

INTRODUCTION
IGH–cytokine receptor–like factor 2 (CRLF2) rearrangement is the most common mechanism of overexpression of CRLF2 in precursor B-cell acute lymphoblastic leukemia (B-ALL); however, P2RY8-CRLF2 rearrangement or alternative alterations leading to CRLF2 overexpression have also been observed, which are associated with increased relapse rate and progression.1 Although many myeloid neoplasms, particularly acute myeloid leukemia (AML), are associated with various recurrent genetic rearrangements and fusions,2 alterations leading to CRLF2 overexpression are exceedingly rare in AML. To date, no patient with myeloid neoplasm with CRLF2 translocation has been reported. One patient with myelodysplastic syndrome with excess blasts-1 (MDS-EB1) with CRLF2 overexpression via multiple copies of an isodicentric Y chromosome, without evidence of CRLF2 rearrangement, has been reported.3 To our knowledge, we report the first patient with AML arising from an MDS, with a P2RY8-CRLF2 fusion, which responded to a myeloid regimen in a clinical trial of hypomethylating agent and immune checkpoint blockade combination therapy. The study was performed in accordance with the Helsinki declaration. Patient consent has been obtained, and the study has been approved by the Memorial Sloan Kettering Cancer Center (MSKCC) Institutional Review Board under protocol 12-245.
CASE REPORT
A 78-year-old man had an initial diagnosis of MDS-EB1 for 1 year, with bone marrow showing 8% blasts by aspirate differential count and 5%-9% blasts by CD34 immunostain (Appendix Fig A1A). He was monitored without any treatment or transfusions before presenting to MSKCC with worsening pancytopenia, including severe neutropenia. Bone marrow examination showed approximately 40% blasts that were medium in size with a rim of vacuolated cytoplasm, dispersed chromatin, and inconspicuous nucleoli (Fig 1A), consistent with progression to AML. Flow cytometry confirmed the presence of an expanded blast population with predominantly abnormal myeloid immunophenotype, but a minute subset of blasts dimly expressed CD19 (Fig 1B). Despite this, the patient is best classified as having AML with myelodysplasia-related changes (MRC) based on the 2016 revision to the WHO classification of myeloid neoplasms and acute leukemias.2
FIG 1.

Acute myeloid leukemia (AML) with myelodysplasia-related changes with P2RY8–cytokine receptor–like factor 2 (CRLF2) fusion. (A) Photomicrographs of the bone marrow biopsy showing AML. (B) Flow cytometry of the bone marrow aspirate showing typical AML markers. (C) Anchored multiplex polymerase chain reaction (Archer FusionPlex) reads showing an in-frame fusion between P2RY8 and CRLF2. (D) Break-apart fluorescence in situ hybridization probes for CRLF2 (top) and P2YR8 (bottom) consistent with interstitial deletion between the 2 genes resulting in fusion.
