

Abstract
Motivation
The proteomics field requires the production and publication of reliable mass spectrometry-based identification and quantification results. Although many tools or algorithms exist, very few consider the importance of combining, in a unique software environment, efficient processing algorithms and a data management system to process and curate hundreds of datasets associated with a single proteomics study.
Results
Here, we present Proline, a robust software suite for analysis of MS-based proteomics data, which collects, processes and allows visualization and publication of proteomics datasets. We illustrate its ease of use for various steps in the validation and quantification workflow, its data curation capabilities and its computational efficiency. The DDA label-free quantification workflow efficiency was assessed by comparing results obtained with Proline to those obtained with a widely used software using a spiked-in sample. This assessment demonstrated Proline’s ability to provide high quantification accuracy in a user-friendly interface for datasets of any size.
Availability and implementation
Proline is available for Windows and Linux under CECILL open-source license. It can be deployed in client–server mode or in standalone mode at http://proline.profiproteomics.fr/#downloads.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
MS-based proteomics, and especially high-throughput bottom-up approaches, have reached a level of maturity compatible with global relative quantification of thousands of proteins in just a few hours (Doll et al., 2017; Mann et al., 2013; Rieckmann et al., 2017). Alongside continuous improvements to instruments, the development of open source and proprietary data-processing software now allows quantitative comparison of proteomes from samples produced in varying biological or physiopathological conditions (Deutsch et al., 2008; Mueller et al., 2008; Nahnsen et al., 2013). However, the intrinsic complexity of bottom-up proteomics experiments requires the use of elaborate algorithms (aggregation of precursor/fragment ion data into protein information, and matching of these data across samples to be compared). These algorithms may introduce inaccuracies and errors throughout the data-processing pipeline. As a result, there is room for improvement in the different steps in identification and quantification pipelines; result reliability must also be carefully assessed, not only by statistically controlled procedures, but also through examination of the underlying data by experts. Thus, although comprehensive and reliable results should be produced through an automated process, it is also very important to be able to manually verify, validate and curate erroneous identification and/or quantification results using a dedicated graphical user interface.
Proteomics experiments are moving toward more complex and ambitious studies, not only based on the analysis of a single fractionated sample, but also composed of multiple datasets with hundreds of samples providing a broad understanding of a biological system from a variety of viewpoints (Aebersold and Mann, 2016). However, despite recent improvements in the field, through the development of free software algorithms and tools to tackle this level of complexity, there is still a need for a laboratory information management system dedicated to MS-based proteomics results that could implement the core data-processing steps (validation, quantification, visualization and submission to repositories), and at the same time could provide an organized and sustainable data persistence system allowing users to explore, compare and automatically or manually validate multiple datasets.
With all these different goals in mind, we developed Proline, a production grade software suite, which provides a unique environment for large-scale MS data management, visualization, analysis and curation with the main objective of promoting the production and sharing of high-quality proteomic datasets. Proline can be used (i) to produce reliable identification and quantification results through robust automated processes, (ii) for data curation, (iii) to systematically save and keep track of metadata from processing steps, parameters and generated data and (iv) to submit highly qualified datasets to public repositories (Vizcaíno et al., 2009, 2014). Altogether, these features make Proline perfectly fitted to the needs of proteomics core facilities and research labs producing a huge amount of data at high throughput.
2 Materials and methods
In the current version of Proline, the standard workflow is mainly focused on validation of peptide and protein identifications, and label-free quantification of those peptides and proteins based on spectral counting or MS1 peak ion intensity. A workflow in Proline is implemented as a collection of tasks (see Fig. 1) that can be performed by the user through the graphical user interface (Supplementary Fig. S1). The more important tasks are detailed in the following paragraphs. In a classical case, users can import multiple identification results corresponding, e.g. to fractions and replicates of a biological sample and combine them before or after validation. The resulting datasets can then be compared or quantified using spectral counting or data dependent acquisition (DDA) label-free quantification, before exporting the results in different file formats. All results are persisted in dedicated relational databases (see Supplementary Section ‘Software architecture’).
Fig. 1.

Schematic representation of the scope of the application: input and output data are represented by gray boxes; tasks which are steps in the data analysis process are represented in blue. Proline provides a set of predefined tasks (dark blue) that can be executed and the paths linking the tasks defines analysis workflows
2.1 Proline main tasks
2.1.1 Import
Identification results files produced by several search engines (Mascot, X! Tandem, OMSSA and Andromeda) can be imported into Proline in their native format. In addition, the mzIdentML format is supported to allow the output from any other search engine compatible with this standard to be imported (e.g. MS-GF+). During this step, no filtering or thresholding is applied: along with the search parameters, all submitted spectra, peptide spectrum matches (PSMs) and protein hits suggested by the search engine are retained in the Proline database to allow subsequent validation of putative identifications. Imported data are organized into search results that can be browsed in Proline’s graphical user interfaces before validation.
2.1.2 Search result validation and protein set inference
Validation can be independently performed at PSM, peptide and protein levels. A set of predefined filters (see Supplementary Table S1) can be applied to automatically accept or reject a PSM or a protein set based on user-defined threshold values applied to various criteria such as score, rank or peptide length for PSM, and score or peptide count for protein sets. Target-decoy validation can also be performed by adjusting the false discovery rate at each level to attain a user-defined value. As identification results can be combined (see Section 2.1.3, below), multiple PSMs matching a single peptide (characterized by a sequence and a list of post-translational modifications and their localization on the sequence) can be grouped together. In this particular case, validation at PSM level is equivalent to validation at peptide level.
