

Abstract
Mild traumatic brain injury (mTBI) can result in permanent impairment in memory and learning and may be a precursor to other neurological sequelae. Clinical treatments to ameliorate the effects of mTBI are lacking. Inhibition of microRNA-181a (miR-181a) is protective in several models of cerebral injury, but its role in mTBI has not been investigated. In the present study, miR-181a-5p antagomir was injected intracerebroventricularly 24 h prior to closed-skull cortical impact in young adult male mice. Paw withdrawal, open field, zero maze, Y maze, object location and novel object recognition tests were performed to assess neurocognitive dysfunction. Brains were assessed immunohistologically for the neuronal marker NeuN, the perineuronal net marker wisteria floribunda lectin (WFA), cFos, and the interneuron marker parvalbumin. Protein quantification was performed with immunoblots for synaptophysin and postsynaptic density 95 (PSD95). Fluorescent in situ hybridization was utilized to localize hippocampal miR-181a expression. MiR-181a antagomir treatment reduced neuronal miR-181a expression after mTBI, restored deficits in novel object recognition and increased hippocampal parvalbumin expression in the dentate gyrus. These changes were associated with decreased dentate gyrus hyperactivity indicated by a relative reduction in PSD95 and cFos expression. These results suggest that miR-181a inhibition may be a therapeutic approach to reduce hippocampal excitotoxicity and prevent cognitive dysfunction following mTBI.
Keywords: Mild traumatic brain injury, Parvalbumin, MicroRNA, Hippocampus, Novel object
Introduction
Traumatic brain injury (TBI) affects up to 42 million people worldwide annually, with total medical costs in the USA estimated at $76.5 billion dollars (Gardner and Yaffe 2015). In addition to the direct sequelae of TBI, recent evidence suggests TBI plays a role as a precursor to other common neurological diseases (Lowenstein et al. 1992). Injury from TBI extends beyond the primary site of injury to internal brain structures such as the hippocampus, which is selectively vulnerable to even mild head trauma (Tang et al. 1997). Learning and memory deficits have been linked to hippocampal dysfunction after a single, mild TBI (mTBI) event (Aungst et al. 2014). In addition to the immediate consequences of mTBI, cognitive and behavioral complications can persist for weeks or months following the initial injury. Interventions that reduce neuronal loss in the hippocampus and preserve cognitive function after mTBI are desperately needed.
One underlying cause of post-injury cognitive dysfunction is loss of fast-spiking inhibitory interneurons that express the calcium binding protein Parvalbumin (PVALB; Hsieh et al. 2017). Loss of PVALB + interneurons results in heightened stress responses (Taylor et al. 2013), reduced novel object exploration, and working memory deficits (Zhang and Reynolds 2002). Perineuronal nets (PNNs) are extracellular matrix proteins that envelop and stabilize a subset of PVALB + interneurons (Cabungcal et al. 2013). Loss of PNNs leads to the subsequent loss of PVALB + neurons (Cabungcal et al. 2013), resulting in impaired learning and memory retention (Hylin et al. 2013; Thompson et al. 2018), and an imbalance of excitatory/inhibitory activity in the hippocampus (Fasulo et al. 2017). TBI severity is directly linked to excessive levels of excitatory neurotransmitters, contributing to excitotoxicity (Palmer et al. 1993). However the underlying molecular mechanisms that regulate post-mTBI hippocampal excitotoxicity and resultant cognitive dysfunction remain a critical knowledge gap impeding the development of novel therapeutic interventions.
MicroRNAs (miRs) are short, noncoding RNAs that inhibit target gene translation via complimentary binding, and their expression has been shown to be altered after TBI (Redell et al. 2009). In particular brain-enriched miR-181a was observed to be acutely down-regulated acutely after mTBI, but elevated by 24 h post-injury (Redell et al. 2009). Elevations in miR-181a expression has been shown to contribute to cerebral injury, including after cerebral ischemia (Xu et al. 2015), epilepsy (Ren et al. 2016), in Parkinson’s disease (Hegarty et al. 2018), and is associated with post-injury behavioral changes in rodents (Chandrasekar and Dreyer 2011). A member of the same miR family, miR-181c, shows the most change of all hippocampal miRs after TBI (Boone et al. 2017). However, whether miR-181a contributes to injury following mTBI, and whether inhibition is protective against mTBI-induced behavioral deficits, has not been previously investigated. Therefore, in the present study we assessed the effects of miR-181a inhibition on acute (6 h and 24 h) and long-term (28 d) hippocampal injury and behavioral outcomes after mTBI.
Methods
All experimental protocols using animals were approved by the Stanford University Animal Care and Use Committee and performed in accordance with NIH guidelines.
Experimental Timeline
Adult male C57/B6 mice (age 8–10 weeks, Jackson Laboratory, Bar Harbor, ME) were sorted into random groups by coin flip and were pre-treated 24 h prior to injury with either miR-181a antagomir or MM control. Tissue samples were collected at 6 h, 24 h, and 28 d after TBI. Behavioral assays and tissue collection for immunoblots were performed prior to, and 28 d after, mTBI (Fig. 1a).
Fig. 1.
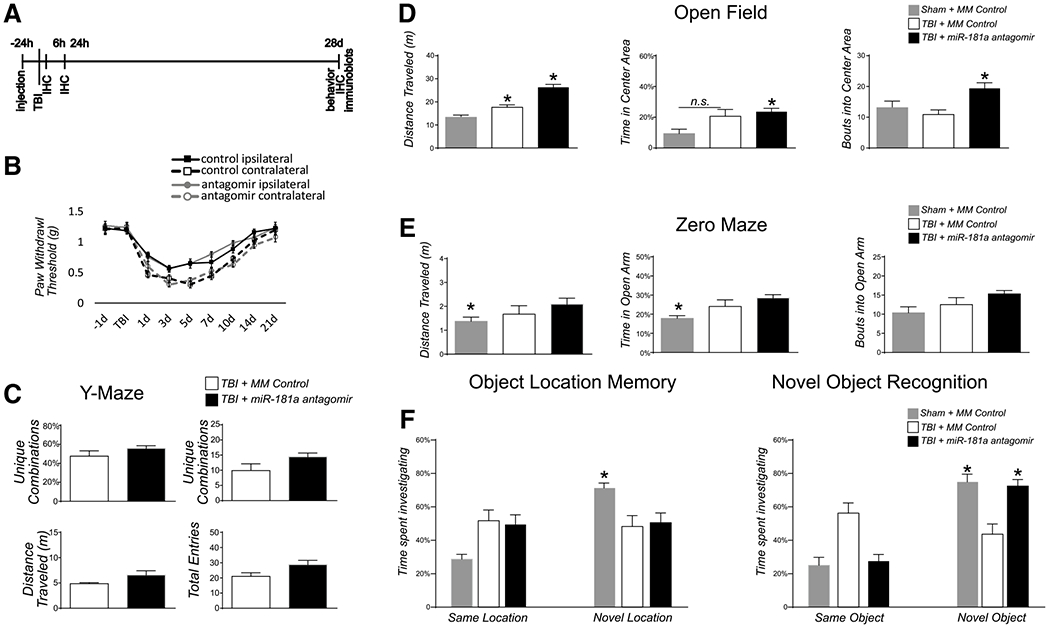
Behavioral tests. a Experimental timeline: Stereotactic intracerebroventricular (ICV) injection of miR-181a-5p antagomir or mismatch control was performed 24 h prior to mild traumatic brain injury (mTBI). Animals were sacrificed at 6 h, and 24 h or 28 d post-mTBI and assessed for histopathological changes in hippocampus. Behavioral testing was performed prior to sacrifice at 28 d. Animals pretreated with miR-181a antagomir or mismatch control were assessed 28 d after mTBI for: b paw withdrawal reflex; c Y maze; d open field test; e zero maze and f novel object recognition and object location memory task. n = 12 per group, *p < .05, Error bars Mean ± SEM
Stereotactic Injection
MiR-181a antagomir or mismatch control was injected intracerebroventricularly (ICV) 24 h prior to injury according to dose/toxicity parameters experimentally determined previously (Ouyang et al. 2012). Briefly, mice were deeply anesthetized and placed in a stereotactic head frame and received a 20-min infusion of 6 μl antagomir (3 pmol/g in 2 μl H2O, 4 μl DOTAP; Roche Applied Science, San Francisco, CA) or mismatch control sequence into the left lateral ventricle (bregma: −0.58 mm; dorsoventral: 2.1 mm; lateral: 1.2 mm; Xiong et al. 2011) .The cannula was removed, and the wound was then closed with bone wax.
