

Abstract
Motivation
Many methods for microbial protein subcellular localization (SCL) prediction exist; however, none is readily available for analysis of metagenomic sequence data, despite growing interest from researchers studying microbial communities in humans, agri-food relevant organisms and in other environments (e.g. for identification of cell-surface biomarkers for rapid protein-based diagnostic tests). We wished to also identify new markers of water quality from freshwater samples collected from pristine versus pollution-impacted watersheds.
Results
We report PSORTm, the first bioinformatics tool designed for prediction of diverse bacterial and archaeal protein SCL from metagenomics data. PSORTm incorporates components of PSORTb, one of the most precise and widely used protein SCL predictors, with an automated classification by cell envelope. An evaluation using 5-fold cross-validation with in silico-fragmented sequences with known localization showed that PSORTm maintains PSORTb’s high precision, while sensitivity increases proportionately with metagenomic sequence fragment length. PSORTm’s read-based analysis was similar to PSORTb-based analysis of metagenome-assembled genomes (MAGs); however, the latter requires non-trivial manual classification of each MAG by cell envelope, and cannot make use of unassembled sequences. Analysis of the watershed samples revealed the importance of normalization and identified potential biomarkers of water quality. This method should be useful for examining a wide range of microbial communities, including human microbiomes, and other microbiomes of medical, environmental or industrial importance.
Availability and implementation
Documentation, source code and docker containers are available for running PSORTm locally at https://www.psort.org/psortm/ (freely available, open-source software under GNU General Public License Version 3).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Ever since, PSORTb was first introduced in 2003, it has remained one of the most precise, widely used protein subcellular localization (SCL) predictors available (Gardy et al., 2003, 2005; Yu et al., 2010). The initial version of PSORTb predicted protein SCL for Gram-negative bacteria. PSORTb 3.0 onwards generates predictions for all the main types of prokaryotic cell structures: archaea, traditional Gram-positive bacteria (Gram-positive without an outer membrane), traditional Gram-negative bacteria (Gram-negative with an outer membrane), Gram-positive bacteria with an outer membrane and Gram-negative bacteria without an outer membrane. PSORTb 3.0 was also the first SCL predictor to include localization subcategories (host-associated, type III secretion, fimbrial, flagellar and spore).
In addition to PSORTb, there are a variety of other SCL prediction tools that have been developed (see https://www.psort.org/). A distinction can be made between tools that perform specialized predictions of one or a few SCLs, such as SignalP (Petersen et al., 2011), or tools that can make broad predictions of multiple SCLs, such as ProteomeAnalyst (Szafron et al., 2004). Several methods like PSORTb, such as Gpos-ECC-mPLoc and Gneg-ECC-mPLoc (Wang et al., 2015), are able to deal with proteins with multiple localizations.
Computational prediction is a relatively rapid and inexpensive alternative to experimental methods for determining microbial protein SCL. It aids in identification of protein function and annotation of genomes, plus the identification of cell surface/secreted proteins for applications, such as the development of ELISA-based diagnostic tests or identification of drug targets or vaccine components. Despite the number of microbial SCL prediction methods that have been developed, there is a notable lack of methods designed specifically to work with metagenomic sequences. One method exists, MetaP (Luo et al., 2009), however, it assumes all sequences are from Gram-negative organisms, and is not made readily available for online use or download except for potentially through contact with the authors. Metagenome-assembled genomes (MAGs) can be analyzed using PSORTb, but each MAG needs to have its cell envelope type known or predicted (i.e. classic Gram-negative, Gram-negative without an outer membrane, etc.), and unassembled sequences are missed in such an analysis (Lau and Maguire et al., 2019; Maguire et al., unpublished data). Thus, we developed PSORTm, a PSORTb-derived program to enable direct-from-reads-based SCL classification, as well as membrane-type (cell envelope type) classification, for metagenomic sequences. Due to the package complexity, Docker images are available (for running via a command line, or a web interface; https://www.psort.org/psortm/). PSORTm maintains the high precision of PSORTb, with increasing sensitivity as input fragment lengths get larger, and is the first bioinformatics tool enabling more automated SCL analysis from metagenomics data, for all main cell envelope types.
