

Abstract
Combined modality therapy incorporating raltitrexed (RTX), a thymidylate synthase inhibitor, and radiation can lead to improved outcome for rectal cancer patients. To increase delivery and treatment efficacy, we formulated a hyaluronic acid (HA) coated nanoparticle encapsulating RTX (HARPs) through layer-by-layer assembly. These particles were determined to have a diameter of ~115 nm, with a polydispersity index of 0.112 and a zeta potential of −22 mV. Cell uptake in CT26 cells determined through flow cytometry showed a ~5-fold increase between untargeted and HA-coated particles. Through viability and DNA damage assays, we assessed the potency of the free RTX and HARPs, and found increased DNA damage in cells treated with the RTX-loaded nanoparticles administered concurrently with radiation. In vivo efficacy through tumor growth inhibition was investigated in a syngeneic murine colorectal cancer model. Nanoparticle treatment showed no acute toxicity in vivo, and all treatments showed survival benefits for their respective groups compared to controls. HARPs alone slowed tumor growth, although not significantly. Radiation alone and in combination with the HARPs showed significant growth delay. Notably, the combination treatment significantly hindered tumor progression relative to the HARPs highlighting the benefit of this multipronged treatment. These results provide a foundation for loading RTX in a nanoparticle formulation, and establish a combined radiation and drug dosing schedule to determine optimal tumor growth delay and subsequent treatment efficacy.
Keywords: raltitrexed, layer-by-layer, tangential flow filtration, radiation therapy, DNA damage
Graphical Absract
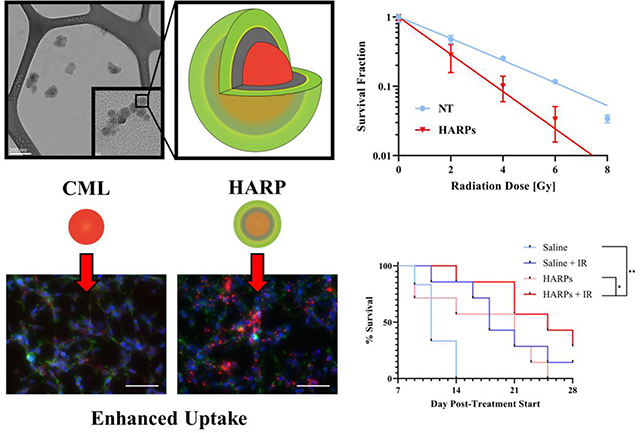
The combination of radiation therapy and nanomedicine allows for improved treatment outcomes in a murine colorectal cancer model.
Introduction
Raltitrexed (RTX),1 is a particularly potent chemotherapeutic used primarily in the treatment of colorectal cancer (CRC).2–5 Also known widely as Tomudex (ZD 1694, AstraZeneca), RTX is an anti-folate compound, owing part of its structure to that of folic acid, an essential component in the synthesis of DNA in both normal and malignant cells.6 Its mechanism of action is the direct inhibition of thymidylate synthase, similar to the therapeutic effect of methotrexate and 5-fluorouracil.7 Thymidylate synthase has varied expression in tumors,8 with expression investigated in relation to various survival outcomes in colorectal,9 breast,10 head and neck,11 non-small cell lung,12 and bladder cancer.13 However, unlike earlier anti-folates, RTX has a specific affinity for the α-folate receptor,6 and directly interferes with thymidylate synthase,7 rather than other constituents in DNA production. This allows for improved uptake, higher potency, less off-target toxicity, and the potential for an increased therapeutic ratio compared to other agents in this class of drugs.
The advent of nanoparticle-based drug carriers presents numerous advantages over free drug analogs when administered intravenously (IV).14 The increased circulation time provided by these nanoscale delivery systems allows for control over release kinetics, as well as a means of cell specific targeting.15 Recently, significant interest has focused on nanocarriers built via layer-by-layer (LbL) deposition. Early systems employing this process utilized inert gold substrates,16,17 with more recent studies showing functionality with liposomes containing RNA payloads.18 Layer-by-layer deposition can be used to attach alternating charged polymers to the surface of substrates, with purification of excess polymer from the layering step generally involving centrifugation.19,20 Alternatively, a tangential flow filtration (TFF) process has been investigated to accelerate the purification process, allowing for efficient production of multilayered assemblies.21 In particular, Correa et al. tested a wide variety of negatively-charged polymers as terminal layers.21 Of particular interest to this work is hyaluronic acid (HA), which has been previously investigated as a targeting agent using LbL techniques on commercial polystyrene substrates.20 Hyaluronic acid, composed of alternating units of D-glucuronic acid and N-acetyl-D-glucosamine,22 is a negatively charged polymer that acts as a targeting agent for CD44.20 CD44 is a cell-surface receptor that plays a role in cancer progression and metastasis,23 and is present in various types of cancer, including breast20,24 and CRC.25
Raltitrexed has been used as an adjuvant therapy versus CRC.26–28 Its combination with radiation often provides a survival benefit to patients.2,5 Radiation therapy (RT) typically involves 1.8 – 2 Gy doses administered 5 days a week combined with bolus injections of chemotherapeutics once every few weeks.5,28 To further improve clinical treatment of RT with adjuvant RTX, preclinical models of CRC can be used, with some limitations. Syngeneic models do not represent the immune microenvironment appropriately, while orthotopic models display increased inflammation,29 which would not be as prevalent in spontaneous tumor formation. For the preclinical modeling with the CT26 murine colorectal cell line, a similar timing schedule for chemoradiation can be employed.30
In this study, we investigated the combination of a targeted nanoparticle containing RTX with RT. Our working hypothesis was that the combination treatment would lead to greater therapeutic benefit and/or survival outcomes than either individual treatment. Here, we present a LbL process that capitalizes on the electrostatic interactions between charged groups on RTX and HA in order to modify the surface of commercially available fluorescent substrates (polystyrene nanoparticles). The enhanced uptake and efficacy of this formulation, HA encapsulating RTX nanoparticles (HARPs), was examined in a CD44+ murine CRC cell line, CT26. DNA damage associated with both RTX and radiation was examined in vitro. A short-term in vivo study was performed to assess tumor growth inhibition for each treatment individually, as well as in combination, to determine the efficacy of the treatment on tumor regression.
Methods
Materials
KR2i tangential flow filtration (TFF) system including all filter modules, pressure transducers and the pump head were purchased from Spectrum Laboratories. Carboxylate-modified latex (CML) polystyrene latex beads with various fluorescent spectra were purchased from ThermoFisher. Raltitrexed was purchased from Sigma Aldrich. Hyaluronan (ultra-low molecular weight, 4 – 15 kDa) was purchased from Fisher Scientific. Poly-L-arginine hydrochloride (PLA, 5–15 kDa) was purchased from Sigma Aldrich. All layering processes were carried out in nuclease-free water (HyClone), purchased from Fisher Scientific.
Layer-by-Layer Nanoparticle Encapsulation
The initial nanoparticle suspension (2 wt% solids) was sonicated and diluted to 0.2 mg/mL. 3 mL of the diluted solution was added to a 15 mL conical tube, then 3 mL of 1 mg/mL PLA was added dropwise to the 3 mL of the nanoparticle solution. Once all PLA was added to the nanoparticle solution, the feed and retentate lines from the TFF setup were placed into the nanoparticle solution. Water was added to the 15 mL conical to fill the conical tube, and the pump was turned on. With 2 mL remaining in the conical tube (excess PLA solution exits through permeate line), water was added to fill the conical tube again, further washing the nanoparticle solution. The nanoparticle solution was concentrated to less than 1 mL, and the feed line was put into a beaker containing water. Water was used to flush the remaining volume out of the system and diluted back to the original starting volume of 3 mL. The positively charged nanoparticles were then mixed with a solution containing both the drug, RTX, and the targeting agent, HA. To create the negative solution for layering, 3 mL of 0.2 mg/mL RTX was mixed with 6 mL of 2 mg/mL ultra-low molecular weight HA. After mixing briefly, the excess drug and targeting reagent were removed by recirculation and washing through a 100 kDa filter module designated for negative polymers. Once washed, the volume was returned to the original nanoparticle solution volume (3 mL) and stored at 4 °C until used for characterization or dosing.
Nanoparticle Characterization
Dynamic light scattering (DLS) measurements were performed in triplicate on nanoparticle suspensions using a Malvern ZS Zetasizer (Malvern Panalytical, UK). Size and zeta potential measurements were carried out in DTS 1070 zeta cells (Malvern), with values presented as mean ± standard deviation of the three measurements. Transmission electron microscopy (TEM) was performed to investigate size and morphology of the bare latex particles and the LbL assembly. TEM images were taken using a Technai F-20 transmission electron microscope operating at 4200 eV. TEM grids were prepared by dropwise addition of 10 μL of nanoparticle solutions to the copper surface of formavar/carbon-backed TEM grids (Ted Pella). The grids were dried in a desiccator for at least 3 hours prior to imaging to ensure complete drying of the grid.
To investigate release of RTX from the nanoparticles, 3 mL of freshly made HARP solution was added to 3.5k MWCO “snakeskin” dialysis tubing (ThermoFisher). The nanoparticle solution was immersed in a beaker containing 60 mL of pH 7.4 phosphate-buffered saline (PBS), with constant agitation from a magnetic stir bar. 100 μL aliquots were drawn in triplicate from the reservoir at 1, 2, 4, 8, 24, 48, and 72 hours. The missing 300 μL of PBS was replenished at each time point. Drug release was quantified using a Shimadzu SPD-20A high performance liquid chromatography (HPLC) instrument (Torrance, CA, USA) equipped with a UV-Vis detector and an Agilent Zorbax Rapid Resolution SBC-18 column (4.6 × 100 mm, 3.5 μm; Santa Clara, CA, USA). Drawn aliquots were run without further dilution. A five-minute gradient method was utilized with a flow rate of 1 mL/min and a 311 nm detection wavelength. The mobile phase increased from 65:35 methanol/water to 70:30 methanol/water over the first 2 mins and held here until the method was complete with elution occurring at 1 min. Samples were compared against aliquots taken 6 days after the start of the experiment to determine the percentage released, assuming complete release after this period (values plateau by 48 hours).
