

Abstract
Objective:
Toll-like receptor (TLR)-mediated catabolic responses are implicated to contribute to osteoarthritis (OA). However, deficiency of TLRs has little chondroprotection in mice in vivo. Here, we studied the effect of deficiency of TLR2 and TLR4 in articular chondrocytes on cellular stress responses in vitro.
Design:
Chondrocytes isolated from TLR2 and TLR4 double knockout (TLR2/4dKO) and wild type (WT) mice and recombinant HMGB1 (rHMGB1) and LPS were used. Expression of antioxidant and DNA repair enzymes including SOD1, SOD2 and OGG1, and phosphorylation of H2AX (a marker for DNA damage) were examined by Western blotting. MitoSOX Red staining was used for assessing mitochondrial superoxide generation. Autophagic activity was monitored by flow cytometry analysis of mean fluorescence intensity (MFI) of GFP and RFP in chondrocytes transfected with a tandem GFP-mRFP-LC3 plasmid, and by Western blot analysis of expression of LC3 and p62, a selective autophagy adaptor.
Results:
Basal expression of SOD2 but not SOD1 was largely reduced in TLR2/4dKO compared to WT chondrocytes, correlated with significantly enhanced menadione-induced mitochondrial superoxide generation (2.85 to 3.92 and 3.39 to 8.97 with mean difference 3.39 and 6.18 for 25 and 50 μM menadione, respectively) and phosphorylation of H2AX. LPS and rHMGB1 induced expression of SOD2, OGG1 and p62 in WT but not TLR2/4dKO chondrocytes. Autophagy flux was impaired in TLR2/4dKO chondrocytes after acute nutrient stress and by LPS and rHMGB1.
Conclusions:
TLR2 and TLR4 deficiency appears to reduce chondrocyte anti-oxidative stress and autophagy flux capacity, which may compromise cartilage homeostasis as a result of chondrocyte dysfunction.
Keywords: Toll-like receptors, chondrocytes, oxidative stress, autophagy flux
Introduction
Osteoarthritis (OA) is a disease of whole joint (1). Progressive articular cartilage degradation is the central feature of OA (1). Articular chondrocytes, the only cells in cartilage, play an important role in cartilage matrix homeostasis (1). Innate immunity has been implicated as an active player to induce low-grade inflammation and catabolic events in articular cartilage, contributing to OA progression (2). Human articular chondrocytes express toll-like receptors (TLRs), which are pattern recognition receptors (PRRs) fundamental in innate immune responses. Expression of TLR2 and TLR4 are increased in human knee OA cartilage lesions (2). Stimulation of chondrocytes in vitro with TLR2 and TLR4 agonists such as microbial products peptidoglycan and LPS, and endogenous molecular products derived from cellular stress and extracellular matrix disruption including high mobility group box protein 1 (HMGB1), S100A8/A9, and low weight hyaluronan (LMW-HA) lead to increased inflammatory and catabolic responses (2). These data indicate that inhibition of TLR2 and TLR4 may be chondroprotective.
However, in vivo studies of deficiency of TLRs in experimental mouse knee OA models yield mixed and mostly unexpected results. TLR2 knockout (KO) mice were shown to have the same cartilage pathology as wild type (WT) mice in the destabilization of medial meniscus (DMM) induced OA model (3). Cartilage damage was more severe in TLR2 KO mice, compared to WT mice in the collagenase-induced OA model that is associated with synovial inflammation (3). Nasi S et al reported deficiency of TLR2, TLR4, or MyD88 that is the signaling adaptor molecule for most of the TLR pathways, did not significantly impact on the severity of OA in the meniscectomy-induced OA model in male mice (4). We observed that TLR2 and TLR4 double knockout (dKO) male mice did not have a significant effect on severity of cartilage degradation in the same model (Supplemental figure). TLR4 deficiency is recently shown to be chondroprotective in obesity (via high-fat diet)-induced OA in aged female mice (5). These mixed results imply that TLRs may have various effects in specific OA phenotypes.