Controlled Cortical Impact
Closed head mTBI and sham procedures were performed as previously described with minor modifications (Luo et al. 2014; Sahbaie et al. 2018). To induce mTBI, a benchmark stereotaxic impactor (MyNeurolab, St. Louis, MO, USA) actuator was mounted on a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA). Mice were placed in a foam mold held in the prone position on the stereotaxic frame after isoflurane anesthesia. The stereotaxic arm was adjusted at a 40° angle, head impact was at a fixed-point relative to the right eye and ear, corresponding to the S1 somatosensory cortex. The force of the impact delivered by the device was 5.8-6.0 m/s (Dwell time = 0.2 s), impact depth of 5 mm with a 5-mm tip. The mice recovered from anesthesia on a warming pad prior to returning to their home cages. No evidence of skull fracture were observed similar to previous reports using comparable head impact force (Luo et al. 2014; Zhang et al. 2016). Furthermore, there were no noticeable effects on lesion size at either 6 h, 24 h or 28 d post-TBI injury. Sham animals received anesthesia and were placed in the stereotactic frame, but the impact device was instead discharged in the air above the animal.
Mechanical Sensitivity Test
Mechanical sensitivity was assessed using nylon von Frey filaments (Stoelting Co., IL, USA) according to the “updown” algorithm developed by Chaplan et al. (1994). We have applied this technique previously to detect 50% withdrawal threshold in mice after injury (Sahbaie et al. 2014; Tajerian et al. 2015). After acclimating mice on the wire mesh platform inside plastic enclosures (10 cm radius), sequential fibers with increasing stiffness ranging from 0.004 to 1.7 g were applied to the plantar surface of a hind limb and left in place 5 s. When 4 fibers had been applied after the first response the testing terminated. Withdrawal of hind paw from the fiber was considered a response. If a response occurred after application of a fiber, then a more flexible fiber was applied; if no response was observed the next stiffest fiber was applied.
Open Field test, Zero Maze, and Memory Tests
All behavioral analysis was conducted by a blinded observer.
The open field arena (40 × 40 × 40 cm) was used to assess locomotion, object location and novel object recognition memory tests. The elevated zero maze was used to measure anxiety according to previously published methods (Tajerian et al. 2014). The zero maze was 60 cm from the floor, had an outer diameter of 60 cm and inner diameter of 50 cm, with two closed (15 cm tall walls) and open quadrants. Mice were randomly placed facing one of the closed quadrants at the beginning of the 5 min test. Total time spent in open and closed quadrants were recorded. Time spent in the open quadrants was compared across groups to measure anxiety.
For object location and novel object recognition memory experiments, mice were initially habituated to two identical objects after being placed in the middle of the open field arena, as previously described (Sahbaie et al. 2018; Zou et al. 2012). Next, during a 5-min trial one of the objects was moved to a novel location and exploratory behavior (investigation time) was recorded. After a 5-min period in home cages, mice were returned to the arena with one of the previous identical objects replaced with a novel one. The new object had a distinct shape and size difference from the identical object sets, and 5-min recordings were performed assessing exploratory behavior directed toward the objects.
Y-maze was used to assess working spatial memory. The arena consisted of 3 symmetrical arms (arms A, B, and C) at 120° angles with a dimension of 20 × 8 × 16 cm (L x W x H) for each arm. The mice were placed in the center of the arena, and arm entry was recorded for 10 min. Unique triad combination of consecutive arm entries was used as a measurement of spontaneous alternation behavior (Hughes 2004). All recordings from the above experiments were analyzed in real time by TopScan software (Clever System, USA).
Fluorescent in situ Hybridization and Immunohistochemical Labeling
Six mice from each group were deeply anesthetized and perfused with ice-cold saline, followed by 4% buffered paraformaldehyde. Brains were then removed and stored in formaldehyde for at least 3 days. A notch in the cortex was used to track the injured hemisphere. Brains were sectioned on a vibrating microtome at 50 μm. Sections were treated with 1% H2O2 for 10 min and blocked with Triton-100X with 5% horse serum overnight at 4 °C. Parvalbumin labeling was performed with 1:500 PBS/anti-sheep antibody (R&D Systems, Minneapolis, Cat. AF5058) and fluorescently stained with Alexa-594 goat-anti sheep secondary antibody (Invitrogen). Perineuronal nets were labeled with 1:500 fluorescein-tagged Wisteria Floribunda Lectin (Vector Labs, Burlingame, Ca, Cat. FL-1351). Six brain slices per animal were mounted on SuperFrost slides with ProLong Glass Antifade Mountant (ThermoFisher, Waltham, MA, Cat. P36980) and #1 coverslips. The expression of miR-181a-5p was determined by fluorescent in situ hybridization (FISH) in combination with IHC labeling for PVALB + neuron colocalization. A double FAM-labeled miR-181a-5p miRCURY LNA miRNA Detection Probe (Qiagen, cat# YD00619309-BED) was employed using a combined protocol described by Chaudhuri et al. and de Planell-Saguer et al. with minor modifications to disrupt the hybridization of miR-181a and antagomir (Datta Chaudhuri et al. 2013; Planell-Saguer et al. 2010). Fluorescent optical sections were obtained using a Zeiss Imager M2 upright microscope at 200x magnification. A Z-stack was captured at steps of 2 μm for stereological counts of parvalbumin-expressing cells and perineuronal nets, as well as their overlap. Exposure and white/black balance were set to the same values in all images captured.
Cell Quantification
Cells were assessed stereologically by an observer blinded to conditions using Neurolucida™ (v2017, MicroBright-Field, Williston, VT) and recorded as belonging to one of three groups based on their location (Fig. 4b): the cornu ammonis-1 (CA1), including the molecular layers above the dentate gyrus (DG); the DG granule cell (GC) layer (including all cells that were only partially overlapping the granule cell layer), and the hilus/CA3c (i.e., cells that were entirely between the two GC layers). The process was repeated with PNNs and then again with both red and green channels to identify PVALB+/PNN colocalization. Neither PVALB or PNN staining showed differences between the contralateral and lateral hippocampal hemispheres, so both hemispheres were counted from four brain sections per animal and were averaged together to represent one animal. cFos showed strong hemisphere bias and was calculated as a ratio of contralateral and ipsilateral activation.
Fig. 4.
