

Summary
Osteoarthritis (OA) is a family of degenerative diseases affecting multiple joint tissues. Despite the diverse etiology and pathogenesis of OA, increasing evidence suggests that macrophages can play a significant role in modulating joint inflammation, and thus OA severity, via various secreted mediators. Recent advances in next-generation sequencing technologies coupled with proteomic and epigenetic tools have greatly facilitated research to elucidate the embryonic origin of macrophages in various tissues including joint synovium. Furthermore, scientists have now begun to appreciate that macrophage polarization can span beyond the conventionally recognized binary states (i.e., pro-inflammatory M1-like vs. anti-inflammatory M2-like) and may encompass a broad spectrum of phenotypes. Although the presence of these cells has been shown in multiple joint tissues, additional mechanistic studies are required to provide a comprehensive understanding of the precise role of these diverse macrophage populations in OA onset and progression. New approaches that can modulate macrophages into desired functional phenotypes may provide novel therapeutic strategies for preventing OA or enhancing cartilage repair and regeneration.
Keywords: Joint synovium, Infrapatellar Fat Pad, Muscle, Ligament, Immunomodulation
Introduction
Osteoarthritis (OA) is a disease of the joint organ system characterized by the degradation of articular cartilage, inflammation of the synovium and joint fat pad, as well as alterations in bone structure. Over 27 million people are estimated to suffer from OA in the US, resulting in a tremendous socioeconomic burden [1]. The etiology of OA has been shown to be heterogenous, and may in fact represent a family of diseases rather than one disease [2, 3]. As such, several risk factors such as genetic predisposition, obesity, aging, and joint trauma have been identified for OA. Irrespective of this multifaceted nature of OA pathophysiology, it is now accepted that joint inflammation plays a major role in OA onset and progression [2].
Inflammation is classically regulated by a variety of immune cells such as T cells, neutrophils, and macrophages. Macrophages are phagocytic cells that can be found in almost every tissue (including brain, liver, skin, and joints). The primary role of macrophages is to maintain tissue homeostasis and protect the host from infection. Although primarily considered to be critical components of innate immunity, macrophages are capable of bridging and instructing the response of the adaptive immune system via various secretory mediators. Based on their interaction with T cells, macrophages have been typically dichotomized into two phenotypes: M1-like macrophages (“classically” activated) are antimicrobial and pro-inflammatory, and are activated in response to stimuli from T helper type 1 (Th1 cell), while M2-like macrophages (“alternatively” activated) are anti-inflammatory and pro-resolving, and are induced by Th2 cells. The dysregulated balance between pro- and anti-inflammatory macrophages may lead to chronic low-grade inflammation and has been suggested to be critical in the development of several musculoskeletal diseases, including OA [3].
Here, we review recent key findings of the origin of tissue macrophages and how these cells modulate anabolic and catabolic responses in OA, with a particular focus on the constituents of the joint organ system. Furthermore, recent advances in next-generation sequencing technologies and transcriptomic analysis have greatly facilitated our understanding of phenotypic identity and heterogeneity of the macrophage populations [4, 5]. Thus, we also summarize the potential contributions of previously unrecognized macrophage subpopulations to OA pathogenesis. Finally, we present novel therapeutic strategies, particularly biomaterial scaffolds and cell reprograming, that have the potential to modulate the specificity and protective function of macrophages for mitigating OA progression or enhancing cartilage repair.
Origin and heterogeneity of macrophages
Conventionally, macrophages were thought to derive solely from circulating bone marrow-derived monocytes [6]. However, the use of single-cell RNA sequencing and flow cytometry for wide-scale analysis of gene and protein expression has improved identification of macrophage origin and subtypes. New evidence suggests that embryonic development of macrophages in mammals, in fact, occurs in three successive waves: 1) the primitive wave, 2) the transient definitive wave, and 3) the definitive wave (Figure 1). A summary of main macrophages markers discussed in the current review is listed in Table 1.
Figure 1.
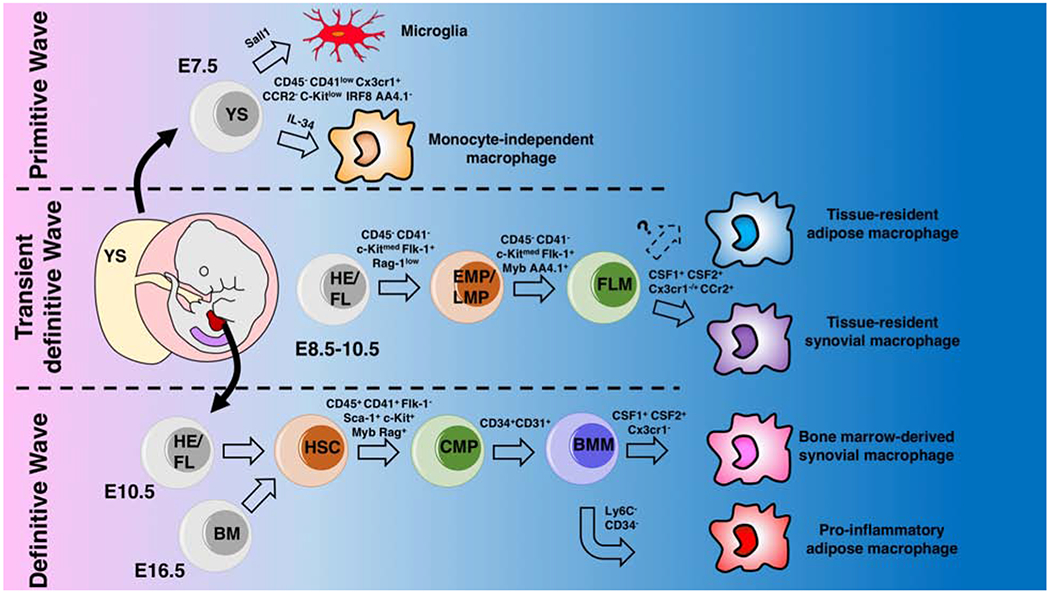
Schematic representation of the origins of embryonic and adult macrophage lineages in the mouse yolk sac (YS), aorta-gonado-mesonephros region (AGM, purple color in the embryo) , and fetal liver (FL, red color in the embryo).