Infrared spectroscopy was performed using a Nicolet iS5 spectrophotometer (ThermoFisher) to investigate functional group presence on the nanoparticle. HARP solution was lyophilized, placed over a diamond filament, and compressed prior to total internal reflectance measurements. The spectrum for the particles was taken at 32X resolution between 4000 to 400 cm−1.
Cell Viability
CT26 murine colorectal carcinoma cells (ATCC) were cultured in RPMI 1640 (Corning) media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. When 90% confluent, the media was aspirated, the cells were washed with PBS, and 0.25% trypsin-EDTA was used to detach the cells from the culture flask. The cells were diluted in media once and counted using a Countess II FL Automated Cell Counter (ThermoFisher). The cells were further diluted to 20,000 cells/mL, then plated at 2,000 cells per well in a 96-well plate and allowed to attach overnight. After seeding, media containing nanoparticle solution was added to the majority of wells, with half-fold serial dilutions across the plate, in triplicate. The final wells were dosed with DMSO and no treatment (i.e. media), in triplicate, for the positive and negative controls. After 72 hours, 10 μL of Alamar Blue cell viability reagent (ThermoFisher) was added to each well. After 1.5 hours, the fluorescent signal was determined at 560/590 excitation/emission wavelength using a Tecan M200 Infinite plate reader (Tecan Trading AG).
Cell Uptake
To evaluate enhanced uptake of the HARPs, uptake in CT26 cells was evaluated by flow cytometry. Utilizing the culture conditions described above, 500,000 cells were plated into each well of a 6-well plate, followed by 2 mL of RPMI media. The cells were allowed to settle overnight. Once settled, bare CML nanoparticles, HA-terminated, and HARPs were added to separate wells. A control well was left untreated. The cells were placed in an incubator for 24 hours to allow uptake of the nanoparticles. Following uptake, the cells were prepared for either fluorescent imaging or flow cytometry.
To prepare the cells for fluorescent imaging, the cells were washed with PBS twice, and 2 mL of 10 % formalin solution was added to each well, and allowed to fix for 30 min. Once fixed, the cells were washed twice with PBS, followed by addition of PBS containing DAPI and AlexaFluor 488-phalloidin dyes (ThermoFisher), and allowed to stain for 30 min. After staining, the cells were washed once, and the wells were filled with 2 mL of PBS. Images could then be acquired using an EVOS FL AUTO II (ThermoFisher), with filter cube sets able to capture the nucleus (blue – DAPI), cell body myelin filaments (green – AlexaFluor 488), and the nanoparticles (red – 660/680 dark red dye).
To prepare the cells for quantitative assessment of cell uptake using flow cytometry, the cells were washed with PBS, detached using trypsin, and diluted in media. The cells were centrifuged at 300 × g for 5 min to form a pellet. The media was aspirated, followed by redispersion of the cells in 1 mL of PBS. The cells were pipetted into polystyrene tubes through cell strainer caps to remove cell aggregates. Using a BD Fortessa LSRII flow cytometer (Becton Dickinson), cellular fluorescence of no treatment control, bare CML nanoparticle-treated, HA-treated, and HARP-treated cells was determined sequentially. The nanoparticle fluorescence was determined using a 640 nm excitation laser line, with a 670/30 nm emission/bandwidth filter. Forward scattering and side scattering were used to determine single cells passing through the detector, and 105 events (or cells) were passed through the detector to establish a distribution for each sample in each treatment group.
γH2AX Flow Cytometry Assay
CT26 cells were cultured in RPMI 1640. Cells were washed, trypsinized, and plated at ~1 × 106 cells per well into each well of a 6-well plate. The cells were allowed to attach overnight in an incubator at 37 °C and 5% CO2. The next day, the media was replaced with either fresh media for the top three wells (NT), or 50 μL of the HARP solution diluted to 2 mL total volume in media for the bottom three wells. A duplicate plate was also made under the same conditions, and the two plates were placed back in the incubator for 24 hours. After 24 hour uptake of the nanoparticles, the media was replaced with fresh media. One of the two plates was then transferred to a CellRad X-Ray irradiation cabinet (Faxitron), and irradiated with 4 Gy at a dose rate of ~0.9 Gy/min. The irradiated plate was returned to the incubator. After 1 hour, both plates were washed with PBS, trypsinized, and dispersed in media. The cells were then spun down to a pellet at 300 × g for 5 min. The media was aspirated, and the cells were dispersed in 1 mL of PBS, and spun down at 300 × g for 5 min. The PBS was aspirated, cells were dispersed in 10% formalin in PBS, and allowed to fix for 15 min at room temperature. After 15 min, the cells were spun down at 300 × g for 5 min, the formalin was aspirated, the cells were dispersed in PBS, and spun down again. After aspiration, the cells were dispersed in blocking buffer (5% goat serum/0.3% Triton-X in PBS), and allowed to incubate for 1 hour at room temperature. After 1 hour, the cells were spun down, washed in PBS, spun down again, aspirated, and dispersed in 1 mL of primary antibody buffer (rabbit H2AX serine 139-targeting antibody diluted 1:500 from stock in PBS, Cell Signaling Technology). The cells were incubated at room temperature for 1 hour, spun down, aspirated, dispersed in PBS, spun down, and aspirated again. The cells were then dispersed in 1 mL of secondary antibody buffer (goat anti-rabbit Cyanine5 dye-labeled antibody at 1:333 dilution from stock in PBS, Cell Signaling Technology), and allowed to incubate at 4 °C overnight. The next day, the cells were spun down, aspirated, dispersed in PBS, spun down, aspirated, and dispersed in 0.5 mL of PBS. The 0.5 mL volume for each sample (12 samples total, 4 treatments in triplicate) was then pipetted into 12 × 75 mm 5 mL volume polystyrene tubes through a filter cap (Falcon). Using a BD Fortessa LSRII flow cytometer, DNA damage of no treatment control, and HARP-treated cells was determined. The antibody fluorescence was determined using a 640 nm excitation laser line, with a 670/30 nm emission/bandwidth filter. Forward scattering and side scattering were used to determine single cells passing through the detector, and 105 events (or cells) were passed through the detector to establish a distribution for each sample in each treatment group.
γH2AX Fluorescence Microscopy
For qualitative visualization of DNA damage, CT26 cells were plated at 105 cells per well in a four well chamber slide after coating the surface of the slides with gelatin (Cell Biologics). To coat the slides, 0.5 mL of the gelatin solution was added to each well for 30 min. The gelatin solution was then aspirated and allowed to dry in the cell culture hood. Once the cells were added to each well of two chamber slides (8 wells total), the slides were placed in an incubator at 37 °C and 5 % CO2 overnight. The next day, 0.5 mL of either media, bare CML particles in media, HA terminated particles in media, and HARPs were added to a separate well in each plate. From this point forward, the same procedure as with flow cytometry was followed. To summarize, the cells were treated with the same concentration of nanoparticles, incubated for 24 hours, and irradiated with 4 Gy. The cells were fixed to the surface of the slide, washed, and treated with the various buffers (0.5 mL instead of 1 mL) in the same manner as the flow cytometry protocol described previously. After the final wash, DAPI and AlexaFluor 488-phalloidin stains were added to the wells to identify the nucleus and cell bodies, respectively. The dyes were incubated for 30 min, then the slides were washed, aspirated, and filled with 0.5 mL of PBS, and kept at 4 °C until imaged. Images could then be acquired using an EVOS FL AUTO II, with filter cube sets able to capture the nucleus (blue – DAPI), cell body myelin filaments (green – AlexaFluor 488), and foci of DNA damage, tagged with primary and secondary antibody (red – Cy5).
Clonogenic Assay
CT26 cells were cultured in RPMI 1640. Cells were washed, trypsinized, and plated at 500,000 cells per well into each well of a 6-well plate. The cells were allowed to settle overnight. The next day, four wells were assigned as each of the following: no treatment, bare, HA-coated, and HARP. The media was removed, and media containing the assigned treatment was added to the respective wells. The cells were allowed to incubate in the media containing the treatment. After 24 hours, the cells were washed with PBS, trypsinized, and dispersed individually (by treatment group) in media in 15 mL conical tubes. The cells were counted and plated in triplicate in two six-well plates for each treatment group at a seeding density of 200 cells per well (a total of 12 wells plated, 3 from each treatment group). The conical tubes were transferred to a CellRad X-Ray cabinet irradiator (Faxitron) and irradiated for 2 Gy. After irradiation, cells were again plated as discussed previously, but at 400 cells per well for the 2 Gy treatment. This process was repeated for 4 Gy (800 cells per well), 6 Gy (1200 cells per well), and 8 Gy (1600 cells per well). After each plating, the six-well plates were placed in an incubator at 37 °C and 5% CO2. After 7 days of incubation, the plates were removed from the incubator. The wells were washed with PBS, fixed with 10 % formalin (in PBS) and allowed to fix for 30 min. After 30 min, the formalin was aspirated, the wells were washed again with PBS, and 0.1 % crystal violet dye (in PBS) was added to each well to stain the cell colonies a deep purple. After 30 min, the crystal violet dye was aspirated, and colonies were counted by hand.