Emerging evidence implicate an important role of TLRs in cellular homeostatic function. Activation of TLR signaling is shown to strongly upregulate protein expression of antioxidant enzymes such as mitochondria-located superoxide dismutase 2 (SOD2) (6), and a number of DNA repair enzymes including OGG1 (7) that is the primary enzyme responsible for the excision of 8-oxoguanine, a mutagenic base byproduct that occurs as a result of exposure to reactive oxygen species (ROS). TLRs also regulate autophagy (8), a specialized biological process involved in maintaining cellular homeostasis through the degradation of protein aggregates and damaged organelles. Since SOD2, OGG1 and autophagy have been shown to involve in regulation of chondrocyte homeostasis, and their dysfunction is associated with OA (9,10), we studied the cellular stress responses in chondrocytes deficient in TLR2 and TLR4 in vitro. We discovered that TLR2 and TLR4 deficiency appears to reduce chondrocyte anti-oxidative stress and autophagy flux capacity, which could potentially compromise cartilage homeostasis as a result of chondrocyte dysfunction.
Method
Reagents
All chemical reagents were obtained from Sigma-Aldrich (St Louis, MO). Purified recombinant HMGB1 (rHMGB1) was from R&D Systems, Inc. (Minneapolis, MN). Antibodies to SOD2, LC3B, p62, phosphorylated and total H2AX (Ser139) were from Cell Signaling Technology, Inc. (Danvers, MA). Antibodies to SOD1 and OGG1 were from Abcam (Cambridge, MA).
Studies of mouse articular chondrocytes
Mouse chondrocytes isolated from 6-8 days old knees (11) of TLR2/4dKO and congenic WT mice were used for the study, which were in compliance with an institutionally reviewed and approved protocol by VA San Diego IACUC. Knee chondrocytes isolated from all mice in an entire litter (average 6 mice/litter) were pooled and cultured in Dulbecco’s modified Eagle’s medium (DMEM) with high glucose, 10% fetal calf serum (FCS), 100 μg/ml streptomycin, and 100 IU/ml penicillin at 37°C. Upon confluence, chondrocytes were re-plated onto 12 well plates at 3 x 105 cells per well for experiments. Real time-PCR analysis confirmed that WT and TLR2/4dKO chondrocytes exhibited similar functional phenotypes, evidenced by similar levels of high expression of type II collagen and aggrecan and low expression of type I collagen. Each experiment was repeated 3 times independently in passage 1 of pooled mouse chondrocytes isolated from 3 different litters.
Detection of mitochondrial superoxide with MitoSOX™ Red reagent
The production of mitochondrial superoxide was assayed by flow cytometry analysis of mean intensity of red fluorescence, because oxidation of MitoSOX™ Red reagent (ThermoFisher) by superoxide produces red fluorescence.
Assessment of autophagy
Chondrocytes were transfected with a tandem fluorescent-tagged LC3 (ptfLC3) plasmid (Addgene, a gift from Tamotsu Yoshimori) in which LC3B is fused to GFP and mRFP for 48 hours. The cells were then subjected to acute nutrient starvation in Hank’s balanced salt solution (HBSS) for the times indicated, followed by flow cytometry analysis of mean fluorescence intensity (MFI) of GFP and RFP. The fluorescence of GFP is acid-sensitive, while the fluorescence of mRFP is relatively stable and retained within lysosomes. The green fluorescence is lost once autophagosomes fuse with lysosomes, because of degradation of GFP by acid lysosomal proteases. Thus, autophagy flux (autophagic degradation) can be determined by the ratio of the RFP to GFP intensity. The endogenous microtubule-associated protein 1 light chain 3 (LC3) conversion (LC3-I to LC3-II) and turnover, and expression of p62, a selective autophagy adaptor, in the presence or absence of choloroquine (CQ), which blocks autophagosome and lysosome fusion, were examined by Western blotting. The turnover of exogenous LC3 was also evaluated by the appearance of higher molecular weight conjugates of mRFP-GFP-LC3 in the same blot.
SDS-PAGE/Western Blot
Cell lysates (10-15 μg) were prepared in RIPA buffer and separated by gradient 4-20% SDS-PAGE, followed by immunoblotting with desired antibodies as described in (9). Densitometry analysis of each Western blot from 3 individual experiments was performed. Expression of each protein was normalized and presented as the ratio of the intensity of the signal for the protein to that of the loading control.