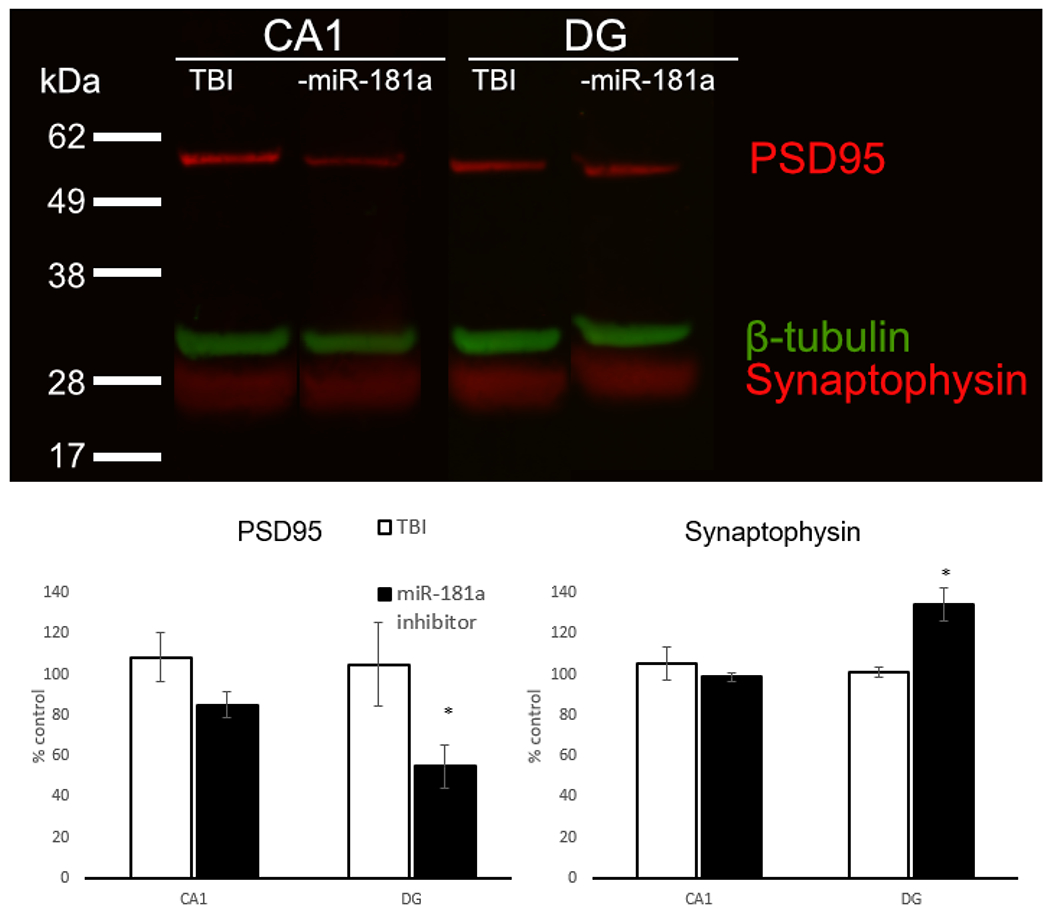
Immunoblots. Hippocampal protein expression of postsynaptic density 95 (PSD95), synaptophysin, and β-Tubulin (a). Quantification of PSD95 (b) and synaptophysin (c) in the dentate gyrus and CA1. n = 4 per group, *p < .05, Error bars Mean ± SEM
Immunoblotting
Protein quantification was performed using 30 μg of protein/sample as previously described (Stary et al. 2017), using primary antibodies to β-tubulin (Abcam, Cat. ab6046), synaptophysin (Abcam, Cat. ab14692), and postsynaptic density 95 (Abcam, Cat. ab18258). Briefly, brain protein was isolated after perfusion with iced cold saline, then 30 μg of protein/sample was separated on a 4-10% Bis-Tris mini-gel (NP0304BOX, ThermoFisher Scientific), and electro-transferred to an Immobilon polyvinylidene fluoride membrane (IPVH00010, Millipore EMD Corp.). Membranes were blocked and incubated with the appropriate primary antibody overnight at 4 °C. Membranes were then washed and incubated with 1:15,000 IRDye conjugated secondary antibodies goat anti-mouse (LiCor, Lincoln, Nebraska, Cat. 925-32210) and goat anti-rabbit (LiCor, Lincoln, Nebraska, Cat. 925-32211). Immunoreactive bands were visualized using the LICOR Odyssey infrared imaging system. Densitometric analysis was performed by an observer blinded to treatment group using Image J software (v1.46, National Institutes of Health). The area under the densitometry curve was normalized to β-tubulin loading control and expressed as percentage of control. Four animals were used for each group.
Statistics
All statistics were performed with SPSS v22.0 (IBM). Group differences were analyzed with ANOVA when there were more than two groups, followed by examination of simple effects with independent-samples t tests. T tests were also used for two comparison groups. Error bars represent mean ± SEM, p < .05 was considered significant.
In Silico Target Predictions
We performed a targeted in silico search for mmu-miR-181a-5p targets (TargetScan v7.2). Only gene targets with cumulative weighted context score ≤ −0.2, or aggregate PCt ≥ 0.5 were included.
We identified 1001 initial targets (1001 transcripts, 1186 conserved sites, 433 poorly conserved sites). Experimentally confirmed miR-181a-5p targets BCL2, BCL2L11, PTPN22 and HIPK2 have low (under – 0.2) context scores, but 0.5 or higher aggregate PCt. We used these criteria to refine our list to 490 putative miR-181a targets (Supplemental Table 1).
Results
Effect of miR-181a Antagomir on Behavioral Outcomes Following mTBI
Sham mice stereotactically injected with antagomir mismatched (MM) control did not differ from non-injected sham animals in any of the tests; therefore, only MM control is presented for comparison. The TBI allodynia profile did not differ between mice pre-treated with miR-181a antagomir (antagomir) from mice that received injections of vehicle (controls; F[1,32] = 0.002, p = .969, partial η2 < 0.001; Fig. 1b). There were no differences between pre-treatment and MM controls for the Y Maze in unique combinations (p = .216), total entries (p = .051), or distance traveled (p = .067; Fig. 1c). In the Open Field Test, there were differences in total distance traveled (F[2,27] = 35.62, p < .001, partial η2 = 0.725) time spent in the center (F[2,27] = 5.226, p = .012, partial η2 = 0.279) and total entries into the center (F[2,27] = 5.799, p = .008, partial η2 = 0.30) between treatment groups (Fig. 1d). Bonferroni post hoc analyses revealed antagomir treated mice traveled more total distance than TBI + MM (p < .001) mice, spent more time in the center than controls (p = .013), but not more than TBI + MM and made more bouts into the center compared to the MM control or TBI + MM groups (p = .009).. There were differences between treatment groups in the Zero maze for time spent in the open arms, F(2,27) = 5.027, p = .014, partial η2 = 0.27, but not for bouts into open arms [F(2,27) = 2.41, p = .018, partial η2 = 0.152]. Bonferroni post hoc analysis showed that antagomir MM control animals spent less time in the open arms than mTBI animals (Fig. 1e). After mTBI, only MM control showed a preference for a familiar object in a novel location (F[2,27] = 4.245, p = .025, partial η2 = 0.239), while mice treated with antagomir showed no increase over TBI + MM (p > .99; Fig. 1f). However, there were differences in the time spent investigating novel objects (F[2,27] = 10.29, p < .001, partial η2 = 0.43). Bonferroni analysis revealed animals treated with miR-181a antagomir spent more time investigating a novel object with no difference to pre-injury levels (p = .99), while mismatch controls showed no such recovery (p = .002; Fig. 1g). Because the hippocampus is implicated in novel object recognition, we assessed for histological evidence of neuronal survivability and synaptic milieu with miR-181a inhibition.