In vivo Treatment Efficacy
All animal procedures were performed in accordance with the guidelines for care and use of laboratory animals of Oregon Health and Science University (OHSU) and experiments were approved by the OHSU Institutional Animal Care and Use Committee. Forty 6-week-old BALB/c mice (Charles River Laboratories, Wilmington, MA) housed in modified barrier animal facilities prior to tumor inoculation. On the day of tumor inoculation, cell suspensions containing 6 × 106 CT26 cells/mL were prepared in sterile PBS for injection. 50 μL injections were made into the right hind flank of isoflurane-anesthetized mice, implanting a total of 3 × 105 cells subcutaneously. The tumors were allowed to grow for 2–3 weeks to reach a threshold size of at least 10 mm3 (measured twice weekly in this 2–3 week period by calipers, using formula 0.5 × long length × short length2).
Once the size threshold had been met, the animals were randomized into four treatment groups. The average difference in size of the tumors was minimized across groups to remove bias from treatment group determination. Once randomized, all animals received a 100 μL tail vein IV injection of either saline or HARP solution while under isoflurane anesthesia. The two radiation treatment groups were immediately transferred to a CellRad irradiator. The animals were shielded with a lead plate with a cutout for the area of the tumor, and 2 Gy radiation (130 keV) was administered to the tumor area, at a dose rate of ~0.9 Gy/min. After irradiation, animals recovered in the presence of a heating lamp and returned to their cages. Caliper measurements continued 3 times per week. Total treatment regimens for the four groups included the following: Saline Only – 100 μL injection of saline via tail vein 3 times per week, separated by 2 days each, for two weeks; Saline with radiation - 100 μL injection of saline via tail vein 3 times per week, separated by 2 days each, for two weeks, with 2 Gy administrations of radiation immediately after the first 3 injections, for a total radiation dose of 6 Gy; Nanoparticle Only - 100 μL injection of HARPs solution via tail vein 3 times per week, separated by 2 days each, for two weeks; Nanoparticle w with radiation - 100 μL injection of HARPs solution via tail vein 3 times per week, separated by 2 days each, for two weeks, with 2 Gy administrations of radiation immediately after the first 3 injections, for a total radiation dose of 6 Gy. Animals were euthanized when meeting one of the following conditions: tumor volume exceeded 2000 mm3, one length measurement by caliper exceeded 20 mm, or ulceration developed superficially on the tumor (weights of the animals were monitored as well, but no animal exhibited weight loss greater than 20% of initial weight to constitute an endpoint by this condition).
Histology
After reaching a humane endpoint, organs and tumors were collected from each animal, weighed, and placed into formalin solution (10% by volume in PBS) at 4°C for 2–3 days. The organs were then transferred to 70% ethanol solution until further processing by the Histopathology Shared Resource at Oregon Health and Science University.
Samples were paraffin embedded, sliced, and prepared according to the required staining protocol, which included either no staining, hematoxylin and eosin (H&E), or caspase-3 (CC3, Promega, 1:1500 dilution). Slides produced from staining were imaged using an EVOS FL AUTO II (ThermoFisher) using brightfield and/or Cy5 filter settings. Representative tumors were taken 21 days post-treatment initiation and 14 days after for the saline control.
Statistical Analysis
All data are represented as mean ± standard deviation. Statistical significance was evaluated using student t-test or logrank (Mantel-Cox) using GraphPad Prism 8 software. P < 0.05 was considered statistically significant, with various asterisks denoting greater confidence (*, P < 0.05, **, P < 0.01, ***, P < 0.001).
Results and Discussion
Nanoparticle Formulation and Characterization
Employing the inherent charge of the functional groups present on various polymers, we deposited PLA (positive due to amines present on the side chain) onto the surface of the negatively charged CML particle. After filtration and purification by TFF, a negative layer containing RTX and HA was deposited on the surface of the amine-functionalized particle. Filtration and washing via TFF removed the excess polyelectrolyte (PE) prior to characterization and biological experiments. The general scheme for this efficient surface assembly process, illustrated in Figure 1, is adopted from other methods reported previously.31 In this formulation, RTX was mixed with the negative polyelectrolyte, HA, in order to be deposited on the surface of the CML-PLA substrate. Others have employed similar schemes, most commonly using centrifugation to purify after each layer addition.15,16,32–35
Figure 1.
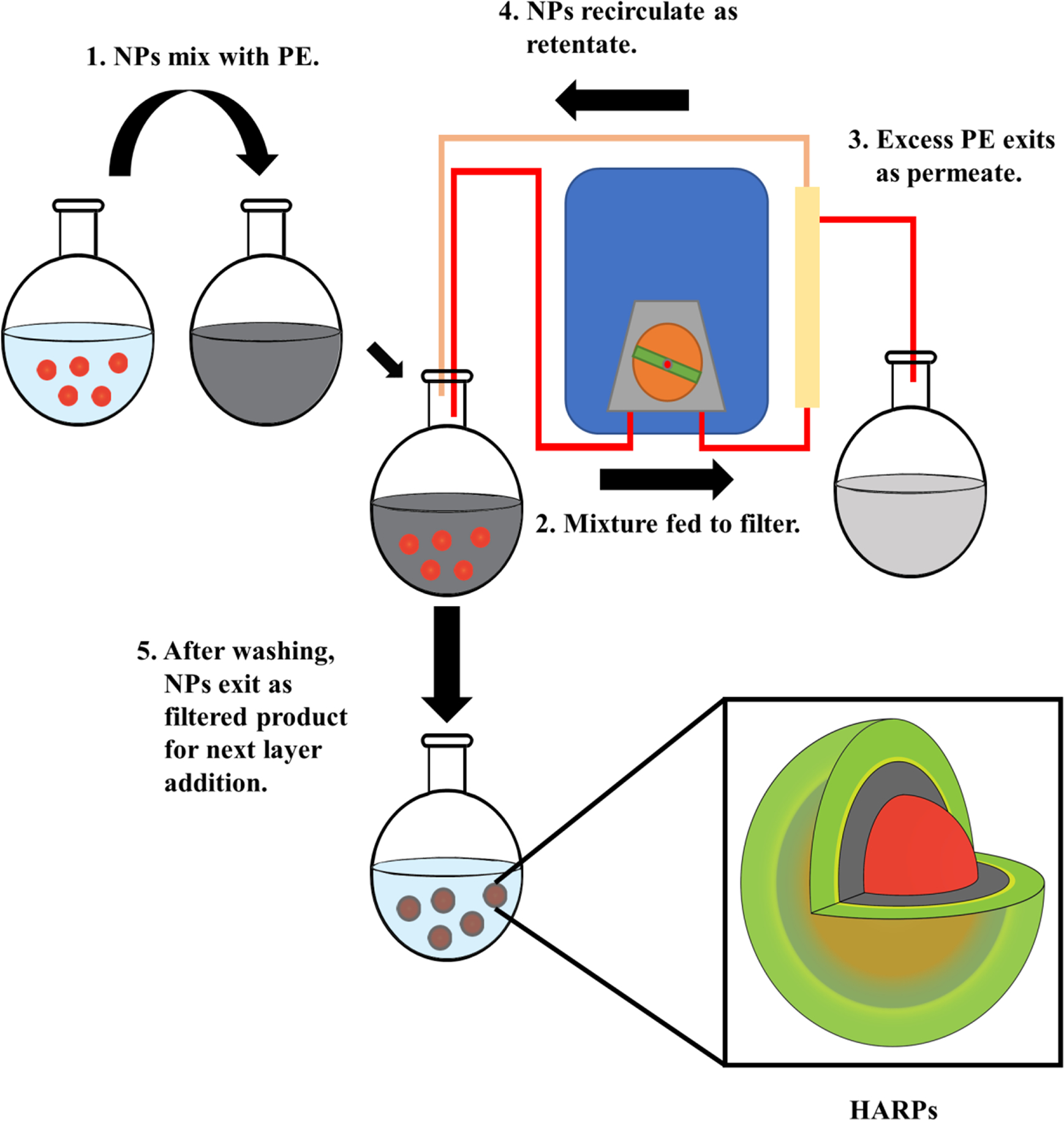
Layer-by-layer formulation process for surface deposition of polyelectrolytes (PE) onto polystyrene substrate. The final formulation is a CML core (red) - poly-L-arginine (gray) - HA/RTX (green/yellow) nanoparticle referred to as HARPs.
Transmission electron microscopy (TEM) was used to visualize the size and morphology of the final formulation (Figure 2A). These electron micrographs show the bare particles as the darker circular shapes, with a lighter, less carbon dense layer of PLA/RTX/HA on its surface. The size as estimated from TEM was confirmed by dynamic light scattering (DLS) as ~100 nm, with a size distribution shown in Figure 2B. The bare particles, prior to the LbL process, were also imaged via TEM (Figure S1). The nanoparticle cores were determined by DLS to have a diameter of 23.7 ± 0.15 nm, with a zeta potential of −51.1 ±1.17 mV. This negative charge present on the cores is due to the presence of carboxylic acid groups on the surface of this commercial product (FluoSpheres, ThermoFisher). These cores also contain a fluorescent dye (either a 365/405 or a 660/680 excitation/emission dye). After addition of PLA, the size increases to 30.2 ± 1.67 nm, and the zeta potential shifts to 51.4 ± 3.70 mV due the presence of the large number of amines on the side chain of the arginine.
Figure 2.