a. Primitive wave. Primitive hematopoiesis starts at E7.5 in the blood islands of the yolk sac (YS) which generates erythro-myeloid progenitors (EMPs) and monocyte independent macrophages that develop prior to the formation of the blood brain barrier.
b. Transient definitive wave. Upon establishment of the blood circulation around E8.5, the YS hemogenic endothelium (HE) generates late EMPs and additional progenitors with lymphoid potentials (LMPs) without long-term reconstitution capacity. These YS generated progenitors seed the fetal liver and rapidly produce myeloid progenitors that can generate fetal monocytes which differentiate into macrophages once circulated into tissue.
c. Definitive wave. Hematopoietic stem cells (HSCs) emerge from the main HE situated in the aorta-gonado-mesonephros (AGM) regions and in the placenta around E10.5. These continue to seed the fetal liver and produce fetal monocytes until E16.5 at which hematopoiesis switches completely from the fetal liver to the bone marrow (BM).
AGM: aorta-gonado -mesonephros region, BM: bone marrow, BMM: bone marrow monocyte, CMP: common myeloid progenitor, EMPs: erythro-myeloid progenitors, FL: fetal liver, FLM: fetal liver monocytes, HE: hemogenic endothelium, LMPs: lymphoid-myeloid progenitors, YS: yolk sac.
Table 1.
Summary of main macrophages markers
| Marker (alternative names) | Role | References |
|---|---|---|
| CX3CR1 (Gpr13) | Immune cells migration, expressed during transient definitive wave | [7, 18] |
| F4/80 (Emr, Adgre1) | Necessary for murine macrophages development, pan macrophages marker | [18] |
| CD11b (Itgam) | Common myeloid marker, mediates leukocytes migration | [10, 15, 19] |
| Ly6C (Gr1) | Marker of classical monocytes, potential marker for bone marrow/monocyte-derived macrophages | [22] |
| CD14 | Macrophage marker in human, co-receptor for TLR4 | [10, 15, 19] |
| CD206 (Mrc1) | Expressed on macrophages and immature dendritic cells, play role in phagocytosis, antigen presentation, and resolution of inflammation | [43, 44] |
| CD11c (Itgax) | Found on dendritic cells, monocytes, and macrophages play a role in phagocytosis, cell migration, and cytokine production | [95] |
| CD115 (Csfr1) | Receptor for Colony stimulating factor 1 (Csf-1), a cytokine which controls the production, differentiation, and function of macrophages. | [91–93] |
| CD163 | Marker for monocytes and macrophages, scavenger receptor | [85] |
While not yet precisely described in humans, during mouse development, the primitive wave occurs around embryonic day 7.5 (E7.5), during which myeloid progenitors arise in the blood islands of the yolk sac and differentiate into primitive macrophages without the need of a monocytic intermediate [7, 8]. For instance, microglia, a subtype of macrophages found in brain, were shown to be definitively derived from yolk sac precursors [9, 10]. However, there is some debate if other tissue-resident macrophages, such as those seen in the epidermis, liver, and lung, are derived from the primitive wave or from migratory erythro-myeloid precursors (EMPs) and lympho-myeloid progenitors (LMPs) in the following transient definitive wave [10, 11].
The transient definitive wave occurs between E8.5 and E10.5. During this stage, the fetal liver of the embryo sequentially acquires C-Myb+/CX3CR1+ EMPs and LMPs that arise from the hemogenic endothelium of yolk sac. These precursors do not exhibit the long-term reconstitution potential of hematopoietic stem cells (HSCs) [12].
The final definitive wave occurs in two overlapping stages and relies on the differentiation of HSCs. In the first stage, HSCs arise in the aorta-gonado-mesonephros (AGM) region and migrate to the fetal liver around E10, initiating definitive hematopoiesis. At the second stage, starting around E16.5, fetal liver hematopoiesis declines and is replaced by bone marrow hematopoiesis, which produces bone marrow monocytes throughout life.
Notably, the heterogeneity of macrophages is related to their origin. For instance, embryonically derived tissue-resident macrophages were shown to be able to self-renew, and their primary role is to maintain tissue homeostasis during adulthood [13]. Upon irradiation, tissue-resident macrophages are less affected than their bone marrow counterparts and are not replenished by circulating monocytes, such as bone marrow-derived macrophages [14, 15]. Generally, tissue-resident macrophages and bone marrow-derived macrophages exhibit distinct genetic and functional signatures, with tissue-resident shown to be more expressive of proangiogenic markers. This observation suggests that tissue-resident macrophages may have a phenotype similar to previously described “M2-like” macrophages [16].
