

Abstract
Challenging and still unsolved problems in kidney transplantation are risk stratification and the treatment of humoral rejection. Antibody-mediated rejection is an important cause of early and chronic rejection. The impact of donor-specific HLA antibodies on antibody-mediated rejection–causing graft damage is well known, but the clinical relevance of non-HLA antibodies remains unclear. Recently, in 2 independent studies, a new correlation was found between the presence of non-HLA anti-Rho guanosine diphosphate dissociation inhibitor 2 (ARHGDIB) antibodies and increased graft failure. RhoGDI2, another name for ARHGDIB, is a negative regulator of the Rho guanosine triphosphate (RhoGTP)ases RhoA, Rac1m, and Cdc42, whose main function is regulating the actin network in a variety of cells. RhoGDI2 is mainly expressed intracellularly, and some expression is observed on the cell surface. Currently, there is no mechanism known to explain this correlation. Additionally, the reason why the antibodies are produced is unknown. In this review, we will address these questions, provide an overview of other diseases in which these antibodies are prevalent, and describe the physiological role of RhoGDI2 itself. If the mechanism and impact of RhoGDI2 antibodies in kidney graft failure are known, improved risk stratification can be provided to decrease the rate of donor kidney graft failure.
For patients with end-stage kidney failure, kidney transplantation is the best form of treatment. Despite improved short-term graft survival, the long-term survival of kidney grafts remains approximately 50%,1 mainly due to the occurrence of chronic rejection. Antibodies contribute to both early and late graft failure in a process termed antibody-mediated rejection (AMR). The standard forms of treatment for acute AMR are currently plasmapheresis and intravenous immunoglobulin, but these are expensive treatments. Despite new treatment options focusing on, for example, depletion of B cells by rituximab or inhibition of complement-dependent endothelial damage, it is a challenge to predict and treat AMR.
AMR is a type of rejection in which antibodies are formed against donor-specific HLA molecules, blood group antigens, and antigens present on the endothelium. Pretransplant anti-HLA antibodies against the donor have been associated with increased occurrence of kidney graft loss, but in a study of identical HLA siblings, it seemed that non-HLA antibodies also played a role in AMR.2,3 Terasaki4 reported in his study that 38% of rejections are due to immunological reactions against non-HLA molecules, 18% are due to HLA antibodies, and 43% are attributed to nonimmunological factors. Non-HLA antibodies are divided into 2 classes: antibodies directed against polymorphic antigens that differ between the recipient and donor, and autoantibodies.5 In vitro, non-HLA antibodies do not induce complement-dependent epithelial damage. It is suggested that they play a role in graft failure as immune mediators.6 Currently, the clinical relevance of non-HLA antibodies and their mechanism are not well studied, but their association with graft loss is a promising feature that can be used for future therapies.
Recently, it was observed that patients who received a kidney from a deceased donor presented decreased graft survival in the presence of a specific non-HLA antibody named anti-Rho guanosine diphosphate (GDP) dissociation inhibitor 2 (ARHGDIB/RhoGDI2).7 Another recent study by Senev et al8 found that kidney transplant recipients with both HLA donor-specific antibodies (DSAs) and pretransplant anti-ARHGDIB/RhoGDI2 antibodies also had an increased risk of graft failure.
ARHGDIB encodes the protein RhoGDI2 (also known as LyGDI, RhoGDIβ, or D4-GDP dissociation inhibitor), which is mainly expressed by hematopoietic cells. It inhibits the dissociation of GDP from Rho guanosine triphosphate (RhoGTP)ases, thereby inactivating them.9,10 A clear overview of the functions of RhoGDI2 is still missing. In addition, the functional relevance of anti-RhoGDI2 antibodies in kidney transplantation is currently unknown. In this review, an overview of the role of anti-RhoGDI2 in kidney transplantation is provided, including possible mechanisms of kidney graft loss by anti-RhoGDI2 autoantibodies.
FUNCTIONS OF THE RhoGDI FAMILY MEMBERS AND THEIR EXPRESSION
RhoGDI2 belongs to the family of RhoGTPases, which is a part of the Ras superfamily and consists of 20 members. RhoGTPases are involved in the regulation of microtubules, cell survival, cell polarity, and gene expression. Additionally, in actin-dependent processes, such as migration, adhesion, and phagocytosis, RhoGTPases play a significant role. Regulation of the RhoGTPase needs to be precisely tuned to correctly respond to environmental stimuli. This regulation is performed by guanine nucleotide exchange factors (GEFs), GTPase-activating proteins, and GDP dissociation inhibitors. RhoGEFs convert the GTPases to the active state by promoting the dissociation of GDP; Rho GTPase-activating proteins promote the conversion of molecules from the GTP-bound to GDP-bound state by increasing hydrolysis activity; and RhoGDIs bind the RhoGTPases to keep them inactive in the cytosol.9
RhoGDIs have 3 biological activities. First, they are able to inhibit the dissociation of GDP from the GTPase and prevent GTPase activation by GEFs. Next, RhoGDIs can interact with Rho in the GTP-bound state to inhibit GTP hydrolysis, prevent interactions with effector molecules and block GTPase activity. Third, they regulate the cycling of the RhoGTPases between the membranes and the cytosol.11 The function that RhoGDI2 performs has been found to be concentration dependent.12
The RhoGDI family consists of 3 members: RhoGDI1, RhoGDI2, and RhoGDI3. RhoGDI consists of 2 domains, a folded C-terminal domain with a length of 134 amino acid residues and a 70-residue-long flexible N-terminal domain. The formation of the complex of a RhoGDI and RhoGTPases occurs via the immunoglobulin-like C terminus of the RhoGDI. This domain also contains an isoprenyl group by which it associates with the membrane of the cell. The N-terminal domain, the regulatory arm of RhoGDIs, is thought to be important in the inhibition of the dissociation of GDP from the GTPase. This part also differs between the 3 members of the RhoGDI family and probably explains the regulatory differences between the molecules.13
The N terminus of RhoGDI2 contains 2 consensus sites for caspase cleavage, suggesting a role in apoptosis.9 After stimulation of the cell, RhoGDI also interacts with other proteins. RhoGDIs can be regulated by GDP dissociation factors, lipids with complex dissociation activities and kinase-mediated phosphorylation. All these actions can result in the dissociation of the RhoGDI from the RhoGTPase. The RhoGTPase becomes active and is able to associate with the cell membrane.11 RhoGDI2 is known to play a role in cell proliferation and survival due to its influence on RhoGTPases. RhoGDI2 is predominantly expressed in hematopoietic cells, of which B and T cells show the highest expression.14,15 Therefore, in the thymus and spleen, elevated levels of RhoGDI2 mRNA expression are observed. Tissues with high vascularity, including lungs, heart, and placenta, also show increased RhoGDI2 mRNA expression.16 The kidney shows low levels of RhoGDI2 expression. Biopsies of transplanted kidneys with no histological abnormalities stained using a RhoGDI2 antibody showed upregulated expression of RhoGDI2 in endothelial cells in interlobular arteries and peritubular and glomerular capillaries. In transplanted kidneys with acute tubular necrosis, these findings were also observed, and in addition, elevated ARHGDIB/RhoGDI2 expression was observed in some podocytes and lymphocytes.7 In biopsies with histology of AMR, there is increased intrarenal expression of ARHGDIB/RhoGDI2 compared with that seen in biopsies without histology of AMR.8
RhoGDI2 is localized in the cytoplasm, where it binds RhoGTPases to keep them in an inactive state. This localization is also observed at the immunological synapse, where the contact sites between antigen-presenting cells and T cells are.17
CELLULAR FUNCTIONS OF RhoGDI2
RhoGTPases play a role in many cellular processes in the body. RhoGDI2 regulates RhoGTPases; therefore, RhoGDI2 also plays a role in many processes, including actin reorganization, vascular remodeling, and cellular processes in lymphocytes. For an overview of the functions of RhoGDI2, see Figure 1.