Histological Observations
All histological observations were made from 6 mice from each group, 6 brain sections per animal, and were counted by blinded observers. Inter-rater reliability was high (R2 > 0.95).
Cornu Ammonis-1 and Cortex
After 28 d of recovery, there was significant healing at the impact site. There were no quantitative differences in expression of NeuN + neurons, PVALB + neurons, or cFos expression between antagomir and mismatch control treatment groups in the CA1 or cortex at the site of impact (Fig. 2).
Fig. 2.
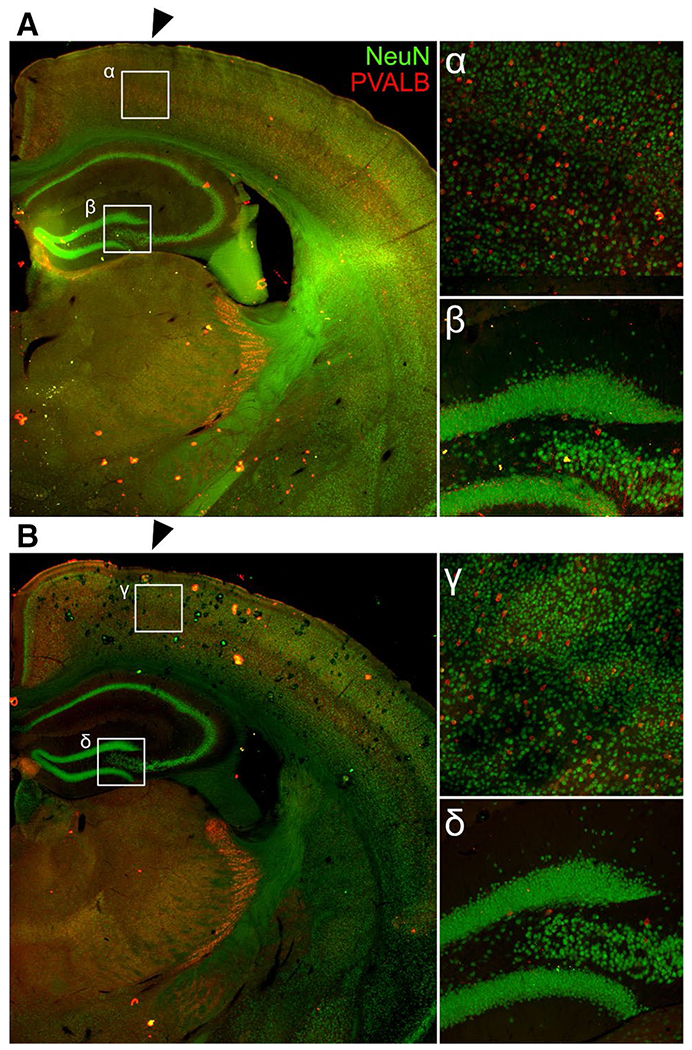
Healing at the cortical impact site. There were no differences after 28 d of healing at the cortical impact site (black arrows) in number of NeuN + neurons, PVALB + neurons, or PNNs in TBI + MM Control (a) or TBI + 181a inhibitor (b). Magnified views of the cortex just below the impact site (α,γ) and the dentate gyrus/CA3c (β,δ)
Dentate Gyrus Granule Cell Layer
Mild TBI resulted in an early, acute loss of PVALB expression from the GC layer at 6 h (t[15] = 3.532, p = .003, Cohen’s d = 1.77) and continued at 24 h, however, had recovered by 28 d after injury (p = .39; Fig. 3c). PVALB + neuron count in the granule cell layer was unaffected by antagomir treatment (F[1,30] = 0.660, p = .423, partial η2 = 0.02). PNN count was decreased by 24 h after mTBI in controls (t[14] = 2.82, p = .012, Cohen’s d = 1.09); however, antagomir-treated animals did not display this loss and had significantly higher PNN expression at 24 h (t[7] = 2.87, p = .019, Cohen’s d = 2.53; Fig. 3d). By 28 d after mTBI, no difference in PNN count was observed between treatments (p = .103). Antagomir-treated animals exhibited a significant decrease in activity (as assessed by cFos expression) in the GC layer from 6 to 24 h (t[9] = 2.60, p = .029, Cohen’s d = 1.64; Fig. 3e), that was not observed in mismatch control-treated animals (p = .107). GC activity in the coup relative to the contracoup hippocampus was lower overall in the antagomir-treated animals at both 6 h (t[7] = 4.43, p = .003, Cohen’s d = 3.70) and 24 h after mTBI (t[6] = 5.28, p = .002, Cohen’s d = 3.27).
Fig. 3.
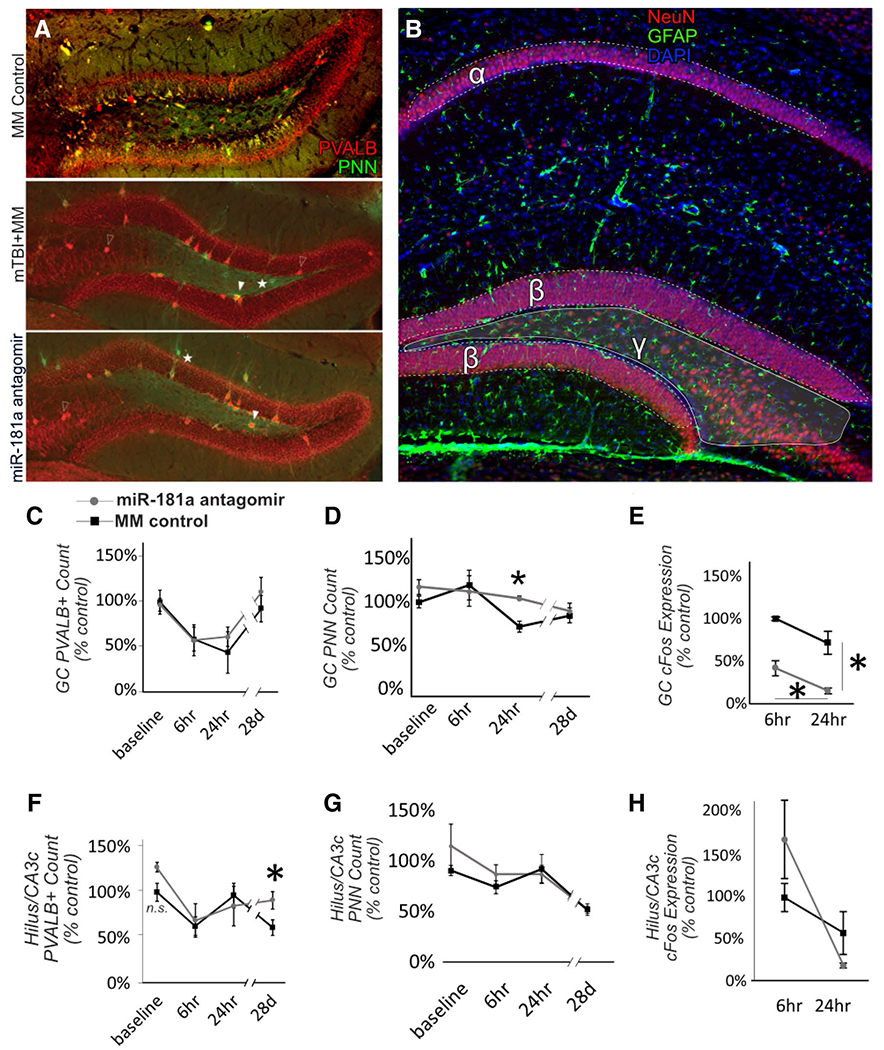
Hippocampal histopathology. Examples of fluorescent immunohistochemistry for assessment of hippocampal parvalbumin positive (PVALB+) neurons and perineuronal nets (PNN) for MM controls, and 28 d after mTBI in animals pre-treated with either miR-181a antagomir or MM control (a). For all analyses, hippocampal subregions were defined as (α) cornu ammonis-1, (β) the granule cell layer of the dentate gyrus and (γ) the dentate gyrus hilus/cornu ammonis-3c (b). Antagomir treatment resulted in a slight increase in PVALB expression in the DG hilus/CA3c. Open arrow = PVALB + interneuron, closed arrow = PVALB + PNN, Star = PNN only. Quantification of PVALB in the dentate gyrus (DG) granule cell (GC) layer (c) and in DG hilus/cornu ammonis-3c (CA3c, f). PNN quantification in GC layer (d) and hilus/CA3c (g) of DG. Neuronal activity in DG measured by cFos expression in GC layer (e) and hilus/CA3c (h). n = 6 per group, *p < .05, Error bars Mean ± SEM
Dentate Gyrus Hilus/CA3c
Similar to the GC layer, we observed an early, acute decrease in PVALB expression at 6 h post-mTBI (t[15] = 3.19, p = .006, Cohen’s d = 1.55) that persisted up to 28 d (t[20] = 2.966, p = .008, Cohen’s d = 1.27; Fig. 3f). Despite this sustained loss, antagomir-treated animals exhibited significantly higher numbers of PVALB + neurons at 28 d compared to controls (t[10] = 2.53, p = .030, Cohen’s d = 1.46). No differences between treatment groups were observed in PNN count or cFos activity (Fig. 3g–h).