COMPREHENSIVE GENOMIC/TRANSCRIPTOMIC PROFILING
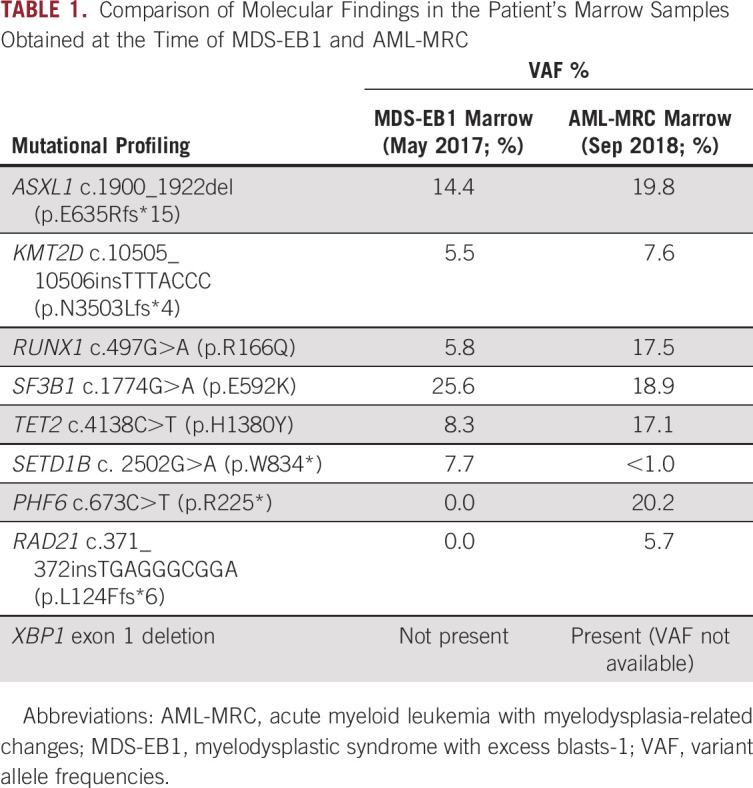
Fluorescence in situ hybridization (FISH) analysis performed on the marrow at the time of the initial diagnosis of MDS-EB1 was negative for recurring MDS-related chromosome abnormalities, including deletion or loss of chromosomes 5, 7, 13, 17, and 20; gain of chromosome 8; and KMT2A (MLL) translocations. At MSKCC, karyotype analysis of the AML bone marrow aspirate specimen showed a normal male karyotype, and FISH analysis was negative for the presence of KMT2A (MLL), EVI1, and CBFB translocations, and TP53 deletion. A targeted next-generation sequencing (NSG) panel (MSK-IMPACT heme panel; MSKCC, New York, NY) was performed on the bone marrow specimen obtained at the time of the AML diagnoses, which showed the alterations listed in Table 1, including mutations in ASXL1, KMT2D, RUNX1, SF3B1, TET2, PHF6, RAD21, and XBP1. Evaluation of this bone marrow sample using an anchored multiplex polymerase chain reaction–based, targeted RNA sequencing assay for detection of fusions (Archer FusionPlex, ArcherDx, Boulder, CO) revealed the presence of a fusion between exon 1 of P2RY8 and exon 1 of CRLF2 (Fig 1C). This fusion was confirmed in 22% of cells using FISH break-apart probes for CRLF2 and P2RY8 (Xp22.33/Yp11.32; Cytocell, Tarrytown, NY; Fig 1D). The signal pattern was consistent with an interstitial deletion between the P2RY8 and CRLF2 genes within the pseudoautosomal region 1 (PAR1).
TABLE 1.
Comparison of Molecular Findings in the Patient's Marrow Samples Obtained at the Time of MDS-EB1 and AML-MRC
To ensure that the P2RY8-CRLF2 rearrangement observed was present in the myeloid blasts rather than cells of other lineages, bone marrow mononuclear cells were separated by fluorescence-activated cell sorting to isolate the leukemic blasts, myelomonocytic cells, and normal lymphocytes. FISH using the CRLF2 break-apart probe revealed the CRLF2 rearrangement in a subset of the myeloid blasts (27.5%) and myelomonocytic cells (19.5%) but not in B cells (Fig 2).
FIG 2.

Confirmation of P2RY8–cytokine receptor–like factor 2 (CRLF2) fusion in myeloid lineage. The bone marrow aspirate sample was sorted by fluorescence-activated cell sorting into the myeloid blast population, myelomonocytic cells, and B lymphocytes. Break-apart fusion probes were applied to the sorted populations showing restriction of the P2RY8-CRLF2 fusion to the myeloid lineage, including the blasts and myelomonocytes.
CONTEXT
Key Objective
Is there utility to fusion discovery of known fusions in novel disease contexts?
Knowledge Generated
P2RY8–cytokine receptor–like factor 2 (CRLF2) fusions have been observed in B-acute lymphoblastic leukemia but we report, to our knowledge, the first detection of this fusion in acute myeloid leukemia arising from myelodysplastic syndrome. Cell sorting, molecular characterization, and immunostaining characterized the myeloid origin of this fusion and subsequent signaling effects.