Macrophages and OA
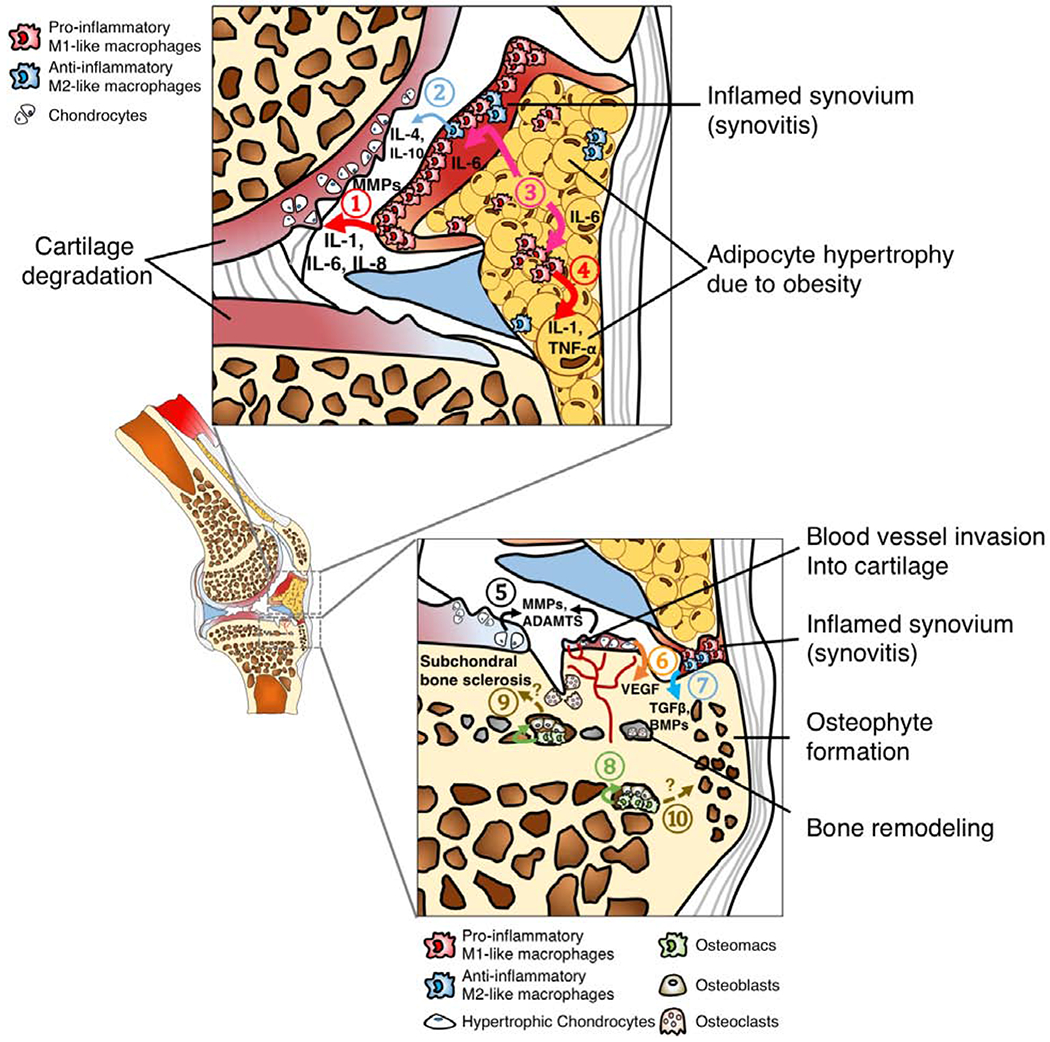
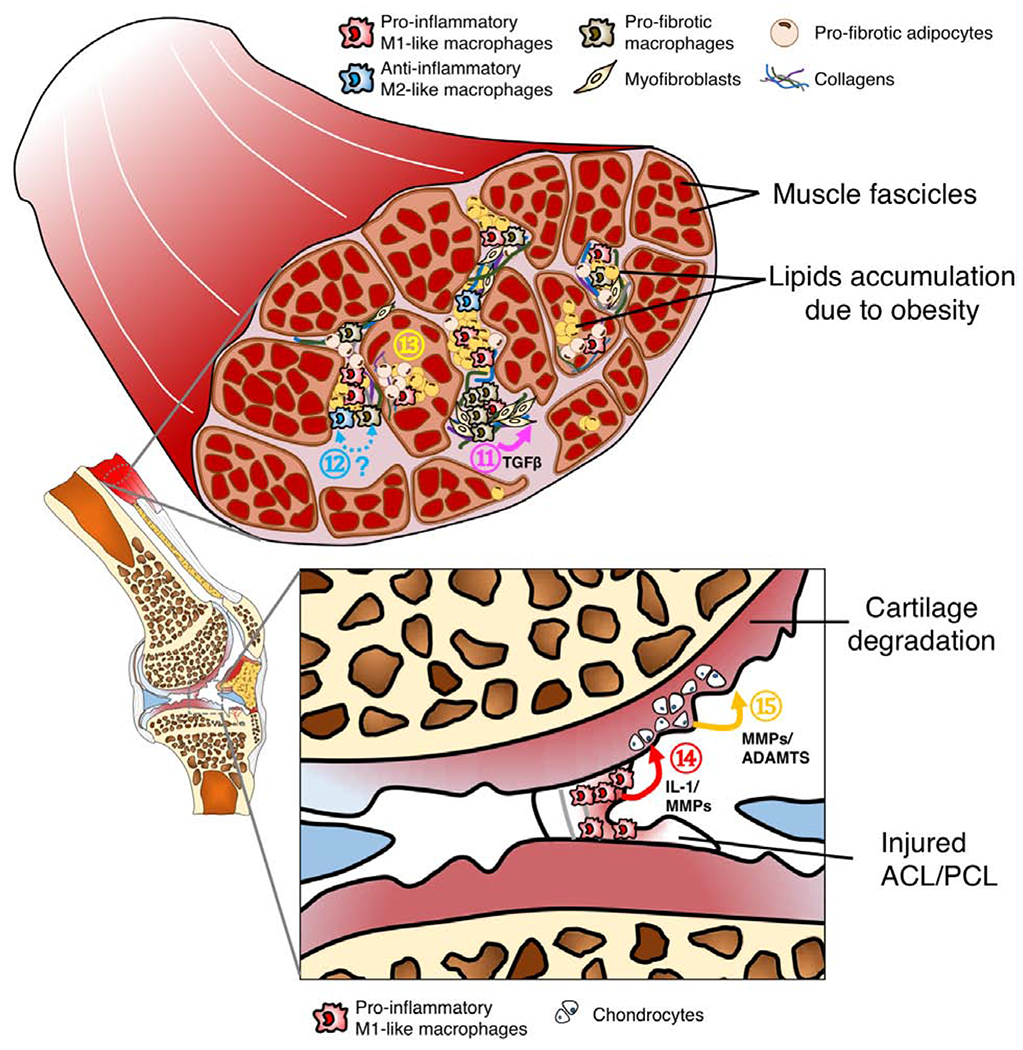
OA is a disease that affects most joint tissues, and increasing evidence suggests that specific phenotypes of macrophages may differentially modulate the anabolic or catabolic responses of different cell types with the onset or progression of OA (Figure 2A–C).
Figure 2.