Synaptic Connectivity
To examine changes in synaptic protein expression in the hippocampus, we quantified the expression of excitatory postsynaptic marker PSD95 and the general presynaptic marker synaptophysin between CA1 and DG (Fig. 4a). Both were unchanged in CA1 in both control and antagomir-treated groups when normalized to β-Tubulin (Fig. 4b-c). The DG demonstrated an overall decrease in PSD95 in the antagomir treated group at 28 d after mTBI compared to controls, suggesting fewer excitatory connections (Fig. 4b). Antagomir treatment also resulted in an increase in synaptophysin at 28 d, suggesting increased overall connectivity (Fig. 4c).
Cellular Localization of miR-181a Expression
The dentate gyrus 24 h post-mTBI exhibited a moderate amount of miR-181a expression (Fig. 5a) which coincides with previously reported findings (Redell et al. 2009). However, miR-181a was undetectable in antagomir-treated animals at 24 h (Fig. 5b). In MM control animals, mTBI resulted did not result in miR-181a / PVALB colocalization in either the granule cell layer or the hilus/CA3c, suggesting miR-181a inhibition within PVALB + interneurons themselves was not the primary mechanism of action in hilar protection from excitotoxicity.
Fig. 5.
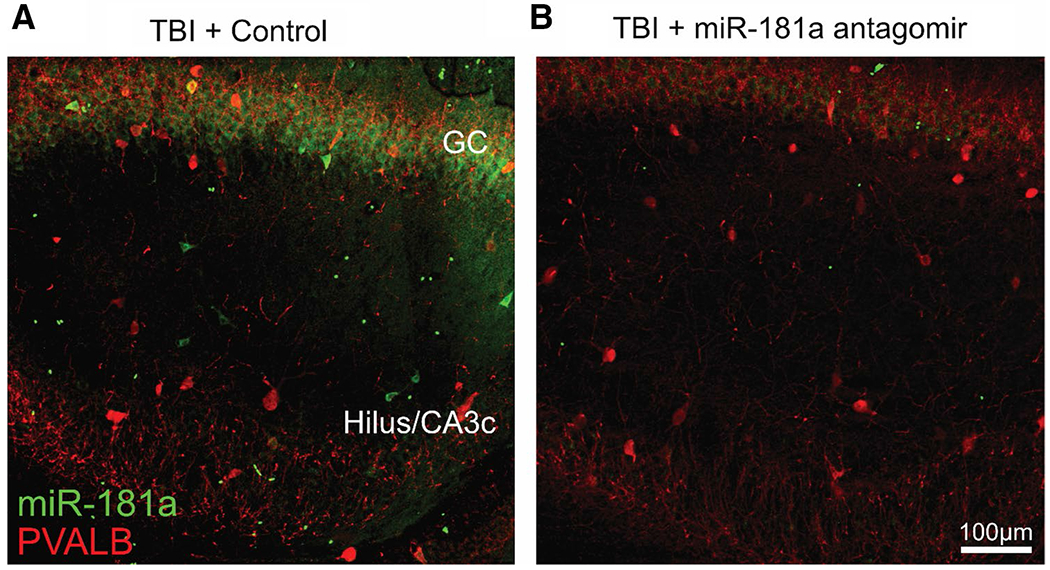
Cellular localization of miR-181a expression in hippocampus. miR-181a expression in the DG GC and Hilus/CA3c at 6 h post-mTBI in MM control (a) and miR-181a-5p antagomir (b) treated animals
Discussion
In the present study, we demonstrate for the first time that inhibition of miR-181a protects against selective behavioral deficits after mTBI and improves the long-term PVALB expression of DG hilar interneurons. The most notable behavioral difference we identified in animals treated with miR-181a antagomir was increased time spent inspecting a novel object. The brain regions associated with novel object recognition remain controversial, although animals with hippocampal lesions perform poorly in the task (Broadbent et al. 2010). DG-specific knockdown of neurogenesis has been shown to disrupt novel object recognition (Jessberger et al. 2009). The DG receives afferent inputs primarily from entorhinal cortex (David G. Amaral et al. 2007), and disruption of μ opioid receptors in the DG impedes signaling from the lateral entorhinal cortex resulting in abolishment of novel object exploration (Hunsaker et al. 2007) and exacerbating injury from TBI (Hayes et al. 2009). Within the DG, the hilar subregion is specifically and particularly sensitive to mTBI, as evidenced by increased inflammation, neuronal loss, and alterations in synaptic transmission detected by electrophysiology (Hicks et al. 1993). Disruption of hilar interneurons’ ability to provide inhibitory input has been linked to many other brain disorders (Lowenstein et al. 1992). Neurons in the hilus do not project to other areas of the hippocampus, but remain localized to the DG and CA3 (Amaral and Witter 1989). Neurons in the CA3c subregion of the hippocampus are heterogeneous in structure and function (Hunsaker et al. 2008), can project back into the DG hilus (Li et al. 1994), and are also susceptible to mTBI (Tang et al. 1997). Because the hilus and CA3c are difficult to differentiate both in fresh tissue and fixed sections, in the present study they were combined. However, it is possible these areas maintain unique responses to mTBI: they share a similar spatial environment, the physical forces from cortical impact are likely similar, and PVALB + interneurons in both regions receive similar afferent inputs from the DG granule cells.