Relevance
This patient was treated with the novel combination of decitabine and ipilimumab with an outstanding response. Immune cell infiltrates in the bone marrow with treatment suggests the response was related to the immune checkpoint inhibition. Additional study of P2RY8-CRLF2 fusions and response to immunotherapy is warranted and could have implications for fusion discovery in novel disease contexts.
FISH evaluation of the prior bone marrow biopsy with MDS-EB1 was also performed, which showed the presence of the P2RY8-CRLF2 rearrangement in 10% of the cells (Appendix Fig A1C). Archer FusionPlex of the prior marrow with MDS-EB1 also identified the same fusion between exon 1 of P2RY8 and exon 1 of CRLF2 (Appendix Fig A1B). OncoScan array (ThermoFisher, Waltham, MA) of the MDS-EB1 biopsy did not reveal any unbalanced genomic changes or copy-neutral loss of heterozygosity (data not shown), consistent with the normal FISH study observed by the outside institution a year prior. Targeted NGS sequencing (MSK-IMPACT heme panel) of the MDS-EB1 bone marrow specimen also showed overlapping mutations with the AML bone marrow sample (Table 1), but with acquisition of new mutations (PHF6, RAD21, XBP1) in the AML sample, suggesting a myeloid primed hematopoietic stem cell/progenitor origin and clonal evolution with expansion of the clone with the P2RY8-CRLF2 rearrangement, likely due to acquisition of a loss-of-function mutation in PHF6, which led to downregulation of genes involved in normal B-cell development.4
Total RNA sequencing of the AML bone marrow specimen further confirmed the presence of the P2RY8-CRLF2 fusion along with overexpression of CRLF2 (Fig 3A). Principal component analysis comparing our patient with cohorts of AML or B-ALL from the TARGET study5 showed that the patient’s neoplasm clustered with other patients with AML, confirming the myeloid differentiation of the blasts (Fig 3B). Gene ontology analysis showed transcriptome-wide increases in cytokine-related signaling pathway genes potentially related to the P2RY8-CRLF2 fusion, as well as genes involved in regulating hematopoietic progenitor cell differentiation and the spliceosome (Fig 3C).