FIGURE 1.
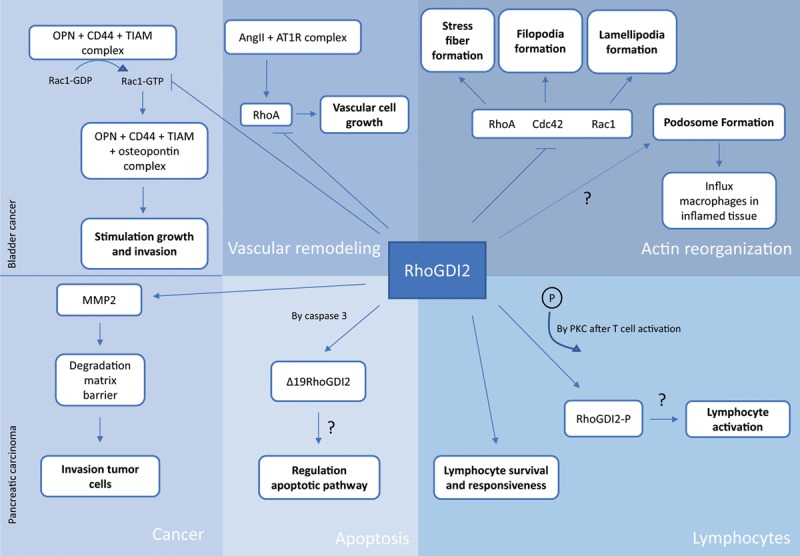
Functions of RhoGDI2 in actin organization, apoptosis, lymphocytes, and cancer. The role of RhoGDI2 is in some mechanisms more clear than in others. AngII, angiotensin II; AT1R, angiotensin II receptor type 1; GDP, guanosine diphosphate; MMP, metalloproteinase; OPN, osteopontin; PKC, protein kinase C; RhoGDI, Rho GDP dissociation inhibitor; TIAM, T-cell lymphoma invasion and metastasis inducing factor.
Function of RhoGDI2 in Actin Reorganization
The best-characterized RhoGTPases are RhoA, Rac1, and Cdc42. They transduce signals from cell surface receptors into intracellular signals, which activate different pathways.9 All 3 GTPases regulate actin cytoskeleton organization, including the formation of stress fibers, lamellipodia, and filopodia. RhoGDI2 keeps the RhoGTPases in an inactive state and is therefore able to inhibit the formation of these structures.
Actin is present in many organelles, which have different functions. Podosomes are actin filament-rich organelles present in cells of the myeloid lineage, including macrophages. Macrophages need to infiltrate into tissues and organs to perform their immunological functions. Penetration between tissues requires degradation of the extracellular matrix, which can be achieved by the actions of podosomes. Inhibition of RhoA and activity of Rac1 are required for podosome formation.18
Rho kinase (ROCK), a downstream target of RhoA, exists in 2 forms, ROCK1 and ROCK2.19 In vitro inhibition of ROCK1 by Y27632 leads to a reduction in the number of podosomes, reduced motility and phagocytosis, and an increased matrix degradation.18,20
RhoA also plays a role in the accumulation of macrophages in blood vessels. Macrophages express the chemokine receptor CX3CR1, which binds the chemokine CX3CL1 produced by the endothelium of the blood vessel wall. CX3CR1 is a G-protein–coupled receptor that functions as GEF for RhoA upon activation. Active RhoA mediates changes in actin polymerization leading to the migration of macrophages to the blood vessel, hyperplasia of the neointima, and differentiation of smooth muscle cells in the vessel wall.
It is known that Rho and ROCK influence T cells and that T cells influence macrophages. Therefore, it is possible that ROCK indirectly affects the mechanisms of macrophages.20 However, it has not yet been elucidated how RhoGDI2 influences the mechanism of podosome formation and therefore the influx of macrophages into tissues during inflammation.
Function of RhoGDI2 in Apoptosis
The 2 consensus sites for caspase cleavage in the N-terminal domain of RhoGDI2 can be bound by caspase proteases.9 RhoGDI2 can be cleaved at Asp19 by the enzyme caspase 3, a member of the cell death-3 subfamily, in response to different stimuli. This family is suggested to be involved in apoptosis. Additionally, cleavage can also occur at Asp55 by caspase 1, a member of the interleukin (IL)-1-β-converting enzyme subfamily, which is involved in inflammation.21,22
The caspase 3–induced cleavage product of RhoGDI2, Δ19RhoGDI2, translocates to the nucleus. What function it has there has still to be elucidated. It is known that the import of proteins into the nucleus is required for nuclear changes during apoptosis. RhoGDI2 was the first product of caspase activity reported to translocate to the nucleus. This could indicate that the cleavage of RhoGDI2 is a regulator of the apoptotic pathway. However, in the phorbol 12-myristate 13-acetate (PMA)-stimulated differentiation of THP1 cells (human model for monocytes) to macrophages, cleavage of RhoGDI2 was also observed in nonapoptotic cells. This study indicated that Δ19RhoGDI2 was also involved in processes other than apoptosis.22,23 In conclusion, it is unknown whether RhoGDI2 plays a role in apoptosis or is just a byproduct.