The observations in the present study are similar to previous reports of PVALB + interneuron loss after TBI in the cortex (Hsieh et al. 2017), Hsieh et al. described post-TBI PVALB + neuron loss at four weeks followed by increased expression in markers of oxidative stress and loss of PNNs (Hsieh et al. 2017). The immediate loss and reappearance of PVALB expression are also consistent with previous observations in a model of cerebral ischemia demonstrating post-injury hippocampal PVALB expression loss and recovery (Johansen et al. 1990), PVALB functions to buffer an influx of calcium ions, and therefore protection by antagomir may have been mediated by calcium-mediated excitotoxic mechanisms. PNNs also work to protect PVALB + neurons from excitotoxicity that follows TBI (Hsieh et al. 2017). However, in the present study (where mTBI did not result in overt cortical lesions by 28 d of recovery) a difference in PNN expression was only observed in the GC layer at 24 h post-injury, when PVALB expression was at its lowest.
The protective effect of antagomir against mTBI may be secondary to decreased excitotoxic-mediated synaptic remodeling. Following TBI, inhibitory synapses are upregulated in the DG and play an important role in balancing excitatory/inhibitory signaling from the DG to CA3 (Reeves et al. 1997). Decreasing excitatory glutamatergic signaling is neuroprotective after TBI (Faden et al. 2001), and there is growing evidence that low doses of ethanol—a GABA channel modulator—can also protect against excitotoxicity after TBI (Janis et al. 1998; Türeci et al. 2004; Wang et al. 2012). Post-injury upregulation of the neuronal activity marker cFos occurs in the hippocampus after TBI in a time- and region-dependent manner (Raghupathi and McIntosh 1996), described as peaking from 2 to 6 h after injury and returning to baseline within 24 h (Yang et al. 1994). TBI-induced expression of cFos is highest in the dentate gyrus hilus (Czigner et al. 2004), and mild trauma results in a larger spike in cFos expression in the hippocampus versus more moderate injury, but this surge is not directly tied to neuronal loss (Phillips and Belardo 1992). In the present study, cFos was low at basal levels and almost undetectable in sham animals. We observed a relative decrease in cFos expression in the GC layer of antagomir treated mice, suggesting that lower activity levels did not trigger a cascade of excitotoxicity that affected PVALB expression, for example in the DG hilus. PNNs were not different in any brain area after 28 d, suggesting that PNNs may only protect against excitotoxicity in the early, acute stages post-injury after mTBI, while long-term PVALB expression differences may be secondary to alternative factors. Supporting this interpretation was our finding that miR-181a is localized in very few PVALB + interneurons themselves and may play a larger role through changing the physiology of afferent inputs to PVALB + interneurons rather than in the neurons themselves.
Inhibitory signaling balances the activity of excitatory input, and imbalances lead to dysfunction after TBI (Mtchedlishvili et al. 2010). In the present study, antagomir pre-treatment may have protected neurons in the hippocampus from excitotoxicity, resulting in long-term synaptic remodeling. In supporting this conclusion, PSD95 was found to be lower in the DG after treatment with antagomir, but synaptophysin expression was higher, suggesting higher overall synaptic density similar to patterns observed in hippocampal neurons after seizure injury (Jackson et al. 2012). PSD95 is a marker of glutamatergic synapses (Keith and El-Husseini 2008), so the increase in synaptic density may be from increased inhibitory connections, coinciding with increased expression of PVALB, though this must be confirmed by future experiments.
Although in the present study activity levels (measured by open field test and bouts into the center of the field) were significantly increased in antagomir-treated animals compared to controls, hyperactivity is a consequence of TBI (Pandey et al. 2009). Furthermore, increased activity and exploration of the center of an open field are used as correlates of non-depressive/anxious behavior in other contexts and are therefore difficult to interpret in this study because other measures of anxious behavior in the present study (elevated plus maze) were not significantly different.
Although we identified a putative therapeutic target from decreased activity and excitotoxicity, we did not explore individual targets of miR-181a. We identified 490 potential miR-181a targets in silico that pass rigorous parameters linked to biological relevance and experimental data (Supplementary Table 1). The regulatory effects of miR-181a are cell type specific (Rippo et al. 2014) and result in myriad cellular changes through its interactions with downstream transcription factors (Kazenwadel et al. 2010) and cytokines (Bhushan et al. 2013; Zhao et al. 2012). The extent of miR-181a’s cognitive effects in the adult brain has not been fully elucidated however, increased expression plays an important role in memory formation (Huang et al. 2015; Zhang et al. 2017), especially those related to fear (Xu et al. 2018) or reward (Chandrasekar and Dreyer 2009).
Another limitation of the present study is that miR-181a antagomir was only administered as pre-treatment, which is useful for elucidating cellular responses, but has little clinical relevance for most mTBI cases. MiR-181a has been shown to be dynamically regulated following TBI, with expression falling shortly after injury but elevating after 24 h (Redell et al. 2009), providing a potential post-injury treatment window to sustain miR-181a suppression. Concordantly, our prior studies in ischemic brain injury models have demonstrated treatment with miR-181a antagomir is an effective treatment when administered both pre-(Ouyang et al. 2012) and post-injury (Xu et al. 2015). Future studies will evaluate the effect of post-injury treatment after mTBI and also assess the effect on synaptic connectivity via electrophysiological studies.
Supplementary Material
Acknowledgments
Funding This study was funded in part by American Heart Association Grants FTF-19970029 to CMS and #18POST33990395 to BBG, the Finnish Cultural Foundation grant #00171200 to OA, NIH #5R01NS053898 to RG, and VA Merit Review Grant #RX001776-01 to DJC.
Footnotes
Conflict of interest The authors declare no competing financial interests.