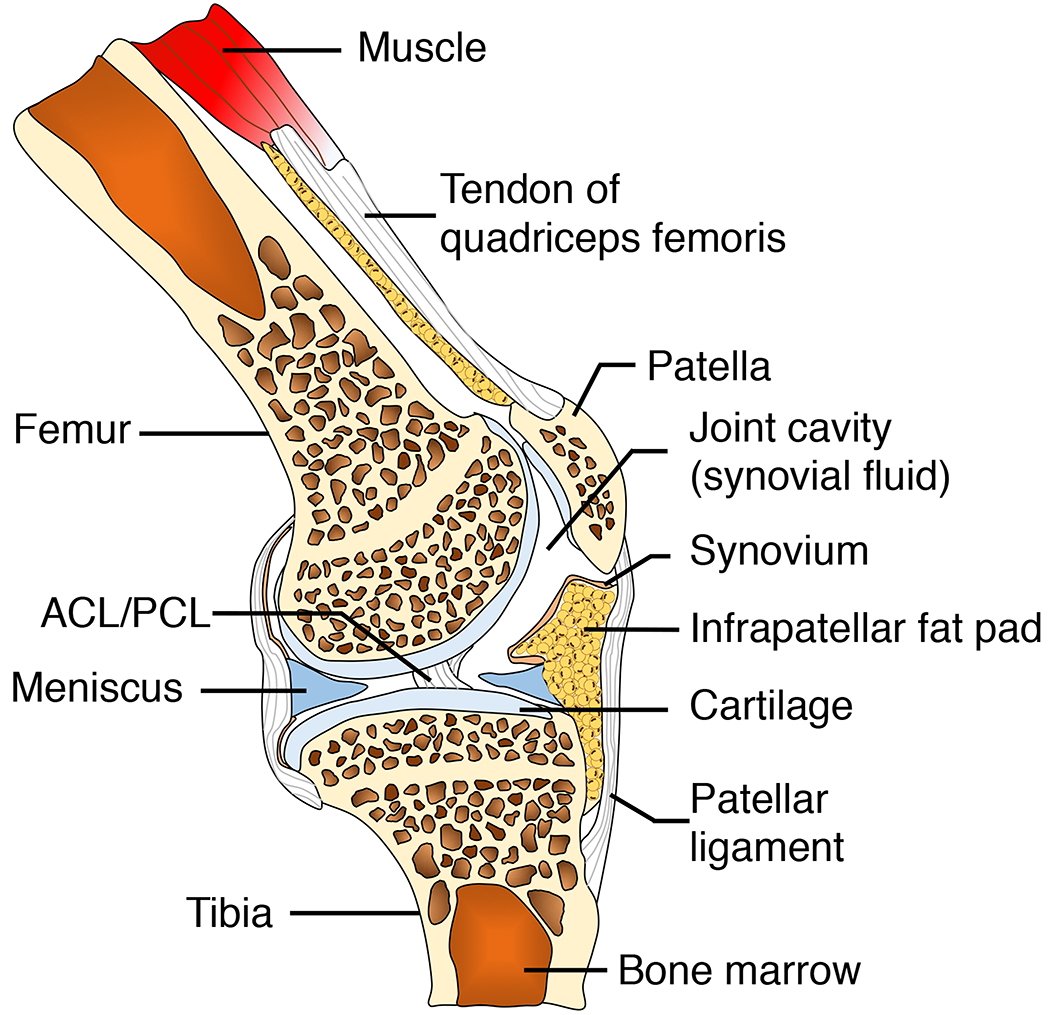
A. Tissue compartments of a healthy knee joints. ACL: anterior cruciate ligament. PCL: posterior cruciate ligament.
B. Tissue compartments of an OA knee joints. ① Pro-inflammatory M1-like macrophages in the synovial lining layer secrete inflammatory cytokines such IL-1, IL-6 and IL-8, as well as cartilage matrix degradation enzymes including MMPs, leading to cartilage degeneration. Although ② Anti-inflammatory M2-like macrophages can release reparative mediators such as IL-4 and IL-10 into joint synovial fluid, these anti-inflammatory molecules are often not sufficient to encounter the catabolic inflammatory response, partially due to high ratio of M1-like to M2-like macrophages [31]. High-fat diet-induced obesity results in adipocyte hypertrophy in infrapatellar fat pad due to increased lipid storage.③ Hypertrophic adipocytes secrete IL-6, activating ④ macrophages in both synovium and joint fat pad to secrete IL-1 and TNF-α, leading to a vicious inflammatory cycle [103]. ⑤ Chondrocytes in OA cartilage also secrete MMPs and disintegrin metalloproteinase with thrombospondin motifs (ADAMTs) [104]. Additionally, ⑥ hypertrophic chondrocytes secrete vascular endothelial growth factor (VEGF) which leads blood vessel invasion into cartilage, an important of step of cartilage and bone remodeling [105]. Bone remodeling in OA can be driven by several factors. For example, ⑦ synovial macrophages, potentially M2-like phenotype, secrete TGFβ and BMPs, facilitating osteophyte formation. Furthermore, ⑧ osteomacs, a recently discovered phenotype of macrophages within bone marrow, form a canopy over osteoblasts, supporting the bone matrix deposition function of osteoblasts. However, whether osteomacs directly contribute ⑨ subchondral bone sclerosis or ⑩ osteophyte formation in OA remains unknown.
C. Tissue compartments of an OA knee joints. It has been suggested that loss of muscle integrity and associated functional deficits due to injury or obesity may alter joint loading, leading to onset of OA. Particularly, ⑪ Pro-fibrotic macrophages activate collagen-producing myofibroblast via the TGFβ signaling pathway. While this reparative process is essential in tissue healing, it may lead to tissue fibrosis if not modulated. ⑫ Currently, the origin and phenotypic plasticity of pro-fibrotic macrophages remains unclear, although some evidence suggests that pro-fibrotic macrophages are associated with anti-inflammatory M2-like macrophages. Interestingly, recent studies demonstrate that bone marrow-derived macrophages can also transdifferentiate into myofibroblasts [106]. Additionally, ⑬ pro-fibrotic adipocytes, a potential phenotypic switch of adipocyte progenitors due to obesity, can also significantly contribute to tissue fibrosis in muscle [107]. Ligament injury often leads to abnormal loading forces on cartilage, predisposing the injured joint to OA development. In addition to altered mechanical loading, macrophage-associated inflammation provides an alternative mechanism for OA pathogenesis post-ligament injury. Specifically, ⑭ Pro-inflammatory M1-like macrophages infiltrate into the injury site, releasing IL-1 and MMPs into joint synovial fluid. ⑮ These pro-inflammatory molecules can activate chondrocytes to secrete more ECM degradation enzymes including MMPs and ADAMTs, further accelerating cartilage degradation.