Function of RhoGDI2 in Lymphocytes
Within T cells, naïve CD4+ and CD8+ T cells have a higher expression of RhoGDI2 than memory cells. This could suggest that RhoGDI2 is involved in the differences between naïve and memory lymphocytes.15
It has been reported that after stimulation of T cells with PMA, RhoGDI2 becomes phosphorylated. PMA is a phorbol ester that is able to mimic lymphocyte activation in vitro. PMA activates protein kinase C and calcium ionophores. RhoGDI2 was phosphorylated only after PMA stimulation, not after stimulation with calcium ionophores, suggesting a role for RhoGDI2 downstream of protein kinase C. It is known that after stimulation of the T-cell receptor, rearrangement of the actin cytoskeleton takes place. This result could indicate that RhoGDI2 is a player in the lymphocyte activation pathway.15 In a study by Barbati et al,14 it was suggested that RhoGDI2 is able to play a role as a negative contributor to lymphocyte activation by dissociating from RhoGTPases upon T-cell receptor activation by anti-CD3.
Function of RhoGDI2 in Cancer
The expression of RhoGDI2 in various types of cancer is variable. Additionally, its role can be protumorigenic or antitumorigenic in different cancer types. For example, in breast, colorectal, gastric, and pancreatic cancers, RhoGDI2 serves as a positive regulator. It affects expression of metalloproteinase 2 resulting in breakdown of matrix barriers.10 In this context, it is be easier for tumor cells to invade other tissues. Elevated RhoGDI2 expression has already been shown in patients with pancreatic carcinoma. In contrast, in Hodgkin lymphoma and bladder cancer, RhoGDI2 functions as a metastasis suppressor10 by binding to and inactivating Rac1 and blocking the metastasis stimulating osteopontin/CD44 pathway.24
Function of RhoGDI2 in Vascular Remodeling and Permeability
Vascular remodeling is involved in pathogenesis of several life-threatening cardiovascular diseases, and also in transplantation and chronic rejection.25 In vascular remodeling, angiotensin II (Ang II) plays an important role. On binding of Ang II to its receptor angiotensin II receptor type 1, small GTP-binding proteins like Ras and RhoA become activated, resulting in for example contraction and proliferation of vascular smooth muscle cells. A recent study shows that Ang II-induced vascular remodeling is regulated via stabilizing RhoGDIs. Proteosomal degradation of RhoGDI1 and RhoGDI2 via increased ubiquitination or SUMOylation results in reduced proliferation of vascular smooth muscle cells, thereby indicating a role for RhoGDIs in vascular remodeling.26
Regarding the role of RhoGTPases in vascular permeability, it has been shown that active RhoA results in endothelial hyperpermeability, which can be abrogated by RhoA/ROCK inhibitors.27,28 However, most studies were performed in mice, and to the best of our knowledge, studies about the specific role of RhoGDI2 in endothelial permeability are not yet available in humans.
ANTI-RhoGDI2 AUTOANTIBODIES
Previously, RhoGDI2 antibodies were found in patients with acute leukemia (AL) and systemic lupus erythematosus (SLE). Recently, 2 studies also found these antibodies in patients who received a donor kidney. An overview of these findings is shown in Figure 2 and described in detail below.
FIGURE 2.
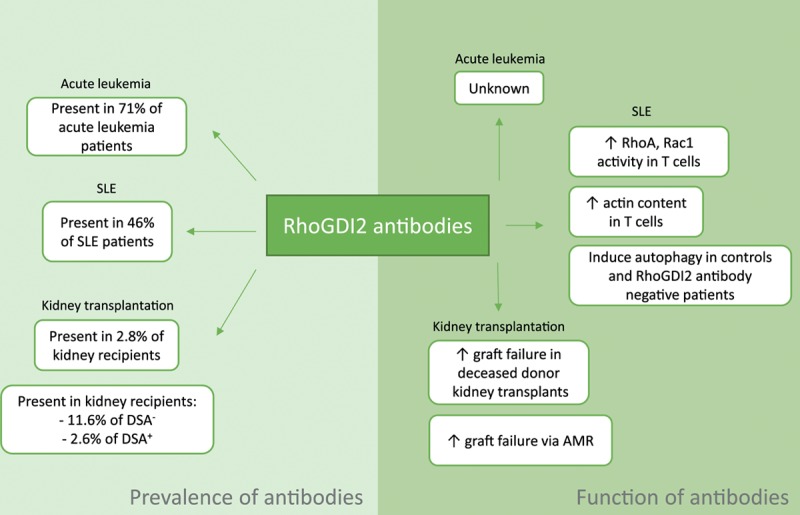
RhoGDI2 antibodies found in SLE, acute leukemia, and kidney transplantation, and their functions. AMR, antibody-mediated rejection; DSA, donor-specific antibodies; RhoGDI, Rho guanosine diphosphate dissociation inhibitor; SLE, systemic lupus erythematosus.
Anti-RhoGDI2 Antibodies in AL
Cui et al29 found antibodies against RhoGDI2 in patients with AL, and this was the first study that found RhoGDI2 antibodies in the human body. The aim of this study was to identify proteins that commonly trigger a humoral response in AL via a proteomic approach. They used leukemic cells derived from the bone marrow of patients with AL and used patients with solid tumors and patients without tumors as controls. Two-dimensional electrophoresis was used to separate the proteins. The sera were screened for any antibody reactivity with Western blot. Finally, the reactive proteins were identified using mass spectrophotometry. In 15 of 21 (71%) AL patients, RhoGDI2 antibodies were reported. In the controls, 20 patients with solid tumors and 22 healthy volunteers, approximately 5% of antibodies were reactive against RhoGDI2. Before this research, autoimmunity in cancer had mainly only been reported in solid tumors.29
Anti-RhoGDI2 Antibodies in SLE
SLE is an autoimmune disease that affects many organs, including the kidneys. One of the features of SLE is the production of autoantibodies by deregulated B cells. Barbati et al14 reported that 46% of SLE patients had RhoGDI2 antibodies present in their sera, whereas no antibodies were observed in the control group.