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s12017-019-08532-y) contains supplementary material, which is available to authorized users
References
- Amaral DG, Scharfman HE, & Lavenex P. (2007). The dentate gyrus: Fundamental neuroanatomical organization (dentate gyrus for dummies). Progress in brain research, 163, 3–22. 10.1016/S0079-6123(07)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral DG, & Witter MP (1989). The three-dimensional organization of the hippocampal formation: A review of anatomical data. Neuroscience, 31(3), 571–591. 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- Aungst SL, Kabadi SV, Thompson SM, Stoica BA, & Faden AI (2014). Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. Journal of Cerebral Blood Flow & Metabolism, 34(7), 1223–1232. 10.1038/jcbfm.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan R, Grünhagen J, Becker J, Robinson PN, Ott C-E, & Knaus P (2013). miR-181a promotes osteoblastic differentiation through repression of TGF-β signaling molecules. The International Journal of Biochemistry & Cell Biology, 45(3), 696–705. 10.1016/j.biocel.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Boone DK, Weisz HA, Bi M, Falduto MT, Torres KEO, Willey HE, et al. (2017). Evidence linking microRNA suppression of essential prosurvival genes with hippocampal cell death after traumatic brain injury. Scientific Reports, 7(1), 6645 10.1038/s41598-017-06341-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbent NJ, Gaskin S, Squire LR, & Clark RE (2010). Object recognition memory and the rodent hippocampus. Learning & Memory, 17(1), 5–11. 10.1101/lm.1650110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabungcal J-H, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK, & Do KQ (2013). Perineuronal nets protect fastspiking interneurons against oxidative stress. Proceedings of the National Academy of Sciences, 110(22), 9130–9135. 10.1073/pnas.1300454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekar V, & Dreyer J-L (2009). microRNAs miR-124, let-7d and miR-181a regulate Cocaine-induced Plasticity. Molecular and Cellular Neuroscience, 42(4), 350–362. 10.1016/j.mcn.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Chandrasekar V, & Dreyer J-L (2011). Regulation of MiR-124, Let-7d, and MiR-181a in the accumbens affects the expression, extinction, and reinstatement of cocaine-induced conditioned place preference. Neuropsychopharmacology, 36(6), 1149–1164. 10.1038/npp.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, & Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. Journal of Neuroscience Methods, 53(1), 55–63. 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Czigner A, Mihály A, Farkas O, Büki A, Krisztin-Péva B, Dobó E, & Barzó P (2004). Dynamics and regional distribution of c-fos protein expression in rat brain after a closed head injury. International Journal of Molecular Medicine, 14(2), 247–252. [PubMed] [Google Scholar]
- Datta Chaudhuri A, Yelamanchili SV, & Fox HS (2013). Combined fluorescent in situ hybridization for detection of micro-RNAs and immunofluorescent labeling for cell-type markers. Frontiers in Cellular Neuroscience. 10.3389/fncel.2013.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Planell-Saguer M, Rodicio MC, & Mourelatos Z (2010). Rapid in situ codetection of noncoding RNAs and proteins in cells and formalin-fixed paraffin-embedded tissue sections without protease treatment. Nature Protocols, 5(6), 1061–1073. 10.1038/nprot.2010.62. [DOI] [PubMed] [Google Scholar]
- Faden AI, O’Leary DM, Fan L, Bao W, Mullins PGM, & Movsesyan VA (2001). Selective blockade of the mGluR1 receptor reduces traumatic neuronal injury in vitro and improves outcome after brain trauma. Experimental Neurology, 167(2), 435–444. 10.1006/exnr.2000.7577. [DOI] [PubMed] [Google Scholar]
- Fasulo L, Brandi R, Arisi I, La Regina F, Berretta N, Capsoni S, et al. (2017). ProNGF drives localized and cell selective parvalbumin interneuron and perineuronal net depletion in the dentate gyrus of transgenic mice. Frontiers in Molecular Neuroscience, 10, 20 10.3389/fnmol.2017.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, & Yaffe K (2015). Epidemiology of mild traumatic brain injury and neurodegenerative disease. Molecular and Cellular Neuroscience, 66, 75–80. 10.1016/j.mcn.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes RL, Lyeth BG, Jenkins LW, Zimmerman R, McIntosh TK, Clifton GL, & Young HF (2009). Possible protective effect of endogenous opioids in traumatic brain injury. Collections, 116(6), 252–261. 10.3171/jns.1990.72.2.0252@col.2012.116.issue-6. [DOI] [PubMed] [Google Scholar]
- Hegarty SV, Sullivan AM, & O’Keeffe GW (2018). Inhibition of miR-181a promotes midbrain neuronal growth through a Smad1/5-dependent mechanism: Implications for Parkinson’s disease. Neuronal Signaling, 2(1), NS20170181 10.1042/NS20170181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks R. r., Smith D. h., Lowenstein D. h., Marie RS, & McINTOSH T.k (1993). Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. Journal of Neurotrauma, 10(4), 405–414. 10.1089/neu.1993.10.405. [DOI] [PubMed] [Google Scholar]
- Hsieh T-H, Lee HHC, Hameed MQ, Pascual-Leone A, Hensch TK, & Rotenberg A (2017). Trajectory of parvalbumin Cell impairment and loss of cortical inhibition in traumatic brain injury. Cerebral Cortex, 27(12), 5509–5524. 10.1093/cercor/bhw318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Liu X, Liao Y, Luo C, Zou D, Wei X, et al. (2015). MiR-181a influences the cognitive function of epileptic rats induced by pentylenetetrazol. International Journal of Clinical and Experimental Pathology, 8(10), 12861–12868. [PMC free article] [PubMed] [Google Scholar]
- Hughes RN (2004). The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neuroscience & Biobehavioral Reviews, 28(5), 497–505. 10.1016/j.neubiorev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Hunsaker MR, Mooy GG, Swift JS, & Kesner RP (2007). Dissociations of the medial and lateral perforant path projections into dorsal DG, CA3, and CA1 for spatial and nonspatial (visual object) information processing. Behavioral Neuroscience, 121(4), 742–750. 10.1037/0735-7044.121.4.742. [DOI] [PubMed] [Google Scholar]
- Hunsaker MR, Rosenberg JS, & Kesner RP (2008). The role of the dentate gyrus, CA3a,b, and CA3c for detecting spatial and environmental novelty. Hippocampus, 18(10), 1064–1073. 10.1002/hipo.20464. [DOI] [PubMed] [Google Scholar]
- Hylin MJ, Orsi SA, Moore AN, & Dash PK (2013). Disruption of the perineuronal net in the hippocampus or medial prefrontal cortex impairs fear conditioning. Learning & Memory, 20(5), 267–273. 10.1101/lm.030197.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson J, Chugh D, Nilsson P, Wood J, Carlström K, Lindvall O, & Ekdahl CT (2012). Altered synaptic properties during integration of adult-born hippocampal neurons following a seizure insult. PLOS ONE, 7(4), e35557 10.1371/journal.pone.0035557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janis LS, Hoane MR, Conde D, Fulop Z, & Stein DG (1998). Acute ethanol administration reduces the cognitive deficits associated with traumatic brain injury in rats. Journal of Neurotrauma, 15(2), 105–115. 10.1089/neu.1998.15.105. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Clark RE, Broadbent NJ, Clemenson GD, Consiglio A, Lie DC, et al. (2009). Dentate gyrus-specific knockdown of adult neurogenesis impairs spatial and object recognition memory in adult rats. Learning & Memory, 16(2), 147–154. 10.1101/lm.1172609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen FF, Tønder N, Zimmer J, Baimbridge KG, & Diemer NH (1990). Short-term changes of parvalbumin and calbindin immunoreactivity in the rat hippocampus following cerebral ischemia. Neuroscience Letters, 120(2), 171–174. 10.1016/0304-3940(90)90030-D. [DOI] [PubMed] [Google Scholar]
- Kazenwadel J, Michael MZ, & Harvey NL (2010). Prox1 expression is negatively regulated by miR-181 in endothelial cells. Blood, 116(13), 2395–2401. 10.1182/blood-2009-12-256297. [DOI] [PubMed] [Google Scholar]