2 Materials and methods
2.1 Software implementation
An overview of PSORTm, comparing it with PSORTb, is schematically shown in Figure 1.
Fig. 1.
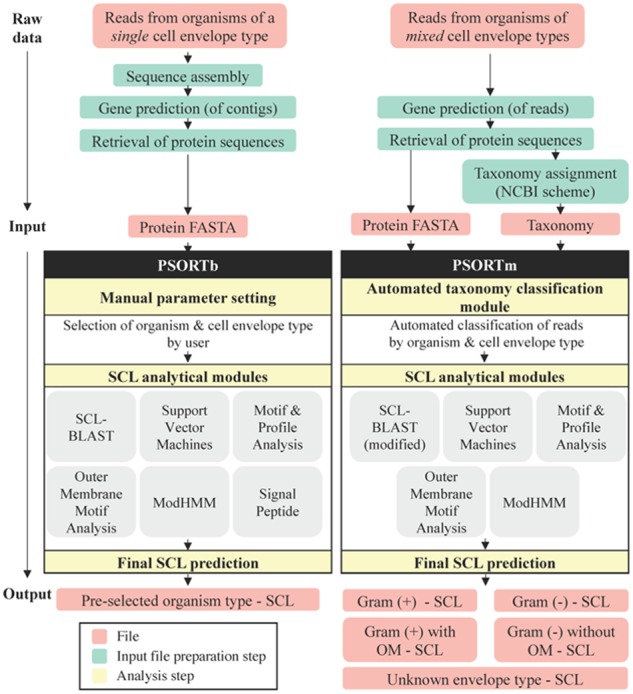
Comparison of PSORTb 3.0.2 and PSORTm 1.0.2 pipelines, from raw data to final SCL prediction. PSORTb (left) requires a protein sequence file, in FASTA format, as input which can be generated from sequence reads of a single genome or multiple genomes from the same organism type with the same membrane structure. It also requires knowledge of cell envelope structure as a manual analysis step. PSORTm (right) is a more automated process, requiring two input files generated from metagenomics reads (can be from genomes with mixed organism and membrane type): (i) a protein sequence file in FASTA and (ii) a taxonomy file containing either the NCBI taxonomic name/ID of the reads in the corresponding protein sequence file. PSORTm contains an additional automated taxonomy classification module to sort input reads by organism type and cell envelope type prior to running the SCL prediction modules. PSORTm implements similar SCL analytical modules used in PSORTb, with the following notable differences: (i) length restriction is removed in the SCL-BLAST module (or else most reads would not have predictions) and (ii) the signal peptide module is removed. While PSORTb generates a single output file of SCL predictions, PSORTm generates five SCL prediction files—one for reads from each of the four cell envelope types and one for reads with an unclassified type
Input files
For SCL prediction, existing widely used programs like PSORTb require a FASTA protein file (usually generated through separately performed annotation of a genome or other sequence), and knowledge of the species associated with the protein(s), including the cell envelope structure (i.e. the user must choose if the species is classic Gram-positive, Gram-negative, etc.). PSORTm similarly requires this information in two input files, a protein sequence file and a taxonomy file, but enables automated classification of cell envelope type. For PSORTm, the protein sequence file, in FASTA format, contains one or more read-derived protein sequence fragments from organisms with different potential cell envelope structure (Gram-positive, Gram-negative, Gram-positive with outer membrane or Gram-negative without an outer membrane). The taxonomy file contains the sequence IDs of each sequence from the corresponding protein sequence fragment file and their associated NCBI taxonomy name (e.g. Pseudomonas) or taxID (e.g. 286).
To generate a read-based protein sequence file for PSORTm, protein-coding sequences first need to be identified from raw reads, using assembly-free gene prediction tools designed for metagenomics sequences, such as MetaProdigal (Hyatt et al., 2012; Joshi and Fass, 2011 ), FragGeneScan (Rho et al., 2010) or Glimmer-MG (Kelley et al., 2012).