FIG 3.
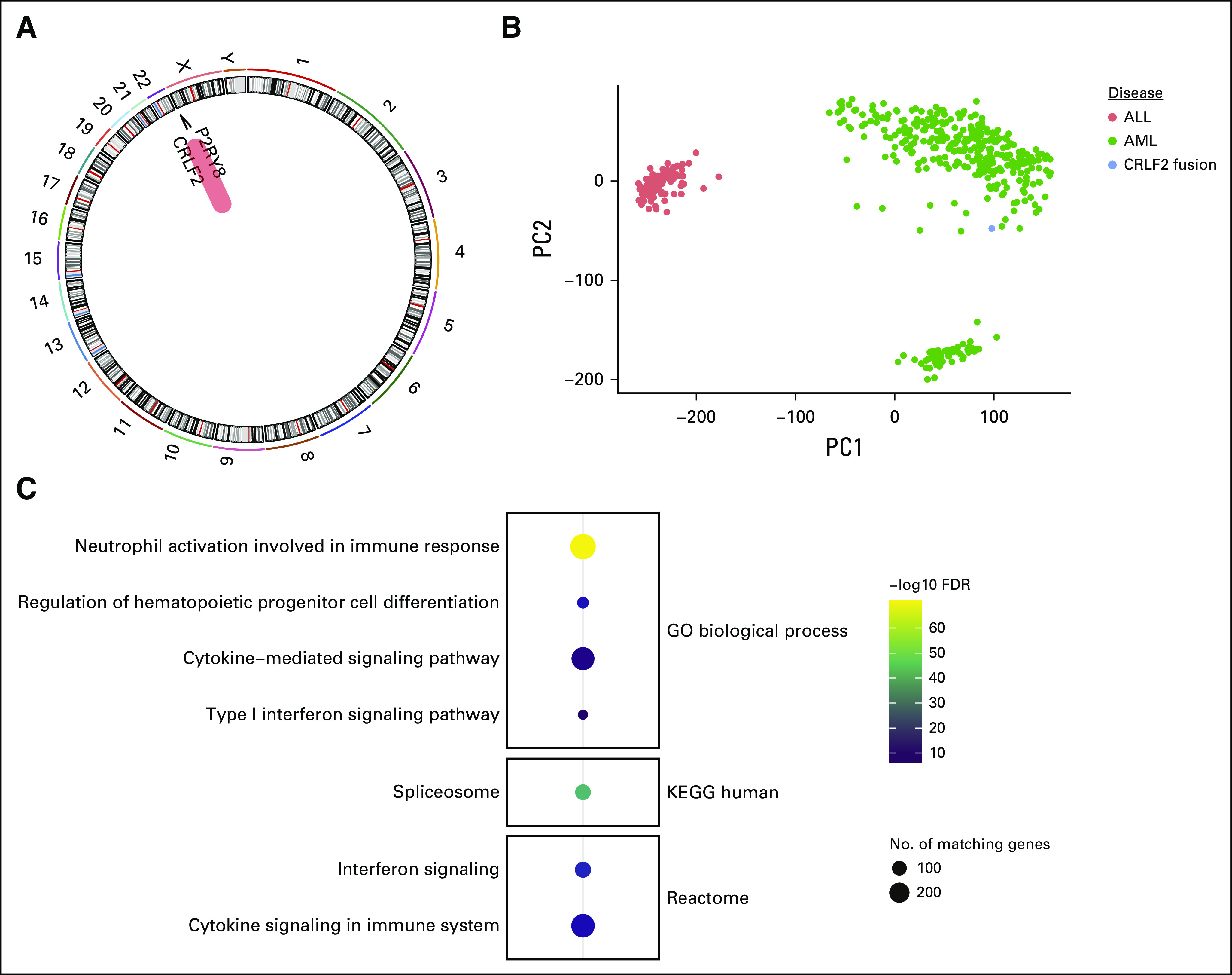
Transcriptomic analysis of P2RY8–cytokine receptor–like factor 2 (CRLF2) fusion AML. (A) Fusion analysis performed from polyA selected mRNA next-generation RNA sequencing. (B) Principal component analysis plot showing 2 strongest principal components (PC1 on x-axis and PC2 on y-axis) from publicly available data from AML (green dots) and B-ALL (red dots) patient samples compared with our patient with P2RY8-CRLF2 fusion (blue dot). (C) Gene ontology analysis of the most highly expressed genes showing enrichment in hematopoietic differentiation, spliceosome, and immune signaling gene pathways. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Because it is known that STAT5 is activated by P2RY8-CRLF2 fusion in B-ALL, we performed immunohistochemistry (IHC) using phosphorylation-specific antibodies that showed increased STAT5 phosphorylation and partial ERK phosphorylation but not STAT3 phosphorylation (Fig 4A). Gene expression and IHC were largely correlated; however, the results are interpreted in the context of the importance of signaling in leukemia overall, and these are not necessarily specific for P2RY8-CRLF2 fusion.
FIG 4.

Immune signaling activation in P2RY8–cytokine receptor–like factor 2 (CRLF2) fusion AML. (A) Immunohistochemical photomicrographs of the bone marrow biopsy showing phospho-specific staining of STAT5 and ERK, but not STAT3. (B) Immunohistochemical photomicrographs of the bone marrow biopsy showing staining of programmed death (PD)-1 in scattered lymphoid cells. PD-ligand 1 (PD-L1) staining was equivocal, whereas PD-L2 was negative. (C) Using multiplex immunofluorescence microscopy, with digital image analysis, we found that the patient’s bone marrow was highly enriched with CD8 plus granzyme B (GZMB) plus T cells. CD34+ staining highlights the myeloblast population at the time of AML transformation. We further identified a population of CD3+CD4+ T cells at baseline.