T cells from healthy subjects and SLE patients showed both intracellular and cell surface expression of RhoGDI2. Upon binding of RhoGDI2 antibodies, T lymphocytes showed the ability to upregulate the GTP-bound form of RhoA and Rac1, suggesting that these antibodies can also influence intact cells. Furthermore, in the presence of these antibodies, T cells showed an increase in F-actin and in 3 actin-binding proteins, namely, ezrin, gelsolin, and vinculin, indicating that RhoGDI2 antibodies influence actin cytoskeleton remodeling.
Recently, it was found that autophagy is a key player in SLE. In patients negative for the presence of RhoGDI2 antibodies and controls, it was shown that RhoGDI2 antibodies were able to induce significant increases in autophagy. However, in RhoGDI antibody-positive SLE patients, this finding was not observed. It has been suggested that overstimulation with RhoGDI2 antibodies could lead to the selection of T cells that are autophagy resistant. This could contribute to the pathogenesis of SLE. Less autophagy will lead to an overload of damaged mitochondria, which can secrete apoptosis inducible factors. More apoptosis would result in an imbalance between autophagy and apoptosis. This imbalance is seen in hematologic manifestations, including leukopenia and lymphopenia. These 2 hematologic manifestations are observed more often in patients with RhoGDI2 antibodies in their serum than in RhoGDI antibody-negative patients.
Anti-ARHGDIB/RhoGDI2 Antibodies in Kidney Transplantation
In 2 independent studies, a correlation between the presence of ARHGDIB/RhoGDI2 antibodies and graft survival was reported. In a national consortium study—Profiling Consortium of Antibody Repertoire and Effector Functions—approximately 5500 kidney transplants from all 8 Dutch university medical centers performed between 1995 and 2005 were studied.30 The researchers investigated, among other things, the associations between the presence of 14 non-HLA antibodies in pretransplant sera and renal graft survival. A total of 2.8% of the patients in the cohort had ARHGDIB/RhoGDI2 antibodies present in their serum. In recipients with serum ARHGDIB/RhoGDI2 antibodies, transplanted with a graft from deceased donors, a decrease in kidney allograft survival was observed; this was not observed in living donors, as shown previously.7 This analysis was corrected for age of the donors and recipients, the type of donor, dialysis years, the ischemia time for deceased donors, history of induction therapy with an IL-2 blocker and the presence of DSAs. No correlation was found with type of end-stage renal disease, sex, and retransplantation which could influence the levels of these autoantibodies (unpublished data). Michielsen et al31 showed that levels of ARHGDIB/RhoGDI2 antibodies—among other non-HLA antibodies—remains stable up to 1 y after transplantation.
Another study found that patients with both HLA-DSAs and anti-ARHGDIB/RhoGDI2 antibodies in their sera had a 19.5 times higher risk for renal graft failure than patients negative for both antibodies. Additionally, it was observed that patients with ARHGDIB/RhoGDI2 antibodies present in their sera but negative for HLA-DSAs had a 4.4-fold increased risk of graft failure.8
Reason for Production of RhoGDI2 Autoantibodies
After kidney transplantation, the donor kidney can suffer from tissue damage induced by ischemia-reperfusion, and vascular injury and graft loss can also occur. All these conditions are able to increase the exposure of autoantigens, resulting in elevated autoantibody formation.32 Chronic kidney inflammation and autoimmune diseases may also trigger sensitization via increased exposure of intracellular autoantigens due to renal damage.
Dieudé et al33 reported in their study that endothelial cells, vascular smooth cells, and tubular epithelial cells release proteasome-active vesicles when injured in vitro. These cells are targets of injury during ischemia-reperfusion and could contribute to the release of apoptotic, immunogenic, exosome-like vesicles in vivo. These vesicles induce B-cell responses in transplanted and naïve mice, resulting in the production of autoantibodies. In vesicles in which proteasome activity was inhibited, a lower production of antibodies in vivo was observed, indicating the importance of the proteasome core in the production of autoantibodies. In conclusion, these apoptotic-exosome–like vesicles can break self-tolerance and induce the production of autoantibodies.33
POSSIBLE PATHOGENIC MECHANISMS OF ANTI-RhoGDI2 ANTIBODIES IN KIDNEY GRAFT FAILURE
RhoGDI2 is mainly expressed in the cytoplasm of endothelial cells in the kidney and podocytes, but some cell surface expression has been observed.7,14 Previous studies have suggested that RhoGDI2 antibodies play an intracellular role. After ischemia-reperfusion injury, intracellular autoantigens could become exposed, and antibodies are able to bind. Possible mechanisms by which the RhoGDI2 antibodies cause renal graft failure are described in this chapter. An overview is given in Figure 3.
FIGURE 3.
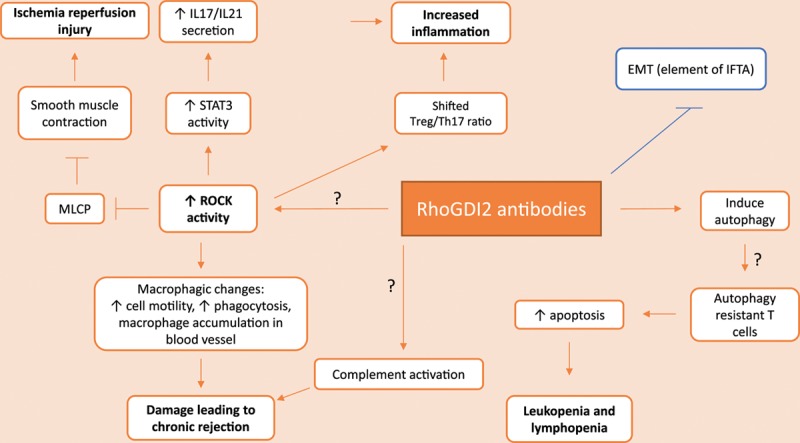
Possible mechanisms of kidney graft rejection by anti-RhoGDI2 antibodies. All mechanisms contribute to the development or maintenance of graft rejection, except the mechanism of epithelial to mesenchymal transformation (displayed in blue). EMT, epithelial to mesenchymal transition; IFTA, interstitial fibrosis and tubular atrophy; IL, interleukin; MLCP, myosin light chain phosphatase; RhoGDI, Rho guanosine diphosphate dissociation inhibitor; ROCK, Rho kinase; STAT3, signal transducer and activator of transcription 3; Treg, regulatory T cell.