Statistical analyses
GraphPad PRISM 8 was used for statistical analyses. Each data point has 3 independent biological replicates (each represented the mean of 2 technical replicates). All data passed QQ plot normality and lognormality tests. Student t-test (expressed as mean±SD) was used to compare two groups. Repeated measures two-way ANOVA (expressed as mean±95%CI) was conducted to compare three or more groups on the effect two independent variables (intervention and genotype). When there was a significant interaction between intervention and genotype, Sidak’s test on the effect of intervention at each concentration or time between genotypes (WT vs TLR2/4dKO) and Tukey’s post-hoc test on the effect of intervention within each genotype were performed. P values less than 0.05 were considered significant.
Results
Reduced anti-oxidative stress capacity in TLR2/4dKO chondrocytes
First, we examined the effect of deficiency of TLR2 and TLR4 on protein expression of antioxidant enzymes such as SOD1 and SOD2. Expression of mitochondria-located SOD2, but not cytosol-located SOD1, was markedly lower at basal levels in TLR2/4dKO chondrocytes (Figure 1A). Menadione, an oxidative stress inducer, significantly induced mitochondrial superoxide generation in both WT and TLR2/4dKO chondrocytes (Figure 1B). Although no significant effect of genotype on basal levels of mitochondrial superoxide was observed, the levels of menadione-induced mitochondrial superoxide were clearly higher in TLR2/4dKO compared to WT chondrocytes (95% CI 2.85 to 3.92 and 3.39 to 8.97 with mean difference 3.39 and 6.18 for 25 and 50 μM menadione, respectively) (Figure 1B), accompanied by augmented DNA damage with an evidence of increased phosphorylation of H2AX (Ser139) (Figure 1C). LPS and rHMGB1 significantly induced expression of SOD2 and DNA repair enzyme OGG1 in WT but not TLR2/4dKO chondrocytes (Figure 1D). TLR2 and TLR4 deficiency did not impact on expression of Ku70, another DNA repair enzyme involved in the repair of DNA double-strand breaks by non-homologous end-joining (Figure 1D,). These data suggest that deficiency of TLR2 and TLR4 may increase DNA damage in response to oxidative stress and reduce DNA repair capacity, particularly for the excision of 8-oxoguanine from damaged DNA in chondrocytes.
Figure 1. Chondrocytes deficient in TLR2 and TLR4 exhibited reduced anti-oxidative stress capacity.
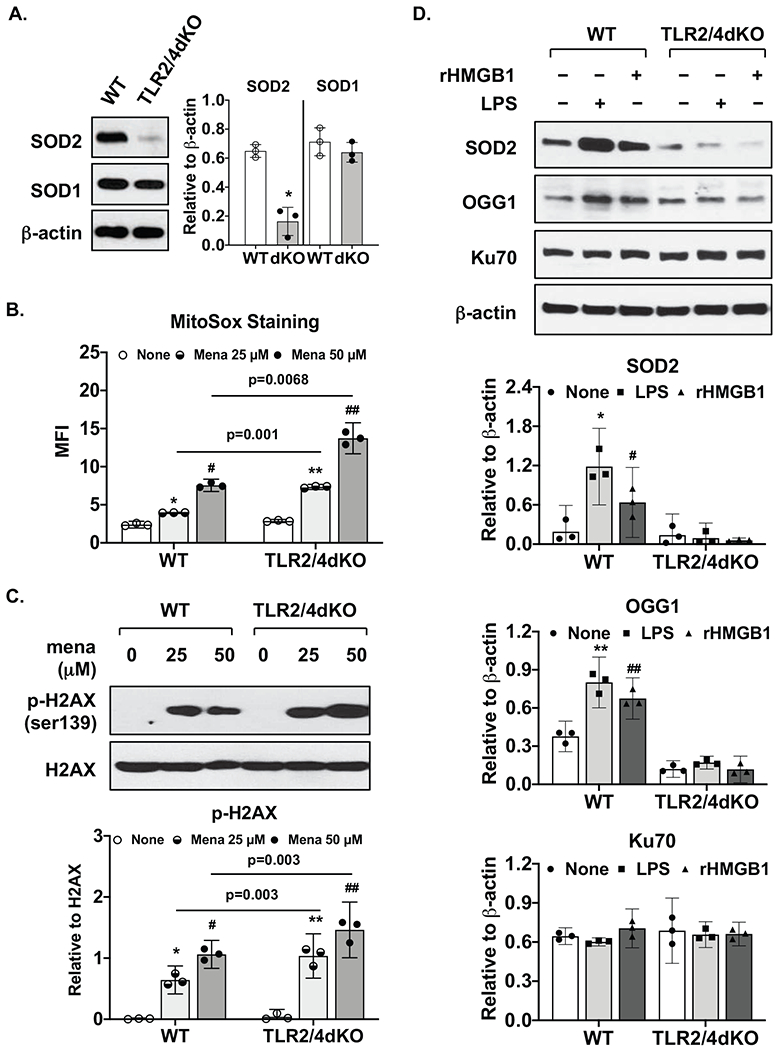
WT and TLR2/4dKO chondrocytes at basal levels (A) or after stimulation with LPS (1 μg/ml) and rHMGB1 (2 μg/ml) for 18 hours (D) were examined for expression of SOD1, SOD2, OGG1 or Ku70 by Western blotting. β-actin was included as a loading control. WT and TLR2/4dKO chondrocytes were treated with menadione at 25 and 50 μM for 1 hour, mitochondrial superoxide generation (B) was assessed by flow cytometry analysis of mean fluorescent intensity (MFI). Phosphorylation and expression of H2AX were examined by Western blotting (C). Densitometry analysis of Western blot data from 3 independent experiments was included. Student t-test (A) and