TREATMENT AND FOLLOW-UP
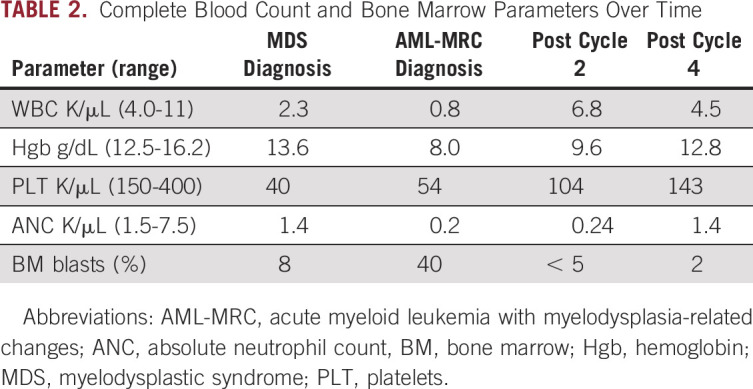
Given the patient’s age and transformation to AML, a clinical trial was recommended. He was consented and enrolled in a study regimen consisting of standard-dose decitabine plus investigational administration of the cytotoxic T-lymphocyte–associated protein-4 (CTLA-4) antagonist, ipilimumab (ClinicalTrials.gov identifier: NCT02890329). After 2 cycles of combination therapy, he achieved complete remission with incomplete count recovery and shortly thereafter a full complete remission (Table 2) with negative measurable residual disease (MRD) status by a flow cytometric assay with a detection limit of 1 in 2,500 cells. This was accompanied by a robust molecular response with undetectable levels of the previously detected PHF6, RUNX1, TET2, and RAD21 mutations, and a marked reduction in the variant allele frequencies for the mutations ASXL1 (0.4%) and SF3B1 (1.4%), likely representing MRD below the detection limit of flow cytometry. The patient continues to be in morphologic remission and in cycle 11 of investigational therapy at the time of this report. IHC was performed on the checkpoint molecules including programmed death (PD)-1, PD-ligand 1 (PD-L1) and PD-L2 (Fig 4B). The PD-1 stain was positive in scattered T cells. Although the PD-L1 stain showed equivocal results, gene expression by RNA sequencing was at the 75th percentile compared with other profiled AML. Multiplexed immunofluorescence with a panel of antibodies against CD34, CD3, CD4, CD8, and granzyme B (GZMB), as previously described,6 was applied to the patient’s pretreatment bone marrow, which revealed the presence of a dense tumor immune infiltrate that was CD8+ GZMB+, suggesting his marrow was enriched with activated CD8 T cells critical for response to checkpoint blockade (Fig 4C).
TABLE 2.
Complete Blood Count and Bone Marrow Parameters Over Time
DISCUSSION
CRLF2 is the protein product of a synonymous gene located in the PAR1 on the short arm of the X chromosome and the Y chromosome. It forms a heterodimer with interleukin 7 receptor alpha and is involved in lymphoid signaling pathways. Activation of this receptor leads to activation of Janus family tyrosine kinases and STAT5. Deregulated expression of this gene has been described as the characteristic alteration in Philadelphia chromosome–like B-ALL in adolescent and adult patients.7,8 Russell et al9 showed that this subgroup of B-ALL has either a translocation juxtaposing the CRLF2 to the IGH gene on chromosome 14 (t(X;14)(p22;q32) or t(Y;14)(p11;q32)) or a deletion of PAR1 region, resulting in overexpression of the CRLF2 gene. Mullighan et al10 reported that the interstitial deletion of PAR1 leads to juxtaposition of CRLF2 to the first, noncoding exon of P2RY8. This event was seen in up to 7% of childhood B-ALLs and in up to half of patients with Down syndrome presenting with ALL.