A taxonomy file is frequently already generated through other metagenomics analyses, but can be generated using DIAMOND (v0.9.25 or higher versions; Buchfink et al., 2015) and Kaiju (Menzel et al., 2016). These tools compare read sequences from metagenomic datasets to a reference database of microbial proteins, such as NCBI RefSeq or nr databases, to assign taxonomy to reads based on sequence similarity. The input taxonomy file should be formatted as a tab-delimited file comprising the read ID followed by the NCBI taxonomy ID or taxa name.
The read-based protein sequence file and the taxonomy file are then analyzed by two sets of modules in PSORTm: (i) a cell envelope classification module, followed by (ii) a set of SCL prediction modules, appropriately chosen for analysis of a set cell envelope type, as described below.
Organism and membrane-type classification module
A taxonomic-based cell envelope classification tool was incorporated into PSORTm as a critical step, since it is usually not feasible to manually identify the cell envelope structure for each taxon in a metagenomics dataset. It automatically sorts input sequences according to type of organism and membrane structure: archaea, Gram-negative bacteria, Gram-positive bacteria, Gram-negative bacteria without an outer membrane and Gram-positive bacteria with an outer membrane. This tool utilizes the two input files of read-based protein sequences along with their associated taxonomic classification to generate a temporary output file of reads sorted into the five aforementioned categories, as well as an additional file of reads that could not be classified.
The cell envelope categorization scheme is derived from an approach, we previously developed and then improved to enable pre-computed PSORTb SCL analyses of complete microbial genomes (Peabody et al., 2016; Rey et al., 2005; Yu et al., 2011 ). This classification tool uses a combination of NCBI genomes with curated phylum taxonomy and marker protein sequences, specific to certain cell envelopes (e.g. the Omp85-type outer membrane protein is the only essential outer membrane protein found in all classic Gram-negative bacteria, and so acts as a marker for that cell envelope structure). The marker sequences are used to categorize newly sequenced complete bacterial and archaeal genomes into organism/cell envelope categories so that the appropriate set of SCL analytical modules could be chosen. These data have now been used to curate the Gram stain and cell membrane structure of taxa with NCBI genomes. Using this resource, this PSORTm module was developed to assign organism and cell membrane/envelope type to each metagenomic read based on the corresponding taxonomy. For reads from which the source organism is novel, PSORTm uses the Omp85 and cutinase protein markers to predict cell structure. Omp85 aids differentiation between classic Gram-negative and Gram-positive bacteria. The cutinase protein is a signature marker for Corynebacteriales, which are found in the classic Gram-positive phylum of Actinobacteria but contain a non-classic waxy outer membrane that is resistant to Gram-staining or is Gram-variable/acid fast. The cutinase detector within this PSORTm module identifies reads from which the source organism contains a Corynebacteriales outer membrane, indicating an unusual Gram-positive structure with an outer membrane and distinguishing it from the classic Gram-positive membrane.
Using this organism and membrane-type classification module, we could globally assign taxa to particular major cell envelope categories. Then, this is used to analyze the read-based protein sequence using the appropriate modules for its deduced cell envelope structure.
SCL prediction modules
PSORTm SCL prediction modules are adapted from PSORTb 3.0 (Yu et al., 2010), with two modules removed or modified (Table 1). PSORTm did not incorporate PSORTb’s signal peptide module, which predicts a protein as cytoplasmic or non-cytoplasmic based on the absence or presence of an N-terminal signal peptide, respectively, Protein sequences derived from metagenomic sequences may start anywhere within the protein, so the first amino acids of a sequence may not reflect the N-terminal amino acids, hence the module would not be effective. The SCL-BLAST module from PSORTb was modified by removing the original restriction that the query sequence must be within 80–120% of the length of the subject protein, to reduce errors due to the domain nature of proteins. This would have been too restrictive for metagenomic fragments, many of which would not meet the 80% length-of-the-protein cut-off, so the length restriction was not implemented in PSORTm.
Table 1.