Validated peptide sequences must be clustered to infer the protein content of the analyzed sample. This protein inference step produces a list of protein sets. Each protein set (a.k.a. protein group) represents a set of potentially identified proteins sharing the same set or a subset of peptides (Nesvizhskii and Aebersold, 2005). Proline uses the widely adopted parsimonious strategy, otherwise known as Occam’s razor (Nesvizhskii et al., 2003), which consists in determining the shortest list of protein sets explaining the list of peptide sequences observed. To do so, Proline compares the sets of peptide sequences that were mapped on FASTA entries by the search engine, and classifies them in supersets and subsets in function of the sequence specificity of the observed peptides. Each set of peptides is finally associated to one or multiple indistinguishable FASTA entries (a.k.a. protein sets). Note that this classification relies only on sequence comparison and that PTM information (such as N-terminal modifications) is not taken into consideration.
In Proline, the whole list of validated PSMs and their corresponding protein sets is called an identification summary. Each summary is persisted in the Proline database for immediate visual inspection. The summary also serves as a checkpoint for subsequent data processing, such as combination of multiple identification summaries into a single dataset, or quantification of the identified and validated species.
2.1.3 Combining identifications
In Proline, identification results can be combined to construct a parent dataset, and create a non-redundant list of identified peptides and proteins. This combination can be performed either before validation (on search results) or after validation (on identification summaries). Since this operation could be recursively performed, it leads to hierarchical structuring of search results and/or identification summaries. On the one hand, combination before validation (taking into account all PSMs identified by the search engine) may, e.g. be relevant when analyzing results obtained after peptide fractionation: in that case, several peptides belonging to the same protein may be spread across different result sets; these sets should be merged before protein validation. On the other hand, merging identification summaries is appropriate when seeking to group the validated results from series of individual samples to be compared or when combining data from different search engines. As Proline does not rank peptides identified using different search engines, unlike other types of software which compute a metascore (Shteynberg et al., 2013; Vaudel et al., 2015), search results from different search engines should not be combined before validation. Except in this latter case, either combining datasets before or after validation could be used depending on the user’s needs.
In both cases, for a given peptide, all PSMs originating from the initial datasets can be conserved (‘union’ mode), or only the PSM of highest score among all the combined search results can be retained (‘aggregation’ mode). This allows the user to validate the merged dataset either at PSM or peptide level (since each unique peptide is represented by a single PSM in aggregation mode, as shown in Supplementary Fig. S3). As a consequence, this will also influence the calculation of the protein standard score (see Supplementary Section ‘Search results validation’).
2.1.4 Spectral counting
Protein sets from different identification datasets can be compared in Proline by counting MS/MS spectra at peptide and protein set levels. Three different spectral counting metrics are calculated for each protein set: the first one considers all identified peptides, the second one examines only specific peptides (peptides identifying a single protein set), and the third takes shared peptides into account (Hesse et al., 2016). In the latter case, shared spectral occurrences are distributed across protein sets based on a weighting factor calculated using the proportion of unique peptides associated with each protein set.
2.1.5 MS1 peptide quantification
More accurate comparisons can also be made by quantifying MS1 signals. Proline detects chromatographic peaks from raw data converted to the mzDB format (Bouyssié et al., 2015). The converter (https://github.com/mzdb/pwiz-mzdb) is based on ProteoWizard, ensuring a compatibility with a wide range of instrument vendors. The following list of file formats can be used as input: Thermo Raw files, AB Sciex Wiff files, Bruker Baf files and mzML files.
After a first signal detection step (see Supplementary Section ‘Signal extraction from mzDB files’), the algorithm associates the chromatographic peaks detected with validated PSMs, first by retrieving the corresponding MS/MS spectra acquired during the peptide elution (i.e. within the detected chromatogram boundaries), and then by matching the precursor m/z value of these spectra to the chromatographic peak m/z value (see Supplementary Section ‘PSM assignment and deisotoping’). After the deisotoping step, the abundance of each ion is estimated from the apex of the chromatographic peak, which corresponds to the theoretically most abundant isotopolog (inferred from the peptide’s atomic composition). The software then aligns the retention time of these annotated ions for all the LC-MS runs to be compared, and uses this information to cross-assign MS/MS data to ions (i.e. chromatographic peaks) that were detected but not identified in other runs (see Supplementary Section ‘Cross-assignment’). The resulting ion abundances are finally stored in the Proline database, making them available for rapid data visualization and further post-processing.
2.1.6 Protein quantification
Finally, peptide ion measurements can be summarized as protein abundances using different computational methods (see Supplementary Section ‘Protein quantification’). The user can opt to perform additional operations such as excluding peptides or ions based on their characteristics (missed cleavages, variable modifications, sequence specificity etc.) or normalizing peptide and protein abundances between runs. These post-processing steps can be executed on-demand using different parameters or methods; there is no need to repeat the whole quantification process when changes are made.