Dysregulated expression of CRLF2 has been shown to occur as both early events (founder or truncal alteration), mostly seen as IGH-CRLF2 fusion, and late events, often observed as P2RY8-CRLF2 fusion. This has been observed in Down syndrome–related ALL as well as non–Down syndrome ALL, supporting the cooperation of CRLF2 events with other genetic alterations in progression of disease.11 However, irrespective of the clone size, presence of this rearrangement is associated with a higher relapse rate.12 There have been suggestions that the increased relapse rate may be due to other concurrent alterations, such as IKZF1 deletion.13 However, other studies have shown the rearrangement as well as the CRLF2 overexpression to be independent predictors of worse outcome in B-ALL.14,15
Until now, only one patient with myeloid neoplasm with CRLF2 overexpression has been reported. However, this patient was associated with multiple copies of an isodicentric Y chromosome and not associated with P2RY8-CRLF2 rearrangement.3 To our knowledge, our patient represents the first P2RY8-CRLF2 rearrangement in myeloid neoplasms, which in our patient was an AML arising from the underlying MDS. Although this rearrangement was previously thought to originate from B lymphocytes because it was only observed in B-ALL, this was not the case for our patient. The flow-sorted FISH analysis has shown that the P2RY8-CRLF2 was not present in the B lymphocytes of this patient but only in the myeloid-lineage cells (myeloid blasts and myelomonocytic cells). In addition, based on the results from total RNA sequencing, our patient’s neoplasm clustered with other patients with AML, providing additional evidence for the myeloid differentiation of the blasts. Furthermore, the alterations observed in this patient, including the P2RY8-CRLF2 rearrangement, were present at the MDS stage, suggesting a stem cell origin with selective pressure leading to development of AML.
PHF6 mutations are rare in de novo AML but relatively common in AML-MRC and exceedingly rare in B-ALL.16,17 PHF6 and DNMT3A are the most common mutations in mixed phenotype acute leukemias (MPAL) with T-lineage differentiation.18 PHF6 is a critical component in lineage determination of precursor cells, especially between T and B lineages, and it is believed that a functioning PHF6 is required for B-cell differentiation leading to selective abundance of this mutation in T-ALL and MPAL with T-lineage differentiation.18,19 This notion is further supported by the observation that PHF6 suppression leads to impaired tumor progression in B-ALL.4 It has also been shown that PAX5-deficient pro-B cells can undergo myeloid-lineage differentiation in the presence of transcription factors, such as GATA or CEBPA.20 In our patient, the immunophenotype and RNA expression data unequivocally point toward myeloid differentiation. Although it is difficult to ascertain the functional consequences of the P2RY8-CRLF2 rearrangement and the acquired PHF6 mutation at the time of AML transformation, we hypothesize that the two may interact in a way that led to the development of AML instead of B-ALL.
Remarkably, this patient with AML-MRC and PHF6, RUNX1, TET2, and RAD21 mutations had an MRD-negative complete response by flow cytometry after just 4 cycles of combination decitabine with ipilimumab. Although the complete results from clinical trials of these agents are needed to evaluate the efficacy and safety of this combination, this patient’s exceptional response could suggest the unique molecular characteristics of his tumor may have been important, namely, the P2RY8-CRLF2 fusion. Recent evidence in head and neck cancers with oncogenic chromosomal rearrangements suggests that gene fusions may be a source of immunogenic neoantigens and stimulate T-cell responses that could be unleashed after immune checkpoint blockade.21 This patient’s T cells were positive for PD-1, suggesting a relation to the response to CTLA-4 blockade. Additional research is needed to define whether this mechanism of action could be at play with other fusions or in the case of subclonal alterations, such as in this patient.
ACKNOWLEDGMENT
We acknowledge the members of the Memorial Sloan Kettering Cancer Center diagnostic molecular laboratory, cytogenetics laboratory, flow cytometry laboratory, and immunohistochemical stain laboratory for their support in performing the relevant assays.
Appendix
FIG A1.