Role in Reperfusion Injury After Transplantation
During ischemia-reperfusion, inflammation is induced. When ROCK is inhibited, less reperfusion damage occurs by limiting the influence of Ang II on the reperfused arteries. Ang II influences vessel smooth muscle contraction. This contraction depends on the level of phosphorylation of the myosin II light chain (MLC), which is regulated by myosin light chain kinase and myosin light chain phosphatase.34,35 ROCK is able to keep myosin light chain phosphatase inactive via phosphorylation. As a result, more phosphorylated than nonphosphorylated MLC will be present, leading to smooth muscle contraction through interacting with actin filaments.36 Inhibiting the ROCK pathway with Y27632 decreases phosphorylated MLC expression thereby reducing renal injury.37
The presence of RhoGDI2 antibodies can lead to increased RhoA activity, resulting in increased ROCK activity.14 RhoGDI2 antibodies might therefore able to indirectly cause reperfusion injury to the arteries after transplantation by the above-mentioned mechanisms. In conclusion, data from kidney allograft rejection models have proven that disruption of the regulation of MLC can lead to increased graft failure.
IL-17 and IL-21 Are Downregulated by a ROCK2 Inhibitor
Th1 and Th2 cells are known to be involved in the loss of kidney grafts. Recently, accumulating evidence has suggested that Th17 cells are also a contributor to the development of chronic and acute kidney rejection.38,39 Th17 cells secrete the proinflammatory cytokines IL-17 and IL-21. Both cytokines play a role in graft survival.
The expression of these cytokines depends on the activity of several transcription factors, including signal transducer and activator of transcription 3 (STAT3), interferon regulatory factor 4, and retinoid-related orphan receptor γt. ROCK2 influences the production of IL-17 and IL-21 by activating STAT3 through phosphorylation. The inhibition of ROCK2 by KD025 has been shown to downregulate STAT3 activity by inhibiting phosphorylation and reducing the levels of interferon regulatory factor 4 and retinoid-related orphan receptor γt. This results in decreased secretion of IL-17 and IL-21 by T cells. The inhibition of ROCK2 also resulted in an increased regulatory T cell (Treg)/Th17 ratio, which was associated with a decrease in renal function.40,41 A similar result was also observed in a rat cardiac model treated with a ROCK1 inhibitor, in which after administration of the inhibitor Y27632, a decrease in cellular infiltration of T cells was observed.42
It is possible that RhoGDI2 antibodies are able to strengthen inflammation via increased ROCK activity, leading to increased IL-17 and IL-21 secretion and a shifted Treg/Th17 ratio.
ROCK Inhibition Results in Macrophage Changes
From literature it has been shown that macrophage infiltration positively correlates to allograft loss,43 which could be abrogated by RhoA/ROCK inhibitors.44 Suppressing the RhoA/ROCK pathway increases matrix degradation and reduces podosome formation resulting in less tissue macrophage infiltration. Currently, the relative role of podosomes in transplant infiltration is not known, as well as upstream regulation of RhoGTPases regarding podosome formation. In addition, crosstalk between other GTPases needs to be further studied.45
T cells have been considered the main players in graft loss. Activated T cells recruit innate cells such as macrophages. When macrophages infiltrate the graft tissue, they begin secreting inflammatory cytokines and damage the graft tissue via the production of reactive oxygen species. Chronically activated macrophages promote interstitial fibrosis in the graft.46 Occlusion of blood vessels by accumulation of macrophages and remodeling of the vessel walls contribute to the development of chronic rejection. These findings have been reported in mice with RhoA-depleted cardiac allografts.47
These data support the possibility that increased RhoA activity due to RhoGDI2-specific antibodies could result in graft failure via macrophage-dependent mechanisms.
Role in Graft Failure Through Complement Activation
Several studies showed that the higher risk to develop AMR and graft loss in stable kidney recipients depends on the ability of HLA antibodies to bind complement.48
Similar to HLA antibodies, some studies indicate that non-HLA antibodies are able to activate the complement cascade upon binding to their antigen.49,50 It has been suggested that the ability to enhance complement-mediated tissue damage depends on the type of non-HLA antibodies and accessibility of non-HLA antigens. We hypothesize that upon cellular damage, intracellular RhoGDI2 may be accessible to antibodies. It must be noted however that no studies are available yet describing the complement activating potential of these antibodies.
RhoGDI2 Induces Epithelial-Mesenchymal Transition
All of the above-mentioned theoretical mechanisms of anti-RhoGDI2 antibodies result in the progression of kidney graft loss. In contrast, in human gastric cancer cells, RhoGDI2 induces epithelial-mesenchymal transition (EMT), an important element of interstitial fibrosis and tubular atrophy that is one of the main causes of late graft failure.51 The mechanism behind this is that Rac1 activates nuclear factor kappa-light-chain-enhancer of activated B cells, which leads to the upregulation of Snail. Snail activity results in the repression of E-cadherin. Functional loss of E-cadherin is required for loss of cell–cell adhesion which is required to achieve EMT. In this model, RhoGDI2 is suggested to act as a positive regulator of Rac1.52 Antibodies against RhoGDI2 will lead to the inhibition of EMT and therefore to protection against graft loss and will not cause progression.
DISCUSSION
Patients on chronic dialysis have kidney transplantation as the only option to improve their quality of life. However, waiting lists are long, and in 50% of the patients, the donor kidney is rejected in the long term. This graft loss is caused by hyperacute, acute, or chronic rejection. Hyperacute rejection is rare now, and cellular acute rejection can be treated, whereas humoral and chronic rejection are the leading causes of kidney failure.
In the processes of AMR and chronic rejection, anti-HLA DSAs are involved, but in recent years, it has been suggested that non-HLA antibodies also contribute. Kidney transplant recipients with chronic rejection and failed allografts do, for example, have increased levels of anti-vimentin53 and anti–angiotensin II receptor type 1 antibodies.54 Furthermore, autoantibodies to agrin were positively correlated with the number of acute rejections and to AMR.55 Finally, increased anti-LG3 antibodies were found in patients with allograft dysfunction compared with the numbers seen in controls.56 These data indicate that the production of non-HLA autoantibodies is an important contributor to the development of chronic rejection.
Recent data show that antibodies specific for RhoGDI2—already reported in patients with AL and SLE—are correlated with decreased graft survival of donor kidneys from deceased donors. In these studies antibodies against other RhoGDIs were not analyzed as autoantibodies only to RhoGDI2 (and not to RhoGDI1/3) were found in patients on dialysis. As far as we know, no literature describes RhoGDI2 autoantibodies associating with graft survival in heart, liver, or lung transplantation.