List of modules used in PSORTb 3.0.2, and whether they were incorporated or modified in PSORTm 1.0.2
| Module | Features used for prediction | SCLs predicted | Incorporated | Modified |
|---|---|---|---|---|
| Signal peptide | N-terminal signal peptide | Non-cytoplasmic | No | — |
| SVMs | Frequent subsequences within protein sequences | All SCLs | Yes | No |
| ModHMM | Transmembrane α-helices | CM | Yes | No |
| Motif | SCL-associated motifs | All SCLs | Yes | No |
| Profile | SCL-associated motifs | All SCLs | Yes | No |
| Outer membrane motifs | Motifs associated with β-barrel OM proteins | OM | Yes | No |
| SCL-BLAST | Homology | All SCLs | Yes | Yes |
SMV, support vector machine; OM, outer membrane; CM, cytoplasmic membrane.
The remaining PSORTb analytical modules were implemented in PSORTm without modifications. The support vector machine (SVM) module consisting of 13 machine learning-based classifiers, one for each Gram-negative and Gram-positive localization site, is included to classify whether a protein belongs to a specific SCL. The hidden Markov model-based ModHMM module identifies proteins spanning the cytoplasmic membrane through the detection of transmembrane alpha helices. The PROSITE module scans the query sequences for the presence of known protein motifs precisely indicative of specific SCL sites. The Profile module similarly detects localization-specific profiles in the query sequences. Finally, the outer membrane motif module classifies a protein as outer membrane or non-outer membrane, based on the presence/absence of motifs associated with beta-barrel proteins that were previously identified using a frequent subsequences data mining approach.
Final SCL output
The final SCL prediction is generated by combining and assessing the results from each of the analytical modules. A naïve Bayes classifier is used, generating a probability score, ranging from 0 to 10, of a protein being at a specific SCL given the prediction of a certain module. A localization can be assigned to a protein given the probability score is 7.5 or above. If the localization site has a lower score, between 4.5 (for Gram-negative) and 5.0 (for Gram-positive) and 7.49, the final prediction will yield ‘Unknown—predicted localization does not exist’. However, prediction outputs from the individual analytical modules can still be examined by the user to draw a conclusion. In some cases, more than one localization site may exhibit high score, indicating the protein may be present in multiple (neighboring) localization sites, such as a protein with domains in periplasm and outer membrane.
The output tab-delimited file is available in either the terse (short) format or the long format. The terse output file returns a list of input sequences, one per row, with their corresponding PSORTm results in the columns. This format contains three columns: sequence read ID, final prediction of localization site and the score for the SCL prediction. The long format contains all the details in the terse format, with the additional localizations and scores from each of the individual prediction modules.
2.2 Training dataset
The training dataset of proteins of experimentally known localization used to evaluate PSORTb 3.0.2 was also applied to PSORTm. The full dataset (available at https://www.psort.org/dataset/datasetv3.html) is comprised of 8230 Gram-negative proteins, 2652 Gram-positive proteins and 810 archaeal proteins, based on experimental data and literature curation (including Rey et al., 2005; Wu et al., 2006). To simulate metagenomics fragments, sequences were randomly fragmented in silico from lengths 60 to 450 in increments of 30. For each of these fragment lengths, fragments were generated 10 times.
2.3 Watershed discovery datasets
Shotgun metagenomics sequencing and bacterial 16S rRNA (V3–V4 hypervariable region) amplicon sequencing datasets from the Watershed Discovery Project (http://www.watersheddiscovery.ca/; Supplementary Table S1) were also used to evaluate PSORTm and demonstrate its utility in an analysis of real data.
2.4 Software evaluation
Five-fold cross-validation
PSORTm 1.0.2 was evaluated by a 5-fold cross-validation approach described in the PSORTdb 3.0 paper (Yu et al., 2010). In brief, the training dataset was randomly split into five subsets, of which four were used for training and construction of the SCL analytical modules and the remaining subset was reserved for testing. Performance metrics used for evaluating PSORTm include precision, defined as TP/(TP+FP), and sensitivity (also known as recall), defined as TP/(TP+FN), where TP, FP and FN represent the number of true positives, false positives and false negatives, respectively.