Presence of the P2RY8-CRLF2 fusion in earlier bone marrow biopsy with myelodysplastic syndrome (MDS). (A) Photomicrographs of the bone marrow biopsy showing MDS with excess blast-1 (MDS-EB1). (B) Anchored multiplex polymerase chain reaction (Archer FusionPlex) reads showing an in-frame fusion between P2RY8 and CRLF2. (C) Breakapart fluorescence in situ hybridization probes for CRLF2 (top) and P2YR8 (bottom) consistent with interstitial deletion between the 2 genes, resulting in fusion.
Footnotes
Supported by the Comprehensive Cancer Center Core Grant (P30 CA008748) at Memorial Sloan Kettering Cancer Center from the National Institutes of Health.
AUTHOR CONTRIBUTIONS
Conception and design: Umut Aypar, Justin Taylor, Jacqueline S. Garcia, Wenbin Xiao, Caleb Ho
Administrative support: Jeeyeon Baik, Allison Sigler
Collection and assembly of data: Umut Aypar, Justin Taylor, Jacqueline S. Garcia, Amir Momeni-Boroujeni, Qi Gao, Dory Londono, Ryma Benayed, Michael Haddadin, Alexander V. Penson, Maria E. Arcila, Kerry Mullaney, Purvil Sukhadia, Andres E. Quesada, Mikhail Roshal, Nicole Cullen, Ana Lako, Scott J. Rodig, Aaron D. Goldberg, Yanming Zhang, Wenbin Xiao, Caleb Ho
Data analysis and interpretation: Umut Aypar, Justin Taylor, Jacqueline S. Garcia, Amir Momeni-Boroujeni, Alexander V. Penson, Scott J. Rodig, Yanming Zhang, Wenbin Xiao, Caleb Ho
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Jacqueline S. Garcia
Consulting or Advisory Role: AbbVie
Research Funding: AbbVie (Inst), Pfizer (Inst), Genentech (Inst), Eli Lilly (Inst)
Maria E. Arcila
Honoraria: Invivoscribe, Biocartis
Consulting or Advisory Role: AstraZeneca
Travel, Accommodations, Expenses: AstraZeneca, Invivoscribe, Raindance Technologies
Mikhail Roshal
Honoraria: Celgene
Consulting or Advisory Role: Agios, Auron
Research Funding: Agios, Roche, Boehringer Ingelheim
Ana Lako
Employment: Bristol-Myers Squibb
Scott J. Rodig
Honoraria: Perkin Elmer, Bristol-Myers Squibb
Consulting or Advisory Role: Bristol-Myers Squibb
Research Funding: Bristol-Myers Squibb, Merck, Affimed Therapeutics, Kite Pharma
Patents, Royalties, Other Intellectual Property: Patent pending for use of anti–galectin-1 antibodies for diagnostic use
Travel, Accommodations, Expenses: Roche, Bristol-Myers Squibb
Aaron D. Goldberg
Honoraria: Dava Oncology
Consulting or Advisory Role: AbbVie, Celgene, Daiichi Sankyo
Research Funding: AbbVie (Inst), ADC Therapeutics (Inst), Pfizer (Inst), Arog (Inst)
Travel, Accommodations, Expenses: Arog
Open Payments Link: https://openpaymentsdata.cms.gov/physician/1195852/summary
Wenbin Xiao
Research Funding: Stemline Therapeutics (Inst)
Caleb Ho
Honoraria: Invivoscribe
Travel, Accommodations, Expenses: Invivoscribe
No other potential conflicts of interest were reported.