repeated measures two-way ANOVA analysis (B,C,D) were used for statistical analysis. There was a significant interaction between the effect of menadione and genotype on mitochondrial superoxide generation in B (F(2, 8)=91.73, p<0.0001) and on normalized phosphorylation of H2AX in C (F(2, 8)=9.91, p=0.0068). There was also a significant interaction between the effect of treatment (LPS or rHMGB1) and genotype on normalized SOD2 expression F(2, 8)=79.18, p<0.0001 and normalized OGG1 expression F(2, 8)= 23.46, p=0.0005, but not normalized Ku70 expression. Sidak’s test on the effect of menadione at each concentration between WT and TLR2/4dKO (B, C) and Tukey’s test on the effect of each treatment within WT or TLR2/4dKO (B,C,D) were followed up. In A, *p=0.002, relative to WT. In B, * p=0.0065, # p<0.0001 relative to none in WT, ** p=0.0003 and ## p=0.003 relative to none in TLR2/4dKO. In C, * p=0.012, # p=0.005 relative to none in WT, **, ## p=0.008 relative to none in TLR2/4dKO. In D. * p=0.0004 and # p=0.03 for SOD2, ** p=0.045 and ## p=0.021 for OGG1, relative to none in WT.
Impaired autophagy flux in TLR2/4dKO chondrocytes
Chondrocytes were transfected with a tandem GFP and mRFP fluorescent-tagged LC3 plasmid for 48 hours followed by acute nutrient starvation, known to rapidly trigger autophagy. Due to the differential sensitivity of GFP and RFP to lysosomal acidity, autophagy flux can be evaluated by the ratio of mean fluorescence intensity (MFI) RFP to GFP. As seen in Figure 2A, RFP/GFP ratio was slightly but not significantly higher in WT compared to TLR2/4dKO chondrocytes at basal levels. However, nutrient starvation significantly increased RFP/GFP ratio in WT (95% CI 0.14 to 0.3 and 0.002 to 0.4 with mean difference 0.22 and 0.2 for 2 and 4 hours, respectively) but not TLR2/4dKO chondrocytes, indicating autophagic degradation was impaired in chondrocytes deficient in TLR2 and TLR4. This result was confirmed by Western blot analysis (Figure 2B) showing that expression of both endogenous LC3B and exogenous LC3B (appeared as higher molecular weight conjugates of mRFP-GFP-LC3 in the same blot) was considerably reduced in response to nutrient starvation in WT but not TLR2/4dKO chondrocytes. Next, chondrocytes were stimulated with LPS and rHMGB1 in the presence or absence of choloroquine (CQ), a known inhibitor of autophagosome and lysosome fusion. As depicted in Figure 2C, expression of p62, which directly binds to LC3 serving as a selective substrate of autophagy (12), was up-regulated robustly by LPS and slightly by rHMGB1 in WT chondrocytes. This was further enhanced by choloroquine, which also increased LC3B-II expression in WT chondrocytes (Figure 2C). In comparison, p62 expression remained low in TLR2/4dKO chondrocytes regardless if LPS and rHMGB1were present. However, the levels of LC3B-II expression were significantly higher with further enhancement by cholorquine in these cells (Figure 2C). These results indicate that TLR-mediated up-regulation of p62 is required for chondrocyte autophagy flux in response to TLR2 and TLR4 agonists.