Performance test
The run time of PSORTm 1.0.2 was assessed as a function of the number of reads using a randomly chosen sample of real metagenomics data from the Watershed Discovery Project. The complete sequence file for the sample was repeatedly split into 2, 4, 6, 8, 16, 32 and 64 parts separately. In each set of split reads, all or some of the subsets were analyzed by PSORTm. For example, while all the halved and quartered read subsets were analyzed by PSORTm, only selected subsets containing 1/64 of the original reads were analyzed by PSORTm. Run time was compared among the various number of input sequences and also between the generation of the short- and long-output formats.
2.5 Comparison of SCL prediction by PSORTm 1.0.2 and PSORTb 3.0.2
A simulated metagenomics dataset was generated by selecting 30 NCBI RefSeq genomes, in April 2019, with one of the following criteria: high-plasmid count, high-genomic island (GI) count or low GI (O’Leary et al., 2016). Genome abundance and plasmid copy number were then randomly assigned a relative abundance following a log-normal distribution and a scaled gamma distribution, respectively. Sequences were subsequently concatenated into a single FASTA file with the appropriate relative abundance. MiSeq v3 250 bp paired-end reads with a mean fragment length of 1000 bp and SD of 50 bp were simulated using art_illumina v2016.06.05 (Huang et al., 2012) at a fold coverage of 2.9, resulting in 31 174 411 read pairs.
In preparation of input reads for PSORTm, the simulated set of metagenomics reads were trimmed and filtered to remove duplicate reads using Trimmomatic with default parameters (Bolger et al., 2014 ). Open-reading frames (ORFs) were directly predicted from the processed reads using Prodigal in metagenome mode (Hyatt et al., 2012). The resultant deduced protein sequences were filtered once again to remove duplicates. Taxonomic assignment of reads was performed by DIAMOND, based on similarity search against NCBI’s nr database. The processed, deduced protein sequences with their taxonomic assignment were inputted into PSORTm for read-based SCL prediction.
To construct MAGs, the simulated metagenomics sequences were trimmed using sickle (v1.33; https://github.com/najoshi/sickle) (Joshi and Fass, 2011), assembled using metaSPAdes (v3.13.0) (Nurk et al., 2017) and binned using DAS Tool (v1.1.1) (Sieber et al., 2018). Prodigal in default mode was used for ORF prediction in MAGs and the predicted protein sequences were inputted into DIAMOND for taxonomic assignment. The MAG-derived protein sequences and their manually determined taxonomic assignment were inputted into PSORTb for MAG-based SCL predictions.
For both MAGs and the reads, antimicrobial resistance genes were predicted using the list of deduced proteins and the Resistance Gene Identifier using the default parameters (Alcock et al., 2020).
3 Results
3.1 Five-fold cross-validation
PSORTm shows similar or substantially higher sensitivity than PSORTb at all the available localization sites for archaea, Gram-negative and Gram-positive bacteria (Fig. 2). This is due in part to the removal of the length restriction in the original SCL-BLAST module in PSORTm. PSORTm enables SCL assignment of proteins which fall outside of 80%–120% of length of the subject proteins from the database of proteins of known SLC used in this module. Although these proteins would likely have the correct SCL prediction (true positives), they would not be assigned an SCL by PSORTb’s SCL-BLAST module due to the failure to meet the sequence-length requirement. Therefore, sensitivity (proportion of identified true positives) of PSORTm will likely be higher than that of PSORTb. Sensitivity tends to increase with increasing fragment length, whereas precision (proportion of true positives in all predicted positives) tends to stay consistently high and not shows a clear trend in relation to fragment length (Fig. 3).
Fig. 2.