2.1.7 Export
Metadata related to all processing steps, such as parameter values or thresholds computed by the algorithms, are recorded in Proline, and included when exporting the results (along with annotated MS/MS spectra) in standard-compliant formats for publication of data in public repositories such as PRIDE and ProteomeXchange. Identification and quantification results can be exported as text or .xlsx files, at peptide ion, peptide or protein level. In the context of PTM analysis, localization confidence values are also exported for peptide ions, if MS/MS searches were performed with Mascot (Savitski et al., 2011).
2.2 Experimental datasets
The dataset used to evaluate the software was prepared by spiking, respectively, 0.01–0.05–0.1–0.250–0.5–1–5–10–25–50 fmol of the UPS1 equimolar mix (Sigma) into 1 μg of yeast lysate (Merck). Trypsin digested samples were then analyzed in quadruplicate by nanoLC–MS/MS using a nanoRS UHPLC system coupled to a Q-Exactive Plus mass spectrometer, to produce 40 raw MS files. A detailed description of the parameters used for data processing with each bioinformatic workflow (Mascot–Proline and Andromeda–MaxQuant) can be found in the Supplementary Material.
3 Results and discussion
To assess the efficiency of Proline for the relative label-free quantification of proteomes based on intensity feature extraction, we compared its performance to that of MaxQuant (Cox and Mann, 2008), a widely used state-of-the-art tool. The comparison was based on a standard (‘ground truth’) dataset generated from a yeast lysate in which 10 different levels of the equimolar UPS1 mix of 48 human proteins were spiked, as described elsewhere (Ramus et al., 2016). Signals for yeast proteins are expected to be constant across all samples, whereas UPS1 signals should vary between samples. Proline was associated with the Mascot search engine for database search, and we thus globally compared the results of the Mascot–Proline workflow to that of Andromeda–MaxQuant. In the first place, we found that these workflows behaved similarly in terms of protein identification and validation, with approximately the same numbers of UPS1 and yeast proteins identified and validated in each case (Supplementary Fig. S7). Subsequently, we performed a detailed comparison of the quantification results produced by each workflow, as well as an evaluation of their processing speed.
3.1 MS1 quantification
3.1.1 Effectiveness of RT alignment and cross-assignment procedures
Matching MS1 signals for the same peptide ion identified across all runs is a core problem in the label-free DDA method. Indeed, because of how MS instruments select peptide ions for fragmentation, numerous peptides present in all samples cannot be systematically identified by MS/MS in every run. Hence, a peptide ion which was not identified in some runs generates missing quantitative values in the final reported intensity matrix, and a cross-assignment procedure (also known as ‘match between runs’) must be applied to recover these intensity values from the RAW data and thus minimize the final proportion of missing values (MVs; America and Cordewener, 2008; Andreev et al., 2007). It is important to highlight that this step is probably the most error-prone in the whole label-free workflow. Indeed, the signals that must be cross-assigned between runs may be very close in m/z and/or time dimensions to signals produced by other peptide species. Consequently, the algorithms used must be able to avoid extraction of abundance by cross-assignment for a PSM present in one sample but absent from another. To achieve this requires, in particular, a thorough retention time alignment between runs to be compared with compensate for reproducibility issues during the chromatographic separation step.
We used our standard dataset (see Section 2) to assess the efficiency of Proline’s cross-assignment procedure. To that aim, we compared the proportion of MVs in the quantification results obtained for yeast peptide ions from the 40 MS runs composing the dataset using both Proline and MaxQuant. When the cross-assignment procedure was not used, the number of MVs across runs mainly reflects the imperfect reproducibility of the LC-MS workflow, notably due to the limited sampling ability of MS instruments, as well as limits of the successive algorithms applied to identify and extract the peptide abundances. Very similar numbers were found when using either Mascot–Proline (56%) or Andromeda–MaxQuant (53%), with more than half of the total number of ions sequenced and quantified in at least 20 out of the 40 runs. Only 21% of yeast ions were fully quantified across all 40 runs when using Proline (Fig. 2). However, as expected, when the cross-assignment procedure was activated, the number of MVs dropped significantly, and 93% of yeast ions were detected in at least 20 runs with Proline, with 57% quantified in all runs. Interestingly, while many MV were also rescued by applying MaxQuant’s match-between-run procedure, a larger number of peptide ions remained incompletely quantified across the runs. Overall, the number of MV fell to 11% in Proline after cross-assignment, whereas it remained around 20% in MaxQuant.
Fig. 2.