Synovium
The joint synovium is comprised of a synovial lining layer and a sublining compartment. Macrophages, one of the most abundant immune cells in the synovium, are mainly located in the synovial lining layer along with fibroblasts and play a critical role in maintaining homeostasis of healthy synovial tissues [17]. Both tissue-resident (i.e., embryonic derived) synovial macrophages and non-tissue resident (i.e., bone marrow-derived) synovial macrophages are capable of self-renewal although tissue-resident synovial macrophages can persist independently of bone marrow hematopoiesis for prolonged periods of time.
In the synovium, tissue-resident macrophages have been proposed to originate from both primitive and transient definitive waves during embryonic development, although predominately from the latter wave. In mice, it is reported that embryonically derived synovial macrophages generated from these two waves are positive for F4/80 (a mouse-specific marker of macrophages) but negative for CD11b (i.e., F4/80+/CD11b−). On the contrary, macrophages both generated from the definitive wave, classified as F4/80lo/CD11b+/Ly6C+ macrophages, and those generated substantially later from bone marrow hematopoiesis, such as CX3CR1+/Ly6Chigh macrophages are relatively short-lived and are categorized as non-tissue resident macrophages. These non-tissue resident macrophages have been shown to differentiate into pro-inflammatoryM1-like or anti-inflammatory M2-like phenotypes after homing to tissues, regulating inflammation and healing [18]. Additionally, macrophages that develop after birth generally represent a mixed phenotype (F4/80+/CD11b+), further complicating the distinction between embryonic and bone marrow-derived precursors [10, 15, 19]. In attempt to elucidate the origin of these differential macrophage populations, Culemann et al. examined the spatiotemporal composition of macrophages in the synovium. The group identified two distinct synovial macrophages subtypes: CX3CR1− interstitial, and CX3CR1+ lining macrophages. The interstitial macrophages, developed from the two early waves, were proposed to make up a pool of proliferating MHCII+ macrophages that could differentiate into additional subpopulations of interstitial macrophages (like RELM-α+) or lining macrophages. Interestingly, CX3CR1+ lining macrophages also displayed a limited response to inflammatory stimulation, conserved their naïve state, and restricted inflammatory progression through a shield of tight junction like structures [20].