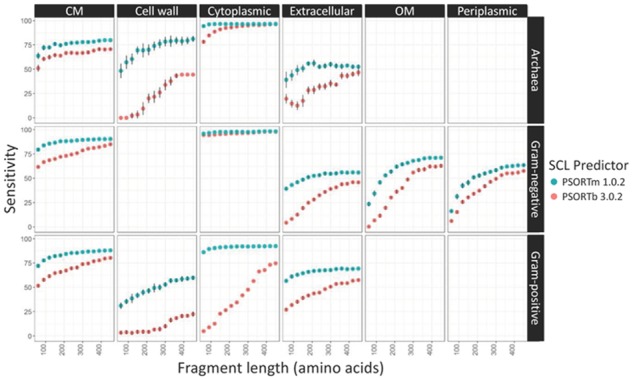
Five-fold cross-validation of PSORTm sensitivity over differing organism type, SCL, and sequence fragment length. PSORTm 1.0.2 has higher sensitivity than PSORTb 3.0.2, and sensitivity tends to increase with increasing fragment length. Error bars show SD, fragment lengths were subsampled 10 times. CM, cytoplasmic membrane; OM, outer membrane
Fig. 3.
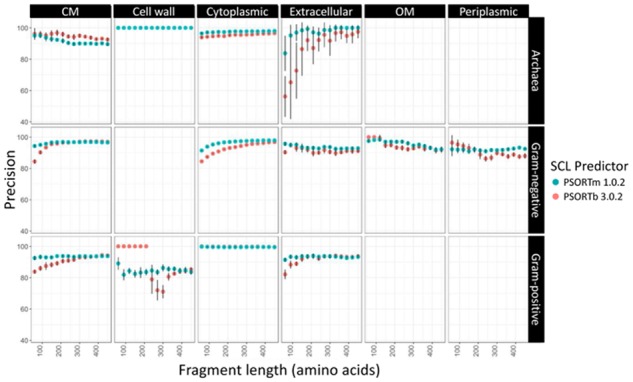
Five-fold cross-validation of PSORTm 1.0.2 precision over differing organism type, SCL, and sequence fragment length. Precision remains consistently high in PSORTm. Error bars for some categories are larger due to the smaller number of these proteins in the test data
3.2 Performance test
PSORTm 1.0.2 performance was compared among different sizes of the input dataset: full dataset (418 500 reads) or a half, a quarter, one-eighth, one-sixteenth, one-thirty-second and one-sixty-fourth of the dataset. Run time increased linearly with the number of input reads (Fig. 4). Also, there was no difference in the time taken to generate the terse (short) or the long-output files, suggesting users can choose to obtain a more detailed analysis report at no additional time cost.
Fig. 4.
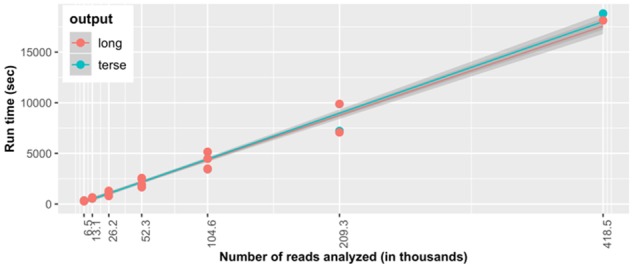
Run time assessment of PSORTm 1.0.2. Comparison of run time in seconds as a function of number of analyzed reads from the randomly selected watershed discovery sample. Each point represents each input dataset (of different numbers of reads). Trend lines indicate run time increases linearly for both long and terse formats
3.3 Comparison of PSORTm 1.0.2 to PSORTb 3.0.2
Using a simulated metagenomic dataset from 30 NCBI RefSeq bacterial genomes, SCL predictions from metagenomic reads using PSORTm 1.0.2 and from MAGs using PSORTb 3.0.2 were compared. The proportion of predicted localizations followed a similar trend between read-based PSORTm and MAG-based PSORTb analyses (Fig. 5). Results were also comparable to the results from PSORTb ran on the reference dataset containing proteins from the 30 genomes used to construct the simulated dataset. This comparison suggests that PSORTm 1.0.2 is able to predict bacterial protein SCLs directly from metagenomic reads while maintaining a similar performance as PSORTb 3.0.2.
Fig. 5.