- Keith DJ, & El-Husseini A (2008). Excitation control: Balancing PSD-95 function at the synapse. Frontiers in Molecular Neuroscience. 10.3389/neuro.02.004.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-G, Somogyi P, Ylinen A, & Buzsáki G (1994). The hippocampal CA3 network: An in vivo intracellular labeling study. Journal of Comparative Neurology, 339(2), 181–208. 10.1002/cne.903390204. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, & McIntosh TK (1992). Selective vulnerability of dentate hilar neurons following traumatic brain injury: A potential mechanistic link between head trauma and disorders of the hippocampus. Journal of Neuroscience, 12(12), 4846–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Nguyen A, Villeda S, Zhang H, Ding Z, Lindsey D, et al. (2014). Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Frontiers in Neurology. 10.3389/fneur.2014.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mtchedlishvili Z, Lepsveridze E, Xu H, Kharlamov EA, Lu B, & Kelly KM (2010). Increase of GABAA receptor-mediated tonic inhibition in dentate granule cells after traumatic brain injury. Neurobiology of Disease, 38(3), 464–475. 10.1016/j.nbd.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Ouyang Y-B, Lu Y, Yue S, & Giffard RG (2012). miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion, 12(2), 213–219. 10.1016/j.mito.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AM, Marion DW, Botscheller ML, Swedlow PE, Styren SD, & DeKosky ST (1993). Traumatic brain injuryinduced excitotoxicity assessed in a controlled cortical impact model. Journal of Neurochemistry, 61(6), 2015–2024. 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- Pandey DK, Yadav SK, Mahesh R, & Rajkumar R (2009). Depression-like and anxiety-like behavioural aftermaths of impact accelerated traumatic brain injury in rats: A model of comorbid depression and anxiety? Behavioural Brain Research, 205(2), 436–442. 10.1016/j.bbr.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Phillips L. l., & Belardo E. t. (1992). Expression of c-fos in the hippocampus following mild and moderate fluid percussion brain injury. Journal of Neurotrauma, 9(4), 323–333. 10.1089/neu.1992.9.323. [DOI] [PubMed] [Google Scholar]
- Raghupathi R, & McIntosh TK (1996). Regionally and temporally distinct patterns of induction of c-fos, c-jun and junB mRNAs following experimental brain injury in the rat. Molecular Brain Research, 37(1), 134–144. 10.1016/0169-328X(95)00289-5. [DOI] [PubMed] [Google Scholar]
- Redell JB, Liu Y, & Dash PK (2009). Traumatic brain injury alters expression of hippocampal microRNAs: Potential regulators of multiple pathophysiological processes. Journal of Neuroscience Research, 87(6), 1435–1448. 10.1002/jnr.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves TM, Lyeth BG, Phillips LL, Hamm RJ, & Povlishock JT (1997). The effects of traumatic brain injury on inhibition in the hippocampus and dentate gyrus. Brain Research, 757(1), 119–132. 10.1016/S0006-8993(97)00170-4. [DOI] [PubMed] [Google Scholar]
- Ren L, Zhu R, & Li X (2016). Silencing miR-181a produces neuroprotection against hippocampus neuron cell apoptosis poststatus epilepticus in a rat model and in children with temporal lobe epilepsy. Genetics and molecular research: GMR. 10.4238/gmr.15017798. [DOI] [PubMed] [Google Scholar]
- Rippo MR, Olivieri F, Monsurrò V, Prattichizzo F, Albertini MC, & Procopio AD (2014). MitomiRs in human inflamm-aging: A hypothesis involving miR-181a, miR-34a and miR-146a. Experimental Gerontology, 56, 154–163. 10.1016/j.exger.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Sahbaie P, Sun Y, Liang D-Y, Shi X-Y, & Clark JD (2014). Curcumin treatment attenuates pain and enhances functional recovery after incision. Anesthesia & Analgesia, 118(6), 1336–1344. 10.1213/ANE.0000000000000189. [DOI] [PubMed] [Google Scholar]
- Sahbaie P, Tajerian M, Yang P, Irvine KA, Huang T-T, Luo J, et al. (2018). Nociceptive and cognitive changes in a murine model of polytrauma. The Journal of Pain. 10.1016/j.jpain.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stary CM, Xu L, Li L, Sun X, Ouyang Y-B, Xiong X, et al. (2017). Inhibition of miR-181a protects female mice from transient focal cerebral ischemia by targeting astrocyte estrogen receptor-α. Molecular and Cellular Neurosciences, 82, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajerian M, Leu D, Zou Y, Sahbaie P, Li W, Khan H, et al. (2014). Brain neuroplastic changes accompany anxiety and memory deficits in a model of complex regional pain syndrome. Anesthesiology : The Journal of the American Society of Anesthesiologists, 121(4), 852–865. 10.1097/ALN.0000000000000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajerian M, Sahbaie P, Sun Y, Leu D, Yang HY, Li W, et al. (2015). Sex differences in a murine model of complex regional pain syndrome. Neurobiology of Learning and Memory, 123, 100–109. 10.1016/j.nlm.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y-P, Noda Y, Hasegawa T, & Nabeshima T (1997). A Concussive-like brain injury model in mice (II): Selective neuronal loss in the cortex and hippocampus. Journal of Neurotrauma, 14(11), 863–873. 10.1089/neu.1997.14.863. [DOI] [PubMed] [Google Scholar]
- Taylor AN, Tio DL, & Sutton RL (2013). Restoration of Neuroendocrine stress response by glucocorticoid receptor or GABAA receptor antagonists after experimental traumatic brain injury. Journal of Neurotrauma, 30(14), 1250–1256. 10.1089/neu.2012.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson EH, Lensjø KK, Wigestrand MB, Malthe-Sørenssen A, Hafting T, & Fyhn M (2018). Removal of perineuronal nets disrupts recall of a remote fear memory. Proceedings of the National Academy of Sciences, 115(3), 607–612. 10.1073/pnas.1713530115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Türeci E, Dashti R, Tanriverdi T, Sanus GZ, Öz B, & Uzan M (2004). Acute ethanol intoxication in a model of traumatic brain injury: The protective role of moderate doses demonstrated by immunoreactivity of synaptophysin in hippocampal neurons. Neurological Research, 26(1), 108–112. 10.1179/016164104773026633. [DOI] [PubMed] [Google Scholar]
- Wang T, Chou DY-T, Ding JY, Fredrickson V, Peng C, Schafer S, et al. (2012). Reduction of brain edema and expression of aquaporins with acute ethanol treatment after traumatic brain injury. Journal of Neurosurgery, 118(2), 390–396. 10.3171/2012.8.JNS12736. [DOI] [PubMed] [Google Scholar]
- Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, & Giffard RG (2011). Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke, 42(7), 2026–2032. 10.1161/strokeaha.110.593772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L-J, Ouyang Y-B, Xiong X, Stary CM, & Giffard RG (2015). Post-stroke treatment with miR-181 antagomir reduces injury and improves long-term behavioral recovery in mice after focal cerebral ischemia. Exp Neurol, 264, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X-F, Jing X, Ma H-X, Yuan R-R, Dong Q, Dong J-L, et al. (2018). miR-181a participates in contextual fear memory formation Via activating mTOR signaling pathway. Cerebral Cortex, 28(9), 3309–3321. 10.1093/cercor/bhx201. [DOI] [PubMed] [Google Scholar]
- Yang K, Mu XS, Xue JJ, Whitson J, Salminen A, Dixon CE, et al. (1994). Increased expression of c-fos mRNA and AP-1 transcription factors after cortical impact injury in rats. Brain Research, 664(1), 141–147. 10.1016/0006-8993(94)91964-X. [DOI] [PubMed] [Google Scholar]
- Zhang S, Chen J, Zhang J, & Xu J (2017). miR-181a involves in the hippocampus-dependent memory formation via targeting PRKAA1. Scientific Reports, 7(1), 8480 10.1038/s41598-017-09095-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X-D, Wang H, Antaris AL, Li L, Diao S, Ma R, et al. (2016). Traumatic brain injury imaging in the second near-infrared window with a molecular fluorophore. Advanced Materials, 28(32), 6872–6879. 10.1002/adma.201600706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZJ, & Reynolds GP (2002). A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophrenia Research, 55(1), 1–10. 10.1016/S0920-9964(01)00188-8. [DOI] [PubMed] [Google Scholar]
- Zhao J, Gong A-Y, Zhou R, Liu J, Eischeid AN, & Chen X-M (2012). Downregulation of PCAF by miR-181a/b provides feedback regulation to TNF-α–induced transcription of proinflammatory genes in liver epithelial cells. The Journal of Immunology, 188(3), 1266–1274. 10.4049/jimmunol.1101976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Corniola R, Leu D, Khan A, Sahbaie P, Chakraborti A, et al. (2012). Extracellular superoxide dismutase is important for hippocampal neurogenesis and preservation of cognitive functions after irradiation. Proceedings of the National Academy of Sciences, 109(52), 21522–21527. 10.1073/pnas.1216913110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.