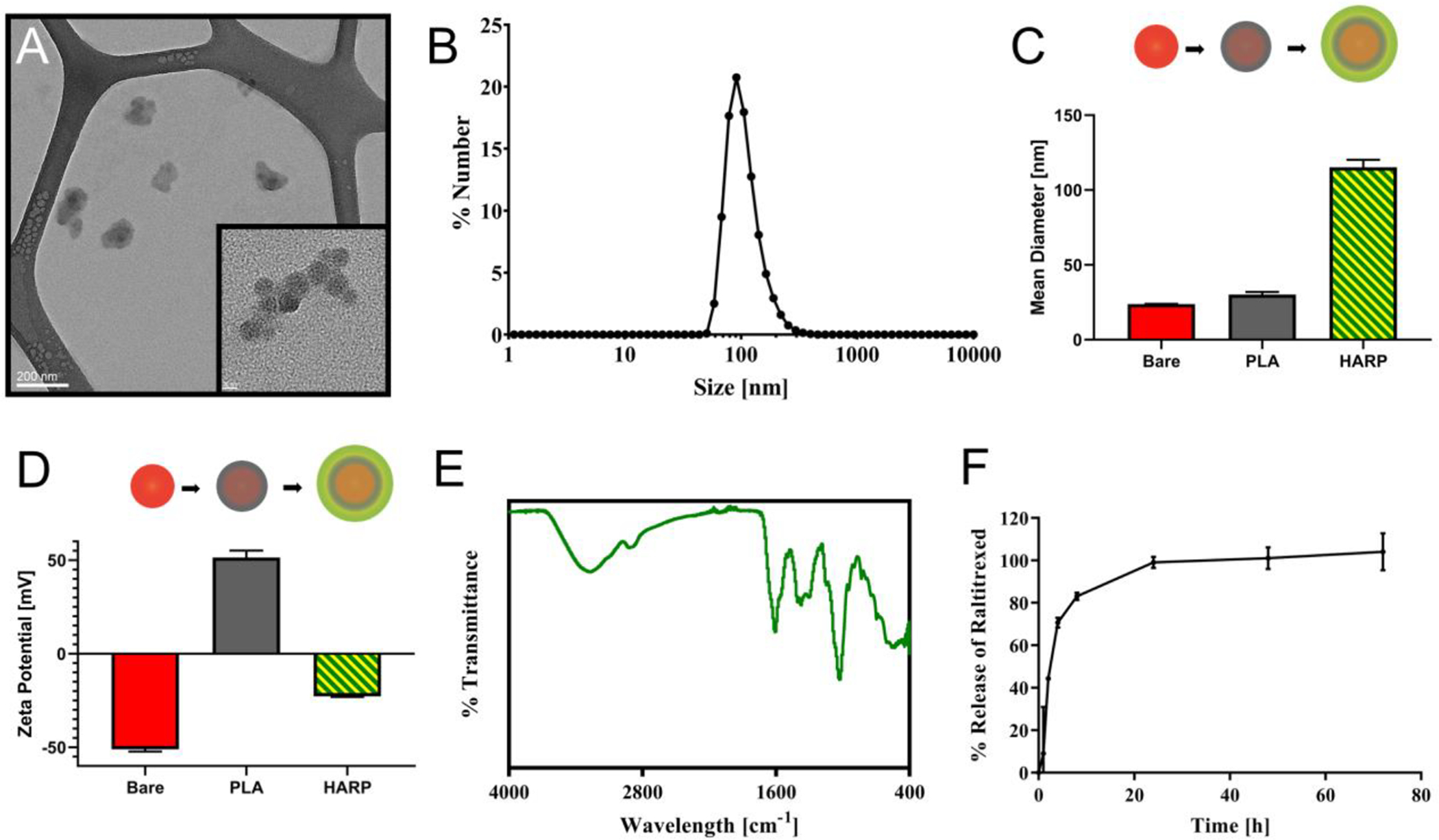
Physiochemical properties of the HARPs platform. (A) Transmission electron micrograph of the HARPs. Lower magnification scale = 200 nm, higher magnification scale = 20 nm. (B) Size distribution by number of the final HARP formulation centered ~100 nm. (C) Mean diameter at each deposition stage shows small size increase with PLA deposition and larger increase with HA/RTX deposition. (D) Zeta potential at each deposition stage. (E) IR spectrum of the HARPs. (F) Release of RTX from the HARPs over 72 hours in pH 7.4 PBS at room temperature.
Finally, after deposition of the RTX and HA, the size increases to 115.2 ± 5.01 nm, with a zeta potential of −22.8 ± 0.26 mV (final polydispersity index was found to be 0.112 ± 0.01). The size and zeta potential changes are shown in Figure 2C/D. HA appears to create a large increase in the average particle size, likely due to multiple particles forming clusters, which can be observed in Figure 2A. This result is consistent with previous work where HA has been shown to add significant size to nanoparticles.18,20,31 Our nanoparticles, in an optimal situation would be smaller and more monodisperse colloidal suspensions. Hyaluronic acid, proved to be a difficult polymer to use as a layering material, and adds substantial size to the nanoparticle suspension. After experimentation with various types of HA available from multiple vendors, the smallest average particle size of ~115 nm was achieved using the stated materials, and was deemed sufficient for pursuing further in vitro and in vivo work. The suspension contained particles that varied around this size, with a proportion of the particles falling closer to more ideal sizes for receptor-mediated uptake.36
To evaluate the surface chemistry, infrared spectroscopy was used to identify functional groups present in the final formulation (Figure 2E). Contributions from PLA would include primary amine peaks (~3500, ~3000 cm−1), and from HA could include carboxylic acid (~3300 cm1 broad beak), secondary amide (1680 cm−1), and hydroxyl peaks (3000–3500 cm−1). Lower peaks likely include amine stretching at 1000–1250 cm−1. Release of RTX into pH 7.4 PBS was determined using dialysis tubing. Triplicate data of reservoir release concentrations was determined at 1, 2, 4, 8, 24, 48, and 72 hours using high-performance liquid chromatography (HPLC). This data presented in Figure 2F, shows fast release of the RTX over the first 24 h in the buffer. The encapsulation efficiency of the HARP, i.e., ratio of the drug present in the final formulation to the drug used to assemble the formulation, was found to be ~11%. Further optimization of synthesis parameters, such as using a higher initial nanoparticle concentration relative to the drug concentration and adjusting the mass ratio of therapeutic to HA targeting ligand, may increase drug loading.
Nanoparticle Uptake
To determine enhanced uptake via CD44-mediated endocytosis, bare CML nanoparticle uptake was compared with uptake of HARPs. Figure 3A shows a qualitative comparison of this nanoparticle uptake utilizing the fluorescence of the CML core (dark red dye, 660/680). From left to right, CT26 cells were treated with media alone (NT), bare CML nanoparticles, and HARPs.
Figure 3.

Nanoparticle uptake increases with HA-coating compared to control particles. (A) Fluorescence microscopy of CT26 cells treated with either no treatment (NT), bare CML nanoparticles, or HARPs for 24 hours. Scale bar is 100 μm. (B) Mean fluorescence intensity measured by flow cytometry of 105 cells (in triplicate for each group) shows an approximate 5-fold increase in average fluorescence. ***, P < 0.001, unpaired t-test. (C) Representative histograms from bare CML and HARPs uptake data.
Nuclei are stained blue (DAPI), cell body structure is stained green (AlexaFluor 488), and nanoparticles are shown in red. The fluorescent signal increases across the images shown, with greatest signal in the cells treated with the HARPs, which contain CD44-targeting HA.20,25 This targeted uptake was also observed with HA-coated nanoparticles without RTX (Figure S2). To quantify the relative number of particles taken up by the CT26 cells, cells dosed with either bare or HARPs were analyzed via flow cytometry. The presence of the 660/680 dark red dye in the NP core was determined from the fixed cell suspensions. Figure 3B shows the mean fluorescence intensity measured from 105 CT26 cells from each treatment group. No treatment (NT) showed minimal signal, while the HARPs displayed approximately 5-fold higher fluorescence. The histograms of the fluorescent signal of the NPs is shown in Figure 3C. The bare nanoparticles have a narrow peak at a low fluorescence intensity, while the HARPs show a broad distribution spanning from the lower fluorescence similar to the bare NPs, while also displaying a large number of cells with higher fluorescence. This validates the qualitative assessment shown in Figure 3A, as the HARPs show higher uptake (via fluorescence detection). The nanoparticles are likely taken up by receptor-mediated endocytosis, with the nanoparticles likely residing in a lysosome or recycling vesicle.37 Charged polymers like poly-l-lysine and poly-l-arginine, especially at higher MW, are capable of aiding in endosomal escape.38 Polystyrene and the polyelectrolytes used are generally biocompatible, and likely do not impart toxic effects while residing in an endosomal compartment or the cytosol, with toxic effects likely attributed to the RTX. While we employ HA as our targeting agent, anti-folate drugs like RTX have been investigated as a targeting agent.6,39,40 It is possible that the presence of the therapeutic alone could target folate receptors on CT26 cells,41 which was not evaluated in this study. In this system, HA primarily serves as a targeting agent, but also provides an encapsulating matrix for slower release of RTX.
DNA Damage, Viability and Clonogenicity
To investigate the DNA damaging effects of the HARPs alone or in combination with radiation, we performed several experiments aimed at quantifying DNA breaks and determining the cell line’s ability to proliferate after treatment. First, the γH2AX assay was used to determine the relative amount of DNA damage on CT26 cells in vitro. Cells in presence of ionizing radiation (IR) are susceptible to DNA damage after which repair pathways may be activated. Following the damage induction, phosphorylation of histone 2A occurs, and can be targeted with an antibody and fluorescent secondary antibody to visualize the double strand DNA breaks. DNA damage can be directly correlated to the fluorescence intensity signal captured from each cell through flow cytometry or fluorescence microscopy (Figure 4A). The top images in Figure 4A show the wells with no radiation treatment, while the bottom images show the irradiated wells. The wells shown in the left column were not treated with HARPs, while the wells shown in the right column were. The DNA damage foci are shown in red. These punctate regions are present on the majority of cell nuclei after irradiation and/or HARPs treatment. The general number and intensity of these red foci increases as follows: NT, IR, HARPs, HARPs + IR. In the HARPs with and without IR wells, the red fluorescence appears to be similar, indicating that the bulk of the DNA damage may be done by the drug treatment alone in vitro. Quantitative assessment beyond the general trend of increasing H2AX signal cannot be assessed via microscopy from our system, due to the presence of pan-nuclear staining of some of the nuclei in the HARPs-treated wells. This staining can arise from sources of irradiation42,43 or genotoxic agents like 5-fluorouracil44. Flow cytometric analysis of identically treated cells indicated that this trend (of increasing H2AX fluorescence) held quantitatively, and that there was a greater effect by the combination of the HARPs with IR in comparison to HARPs alone (Figure 4B).