Recent studies have demonstrated that with rheumatoid arthritis (RA), macrophages that infiltrate into the synovium are mainly migratory bone marrow-derived macrophages, rather than tissue-resident macrophages. Supporting this evidence, bone marrow-derived Ly6Chigh monocytes are recruited and differentiate into macrophages at inflammatory sites in RA [21]. Furthermore, bone marrow-derived macrophages express CD80 and CD86, and secrete high levels of pro-inflammatory mediators such as interleukin (IL)-1β and IL-12, as compared with tissue-resident macrophages. Additionally, bone marrow-derived macrophages are located near blood vessels, in contrast to F4/80+ tissue-resident macrophages, implying that bone marrow-derived macrophages are more likely to be newly recruited cells upon injury [15, 22]. Interestingly, Wood et al. reported 2 subtypes of macrophages in human OA synovium: inflammatory like macrophages (iOA) and classical macrophages (cOA) are both present in OA synovium and possess different proliferation abilities. The cOA were characterized by a remodeling phenotype and expression of the IGFBP5 gene, while iOA were more proliferative and were enriched in MK167 and CTRL genes [23].
Regardless their origin, it is well recognized that synovial macrophages are an important source of post-joint injury pro-inflammatory signaling molecules, including alarmins (e.g., s100A9) [24, 25] and cytokines such as IL-1 and tumor necrosis factor (TNF)-α [26, 27]. Moreover, human CD14+ synovial macrophages are reported to activate the production of matrix metalloproteinases (MMPs), destructive matrix-degrading enzymes, from synovial fibroblasts [28, 29]. Although some macrophages, particularly the M2-like phenotype, can release anti-inflammatory cytokines including IL-4 and IL-10, these anti-inflammatory molecules are often not sufficient to counter the catabolic inflammatory response, particularly in the presence of a high pro-inflammatory (M1-like) to anti-inflammatory (M2-like) ratio [30]. As a result, these MMPs and inflammatory mediators shift the cytokine profile of joint synovial fluid in favor of inflammation with high levels of IL-1β, IL-6, and IL-8, stimulating chondrocytes to produce more ECM-degradative enzymes and further aggravating cartilage matrix destruction. Macrophages and monocytes have also been detected in the synovial fluid of OA joints. Their presence positively correlates with joint stiffness, pain [32], and reduced quality of life [31, 32].
Individuals with obesity and metabolic disorders such as hypercholesterolemia exhibit chronic low-grade systemic inflammation. Macrophages, particularly those accumulated in visceral fat, are involved in dysregulated systemic inflammation and may be essential contributors for obesity-associated OA. In obesity driven OA, scientists reported an increase in macrophage content in the synovium both with injury [33–35] and with spontaneous OA [30] in mice. For instance, Sun et al. demonstrated an increase in M1-like (NOS2+) macrophages in OA synovium from obese mice [36]. In patients with obesity, increased macrophage content in the synovium has also been described [17].
Infrapatellar Fat Pad (IPFP)
The infrapatellar fat pad (IPFP), or Hoffa’s pad, is an adipose tissue depot found in the knee joint. It is an active endocrine organ within the joint and a potent producer of adipokines, including adiponectin and leptin [37], and has been proposed to have an important role in the pathogenesis of OA [38]. The IPFP also contains macrophages [39, 40] with an immune cell profile comparable but not identical to that of the synovium [17] and subcutaneous adipose tissue [41]. The number of macrophages in IPFP was reported to increase during OA and other in joint pathologies [42, 43]. Both M1-like (CD11c+) and M2-like (CD206+) macrophage markers have been reported in this tissue [44]. Wei and co-workers suggest that macrophages harvested from the IPFP of diseased joints inhibit chondrogenesis of mesenchymal stem cells (MSCs) [45], supporting the notion that IPFP macrophages play a potentially detrimental role in cartilage regeneration. However, the precise function and origin of macrophages in the IPFP remain to be elucidated.
The IPFP has been shown to contain multipotent cells [46] and to increase in size and develop fibrosis in obese mice [44]. In humans, the IPFP also increases in size with age and OA [47], potentially due to adipocyte hypertrophy [43]. Interestingly, in early OA, high-fat diet induced obese mice do not have increased numbers of M1-like macrophages in IPFP relative to lean mice [44], while the study on obese OA patients have reported either no differences [39] or an increased content of M2-like (CD206+) macrophages in the IPFP as compared to lean patients [43]. Thus, IPFP is a potent reservoir of macrophages, whose roles OA development remain unknown.