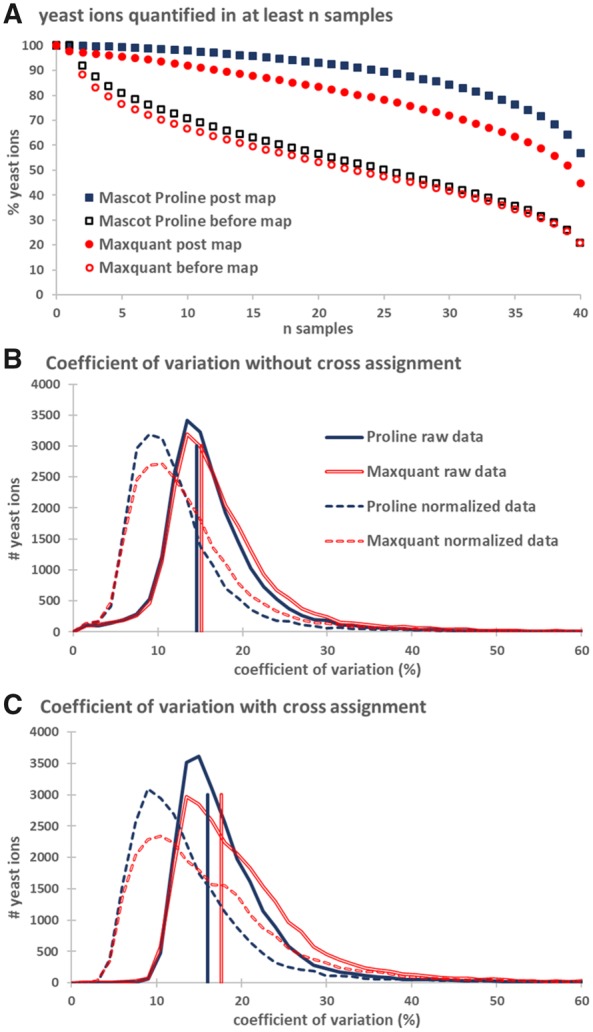
Missing values and CV distributions of yeast ions. (A) Proportions of MVs were represented as percentages of ions matching a yeast protein for which an abundance value was defined in more than n samples. The proportion of ions quantified in the 40 runs was different for Proline (57%) and MaxQuant (45%). (B) CV distribution of yeast ions before applying the cross-assignment procedure. Vertical lines indicate median CV values, 15.2% and 14.6%, respectively, for MaxQuant and Proline. (C) CV distribution of yeast ions after cross-assignment. Median CV values were increased to 17.6% and 16% for MaxQuant and Proline, respectively. In both (B) and (C), solid lines represent CV values calculated from raw intensities, whereas dashed lines represent CV values after median normalization
Although reaching a low MV proportion is a laudable goal, it must be achieved carefully to avoid incorrect assignment of abundances. We thus evaluated the correctness of the values recovered in Proline and MaxQuant by measuring the coefficient of variation (CV) for ion abundances before and after cross-assignment. The upper panel in Figure 2B shows the distribution of CV for ions for which no cross-assignment was performed, whatever the number of runs in which they were quantified. The median CV before cross-assignment was quite similar between MaxQuant and Proline, at around 15%. As a comparison, the median CV for ions from highly abundant yeast proteins, systematically sequenced by MS/MS across all the runs and quantified across the 40 runs by both Proline and MaxQuant was around 14% (data not shown). This value is representative of the accuracy of the label-free measurement. Conversely, the lower panel in Figure 2B shows the CVs for ions after cross-assignment. The distributions of CVs calculated for these ions before and after cross-assignment remain very similar, with a median CV shifting only from 14.6% to 16% using Proline (15.2–17.6% using Maxquant), indicating that the process probably does not induce major assignment errors nor increase the variability of the final number of matched peptide ion abundances. This conclusion remains unchanged when CVs are calculated from median normalized abundances.
In conclusion, the cross-assignment procedure can be used to reduce the proportion of MVs by a factor of 4 using Proline (versus a factor of 2.3 using MaxQuant). A higher number of ion values were recovered by Proline for this dataset. Moreover, these additional values are consistent since they don’t change significantly the CV measured before and after the cross-assignment operation.
3.1.2 Accuracy of protein abundance measurements
To investigate the accuracy of the protein abundances measured by Proline we used the same dataset, but focused our attention on the spiked UPS1 proteins, for which the theoretical relative abundances in the different samples were known. We first used a simple sum of peptide abundances obtained by summing ion abundances to infer protein abundances for each of the different concentrations of UPS1 proteins. The comparison of measured and theoretical ratios between the highest UPS1 concentration (50 fmol/µg of yeast lysate) and the lower concentration spikes revealed that for most UPS1 proteins, both Proline and MaxQuant accurately estimated the ratios for concentrations down to 1 fmol/µg (Fig. 3, upper panel). For lower concentration points, for which some peptides were below the detection limit of the mass spectrometer, the two software behaved differently. MaxQuant tended to overestimate the ratio, whereas Proline slightly underestimated the ratio.
Fig. 3.