Figure 4.
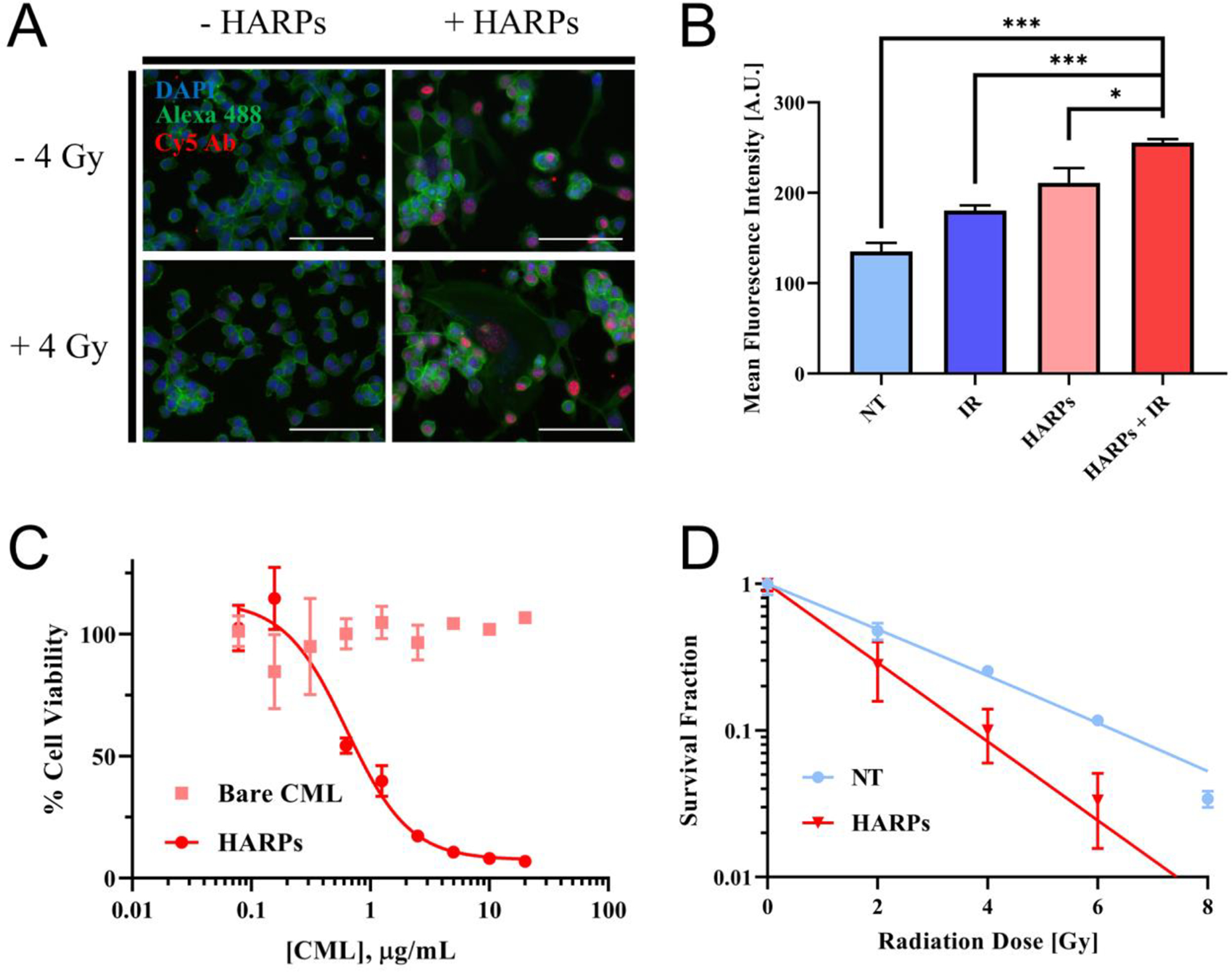
In vitro efficacy of HARP and combination therapy. (A) Fluorescence microscopy of γH2AX assay. IR (4 Gy), HARPs (24 hour incubation, 0.55 μg/mL RTX), and a combination shows brighter fluorescence, indicating DNA damage at these “foci”. Scale bar = 100 μm. (B) Mean fluorescence intensity measured by flow cytometry of 105 cells (in triplicate for each group, mean ± standard deviation shown) shows an increase in DNA damage when either IR or HARPs are used, with further damage from the combination of the two treatments. Data significance evaluated with unpaired student t-test, *, P < 0.05, **, P < 0.01, ***, P < 0.001. (C) Cell viability at 72 hours shows little to no toxicity for bare nanoparticles, with a large increase in toxicity as HARPs approaches 1 μg/mL. (D) Survival fractions show reduced clonogenicity of cells treated with 0.55 μg/mL HARPs for 24 hours before exposure to IR (compared to those exposed to IR alone).
Mean fluorescence intensities of the distributions (and the two sets of replicate data for each treatment group) show an increasing signal in the same order as the fluorescence microscopy images. (Representative histograms of the fluorescence intensity signal distributions are shown in Figure S4). The combination of HARPs and IR shows statistically greater DNA damage than either individual treatment or no treatment, in terms of the mean fluorescence intensity detected over the measured cells. Raltitrexed is known to cause DNA damage.45,46 At the dose administered in this assay (0.55 μg/mL, 24 h), it appears to be the major source of DNA damage.
A second experiment was used to determine the toxicity of the HARP and combined treatments with CT26 cells. Cell viability was determined after 72 hours exposure to various concentrations of either bare nanoparticles or HARPs (Figure 4C). Over the entire concentration range, the bare nanoparticle-treated cells showed minimal toxicity after 72 hour incubation. The HARPs treated cells showed toxicity over most of the similar concentration range, with an IC50 value of 0.64 μg/mL. Raltitrexed alone was determined to have a 72 hour IC50 value of 14.44 nM vs. the same CT26 cells (Figure S2). Raltitrexed is known to be a particularly potent thymidylate synthase inhibitor7, so a low IC50 was anticipated.
To evaluate radiobiological cell death, the clonogenic assay was used to test for colony formation ability, or “clonogenicity”, of the CT26 cells after treatment with HARPs and IR. This assay works by comparing proliferation of individual CT26 cells after exposure to some form of treatment to untreated cells across a range of radiation doses. The survival fraction for each treatment group was determined by dividing the plating efficiency at a particular radiation dose by the plating efficiency at 0 Gy (no IR). The survival fractions are plotted as a function of radiation dose applied in Figure 4D. Each plate, corresponding to a radiation dose, gives a single data point (as mean ± standard deviation of triplicate wells) for each treatment group. These data points can be fitted to a curve (linear-quadratic model survival fraction = e(-α*dose-β*dose2), as seen in previous work),47 also shown in Figure 4D. The surviving fraction of the HARPs-treated cells declines faster with increasing radiation dose than the untreated cells. The 8 Gy wells for the HARP-treated cells showed no clonogenicity; hence, no data shown for this point. The combination of RTX with radiation leads to greater inhibition of colony formation. This was maintained across the entire range of IR doses for the concentration of HARPs dosed. At 0 Gy, where the survival fraction is unity, the number of colonies present in the HARPs-treated wells was roughly half the number of colonies formed in the untreated wells, further indicating the colony formation inhibition of the HARPs treatment (data not shown). This data, combined with the viability data described above, shows that the HARPs are potent drug carriers capable of inhibiting cancer cell growth and proliferation with and without radiation in vitro.
In Vivo Tumor Efficacy
To determine the in vivo efficacy of the HARPs, CT26 flank tumors were grown over a 2–3 week period in BALB/c mice (6–8 weeks old). Treatment involved tail vein administration of saline control or HARPs six times over two weeks, either with or without three 2 Gy doses of IR with the first three injections (Figure 5A). The tumor growth curves for all animals, organized by treatment group, is shown in Figure 5B. To investigate the efficacy of the various treatments, we employ a rate-based analysis of tumor growth, described previously.48 Here we note that the rapidly growing CT26 tumors reach a humane endpoint before the desired end of the study. Thus, instead of terminating treatment groups prematurely or censoring a large number of animals from the study, we chose to analyze the change in the tumor growth for the period that the animal survives for the experiment. The tumor volumes for each animal are converted to a base-10 logarithmic scale, and the slope of a linear fit to the tumor volumes is calculated, giving a “growth rate” for each animal in each treatment group (Figure 5C).
Figure 5.