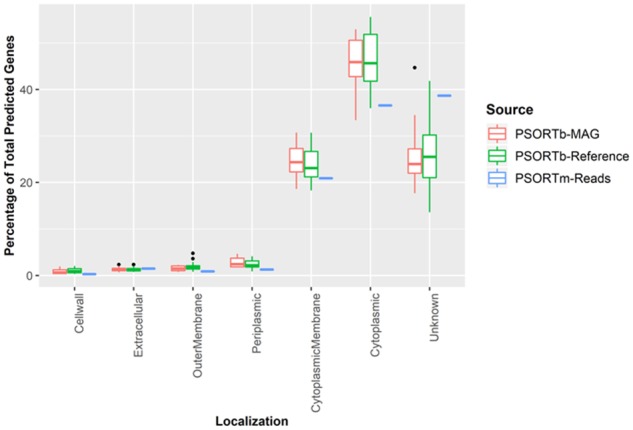
Distribution of SCL predictions from simulated metagenomic reads by PSORTm 1.0.2 and MAGs by PSORTb 3.0.2. PSORTb-MAG refers to PSORTb localization results from protein sequences predicted in the simulated MAGs. PSORTb-Reference refers to PSORTb localization results from protein sequences predicted from the 30 RefSeq bacterial genomes used in the simulated metagenomic dataset. PSORTm-Reads refer to PSORTm prediction from protein sequences directly deduced from the simulated metagenomic reads. Comparison of run time in seconds (top) and number of taxonomic assignment (bottom) as functions of the fraction of reads from the randomly selected watershed discovery sample. Number of taxonomic assignments corresponds to the number of reads successfully analyzed by PSORTm
3.4 Analysis of watershed datasets
Analysis of the watershed samples, to demonstrate an analysis with real-read data, revealed the importance of normalization and identified potential biomarkers of water quality. A full analysis of this watershed dataset using PSORTm can be found in the Supplementary Materials.
4 Discussion
We have developed PSORTm, a novel SCL prediction tool derived from PSORTb 3.0.2, with implementation of an automated cell envelope classifier, to enable automated analysis of proteins encoded by metagenomic sequences for the first time. PSORTm is unique in that it is both optimized for unassembled reads, and is able to automatically predict SCL from mixed organisms of different envelope types, as is characteristic in a metagenomics dataset. PSORTb analyzes complete protein sequences from organisms of one cell envelope type at a time, and the appropriate cell envelope type must be manually chosen to enable the analysis. PSORTm performs well versus PSORTb, maintaining a high level of precision over a range of fragment lengths. Sensitivity also generally remains high, tending to show a modest improvement as input fragment length increases. However, certain categories of localization benefited much more from increased fragment lengths, such as outer membrane proteins in Gram-negative bacteria. Sensitivity increased from 25% to almost 75% as input fragment length increased from 60 to 450 amino acids, demonstrating the value of longer sequence reads—which can be reasonably achieved with the improved lengths of single or paired-end reads generated by current next-generation sequencing platforms. For MAGs (Parks et al., 2017), PSORTb or other sequence-assembly-dependent protein SCL predictors can be used. However, it must be emphasized that PSORTm, as a read-based SCL predictor, provides an important complement to MAG-based PSORTb analysis. Assembly-based methods can miss SCL or gene predictions in unassembled reads, and MAGs are prone to false predictions from chimeras resulting from incorrect assembly during MAG construction (Lau et al., 2019; Maguire et al., unpublished data). For example, we identified the gene EmrA in our synthetic reads but not MAGs. EmrA is a membrane localized efflux pump subunit responsible for macrolide resistance. This example highlights the importance of using a read-based method (e.g. PSORTm) to complement draft genome/MAG-based methods (e.g. PSORTb applied to MAGs manually classified by cell envelope). We foresee the need in the future to make a separate cell envelope-prediction tool to enable more automated classification of MAGs as well, as combinations of MAGs and read-based analyses, using both long- and short-read sequence technologies, become more commonly used for robust metagenomic analyses.