It is not known yet if the observed upregulation of RhoGDI2 in kidney grafts is beneficial or detrimental in relation to transplant outcome. It has been suggested that cell contents including antigens like LG3 and RhoGDI2 are released or become expressed on the cell surface of apoptotic cells after ischemia-reperfusion injury and chronic inflammation.3,32 In addition, during the period of hypoxia, the expression of autoantigens, including RhoGDI2, can be upregulated. The release of autoantigens into the circulation may be a stimulus causing the increased production of antibodies against RhoGDI2.
Apoptotic-exosome–like vesicles are also able to trigger increased production of autoantibodies by B cells. Exosomes—released from the intracellular compartment—contain proteins involved in cytoskeletal organization like Rho GTPases,57 and RhoGDI2 was also found in extracellular vesicles derived from human colorectal colon cancer cells.58 Although apoptotic bodies differ from exosomes in size, RNA, and protein contents,59,60 some research have identified proteins involved in actin cytoskeleton signaling.61 However, hard evidence is missing whether apoptotic vesicles also express RhoGDI2.
Although the mechanism and reason for the production of these antibodies in kidney transplant recipients are unclear, some hypothetical mechanisms could explain the correlation found in kidney transplant recipients. Studies on the inhibition of ROCK1 or ROCK2, the downstream effectors of RhoA, have shown significant results in the reduction of graft failure via numerous ways. In the presence of RhoGDI2 antibodies, the activities of ROCK and RhoA are increased because of the inhibition of the negative regulator of RhoA. This increased activity is theoretically able to lead to macrophage accumulation, increased phagocytosis by macrophages and macrophage cell motility, increased secretion of the inflammatory cytokines IL-17 and IL-21 by T cells, a shifted Treg/Th17 ratio and an increased influence of Ang II on arteries, resulting in more ischemia-reperfusion injury. These possible mechanisms focus on the effect of downstream effectors of RhoGDI2. Another mechanism could be that increased activity of RhoGTPases by RhoGDI2 antibodies influences the induction of autophagy, as observed in patients with SLE. This last mechanism specifically includes the direct working of RhoGDI2 antibodies.
Data about the pathophysiological role of RhoGDI2 in transplantation are limited, and provided mechanisms could be potential explanations for the function of anti-RhoGDI2 antibodies in relation to graft loss in solid organ transplantation. To get better insight in the role of RhoGDI2 autoantibodies, we need to study the expression of this autoantigen in a transplantation model to elucidate the reason why present antibodies could bind to intracellular RhoGDI2.
Furthermore, more studies are required to focus on the ability of anti-RhoGDI2 antibodies to fix complement to induce cell apoptosis. Analyses of their subclasses and levels are also required, like the development of functional assays.
The mechanism by which RhoGDI2 antibodies cause harm in transplant recipients is still unclear and needs to be revealed. Therefore, therapies that reduce the antibody levels are the best option for treatment currently. However, if in the future one of the above-mentioned mechanisms or a completely new mechanism is confirmed in relation to non-HLA antibodies, more specific therapies targeting a specific pathway could be used.
Footnotes
Published online 9 April, 2020.
T. Kardol-Hoefnagel and S.A.L.M. van Logtestijn contributed equally to this study.
S.A.L.M.v.L., T.K.-H., and H.G.O. wrote the article. All authors provided final approval of the version to be published.
The authors declare no funding or conflicts of interest.
REFERENCES
- 1.Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: a critical reappraisal. Am J Transplant. 2011; 11:450–462. doi:10.1111/j.1600-6143.2010.03283.x [DOI] [PubMed] [Google Scholar]
- 2.Opelz G. Non-HLA transplantation immunity revealed by lymphocytotoxic antibodies. Lancet. 2005; 365:1570–1576. doi:10.1016/S0140-6736(05)66458-6 [DOI] [PubMed] [Google Scholar]
- 3.Zhang Q, Reed EF. The importance of non-HLA antibodies in transplantation. Nat Rev Nephrol. 2016; 12:484–495. doi:10.1038/nrneph.2016.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Terasaki PI. Deduction of the fraction of immunologic and non-immunologic failure in cadaver donor transplants. Clin Transpl. 2003; 449 [PubMed] [Google Scholar]
- 5.Garces JC, Giusti S, Staffeld-Coit C, et al. Antibody-mediated rejection: a review. Ochsner J. 2017; 17:46–55 [PMC free article] [PubMed] [Google Scholar]
- 6.Sumitran-Holgersson S. Relevance of MICA and other non-HLA antibodies in clinical transplantation. Curr Opin Immunol. 2008; 20:607–613. doi:10.1016/j.coi.2008.07.005 [DOI] [PubMed] [Google Scholar]
- 7.Kamburova EG, Gruijters ML, Kardol-Hoefnagel T, et al. Antibodies against ARHGDIB are associated with long-term kidney graft loss. Am J Transplant. 2019; 19:3335–3344. doi:10.1111/ajt.15493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Senev A, Otten HG, Kamburova EG, et al. Antibodies against ARHGDIB and ARHGDIB gene expression associate with kidney allograft outcome. Transplantation. 2019. doi:10.1097/tp.0000000000003005 [DOI] [PubMed] [Google Scholar]
- 9.Griner EM, Theodorescu D. The faces and friends of RhoGDI2. Cancer Metastasis Rev. 2012; 31:519–528. doi:10.1007/s10555-012-9376-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi B, Hu Y, Qin G, et al. Depletion of RhoGDI2 expression inhibits the ability of invasion and migration in pancreatic carcinoma. Int J Mol Med. 2014; 34:205–212. doi:10.3892/ijmm.2014.1765 [DOI] [PubMed] [Google Scholar]
- 11.DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005; 15:356–363. doi:10.1016/j.tcb.2005.05.001 [DOI] [PubMed] [Google Scholar]