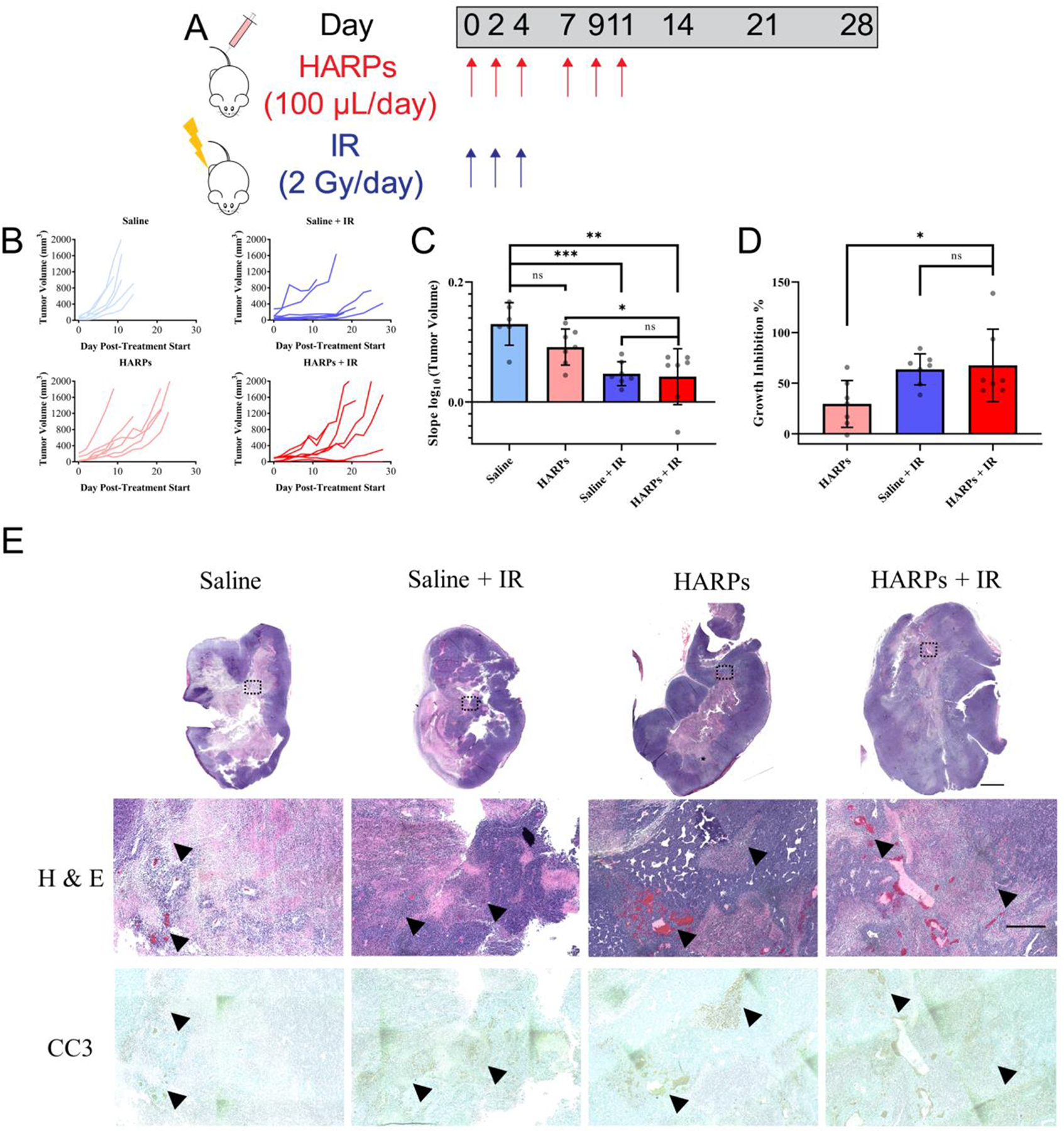
Tumor treatment schedule and outcomes. (A) Schematic showing dosing schedule for both saline/HARPs and IR. (B) Individual tumor growth curves for each animal divided into treatment groups. (C) Slope of logarithmic converted tumor volumes, or “tumor growth rate”, for each treatment group presented as mean ± standard deviation. (D) Growth rate inhibition for each treatment group (relative to saline alone control) presented as mean ± standard deviation. Data significance evaluated with unpaired student t-test, *, P < 0.05, **, P < 0.01, ***, P < 0.001. (E) Images of histology slides highlighting necrotic/apoptotic regions in tumors excised from various treatment groups 21 days after initiating treatment. Low magnification scale bar indicates 2 mm; high magnification scale bar indicates 500 μm.
The mean and standard deviation is displayed for the data from each group. Relative higher values indicate a faster average growth rate of the tumors in a particular treatment group. Saline injected control animals showed the fastest tumor growth, while slowest growth occurred in animals treated with both HARPs and IR. HARPs alone showed reduced average growth rate of the tumors versus saline alone, although not statistically significant. Greater growth inhibition may be achieved through further optimization to the formulation or dosing schedule, as the current dose appears to be well below the maximum tolerated dose.49 HARPs + IR treatment animals showed a significant decrease in tumor growth compared to HARP only treatment animals. This shows a desired trend, in that IR treatment in combination with RTX leading to tumor growth inhibition. However, there was no significant difference between the IR and HARPs + IR group, indicating that the IR delivered likely saturated the potential dose-response of the tumors for this treatment regimen. Further investigation of schedule and lower IR dose may be able to achieve greater separation between these groups. The clonogenic data presented in Figure 4D indicates, in vitro, that there should be a difference between the cancer cell proliferation for IR and HARPs + IR treatment groups (NT indicates an IR only treatment for the cells in vitro). With a high sample size, under the right set of treatment conditions, this effect should exist in vivo as well.
One issue with interpreting the “growth rate” from the method shown here is that the values derived vary among cell types. To normalize the values gained from this analysis, Hather et al. suggest a definition of the term “growth rate inhibition”,48 which is defined as (1-(growth rate of treatment group)/(growth rate of control group)) × 100%. Between 0 and 100%, this term indicates growth inhibition, 0% indicates no effect from the treatment, and > 100% indicates regression from the treatment. It may also indicate, in rare cases of < 0%, an increase in growth from the treatment, in instances where the tumor did not respond to the treatment and grew faster than the average of the control tumors. In our analysis, we calculate the growth rate inhibition for each animal in the treatment groups versus the average growth rate of the saline control (Figure 5D). This shows the same trend as the growth rate statistics shown in Figure 5C, but inverted. HARPs inhibit tumor growth ~30%, IR inhibits tumor growth ~64%, and the combined treatment inhibits tumor growth ~68%, on average. The IR and HARPs + IR treatments were not significantly different, while the HARPs + IR showed a significantly greater growth inhibition (P < 0.05) than HARPs alone. Interestingly, one animal showed complete tumor regression and no recurrence of the tumor after 39 days. Another animal appeared to show a similar trend initially, but showed recurrence of the tumor towards the end of the trial period (day 21), and its growth rate inhibition fell back slightly under 100%.
Tumors were collected from each animal from each treatment group as endpoints were met, and processed for sectioning and staining. Representative tumor sections stained with hematoxylin and eosin (H&E) and caspase-3 (CC3) are shown in Figure 5E. Low magnification images of the entire tumor cross-section are shown at the top, with increased magnification of particular parts shown below. Generally, these tumors appear to have less dense nuclear regions at the center, due to reduced oxygen and nutrients present. These regions are necrotic, and generally show apoptotic markers (CC3 – brown). All tumor sections showed apoptotic regions clustered around necrotic tissue. The tumors treated with IR showed more diffuse apoptotic regions spread across larger areas of the tissue, perhaps indicating a larger cell-killing effect due to the accumulated radiation dose. Tumor sections separated by at least 500 μm were left unstained to investigate any detectable fluorescence from the HARPs within the tissue. The unstained sections were scanned for fluorescence using an infrared imaging scanner (LI-COR, Odyssey). The sections were scanned at the 700 nm setting in the software for the system, as seen in Figure 6A.
Figure 6.
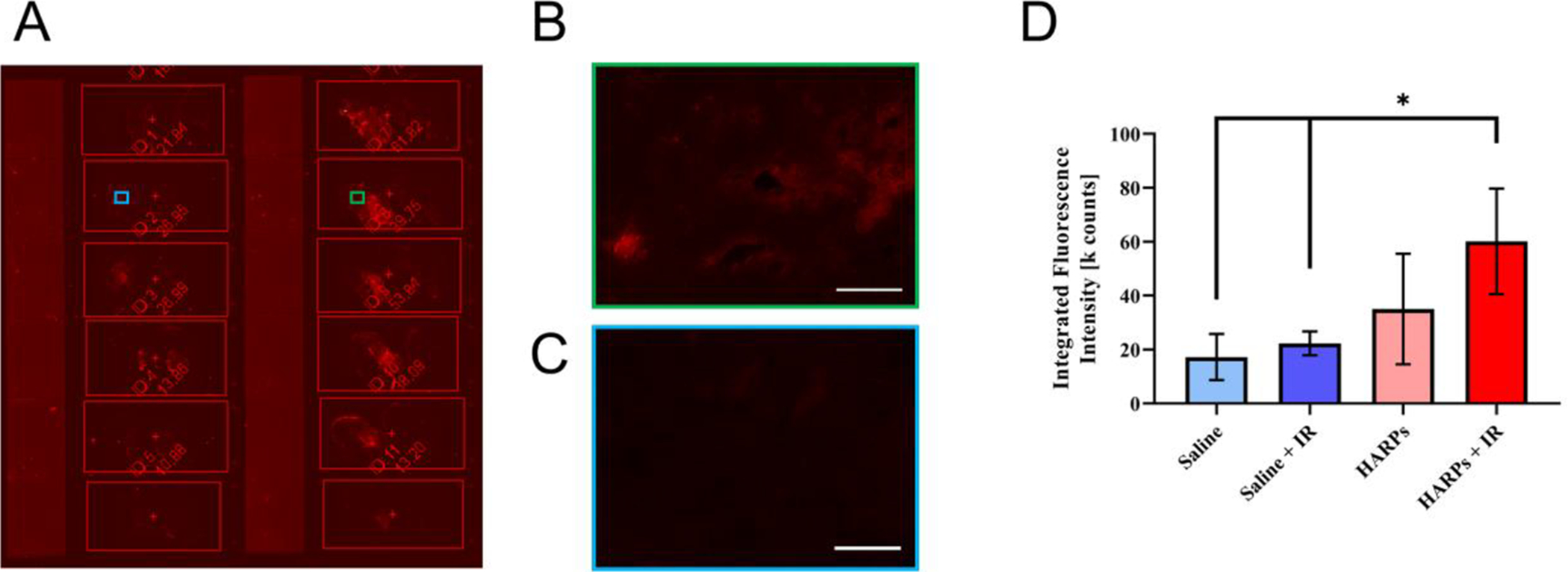
Increased fluorescence signal in tumor sections from HARPs-treated animals. (A) Image of unstained tumor sections in the fluorescent scanner (3 slides per treatment group, left column are saline treated, right column are HARPs treated). Higher magnification images of representative sections from (B) HARPs + IR and (C) saline + IR treatment groups. Scale bars indicate 100 μm. (D) Fluorescence intensity data collected from unstained tumor sections from representative tumors from each treatment group. Bars indicate mean ± standard deviation. Data significance evaluated with unpaired student t-test, *, P < 0.05.