Figure 2. Autophagy flux was impaired in chondrocytes deficient in TLR2 and TLR4.
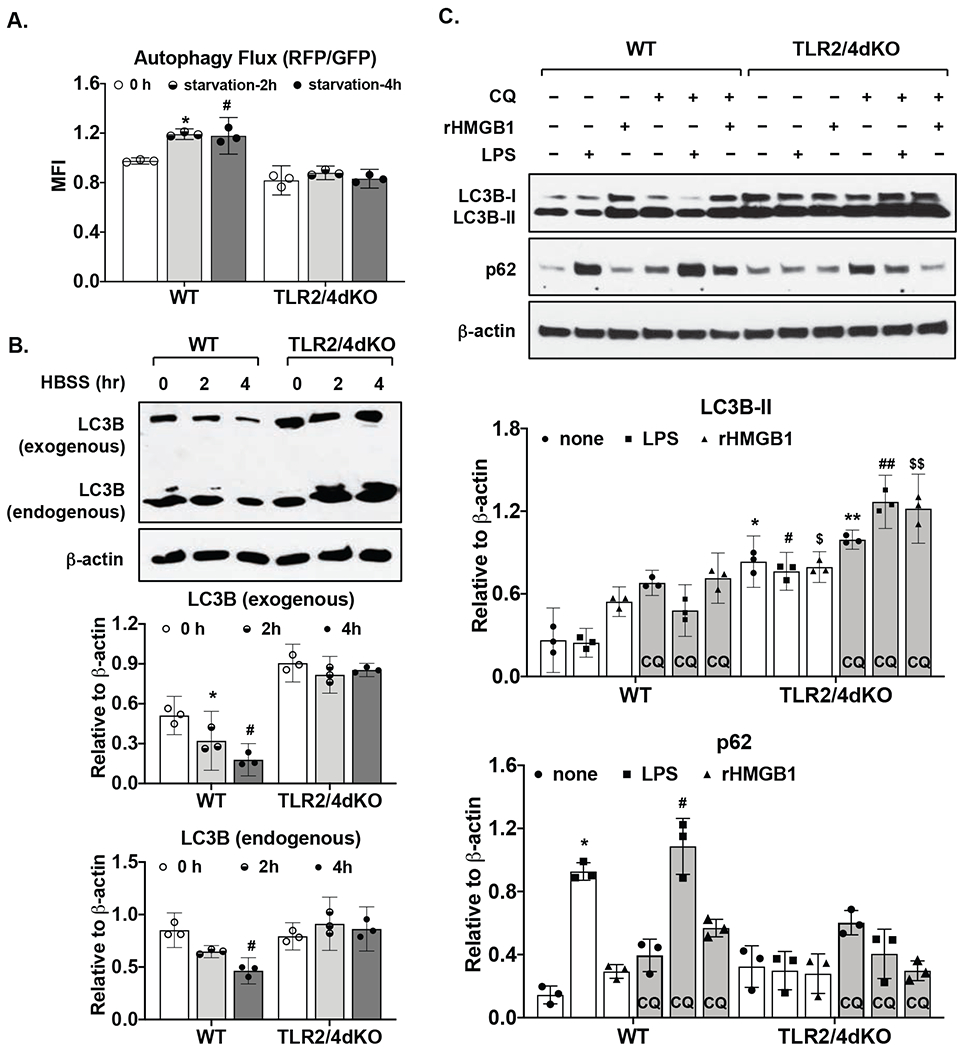
Both WT and TLR2/4dKO chondrocytes were transfected with a tandem mRFP and GFP fluorescent-tagged LC3 (ptfLC3) plasmid for 48 hours. The cells were then subjected to nutrient starvation in HBSS for 2 and 4 hours, followed by flow cytometry analysis of mean fluorescence intensity of RFP and GFP. Autophagy flux was determined by the ratio of RFP/GFP (A). Expression of endogenous and exogenous LC3B that was appeared as higher molecular weight conjugates of mRFP-GFP-LC3 on the same blot was examined by Western blotting (B). WT and TLR2/4dKO chondrocytes were stimulated with LPS (1 μg/ml) and rHMGB1 (2 μg/ml) for 18 hours in the presence or absence of choloroquine (CQ, 50 μM). Expression of p62 and LC3B was examined by Western blotting (C). β-actin was included as a loading control. Densitometry analysis of Western blot data from 3 independent experiments was included. Repeated measures two-way ANOVA was used for statistical analysis. There was a significant interaction between the effect nutrient starvation and genotype on autophagy flux (RFP/GFP) F(2, 8)=18.85, p=0.0009) in A, on normalized exogenous LC3 expression F(2, 8)=33.18, p=0.0001 and normalized endogenous LC3 expression F(2, 8)=16.7, p=0.001) in B. There was also a significant interaction between treatment (LPS or rHMGB1) and genotype on normalized LC3B-II expression F(5, 20)=11.46, p<0.0001 and normalized p62 expression F(5, 20)=48, p<0.0001 in C. Tukey’s test on the effect of nutrient starvation or treatment within WT or TLR2/4dKO (A,B,C) and Sidak’s test on the effect of each treatment between WT and TLR2/4dKO (C) were followed up. In A, *p=0.007, #p=0.049, relative to 0 h. In B, * p=0.04, # p=0.005 relative to 0 hour in WT for exogenous LC3B, #p=0.017 relative to 0 hour in WT for endogenous LC3B. For LC3B-II in C, * p=0.0009, # p=0.0002, $ p=0.014 relative to none, LPS and rHMGB1, respectively, in WT, ** p=0.003, ## p=0.001, $$ p=0.018 relative to none, LPS and rHMGB1 respectively, in WT in the presence of choloroquine. For p62 in C, * p=0.01 and # p=0.018 relative to none in WT and WT in the presence of choloroquine, respectively.
Discussion
The paradoxical results of in vitro and in vivo studies of deficiency of TLR2 and/or TLR4 imply that TLR2 and TLR4 may function as a double-edged sword in OA progression. In this study, we found that TLR2/4dKO chondrocytes, compared to WT chondrocytes, were more vulnerable to oxidative stress and DNA damage. This may partly attribute to impaired expression of SOD2 and OGG1, as TLR2 and TLR4 agonists