Estimated versus expected ratios for UPS1 proteins. The abundances of the 48 UPS1 proteins were extracted by Proline (left panel) and MaxQuant (right panel) in each sample from the standard dataset, using either a sum aggregation (upper panels) or ratio-fitting algorithms (bottom panels). The ratios determined, calculated relative to the 50 fmol/µg concentration, were plotted against the expected ratios for the UPS1 proteins across the 10 different concentration points. Both Proline and MaxQuant accurately estimated the ratios calculated for concentration spikes down to 1 fmol/µg (expected ratio 5:6) for most UPS1 proteins. For lower concentration points, when some peptides fell below their limit of detection, the two software behaved differently, with a trend for overestimation of the ratio for MaxQuant, while Proline ratios were still well-fitted down to 250 amol/µg (expected ratio 7:6). When the ratios were calculated using MRF in Proline (bottom left), ratio variability around expected values was reduced compared with the sum method; variability was increased when MaxQuant MaxLFQ was applied (bottom right)
Inferred protein abundances are affected by the accuracy of ion measurements, but also by the peptide aggregation method used. To select the most appropriate ion signals to more accurately infer protein abundances, more complex algorithms such as MaxLFQ in MaxQuant (Cox et al., 2014) or Median Ratio Fitting (MRF) in Proline (see section ‘MS1 Quantification’ in the Supplementary Material) have been developed. When the UPS1 ratios were calculated using the MRF aggregation method in Proline (Fig. 3, bottom left panel), the smaller ratios were more accurately determined. Notably, these ratios were quite consistent across all 48 spiked proteins. In contrast, a substantial dispersion of the ratios around the expected value was observed with MaxQuant when using the MaxLFQ aggregation method (Fig. 3, bottom right panel). As illustrated on individual plots for the 48 UPS1 proteins (Supplementary Fig. S8), this dispersion results from MaxQuant’s tendency to overestimate the ratio for a subset of proteins, i.e. to underestimate the abundance of proteins spiked at low concentrations, possibly because it has missed some low-intensity signals. Conversely, more low-abundance peptide signals were generally extracted by Proline, providing a more accurate estimation of the ratios down to a spiked concentration of 250 amol/µg for most proteins. For lower concentrations, the ratios estimated by Proline remained at a constant value, due to the extraction of background noise signals when the peptides fell under the limit of detection. Importantly, the graphical interface available in Proline allows users to easily visualize the extracted signals, and check if they actually correspond to the peptide ion of interest. As shown for one peptide from the UPS1 spiked interferon g protein (Supplementary Fig. S9), the software can indeed retrieve ‘true’ peptide signals that are detectable down to 250 amol/µg. Altogether, these results demonstrate that the signal extraction procedure in Proline is highly sensitive, and thus the number of MVs is reduced and the accuracy of protein quantification at low concentration is improved compared with MaxQuant.
3.2 Differential analysis
3.2.1 MS1 quantification
The ultimate goal of most quantitative proteomics studies is to find proteins whose relative abundance is significantly increased or decreased in complex samples. Among the numerous samples or datasets allowing to assess the performance of quantitation workflows, the one provided to volunteer participants of the 2015 study (Choi et al., 2017) by the Association of Biomolecular Resource Facilities is interesting since it gives the possibility to compare Proline’s performances with the 51 submissions reported (see Supplementary Section ‘MS1 quantification’). However, to enable an accurate comparison of performances a sufficiently large number of differentially abundant proteins are required. Similar to what was described in (Ramus et al., 2016) we gathered together the quantitative results of three pairwise comparisons of four different spiked levels of UPS1 in a yeast lysate (50 versus 25 fmol/µg, 50 versus 5 fmol/µg and 50 fmol/µg versus 500 amol/µg), to obtain differentially abundant proteins at various concentrations (expected fold changes of respectively 2, 10 and 100). Both software successfully discriminated UPS1 proteins with a theoretical fold change of 10 and 100 from the yeast background, either based on fold change or P-value, even when only the simple sum aggregation method was used (Fig. 4, upper panels). The separation of the expected variant and invariant populations was less obvious in the 50 versus 25 fmol/µg comparison when using sum aggregation, but became more so when the ratio-fitting method was applied. This enhancement was mainly due to improvement of the P-values for expected variant proteins (Fig. 4, compare upper and lower panels). Furthermore, application of the MRF or MaxLFQ methods had a similar impact on the quantification of invariant yeast proteins (black circles) but to a more limited extent on the P-value axis when using Proline. Importantly, the fold changes measured for UPS1 proteins were closer to the expected values with Proline than with MaxQuant, whether the corresponding ratio-fitting algorithm was used or not, especially for higher theoretical fold change (Fig. 4).
Fig. 4.