Higher magnification images of particular portions of a saline + IR section (Figure 6B) and a HARPs + IR section (Figure 6C) are also shown. In these figures, the red color indicates the presence of a fluorophore that is detected by the instrument. The slides are unstained, and thus the brighter red sections of the slides containing the HARPs likely indicate the presence of the nanoparticles in the tissue. The amount of fluorescence detected is collected as the integrated fluorescence intensity. In Figure 6D, the means and standard deviations of the three sections from each tumor are shown. The sections from saline treatment groups show lower signal than the HARPs treatment groups, with only the HARPs + IR sections showing a significantly higher integrated signal from the regions of interest. This likely indicates that the nanoparticles are getting to the tumor, but is only representative of an overall trend. This could be probed in future experiments with a further infrared dye in the nanoparticles, but is beyond the scope of the current work.
Animal survival times were collected as animals met their humane endpoints. The survival curves for each treatment group are shown in Figure 7A.
Figure 7.
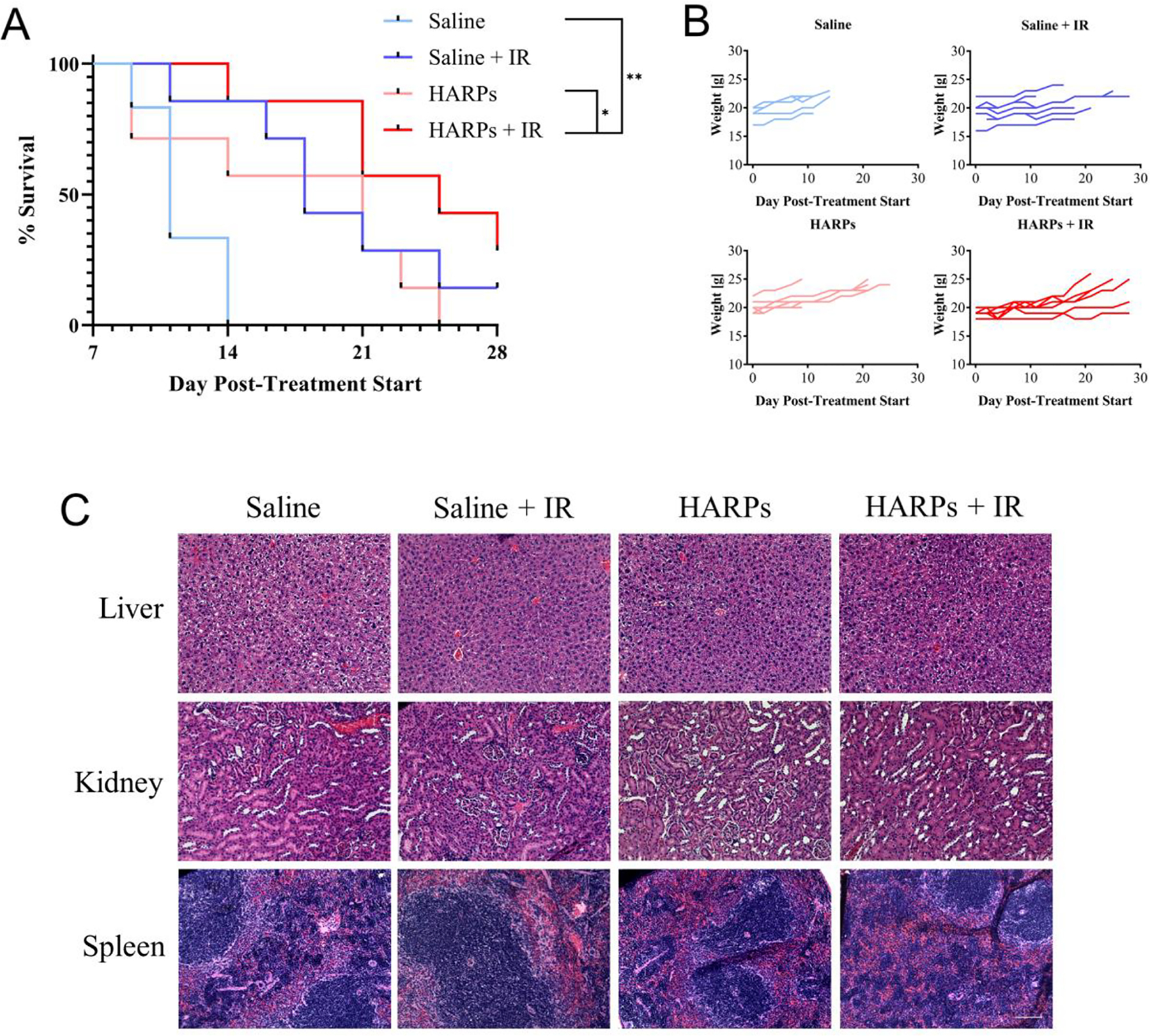
Survival curves and relative safety of various treatments. (A) Kaplan-Meier survival curves for each treatment group. Data significance evaluated using log rank (Mantel-Cox) test, *, P < 0.05. Sample size = 7 animals for each group, except for saline alone, for which sample size = 6 animals. (B) Individual animal weights for the experiment duration, divided into treatment groups. (C) Images from clearance organs from indicated treatment groups. Scale bar indicates 100 μm.
Median survival values were as follows: saline: 11 days, saline + IR: 18 days, HARPs: 21 days, HARPs + IR: 25 days. All of the treatment groups showed survival benefit over saline alone. The HARPs + IR treatment showed significantly greater survival over both the saline alone and HARPs alone groups. To evaluate relative safety of the treatments, animal weights were monitored on the same days as size measurements, with no weight loss detected over the course of the experiment. The weight curves for all animals is shown in Figure 7B. The amount of RTX administered over the course of the treatment duration (~0.11 mg/kg of drug each treatment day, ~0.66 mg/kg accumulated dose over 11 days) is a low dose (maximum tolerated dose > 30 mg/kg/day for free drug on shorter schedule50). The animals showed no distress or weight loss in any treatment group, indicating relative safety of the formulation. The marked weight gain observed in some of the animals can be explained by the rapid growth of the associated tumor. Clearance organs were excised as animals met their endpoints and were sectioned and stained with H&E. The representative sections from organs from each treatment group are shown in Figure 7C. Across the images from the liver, spleen, and kidney, there was little to no variance between treatment groups in structure or morphology, perhaps indicating that there was negligible off-target toxicity of the HARPs and/or IR treatments. Raltitrexed has been investigated in combination with RT in the treatment of CRC5,51 and recently for preclinical treatment of esophageal cancer.45 These results demonstrate the potential to improve multimodal therapy. Further collection of data for various treatment regimens will provide a foundation for establishing synergistic treatment paradigms.
Conclusions
HARPs proved to be an effective treatment against CT26 tumors in BALB/c mice. Furthermore, radiation in addition to the chemotherapeutic-loaded nanoparticle showed significant increase in tumor growth inhibition as compared to the formulation alone. In this initial trial, the combination treatment led to greater survival benefit (median survival) than either individual treatment. In each of the individual treatment groups, multiple animals showed no response to the treatment, whereas in the combined treatment group, all the animals appeared to respond to some component of the therapy.
The formulation process presented here highlights the potential for LbL assembly of chemotherapeutic nanoparticles. As the field of nanotechnology rapidly expands and moves into the clinic, there is a need for high-throughput synthesis methods. LbL synthesis is highly modular and allows for production of optimally-sized dispersions. There is limited evidence for successful delivery of chemotherapeutic nanoparticles in combination with RT. Here, we provide in vitro and in vivo validation of HARPs against a colorectal cancer model. Upon further optimization of IR and HARPs dosing and timing, this work has potential for translation to clinical trials.
Supplementary Material
Figure S1. TEM of bare CML nanoparticles prior to layer-by-layer deposition.
Figure S2. Fluorescence microscopy images of no treatment and CML-PLA-HA treated wells after 24 hour exposure. The no treatment well is the same as presented in Figure 3. The hyaluronic acid only nanoparticles showed similar fluorescence to the HARPs.
Figure S3. 24, 48, and 72 hour viability curves for raltitrexed and HARPs.
Figure S4. Stacked histograms showing the changing fluorescent signal with different treatments.
Acknowledgments
We gratefully acknowledge the laboratory facilities and equipment support provided by the Portland State University Center for Electron Microscopy & Nanofabrication and Mr. Hayden Winter for acquiring TEM images, the Oregon Health and Science University Flow Cytometry Core, and the Oregon Health and Science University Histopathology Shared Resource.
Funding Sources
This work was supported by the NIH NIGMS as a Maximizing Investigators’ Research Award, 1R35GM119839-01 (C.S.), and Oregon State University College of Pharmacy Start-up Funds.
Abbreviations
- RTX
Raltitrexed
- CRC
Colorectal Cancer
- IV
Intravenous
- LbL
Layer-by-Layer
- RT
Radiation Therapy
- PE
Polyelectrolyte
- NT
No Treatment
- CML
Carboxylate-Modified polystyrene Latex
- PLA
Poly-L-Arginine
- HA
Hyaluronic Acid
- HARPs
Hyaluronic Acid Raltitrexed Nanoparticles
- IR
Ionizing Radiation
- PBS
Phosphate-Buffered Saline
- TFF
Tangential-Flow Filtration
- DAPI
4’,6-diamidino-2-phenylindole
- TEM
Transmission Electron Microscopy
- MWCO
Molecular Weight Cutoff
- HPLC
High-Performance Liquid Chromatography
- PES
Polyethersulfone
- H&E
Hematoxylin and Eosin
- CC3
Cleave Caspase-3
Footnotes
Supporting Information
The following files are available free of charge.