Bone
During OA development, the subchondral bone compartment undergoes active remodeling, a process that is partially influenced by macrophages. Although some studies implied that bone microdamage might be the first sign of OA-related bone remodeling, followed by increased bone thickening and ultimately resulting in bone sclerosis, the sequence of cartilage degradation and subchondral bone alteration remains controversial and may likely depend on the subtypes of OA [48]. During OA progression, CD68+ cells have been shown to localize in sclerotic regions of subchondral bone [49]. Increased subchondral bone turnover is also accompanied by abnormal osteophyte formation, one of the hallmarks of OA that is positively correlated with immobility and increased pain. Synovial lining macrophages have been shown to mediate formation of osteophytes in OA. The depletion of synovial macrophages by clodronate-laden liposomes (which lead to macrophage apoptosis when being phagocytized) significantly reduced the onset of osteophyte formation [50], and it is believed that TGFβ produced by synovial macrophages plays a major role in osteophyte formation [51]. The effect of macrophage depletion on OA development will be discussed in detail in a later section of this review.
Bone remodeling is tightly regulated by osteoblasts originating from the mesenchymal lineage and osteoclasts derived from myeloid lineage. Recent studies show that in addition to resident tartrate-resistant acid phosphatase (TRAP+) osteoclasts, two other monocyte-derived cell populations may be involved: 1) osteal macrophages (osteomacs) and 2) bone marrow macrophages. Bone marrow macrophages are located in the bone marrow stroma and are important in regulating hematopoiesis throughout adulthood [52]. Osteomacs, reported to be F4/80+/CD115+/Mac3+/CD169+/TRAP− cells, reside within periosteal and endosteal compartments of the bone and may play an anabolic role through stimulating osteoblasts both to increase the mineralization process as well as to promote bone healing and remodeling following fracture [53]. However, whether osteomacs respond to mechanical stimuli such as joint loading, as well as whether they directly contribute to subchondral bone sclerosis and osteophyte formation in OA development, require further investigation.
Ligament
Ligaments are an essential component for joint formation as they connect two joint bones together and provide joint stability. Hence, ligament injuries are among the risk factors for developing OA. For instance, around 50% of patients with anterior cruciate ligaments (ACL) injuries show evidence of OA within 10-20 years post-trauma [54]. Immediately post-ligament injury, it is the malaligned joint results in abnormal loading forces on cartilage, leading to chondrocyte apoptosis, a potential mechanism initiating the onset of OA. In addition to mechanical factors, several lines of evidence suggest that macrophages infiltrate into the injury site and regulate inflammation and healing of the ligament by secreting various mediators, providing an alternative mechanism that is driven by cytokines in modulating OA pathogenesis. Muir et al. reported that ACL diseases and rupture significantly increased the number of pro-inflammatory macrophages expressing degradative enzymes including TRAP and cathepsin K (a potent collagenolytic protease) within torn canine ligament tissue, compared to dogs with intact ACL [55, 56]. For humans, infiltration of TRAP+ macrophages, but not cathepsin K+ macrophages, was observed in the injured ACL tissue of patients [57], implying distinct differences between dogs and humans in proteolytic activities of injured ligaments. Nevertheless, the infiltration of macrophages plays an important role in the acute inflammatory phase post-injury (about 24-28 hours), by removing tissue debris and remodeling matrix via secreted proteases and mannose receptor-mediated phagocytic route [58, 59]. However, macrophages may also decrease the mechanical properties of the injured ligament due to excess matrix degradation if the resolution of inflammation is not regulated. Importantly, ligamentous injury may result in macrophage infiltration of other surrounding joint tissues such as the meniscus [60], leading to a local pro-inflammatory response, particularly in the vascularized zone [61].
Tendon
Tendon is a fibrous, connective tissue bridging muscles to bone. Tendons are composed mainly of collagen fibrils, and their main function is to respond to mechanical forces, providing joint flexion and stability. Similar to ligaments, the instability of tendons due to injury is among the risk factors for OA onset and contributors to OA progression, but the direct influence of inflammatory tendon changes on OA remains a largely open area for investigation. The presence of M1-like macrophages in conjunction with loading may facilitate tendon repair compared to injured tendon without loading [62]. However, in IKKβCAScx mice (a cre-mediated mouse model in which IKKβ is constitutively active in scleraxis (Scx) positive tendon fibroblasts, an overabundance of pro-inflammatory macrophages in tendon induced by increased NF-κB activity negatively affected tendon healing, the surrounding attachment site, and bone integrity [63]. Therefore, determining the time-course and balance of pro and anti-inflammatory macrophages is likely critical in maintaining homeostasis and joint integrity. When macrophages are modulated using extracellular exosome vesicles to encourage a lower ratio of M1-like to M2-like macrophages, healing is improved in the Achilles tendon [64]. While some pro-inflammatory mediators potentially driven by M1-like macrophages have been identified as detrimental to tendon healing, adipose-derived mesenchymal stromal cells can modulate the tendon inflammatory response in vitro [65]. In obesity/type II diabetes, a higher incidence of M1-like macrophages has been reported, coupled with elevated and prolonged expression of M2-like markers, resulting in increased extracellular matrix deposition and stiffer tendons [66]. Together with altered muscle integrity, stiffer tendons could contribute to alterations in joint loading that may antagonize OA with obesity. While the ideal ratio of pro- and anti-inflammatory macrophages for tendon repair remains unknown, it appears that a certain threshold of M1-like macrophages may be beneficial. Nevertheless, chronic exposure to overabundant M1-like macrophages may be deleterious for tendon repair. Identifying threshold for unique macrophage subtypes in tendon injuries may offer therapeutic opportunities for tendon repair.