were no longer able to up-regulate expression of these antioxidant and DNA repair enzymes in TLR2/4dKO chondrocytes. Knockdown of SOD2 expression in chondrocytes results in oxidative damage and mitochondrial dysfunction in chondrocyte in vitro (13). Our recent study revealed an important role of SOD2 and OGG1 in maintaining mitochondrial DNA integrity and function (9). The promoter regions of both SOD2 and OGG1 genes contain DNA binding site for AP1 (6,7), and SOD2 gene promoter also has DNA binding site for NF-κB (6). Given that TLR2 and TLR4 signaling activates NF-κB and AP-1, the endogenous TLR2 and TLR4 agonists generated after injury in the joint would not be able to induce expression of SOD2 and OGG1 in mice deficient in TLR2 and/or TLR4. In this context, cartilage homeostasis could be compromised as a result of chondrocyte dysfunction.
Although TLR2/4dKO chondrocytes had slightly reduced basal capacity of autophagy flux, they caused little cellular senescence at basal levels. These were supported by non-detectable of basal p16INK4a (data not shown) and phosphorylation of histone H2AX (Ser139), the second most common marker of cellular senescence, in either WT or TLR2/4dKO chondrocytes. TLR2/4dKO chondrocytes exhibited impaired autophagic degradation in response to acute nutrient starvation and TLR2 and TLR4 agonists. Previous studies revealed that TLR activation is required for p62 upregulation (14), and p62 plays an essential role in autophagic selective degradation (12,14). We observed that LPS and rHMGB1 failed to up-regulate p62 expression in TLR2/4dKO chondrocytes. Interestingly, TLR4 induces p62 expression via its downstream signaling that leads to activation of nuclear factor erythroid 2-related factor 2 (Nrf2), a master regulator of cellular redox homeostasis, which binds to p62 promoter (14). p62-mediated induction of autophagy is also crucial for Nrf2 activation and elimination of mitochondrial dysfunction and oxidative stress (15). Given that autophagy is a highly conserved recycling process that involves degradation of cellular constituents in lysosome, the diminished efflux of products from autolysosomes is likely to induce a state of metabolic insufficiency and accumulate dysfunctional organelles and aggregates of macromolecules, therefore compromising cellular homeostasis. Because TLR signaling is required for upregulation of p62, the endogenous TLR2 and TLR4 agonists generated after injury in the joint would not be able to do so in mice deficient in TLR2 and/or TLR4. As such, chondrocyte capacity of autophagy flux would be impaired, which could lead to disturbance of cartilage homeostasis.