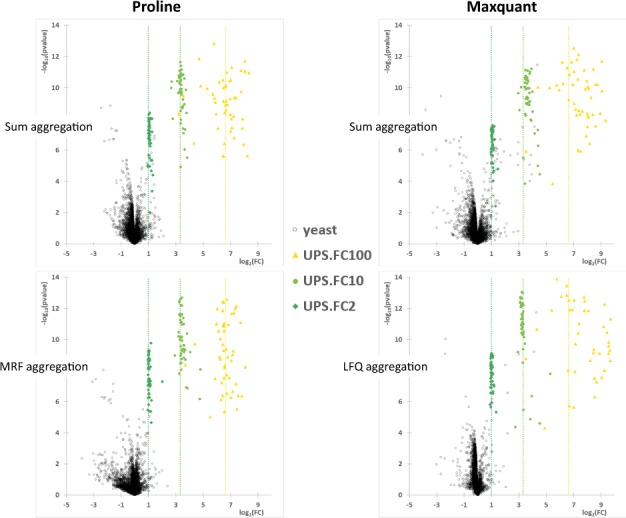
Volcano plots of the mixed dataset differential analysis. Each protein in the mixed dataset obtained from the quantitative output of three different pairwise comparisons was plotted in a cartesian coordinate defined by the fold change (FC, in log2) on the horizontal axis and the inverse of the P-value (log10) on the vertical axis. The graphs illustrate the quantitative results for the UPS1 proteins quantified in each binary comparison (dark green: comparison of 25 versus 50 fmol/µg, theoretical fold change of 2; light green: comparison of 5 versus 50 fmol/µg, theoretical fold change of 10; yellow: comparison of 500 amol/µg versus 50 fmol/µg, theoretical fold change of 100). Black circles correspond to yeast proteins. The expected ratios of the different concentration points are represented by the dashed vertical lines. For each software, two different peptide-to-protein aggregation methods were implemented: the simplest one consists in an aggregation of non-shared peptides abundances by a sum function (upper part), whereas the second one determines the protein abundances by fitting protein ratios to all observed peptide ratios (MRF or MaxLFQ methods, lower part)
The q-values were then used to classify variant and invariant proteins from this mixed dataset, and performance was assessed using receiver operating characteristic curves (ROC curves, Fig. 5). Results obtained by the most straightforward aggregation approach (sum aggregation, solid lines) demonstrated that Proline retrieves a high proportion of true positive (TP) UPS1 proteins while maintaining a low rate of false positive (FP) yeast proteins. Interestingly, although the number of proteins identified and quantified by the Mascot–Proline workflow was slightly higher, the number of FP was always lower in Proline results than in MaxQuant results (e.g. as shown in Supplementary Table S3, a q-value < 10−3 generated 26 FP in Proline versus 44 in Maxquant). These results corroborate the observed higher precision and accuracy of signal measurements and the lower rate of MVs returned by Proline. The use of ratio-fitting algorithms to aggregate peptide abundances—MRF and MaxLFQ in Proline and MaxQuant, respectively—allows better discrimination of expected variant and invariant proteins (dashed lines in Fig. 5). Indeed, this approach lowers the q-values obtained, especially for differentially abundant proteins, since it is less sensitive to outlier peptides than a simple sum aggregation. With our test dataset, a better balance between true positive rate (TPR) and false discovery proportion (FDP) was obtained using MRF in Proline (from 99% TPR, 28% FDP to 100% TPR and 20% FDP at a q-value threshold < 10−2). The balance appears to be less affected when comparing MaxQuant MaxLFQ to MaxQuant Intensity, as the FDP remains relatively high at the same q-value threshold (from 99% TPR, 32% FDP to 100% TPR and 26% FDP). Interestingly, it is possible to reach no yeast as FP without affecting too much sensitivity with Proline MRF (TPR 65%) whereas with Maxquant MaxLFQ, sensitivity is significantly degraded (TPR 22%). Moreover, Proline’s MRF aggregation method allows a higher number of proteins to be quantified compared with MaxQuant MaxLFQ (see Supplementary Table S3). Taken together, these results demonstrate that Proline performs well in differential analyses involving label-free quantification of MS1 signals.
Fig. 5.
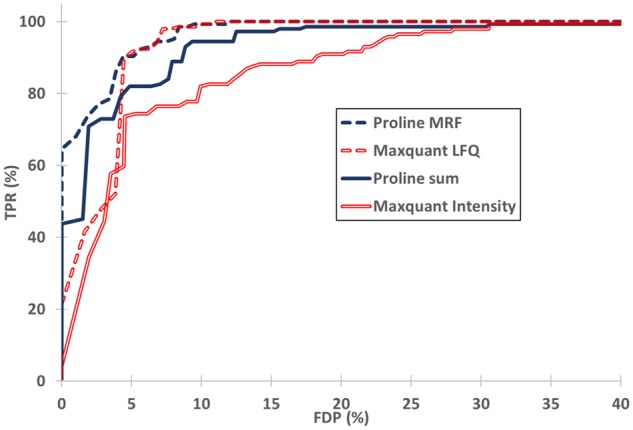
Differential analysis results in terms of sensitivity and FDP. For each software, proteins from the mixed dataset were classified as variant through the application of q-value thresholding. Sensitivity (TPR = TP/144, TP UPS1 proteins) was plotted as a function of FDP [FDP = FP/(TP+ FP), FP yeast proteins]
3.2.2 Spectral counting
Quantification based on spectral counting approaches are known to be less sensitive than workflows based on MS1 quantification (Ramus et al., 2016). Nevertheless, spectral counting metrics can be extracted almost instantly from identification data and work quite well to identify proteins with medium to high fold changes. It thus remains a powerful tool to give a preliminary overview of a dataset. The algorithm implemented in Proline is similar to the one used in a previously published software (Hesse et al., 2016) and can easily discriminate between proteins with a theoretical fold change of 10 and 100, while most UPS1 proteins with an expected fold change of 2 cannot be distinguished from the yeast invariant background (Supplementary Fig. S11).
3.3 Proteomics data curation
In addition to the intrinsic quality of any results generated automatically, a pivotal feature of Proline is that it provides an interactive interface allowing users to examine and manually curate all results. Thus, details of the identification features and abundance measurements for a protein can be accessed through a synchronized panel. This user interface represents a powerful tool to check for and eliminate errors which may have occurred during the successive data-processing steps. In the case of relative quantification based on the extraction of MS1 signals, peptides measured across MS runs are displayed in the same panel. Furthermore, for each peptide, the ions quantified and their extracted chromatographic peaks can be viewed (Supplementary Figs S9d and S12b). Erroneous or suspect profiles can be individually discarded by the user, and protein quantification can then be recomputed with the remaining validated measurements. Technically, all data are stored in an embedded relational database where each module in the suite reads the required information and writes the produced information with its associated metadata. This type of centralized persistent storage allows relevant information to be extracted at any level of detail upon request from an algorithm or end user. This architecture (see Supplementary Section ‘Software architecture’) facilitates data visualization and exploration compared with the tabulated text files typically produced as output by other similar software. It is also compatible with the incremental addition of newly acquired MS runs which form part of an ongoing study, or with performing a time-consuming process in stages, as it is possible, e.g. to modify and recompute the summarization of peptide abundances into proteins without repeating the whole quantification process.
