee H-H; Li C-W; Chou C-K; Lim S-O; Chang S-S; Litton J; Arun B; Hortobagyi GN; Hung M-C PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clinical Cancer Research 2017, 23, 3711–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Fruman DA; Chiu H; Hopkins BD; Bagrodia S; Cantley LC; Abraham RT The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Qin H; Liu L; Sun S; Zhang D; Sheng J; Li B; Yang W The Impact of PI3K Inhibitors on Breast Cancer Cell and Its Tumor Microenvironment. PeerJ 2018, 6, e5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Information for Consumers (Drugs) - The FDA’s Drug Review Process: Ensuring Drugs Are Safe and Effective; U.S. Food and Drug Administration: Silver Spring, MD, 2017. [Google Scholar]
- (62).Clinical Pathology Data for BALB/c Mouse Colonies in North America for January 2008 - December 2012; Charles River: Wilmington, MA, 2012. [Google Scholar]
- (63).Highlights of Talazoparib Prescribing Information; U.S. Food and Drug Administration: Silver Spring, MD, 2018. [Google Scholar]
- (64).Zahedi R; Bakhshandeh M; Sabouri H; Ahmadi MY; Roshani D Early Effect of Radiation on the Liver Function Tests of Patients with Thoracic and Abdominal Tumors during Radiotherapy. Journal of Paramedical Sciences 2016, 7, 8–12. [Google Scholar]
- (65).Dönmez M; Uysal B; Poyrazoğlu Y; Er Öztaş Y; Türker T; Kaldirim Ü; Korkmaz A PARP Inhibition Prevents Acetaminophen-Induced Liver Injury Andincreases Survival Rate in Rats. TURKISH JOURNAL OF MEDICAL SCIENCES 2015, 45, 18–26. [DOI] [PubMed] [Google Scholar]
- (66).Deng L; Virts EL; Kapur R; Chan RJ Pharmacologic Inhibition of PI3K P110δ in Mutant Shp2E76K-Expressing Mice. Oncotarget 2017, 8, 84776–84781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Zaorsky NG; Williams GR; Barta SK; Esnaola NF; Kropf PL; Hayes SB; Meyer JE Splenic Irradiation for Splenomegaly: A Systematic Review. Cancer Treat. Rev 2017, 53, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Fessi H; Puisieux F; Devissaguet JP; Ammoury N; Benita S Nanocapsule Formation by Interfacial Polymer Deposition Following Solvent Displacement. International Journal of Pharmaceutics 1989, 55, R1–R4. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.