- 12.Chuang TH, Xu X, Knaus UG, et al. GDP dissociation inhibitor prevents intrinsic and GTPase activating protein-stimulated GTP hydrolysis by the Rac GTP-binding protein. J Biol Chem. 1993; 268:775–778 [PubMed] [Google Scholar]
- 13.Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011; 12:493–504. doi:10.1038/nrm3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barbati C, Alessandri C, Vomero M, et al. Autoantibodies specific to D4GDI modulate rho gtpase mediated cytoskeleton remodeling and induce autophagy in T lymphocytes. J Autoimmun. 2015; 58:78–89. doi:10.1016/j.jaut.2015.01.005 [DOI] [PubMed] [Google Scholar]
- 15.Scherle P, Behrens T, Staudt LM. Ly-GDI, a GDP-dissociation inhibitor of the RhoA GTP-binding protein, is expressed preferentially in lymphocytes. Proc Natl Acad Sci U S A. 1993; 90:7568–7572. doi:10.1073/pnas.90.16.7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Theodorescu D, Sapinoso LM, Conaway MR, et al. Reduced expression of metastasis suppressor RhoGDI2 is associated with decreased survival for patients with bladder cancer. Clin Cancer Res. 2004; 10:3800–3806. doi:10.1158/1078-0432.CCR-03-0653 [DOI] [PubMed] [Google Scholar]
- 17.Groysman M, Hornstein I, Alcover A, et al. Vav1 and Ly-GDI two regulators of Rho GTPases, function cooperatively as signal transducers in T cell antigen receptor-induced pathways. J Biol Chem. 2002; 277:50121–50130. doi:10.1074/jbc.M204299200 [DOI] [PubMed] [Google Scholar]
- 18.Chen W, Ghobrial RM, Li XC, et al. Inhibition of RhoA and mTORC2/Rictor by Fingolimod (FTY720) induces p21-activated kinase 1, PAK-1 and amplifies podosomes in mouse peritoneal macrophages. Immunobiology. 2018; 223:634–647. doi:10.1016/j.imbio.2018.07.009 [DOI] [PubMed] [Google Scholar]
- 19.Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken). 2010; 67:545–554. doi:10.1002/cm.20472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Tejpal N, You J, et al. ROCK inhibition impedes macrophage polarity and functions. Cell Immunol. 2016; 300:54–62. doi:10.1016/j.cellimm.2015.12.005 [DOI] [PubMed] [Google Scholar]
- 21.Olofsson B. Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell Signal. 1999; 11:545–554. doi:10.1016/s0898-6568(98)00063-1 [DOI] [PubMed] [Google Scholar]
- 22.Ota T, Jiang YS, Fujiwara M, et al. Apoptosis-independent cleavage of RhoGDIβ at Asp19 during PMA-stimulated differentiation of THP-1 cells to macrophages. Mol Med Rep. 2017; 15:1722–1726. doi:10.3892/mmr.2017.6199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krieser RJ, Eastman A. Cleavage and nuclear translocation of the caspase 3 substrate Rho GDP-dissociation inhibitor, D4-GDI, during apoptosis. Cell Death Differ. 1999; 6:412–419. doi:10.1038/sj.cdd.4400515 [DOI] [PubMed] [Google Scholar]
- 24.Ahmed M, Sottnik JL, Dancik GM, et al. An osteopontin/CD44 axis in RhoGDI2-mediated metastasis suppression. Cancer Cell. 2016; 30:432–443. doi:10.1016/j.ccell.2016.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kloc M, Ghobrial RM. Chronic allograft rejection: a significant hurdle to transplant success. Burns Trauma. 2014; 2:3–10. doi:10.4103/2321-3868.121646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dai F, Qi Y, Guan W, et al. RhoGDI stability is regulated by SUMOylation and ubiquitination via the AT1 receptor and participates in Ang II-induced smooth muscle proliferation and vascular remodeling. Atherosclerosis. 2019; 288:124–136. doi:10.1016/j.atherosclerosis.2019.07.010 [DOI] [PubMed] [Google Scholar]
- 27.Gorovoy M, Neamu R, Niu J, et al. RhoGDI-1 modulation of the activity of monomeric RhoGTPase RhoA regulates endothelial barrier function in mouse lungs. Circ Res. 2007; 101:50–58. doi:10.1161/CIRCRESAHA.106.145847 [DOI] [PubMed] [Google Scholar]
- 28.Karki P, Birukov KG. Rho and reactive oxygen species at crossroads of endothelial permeability and inflammation. Antioxid Redox Signal. 2019; 31:1009–1022. doi:10.1089/ars.2019.7798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui JW, Li WH, Wang J, et al. Proteomics-based identification of human acute leukemia antigens that induce humoral immune response. Mol Cell Proteomics. 2005; 4:1718–1724. doi:10.1074/mcp.M400165-MCP200 [DOI] [PubMed] [Google Scholar]
- 30.Kamburova EG, Hoitsma A, Claas FH, et al. Results and reflections from the PROfiling Consortium on Antibody Repertoire and Effector functions in kidney transplantation: a mini-review. HLA. 2019; 94:129–140. doi:10.1111/tan.13581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michielsen LA, van Zuilen AD, Kardol-Hoefnagel T, et al. Michielsen LA. Sensitization against non-HLA antigens in renal transplantation. In: Improving Renal Graft Survival, a Characterization of Antibodies and Complement Regulatory Proteins. 2019, Utrecht, the Netherlands: Utrecht University, 75–85 [Google Scholar]
- 32.Cardinal H, Dieudé M, Hébert MJ. The emerging importance of non-HLA autoantibodies in kidney transplant complications. J Am Soc Nephrol. 2017; 28:400–406. doi:10.1681/ASN.2016070756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dieudé M, Bell C, Turgeon J, et al. The 20S proteasome core, active within apoptotic exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci Transl Med. 2015; 7:318ra200 doi:10.1126/scitranslmed.aac9816 [DOI] [PubMed] [Google Scholar]
- 34.Giral M, Foucher Y, Dufay A, et al. Pretransplant sensitization against angiotensin II type 1 receptor is a risk factor for acute rejection and graft loss. Am J Transplant. 2013; 13:2567–2576. doi:10.1111/ajt.12397 [DOI] [PubMed] [Google Scholar]
- 35.Szadujkis-Szadurski R, Slupski M, Szadujkis-Szadurska K, et al. Rho-kinase inhibitor reduces hypersensitivity to ANG II in human mesenteric arteries retrieved and conserved under the same conditions as transplanted organs. Postepy Hig Med Dosw (Online). 2014; 68:1022–1027. doi:10.5604/17322693.1118217 [DOI] [PubMed] [Google Scholar]