3.4 Computational efficiency
Data-processing efficiency is today a major concern for many labs, and the core facilities that routinely deal with several ongoing large-scale label-free studies in parallel. Moreover, as the number of studies increases, the dataset scaling (number of runs to be compared) has also expanded. Consequently, it is now relatively common to generate and analyze datasets containing tens or even hundreds of LC-MS/MS runs. Thus, to assess the relative computational efficiency of Proline and MaxQuant, we benchmarked them on the same computer (see Supplementary Section ‘Hardware used to assess computational speed’) and recorded the processing time for the corresponding label-free workflows, using Mascot and Andromeda, respectively, for the MS/MS search. The processing speed was measured for two different datasets: one composed of eight LC-MS/MS runs from the standard dataset (UPS1 spiked at 50 and 25 fmol/µg, four replicates for each), and a larger one corresponding to the whole standard dataset (40 LC-MS/MS runs).
Data processing in MaxQuant starts with feature detection and extraction of MS signal from the raw file, whereas the Proline workflow begins with MS/MS data processing (including database search with Mascot, as shown here, or with another search engine). Whatever the order of the different steps, this comparative analysis clearly shows that many steps (peaklist creation, search result validation, quantification) are performed faster by Proline and also that the Mascot search is faster than the Andromeda search (Fig. 6). It must be highlighted that the search results import step, absent from the MaxQuant workflow, negatively impacts the final Proline processing time, and that writing of the results in Proline also longer than in MaxQuant. However, even with these drawbacks, Proline remains faster than MaxQuant when processing the two datasets tested, as the time spent on the other steps is significantly reduced, particularly for peaklist creation and extraction of quantitative data. These two steps avail of the mzDB file format, which provides optimal data access thanks to precise indexation of the MS information (Bouyssié et al., 2015; Handy et al., 2017). Interestingly, previous studies showed that not only the characteristics of the hardware setup are important to speed up the quantitative workflow, but that IO operations are also a major bottleneck in data processing with MaxQuant (Neuhauser et al., 2013). To overcome this bottleneck, these authors suggested the use of optimized computers equipped with solid state drives as an efficient way to alleviate memory constraints when accessing raw data. In the evaluation presented here, we used this type of hardware to compare the software in the best conditions. Nevertheless, we still observed a significant increase in speed with Proline, indicating that the use of indexed, dedicated file formats such as mzDB also represents a powerful solution for optimal access to the MS data. To date, Proline requires a manual execution of the different steps of the workflow, but offers the possibility to save processing parameters at each step. Although this clearly increases the amount of manual work, it also allows intermediate quality control all along the workflow. However, in order to speed up the process, we will soon release two additional tools: one to run Proline through command line interface from a single configuration file, and a second one embedding several open-source search engines and enabling the definition of fully automated workflows starting from raw files.
Fig. 6.
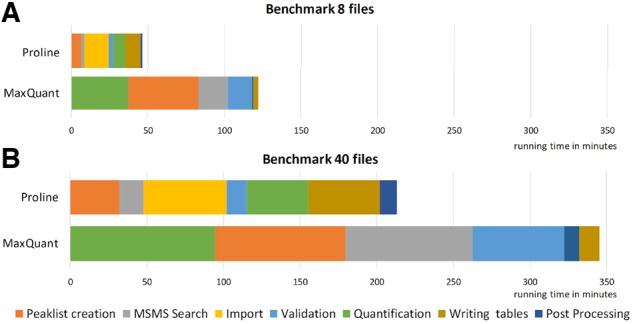
Workflows computation time. Performance of the Mascot–Proline and Andromeda–MaxQuant label-free workflows were compared on two datasets of different sizes. The main steps of the compared workflows are shown in the same color when possible. Time values were taken from the ‘runningTimes.txt’ output file for MaxQuant and from the log files for Proline. (A) Performance observed for a dataset containing eight UPS1-Yeast LC-MS/MS runs: Total processing time was 122 min for MaxQuant and 46 min for Proline (average time per file 15.26 and 5.79 min, respectively). (B) Performance observed for the whole UPS1-Yeast dataset (40 LC-MS/MS runs): Total processing time 346 min for MaxQuant and 214 min for Proline (average time per file: 8.63 and 5.34 min, respectively)
4 Conclusion
Proline is an open-source, cross-platform software (running on MacOS, Linux and Windows), written in the Java and Scala programing languages. It is distributed under the CECILL license (https://github.com/profiproteomics). Its core libraries constitute a java virtual machine-based framework that can be used as a starting point for the development of new tools. Proline accurately and efficiently processes MS-based proteomics data, and can handle both small and very large datasets. The highly detailed data and metadata stored and made available combined with its ease of use perfectly fits the needs of mass spectrometry experts working in proteomics as well as core proteomics facilities. The software was designed to be computationally efficient and capable of organizing, linking and storing all MS data in a centralized database. MS-based proteomics results associated with a particular project can then be navigated and browsed by users through the graphical user interfaces provided; alternatively, the data can be extracted by querying the database. Usability, algorithms and validation rules implemented in Proline were developed in close collaboration with MS researchers to ensure a consistent, reliable and efficient end-user experience. The label-free MS1 quantification algorithm combines a novel signal detection procedure starting from chromatographic peak apexes with a cross-assignment method based on efficient RT alignment computation and identification-based deisotoping. This algorithm demonstrates very good performance levels, with a low MVs rate and a good accuracy of observed versus expected ratios on a standard spiked dataset. The tool is thus very competitive with respect to existing solutions such as MaxQuant.