5 Conclusion
PSORTm is the first readily available protein SCL predictor designed for metagenomic sequences for all the main cell envelope types, with open-source code freely available, and Docker images for running locally (through the command line or a web interface) due to the package complexity and large size of metagenomics datasets commonly analyzed. It maintains high precision across a wide range of sequence lengths. The primary utility of this assembly-free tool is to enable SCL prediction from short reads (i.e. Illumina sequences), which are currently most commonly used by public health agencies worldwide, and to enable analysis of more complex microbiome environments where MAGs may be challenging to assemble. MAG-based analysis can also miss key genes in their assemblies, and requires manual assignment of classification of cell envelope type as is necessary for protein localization prediction. PSORTm has many potential applications, such as in the identification of cell-surface based biomarkers for protein-based diagnostic tests, or to aid annotation or identification of potential vaccine components. PSORTm should complement PSORTb, aiding in a wide range of microbial community analyses of medical, agricultural (i.e. agri-foods pathogen monitoring) or environmental interest.
Supplementary Material
Acknowledgements
The authors acknowledge the Simon Fraser University (SFU) Research Computing Group for IT support and the researchers involved in the Watershed Discovery Project http://www.watersheddiscovery.ca/.
Funding
This work was primarily supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) RGPIN grant to F.S.L.B., with additional financial support by Genome Canada/Genome BC and Simon Fraser University.
Conflict of Interest: none declared.
References
- Alcock B. P. et al. (2020) CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acid Research, 48, D517–D525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M. et al. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B. et al. (2015) Fast and sensitive protein alignment using DIAMOND. Nat. Methods, 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Gardy J.L. et al. (2003) PSORT-B: improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res., 31, 3613–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardy J.L. et al. (2005) PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics, 21, 617–623. [DOI] [PubMed] [Google Scholar]
- Huang W. et al. (2012) ART: a next-generation sequencing read simulator. Bioinformatics, 28, 593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D. et al. (2012) Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics, 28, 2223–2230. [DOI] [PubMed] [Google Scholar]
- Joshi,N.A. and Fass,J.N. (2011) Sickle: a sliding-window, adaptive, quality-based trimming tool for FASTQ files (version 1.33). Available at: https://github.com/najoshi/sickle. [Google Scholar]
- Kelley D.R. et al. (2012) Gene prediction with Glimmer on metagenomic sequences augmented by phylogenetic classification and clustering. Nucleic Acids Res., 40, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau W.Y., Maguire F.. et al. (2019) Adapting predictive genome-scale bioinformatic approaches to the challenges of metagenomic data. In: Applied Bioinformatics for Public Health Microbiology Conference. 5–7 June 2019. Hinxton, UK.
- Luo H. et al. (2009) Subcellular localization of marine bacterial alkaline phosphatases. Proc. Natl. Acad. Sci. USA, 106, 21219–21223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel P. et al. (2016) Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun., 7, 11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S. et al. (2017) metaSPAdes: a new versatile metagenomic assembler. Genome Res., 27, 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary N.A. et al. (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res., 44, D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks D.H. et al. (2017) Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol., 2, 1533–1542. [DOI] [PubMed] [Google Scholar]
- Peabody M.A. et al. (2016) PSORTdb: expanding the bacteria and archaea protein subcellular localization database to better reflect diversity in cell envelope structures. Nucleic Acids Res., 44, D663–D668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen T.N. et al. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods, 8, 785–786. [DOI] [PubMed] [Google Scholar]
- Rey S. et al. (2005) PSORTdb: a database of subcellular localizations for bacteria. Nucleic Acids Res., 33, D164–D168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho M. et al. (2010) FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res., 38, e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber C.M.K. et al. (2018) Recovery of genomes from metagenomics via a dereplication, aggregation and scoring strategy. Nat. Microbiol., 3, 836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafron D. et al. (2004) Proteome Analyst: custom predictions with explanations in a web-based tool for high-throughput proteome annotations. Nucleic Acids Res., 32, W365–W371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. et al. (2015) Multi-location gram-positive and gram-negative bacterial protein subcellular localization using gene ontology and multi-label classifier ensemble. BMC Bioinformatics, 16 (Suppl. 12), S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.H. et al. (2006) The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res., 34, D187–D191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu N.Y. et al. (2010) PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics, 26, 1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu N.Y. et al. (2011) PSORTdb–an expanded, auto-updated, user-friendly protein subcellular localization database for Bacteria and Archaea. Nucleic Acids Res., 39, D241–D244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.