- 36.Budzyn K, Marley PD, Sobey CG. Targeting Rho and Rho-kinase in the treatment of cardiovascular disease. Trends Pharmacol Sci. 2006; 27(2):97–104. doi:10.1016/j.tips.2005.12.002 [DOI] [PubMed] [Google Scholar]
- 37.Prakash J, de Borst MH, Lacombe M, et al. Inhibition of renal Rho kinase attenuates ischemia/reperfusion-induced injury. J Am Soc Nephrol. 2008; 19:2086–2097. doi:10.1681/ASN.2007070794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crispim JC, Grespan R, Martelli-Palomino G, et al. Interleukin-17 and kidney allograft outcome. Transplant Proc. 2009; 41:1562–1564. doi:10.1016/j.transproceed.2009.01.092 [DOI] [PubMed] [Google Scholar]
- 39.Deteix C, Attuil-Audenis V, Duthey A, et al. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol. 2010; 184:5344–5351. doi:10.4049/jimmunol.0902999 [DOI] [PubMed] [Google Scholar]
- 40.Chung BH, Yang CW, Cho ML. Clinical significance of Th17 cells in kidney transplantation. Korean J Intern Med. 2018; 33:860–866. doi:10.3904/kjim.2018.095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zanin-Zhorov A, Weiss JM, Nyuydzefe MS, et al. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc Natl Acad Sci U S A. 2014; 111:16814–16819. doi:10.1073/pnas.1414189111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Kloc M, Tejpal N, et al. Rock1 inhibitor abrogates chronic rejection in rat cardiac model system. Open J Organ Transplant Surg. 2012; 2:46–51. doi:10.4236/ojots.2012.24012 [Google Scholar]
- 43.Bergler T, Jung B, Bourier F, et al. Infiltration of macrophages correlates with severity of allograft rejection and outcome in human kidney transplantation. PLoS One. 2016; 11:e0156900 doi:10.1371/journal.pone.0156900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen W, Chen S, Chen W, et al. Screening RhoA/ROCK inhibitors for the ability to prevent chronic rejection of mouse cardiac allografts. Transpl Immunol. 2018; 50:15–25. doi:10.1016/j.trim.2018.06.002 [DOI] [PubMed] [Google Scholar]
- 45.van Helden SF, Hordijk PL. Podosome regulation by Rho GTPases in myeloid cells. Eur J Cell Biol. 2011; 90:189–197. doi:10.1016/j.ejcb.2010.05.008 [DOI] [PubMed] [Google Scholar]
- 46.Liu Y, Kloc M, Li XC. Macrophages as effectors of acute and chronic allograft injury. Curr Transplant Rep. 2016; 3:303–312. doi:10.1007/s40472-016-0130-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Chen W, Wu C, et al. Macrophage/monocyte-specific deletion of Ras homolog gene family member A (RhoA) downregulates fractalkine receptor and inhibits chronic rejection of mouse cardiac allografts. J Heart Lung Transplant. 2017; 36:340–354. doi:10.1016/j.healun.2016.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee DR, Kim BC, Kim JP, et al. C3d-binding donor-specific HLA antibody is associated with a high risk of antibody-mediated rejection and graft loss in stable kidney transplant recipients: a single-center cohort study. Transplant Proc. 2018; 50:3452–3459. doi:10.1016/j.transproceed.2018.06.037 [DOI] [PubMed] [Google Scholar]
- 49.Fleming SD. Natural antibodies, autoantibodies and complementactivation in tissue injury. Autoimmunity. 2006; 39:379–386. doi:10.1080/08916930600739381 [DOI] [PubMed] [Google Scholar]
- 50.Mahesh B, Leong HS, McCormack A, et al. Autoantibodies to vimentin cause accelerated rejection of cardiac allografts. Am J Pathol. 2007; 170:1415–1427. doi:10.2353/ajpath.2007.060728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chand S, Atkinson D, Collins C, et al. The spectrum of renal allograft failure. PLoS One. 2016; 11:e0162278 doi:10.1371/journal.pone.0162278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cho HJ, Park SM, Kim IK, et al. RhoGDI2 promotes epithelial-mesenchymal transition via induction of Snail in gastric cancer cells. Oncotarget. 2014; 5:1554–1564. doi:10.18632/oncotarget.1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter V, Shenton BK, Jaques B, et al. Vimentin antibodies: a non-HLA antibody as a potential risk factor in renal transplantation. Transplant Proc. 2005; 37:654–657. doi:10.1016/j.transproceed.2004.12.043 [DOI] [PubMed] [Google Scholar]
- 54.Dragun D, Müller DN, Bräsen JH, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med. 2005; 352:558–569. doi:10.1056/NEJMoa035717 [DOI] [PubMed] [Google Scholar]
- 55.Joosten SA, Sijpkens YW, van Ham V, et al. Antibody response against the glomerular basement membrane protein agrin in patients with transplant glomerulopathy. Am J Transplant. 2005; 5:383–393. doi:10.1111/j.1600-6143.2005.00690.x [DOI] [PubMed] [Google Scholar]
- 56.Cardinal H, Dieudé M, Brassard N, et al. Antiperlecan antibodies are novel accelerators of immune-mediated vascular injury. Am J Transplant. 2013; 13:861–874. doi:10.1111/ajt.12168 [DOI] [PubMed] [Google Scholar]
- 57.Choi DS, Kim DK, Kim YK, et al. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics. 2013; 13:1554–1571. doi:10.1002/pmic.201200329 [DOI] [PubMed] [Google Scholar]
- 58.Choi DS, Yang JS, Choi EJ, et al. The protein interaction network of extracellular vesicles derived from human colorectal cancer cells. J Proteome Res. 2012; 11:1144–1151. doi:10.1021/pr200842h [DOI] [PubMed] [Google Scholar]
- 59.Battistelli M, Falcieri E. Apoptotic bodies: particular extracellular vesicles involved in intercellular communication. Biology (Basel). 2020; 9:E21 doi:10.3390/biology9010021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zaborowski MP, Balaj L, Breakefield XO, et al. Extracellular vesicles: composition, biological relevance, and methods of study. Bioscience. 2015; 65:783–797. doi:10.1093/biosci/biv084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turiák L, Misják P, Szabó TG, et al. Proteomic characterization of thymocyte-derived microvesicles and apoptotic bodies in BALB/c mice. J Proteomics. 2011; 74:2025–2033. doi:10.1016/j.jprot.2011.05.023 [DOI] [PubMed] [Google Scholar]