It is also important to highlight that Proline’s architecture, based on a message-oriented middleware, is compatible with moving to a more distributed architecture in which Proline components could be executed across multiple computers to complete a single processing request. We believe that this future capability could be of interest when processing very large-scale datasets, as it would allow the advantages of a cloud infrastructure to be exploited. Proline version 2.0 is the seventh publicly available release of the software. It can be downloaded from http://www.profiproteomics.fr/proline/#downloads. On the same web page, a user forum, an online documentation and tutorials using a published dataset (Ramus et al. 2016) are also provided to help new users to discover the Proline suite.
Supplementary Material
Acknowledgements
The authors thank Virginie Brun and Myriam Ferro for constructive criticism of the article and Maighread Gallagher-Gambarelli for advice on English usage and editing suggestions.
Funding
This work was supported by the French National Agency for Research (ANR) [ANR-10-INBS-08]; ProFI project, ‘Infrastructures Nationales en Biologie et Santé’; ‘Investissements d’Avenir’call.
Conflict of Interest: none declared.
References
- Aebersold R., Mann M. (2016) Mass-spectrometric exploration of proteome structure and function. Nature, 537, 347–355. [DOI] [PubMed] [Google Scholar]
- America A.H.P., Cordewener J.H.G. (2008) Comparative LC-MS: a landscape of peaks and valleys. Proteomics, 8, 731–749. [DOI] [PubMed] [Google Scholar]
- Andreev V.P. et al. (2007) A new algorithm using cross-assignment for label-free quantitation with LC-LTQ-FT MS. J. Proteome Res., 6, 2186–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyssié D. et al. (2015) mzDB: a file format using multiple indexing strategies for the efficient analysis of large LC-MS/MS and SWATH-MS data sets. Mol. Cell. Proteomics, 14, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M. et al. (2017) ABRF Proteome Informatics Research Group (iPRG) 2015 Study: detection of differentially abundant proteins in label-free quantitative LC-MS/MS Experiments. J. Proteome Res., 16, 945–957. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol., 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J. et al. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics, 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E.W. et al. (2008) Data analysis and bioinformatics tools for tandem mass spectrometry in proteomics. Physiol. Genomics, 33, 18–25. [DOI] [PubMed] [Google Scholar]
- Doll S. et al. (2017) Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun., 8, 1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handy K. et al. (2017) Fast, axis-agnostic, dynamically summarized storage and retrieval for mass spectrometry data. PLoS One, 12, e0188059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse A.-M. et al. (2016) hEIDI: an intuitive application tool to organize and treat large-scale proteomics data. J. Proteome Res., 15, 3896–3903. [DOI] [PubMed] [Google Scholar]
- Mann M. et al. (2013) The coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell, 49, 583–590. [DOI] [PubMed] [Google Scholar]
- Mueller L.N. et al. (2008) An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res., 7, 51–61. [DOI] [PubMed] [Google Scholar]
- Nahnsen S. et al. (2013) Tools for label-free peptide quantification. Mol. Cell. Proteomics, 12, 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A.I., Aebersold R. (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics, 4, 1419–1440. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A.I. et al. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem., 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Neuhauser, N. et al. (2013) High performance computational analysis of large-scale proteome data sets to assess incremental contribution to coverage of the human genome.J Proteome Res., 12, 2858–2868. [DOI] [PubMed] [Google Scholar]
- Ramus C. et al. (2016) Benchmarking quantitative label-free LC-MS data processing workflows using a complex spiked proteomic standard dataset. J. Proteomics, 132, 51–62. [DOI] [PubMed] [Google Scholar]
- Rieckmann J.C. et al. (2017) Social network architecture of human immune cells unveiled by quantitative proteomics. Nat. Immunol., 18, 583–593. [DOI] [PubMed] [Google Scholar]
- Savitski M.M. et al. (2011) Confident phosphorylation site localization using the mascot delta score. Mol. Cell. Proteomics, 10. doi: 10.1074/mcp.M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteynberg D. et al. (2013) Combining results of multiple search engines in proteomics. Mol. Cell. Proteomics, 12, 2383–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaudel M. et al. (2015) PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol., 33, 22–24. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J.A. et al. (2009) A guide to the proteomics identifications database proteomics data repository. Proteomics, 9, 4276–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J.A. et al. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol., 32, 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.