In conclusion, TLR2 and TLR4 signaling has a dual role in articular chondrocytes in vitro, regulating not only innate immunity-mediated catabolic activity, but also antioxidant, DNA repair and autophagy flux activity. Further in vivo study is needed to verify the role of TLR signaling in cellular stress responses and quality control that are critical for chondrocyte/cartilage homeostasis. Nevertheless, the findings of our study pointed out that caution must be taken in considering new therapeutic strategies involving manipulation of TLR pathways for OA, because finding the balance between TLR-mediated tissue destructive and tissue protective events in the joint could be very challenging.
Supplementary Material
Acknowledgments
Studies were supported by the Department of Veterans Affairs grant I01BX007080 (RLB) and NIH grant AR106796 (RLB).
Role of the funding source:
This study was supported by Department of Veterans Affairs Merit Review grant 1I01BX002234 (PI: Liu-Bryan) and NIH grant AR106796 (PI: Liu-Bryan).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: None
References
- 1.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu-Bryan R. Synovium and the innate inflammatory network in osteoarthritis progression. Curr Rheumatol Rep. 2013;15:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blom AB, van Lent PL, Abdollahi-Roodsaz S, van der Kraan P, van den Berg W. Elusive role for toll like receptor 2 in joint pathology during experimental osteoarthritis. Osteoarthritis and Cartilage. 2011;19(Suppl 1):25. [Google Scholar]
- 4.Nasi S, Ea HK, Chobaz V, van Lent P, Lioté F, So A, et al. Dispensable role of myeloid differentiation primary response gene 88 (MyD88) and MyD88-dependent toll-like receptors (TLRs) in a murine model of osteoarthritis. Joint Bone Spine. 2014;81:320–4. [DOI] [PubMed] [Google Scholar]
- 5.Kalaitzoglou E, Lopes EBP, Fu Y, Herron JC, Flaming JM, Donovan EL, Hu Y, Filiberti A, Griffin TM, Humphrey MB. TLR4 Promotes and DAP12 Limits Obesity-Induced Osteoarthritis in Aged Female Mice. JBMR Plus. 2018;3:e10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rakkola R, Matikainen S, Nyman TA. Proteome analysis of human macrophages reveals the upregulation of manganese-containing superoxide dismutase after toll-like receptor activation. Proteomics. 2007;7:378–84. [DOI] [PubMed] [Google Scholar]
- 7.Kutikhin AG, Yuzhalin AE, Tsitko EA, Brusina EB. Pattern recognition receptors and DNA repair: starting to put a jigsaw puzzle together. Front Immunol. 2014;5:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen LY, Wang Y, Terkeltaub R, Liu-Bryan R. Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthritis Cartilage. 2018;26:1539–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lotz MK, Caramés B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat Rev Rheumatol. 2011;7:579–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc. 2008;3:1253–60. [DOI] [PubMed] [Google Scholar]
- 12.Komatsu M, Kageyama S, Ichimura Y. p62/SQSTM1/A170: physiology and pathology. Pharmacol Res. 2012;66:457–62. [DOI] [PubMed] [Google Scholar]
- 13.Gavriilidis C, Miwa S, von Zglinicki T, Taylor RW, Young DA. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheum. 2013;65:378–87. [DOI] [PubMed] [Google Scholar]
- 14.Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A. 2011;108:1427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah SZA, Zhao D, Hussain T, Sabir N, Mangi MH, Yang L. p62-Keap1-NRF2-ARE Pathway: A Contentious Player for Selective Targeting of Autophagy, Oxidative Stress and Mitochondrial Dysfunction in Prion Diseases. Front Mol Neurosci. 2018;11:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.