REFERENCES
- 1.Schmäh J, Fedders B, Panzer-Grümayer R, et al. Molecular characterization of acute lymphoblastic leukemia with high CRLF2 gene expression in childhood. Pediatr Blood Cancer. 2017;64:e26539. doi: 10.1002/pbc.26539. [DOI] [PubMed] [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 3.Mangaonkar AA, Patnaik MM, Oliver GR, et al. Multiple isodicentric Y chromosomes in myeloid malignancies: A unique cytogenetic entity and potential therapeutic target. Leuk Lymphoma. 2019;60:821–824. doi: 10.1080/10428194.2018.1498492. [DOI] [PubMed] [Google Scholar]
- 4.Meacham CE, Lawton LN, Soto-Feliciano YM, et al. A genome-scale in vivo loss-of-function screen identifies Phf6 as a lineage-specific regulator of leukemia cell growth. Genes Dev. 2015;29:483–488. doi: 10.1101/gad.254151.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371–376. doi: 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel SS, Weirather JL, Lipschitz M, et al. The microenvironmental niche in classic Hodgkin lymphoma is enriched for CTLA-4-positive T cells that are PD-1-negative. Blood. 2019;134:2059–2069. doi: 10.1182/blood.2019002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rochman Y, Leonard WJ. Thymic stromal lymphopoietin: A new cytokine in asthma. Curr Opin Pharmacol. 2008;8:249–254. doi: 10.1016/j.coph.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vesely C, Frech C, Eckert C, et al. Genomic and transcriptional landscape of P2RY8-CRLF2-positive childhood acute lymphoblastic leukemia. Leukemia. 2017;31:1491–1501. doi: 10.1038/leu.2016.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114:2688–2698. doi: 10.1182/blood-2009-03-208397. [DOI] [PubMed] [Google Scholar]
- 10.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41:1243–1246. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Potter N, Jones L, Blair H, et al. Single-cell analysis identifies CRLF2 rearrangements as both early and late events in Down syndrome and non-Down syndrome acute lymphoblastic leukaemia. Leukemia. 2019;33:893–904. doi: 10.1038/s41375-018-0297-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Attarbaschi A, Morak M, Cario G, et al. Treatment outcome of CRLF2-rearranged childhood acute lymphoblastic leukaemia: A comparative analysis of the AIEOP-BFM and UK NCRI-CCLG study groups. Br J Haematol. 2012;158:772–777. doi: 10.1111/j.1365-2141.2012.09221.x. [DOI] [PubMed] [Google Scholar]
- 13.Morak M, Attarbaschi A, Fischer S, et al. Small sizes and indolent evolutionary dynamics challenge the potential role of P2RY8-CRLF2-harboring clones as main relapse-driving force in childhood ALL. Blood. 2012;120:5134–5142. doi: 10.1182/blood-2012-07-443218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dou H, Chen X, Huang Y, et al. Prognostic significance of P2RY8-CRLF2 and CRLF2 overexpression may vary across risk subgroups of childhood B-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2017;56:135–146. doi: 10.1002/gcc.22421. [DOI] [PubMed] [Google Scholar]
- 15.Fang Q, Zhao X, Li Q, et al. IKZF1 alterations and expression of CRLF2 predict prognosis in adult Chinese patients with B-cell precursor acute lymphoblastic leukemia. Leuk Lymphoma. 2017;58:127–137. doi: 10.1080/10428194.2016.1180682. [DOI] [PubMed] [Google Scholar]
- 16.Van Vlierberghe P, Patel J, Abdel-Wahab O, et al. PHF6 mutations in adult acute myeloid leukemia. Leukemia. 2011;25:130–134. doi: 10.1038/leu.2010.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mori T, Nagata Y, Makishima H, et al. Somatic PHF6 mutations in 1760 cases with various myeloid neoplasms. Leukemia. 2016;30:2270–2273. doi: 10.1038/leu.2016.212. [DOI] [PubMed] [Google Scholar]
- 18.Xiao W, Bharadwaj M, Levine M, et al. PHF6 and DNMT3A mutations are enriched in distinct subgroups of mixed phenotype acute leukemia with T-lineage differentiation. Blood Adv. 2018;2:3526–3539. doi: 10.1182/bloodadvances.2018023531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heavey B, Charalambous C, Cobaleda C, et al. Myeloid lineage switch of Pax5 mutant but not wild-type B cell progenitors by C/EBPalpha and GATA factors. EMBO J. 2003;22:3887–3897. doi: 10.1093/emboj/cdg380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang W, Lee K-W, Srivastava RM, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25:767–775. doi: 10.1038/s41591-019-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]