Competing Interests
The authors declare that they have no competing interests.
References
- 1.Valentini V, Doglietto GB, Morganti AG, Turriziani A, Smaniotto D, De Santis M, Ratto C, Sofo L and Cellini N, Eur. J. Cancer, 2001, 37, 2050–2055. [DOI] [PubMed] [Google Scholar]
- 2.Avallone A, Delrio P, Guida C, Tatangelo F, Petrillo A, Marone P, Cascini LG, Morrica B, Lastoria S, Parisi V, Budillon A and Comella P, Br. J. Cancer, 2006, 94, 1809–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, Wu W, Hong W, Sun X, Wu J and Huang Q, Clin. Res. Hepatol. Gastroenterol, 2014, 38, 219–225. [DOI] [PubMed] [Google Scholar]
- 4.Wilson KS, Fitzgerald CA, Barnett JB, Gill S and Khoo KE, Cancer Invest, 2007, 25, 711–714. [DOI] [PubMed] [Google Scholar]
- 5.Gambacorta MA, Valentini V, Coco C, Morganti AG, Smaniotto D, Miccichè F, Mantini G, Barbaro B, Garcia-Vargas JE, Magistrelli P, Picciocchi A and Cellini N, Int. J. Radiat. Oncol. Biol. Phys, 2004, 60, 139–148. [DOI] [PubMed] [Google Scholar]
- 6.Jackman AL, Theti DS and Gibbs DD, Adv. Drug Deliv. Rev, 2004, 56, 1111–1125. [DOI] [PubMed] [Google Scholar]
- 7.Zalcberg JR, Cunningham D, Van Cutsem E, Francois E, Schornagel J, Adenis A, Green M, Iveson A, Azab M and Seymour I, J. Clin. Oncol, 1996, 14, 716–721. [DOI] [PubMed] [Google Scholar]
- 8.Popat S, Matakidou A and Houlston RS, J. Clin. Oncol, 2004, 22, 529–536. [DOI] [PubMed] [Google Scholar]
- 9.Aschele C, Lonardi S and Monfardini S, Cancer Treat. Rev, 2002, 28, 27–47. [DOI] [PubMed] [Google Scholar]
- 10.Pestalozzi BC, Peterson HF, Gelber RD, Goldhirsch A, Gusterson BA, Trihia H, Lindtner J, Cortés-Funes H, Simmoncini E, Byrne MJ, Golouh R, Rudenstam CM, Castiglione-Gertsch M, Allegra CJ and Johnston PG, J. Clin. Oncol, 1997, 15, 1923–1931. [DOI] [PubMed] [Google Scholar]
- 11.Johnston PG, Recant W, Behan A, Dolan ME, Ratain MJ, Ralph R, Carmen J and Vokes EE, J. Natl. Cancer Inst [DOI] [PubMed] [Google Scholar]
- 12.Chang MH, Ahn JS, Lee J, Kim KH, Park YH, Han J, Ahn MJ and Park K, Lung Cancer, 2010, 69, 323–329. [DOI] [PubMed] [Google Scholar]
- 13.T. N, M. N, Y. F, T. H, H. M and Y. N, Int. J. Urol, 2002, 9, 368–376.12165018 [Google Scholar]
- 14.Allen TM and Cullis PR, Science, 2004, 303, 1818–22. [DOI] [PubMed] [Google Scholar]
- 15.Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE and Hammond PT, ACS Nano, 2013, 7, 9571–9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elbakry A, Zaky A, Liebl R, Rachel R, Goepferich A and Breunig M, Nano Lett, 2009, 9, 2059–2064. [DOI] [PubMed] [Google Scholar]
- 17.Schneider G and Decher G, Nano Lett, 2004, 4, 1833–1839. [Google Scholar]
- 18.Gu L, Deng ZJ, Roy S and Hammond PT, Clin. Cancer Res, 2017, 23, 7312–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morton SW, Poon Z and Hammond PT, Biomaterials, 2013, 34, 5328–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dreaden EC, Morton SW, Shopsowitz KE, Choi JH, Deng ZJ, Cho NJ and Hammond PT, ACS Nano, 2014, 8, 8374–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Correa S, Choi KY, Dreaden EC, Renggli K, Shi A, Gu L, Shopsowitz KE, Quadir MA, Ben-Akiva E and Hammond PT, Adv. Funct. Mater, 2016, 26, 991–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mero A and Campisi M, Polymers (Basel)., 2014, 6, 346–369. [Google Scholar]
- 23.Senbanjo LT and Chellaiah MA, Front. Cell Dev. Biol, DOI: 10.3389/fcell.2017.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogus DR, Evans MA, Pusuluri A, Barajas A, Zhang M, Krishnan V, Nowak M, Menegatti S, Helgeson ME, Squires TM and Mitragotri S, J. Control. Release, 2017, 267, 191–202. [DOI] [PubMed] [Google Scholar]
- 25.Seok HY, Sanoj Rejinold N, Lekshmi KM, Cherukula K, Park IK and Kim YC, J. Control. Release, 2018, 280, 20–30. [DOI] [PubMed] [Google Scholar]
- 26.Van Cutsem E, Eur. J. Cancer, 1999, 35, 2–3. [DOI] [PubMed] [Google Scholar]
- 27.Wils J, Br. J. Cancer, 1998, 77, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.James R, Price P and Valentini V, Eur. J. Cancer, 1999, 35, 19–22. [DOI] [PubMed] [Google Scholar]
- 29.Zhao X, Li L, Starr T and Subramanian S, Oncotarget, 2017, 8, 54775–54787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin WC, Wang WH, Lin YH, Der Leu J, Cheng SY, Chen YJ and Hwang JJ, Oncol. Rep, 2018, 40, 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Correa S, Choi KY, Dreaden EC, Renggli K, Shi A, Gu L, Shopsowitz KE, Quadir MA, Ben-Akiva E and Hammond PT, Adv. Funct. Mater, 2016, 26, 991–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan YF, Mundargi RC, Chen MHA, Lessig J, Neu B, Venkatraman SS and Wong TT, Small, 2014, 10, 1790–1798. [DOI] [PubMed] [Google Scholar]
- 33.Morton SW, Poon Z and Hammond PT, Biomaterials, 2013, 34, 5328–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou J, Romero G, Rojas E, Ma L, Moya S and Gao C, J. Colloid Interface Sci, 2010, 345, 241–247. [DOI] [PubMed] [Google Scholar]
- 35.Poon Z, Chang D, Zhao X and Hammond PT, ACS Nano, 2011, 5, 4284–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang W, Kim BYS, Rutka JT and Chan WCW, Nat. Nanotechnol, 2008, 3, 145–150. [DOI] [PubMed] [Google Scholar]
- 37.Ekkapongpisit M, Giovia A, Follo C, Caputo G and Isidoro C, Int. J. Nanomedicine, 2012, 7, 4147–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng ZJ, Morton SW, Bonner DK, Gu L, Ow H and Hammond PT, Biomaterials, 2015, 51, 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao C, Fan L, Qi F, Ou S, Yu L, Yi X, Ni B, Zheng Z, Lu J, Zhang C, Chen C, Lu X, Cheng L, Hu T and Ma Y, Anticancer. Drugs, 2016, 27, 689–694. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Cheng M, Cao J, Zhang Y, Yuan Z, Wu Q and Wang W, Nanoscale, 2019, 11, 5005–5013. [DOI] [PubMed] [Google Scholar]
- 41.Varshosaz J, Hassanzadeh F, Sadeghi-Aliabadi H and Firozian F, Biomed Res. Int, 2014, 2014, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marti TM, Hefner E, Feeney L, Natale V and Cleaver JE, Proc. Natl. Acad. Sci, 2006, 103, 9891–9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding D, Zhang Y, Wang J, Zhang X, Gao Y, Yin L, Li Q, Li J and Chen H, Cell Death Discov, DOI: 10.1038/cddiscovery.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuzaki K, Harada A, Takeiri A, Tanaka K and Mishima M, Mutat. Res. - Genet. Toxicol. Environ. Mutagen, 2010, 700, 71–79. [Google Scholar]
- 45.Ding W-X, Liu S, Ma J-X, Pu J, Wang H-J, Zhang S and Sun X, Cancer Cell Int, 2019, 19, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schöber C, Gibbs JF, Yin MB, Slocum HK and Rustum YM, Biochem. Pharmacol, 1994, 48, 997–1002. [DOI] [PubMed] [Google Scholar]
- 47.Briggs A and Brown R, Nanomedicine Nanotechnology, Biol. Med, 2013, 9, 1098–1105. [DOI] [PubMed] [Google Scholar]
- 48.Hather G, Donelan J, Liu R, Bandi S, Mettetal J, Manfredi M, Shyu W and Chakravarty A, Cancer Inform., 2014, 13, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clarke SJ, Farrugia DC, Aherne GW, Pritchard DM, Benstead J and Jackman AL, Clin. Cancer Res, 2000, 6, 285–296. [PubMed] [Google Scholar]
- 50.Cao S, McGuire JJ and Rustum YM, Clin. Cancer Res, 1999, 5, 1925–1934. [PubMed] [Google Scholar]
- 51.Botwood N, James R, Vernon C and Price P, Ann. Oncol, 2000, 11, 1023–1028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. TEM of bare CML nanoparticles prior to layer-by-layer deposition.
Figure S2. Fluorescence microscopy images of no treatment and CML-PLA-HA treated wells after 24 hour exposure. The no treatment well is the same as presented in Figure 3. The hyaluronic acid only nanoparticles showed similar fluorescence to the HARPs.
Figure S3. 24, 48, and 72 hour viability curves for raltitrexed and HARPs.
Figure S4. Stacked histograms showing the changing fluorescent signal with different treatments.