Muscle
Muscle integrity, particularly strength and structure, play a role in the onset and progression of OA. It is plausible that altered muscle integrity and associated functional deficits would alter joint loading, leading to joint degeneration. Macrophages are one of the most active immune cells in skeletal muscle and, together with satellite cells, are involved in maintaining muscle homeostasis [67]. For example, macrophages protect against muscle atrophy by releasing insulin-like growth factor 1 in repair scenarios [68]. The tissue resident macrophage population in muscle has been described as CD11b+/F4/80+/CD11c−/Ly6C−/CX3CR1−[69], a phenotype which activates the innate immune response to injury. Interestingly, both Ly6C+ and Ly6C− monocytes migrate to muscle post-muscle injury [70]. It is postulated that incomplete or maladaptive repair may induce muscle weakness in a low-level systemic inflammatory environment, and macrophages are implicated as one of the key cell types in this process [71].
OA patients with obesity consistently demonstrate muscle weakness [72]. It appears that human patients with composition-based obesity and sarcopenic obesity, but not sarcopenia or muscle wasting alone, are positively associated with knee OA [73]. In muscle of lean athletes, lipid and adipose stores can be found within and between muscle fibers, presumably to provide a readily-available fuel source [74]. With obesity, pro-inflammatory M1-like macrophages are present in the quadriceps muscle of mice [75], rats [76, 77], and humans [75, 78]. Other inflammatory alterations in skeletal muscle with obesity are reviewed in detail by Wu and colleagues [79].
Nutrient overload results in the development of additional lipid stores within and between muscle fibers [71], and intracellular lipid accumulation is associated with metabolic disturbance and insulin resistance [80]. Macrophages and T-cells can migrate and infiltrate local fat and/or lipid stores within the muscle [79] and alter the inflammatory status of the lipid and muscle. In fact, T-cells and macrophages are located predominately in intramuscular and perimuscular adipose tissue in skeletal muscle of individuals with obesity, supporting the notion that intramuscular adipose depots can become active inflammatory sites within this tissue [78]. There is known lipid-muscle cross-talk in vitro, where inflammation from obese adipocytes can induce muscle atrophy [81]. Furthermore, our group demonstrated that high-fat diet induced obese mice receiving muscle-target gene therapy for follistatin, an activin binding protein, showed increased muscle mass coupled with both decreased OA severity and numbers of M1-like (CD11b+/CD11c+) macrophages in visceral fat compared to wild type obese mice [82].
While the direct relationship between muscle inflammation, muscle macrophage populations, and OA is yet to be established, the ability of muscle macrophages to directly and indirectly influence the homeostasis of joint organ system is an interesting therapeutic concept for future studies.
Modulation of macrophages and their associated phenotypes as immunotherapies for OA and cartilage repair
Macrophage depletion as a therapeutic intervention
As macrophages play a crucial role in orchestrating other cells in initiation of OA immunopathogenesis, numerous studies have investigated whether cartilage health and joint integrity would benefit from ablation of macrophages. For example, the removal of synovial lining macrophages by intra-articular injection of clodronate-laden liposomes significantly decreased gene expression of MMP-3 and MMP-9 in the synovium (but not in cartilage), along with reducing TGF-β-mediated osteophyte formation in a collagenase-induced mouse model [50, 83, 84]. Additionally, to understand the effect of macrophage depletion on ligament healing, Chamberlain and co-workers systemically depleted macrophages via intravenous injection of clodronate-laden liposomes two days prior to injury induction in medial collateral ligament (MCL) in rats. The authors observed that the MCL of the rats receiving macrophage depletion deteriorated mechanical strength along with decreased number of M1-like (CD68+) and M2-like (CD163+) macrophages compared to the MCL of the rats without treatment [85].
Interestingly, systemic macrophage depletion in mice using clodronate-laden liposomes (4 hr prior and then daily post-injury for 4 days) appeared to be beneficial for healing process of injured Achilles tendon by enhancing its ultimate tensile strength and Young’s modulus [86]. Similarly, using tendon as a graft, weekly systemic macrophage depletion by liposomal clodronate (up to 42 days) was found to improve the healing and mechanical strength at tendon-bone interface in a model of ACL reconstruction in rats [87]. The disparities in the results of these studies imply that to achieve a beneficial effect of macrophage depletion on wound healing, depletion strategies, particularly routes (systemic vs. local) and frequency (short-term vs. long-term), could be tissue-dependent. The reasons for the requirement of tissue-specific depletion approaches may be associated with the phenotypic identities of macrophages within a given joint issue. Moreover, the degree and duration of vascularity of the tissues during the healing process may influence tissue-specific macrophage depletion outcomes, as the vessel channels serve as the entry sites of monocytes/macrophages.
These earlier findings have led to the hypothesis that depletion of macrophages could mitigate OA in obesity. In our recent study, macrophage Fas-induced apoptosis (MaFIA) mice – a transgenic mouse model that allows conditional depletion of macrophages expressing colony stimulating factor 1 receptor (CSF1R) upon administration of the small molecule AP20187 – were placed on a high-fat diet and underwent DMM surgery to induce knee OA [88]. CSF1R+ macrophages were systemically depleted two weeks prior to DMM surgery in obese MaFIA mice. Surprisingly, systemic depletion of CSF1R+ macrophage did not attenuate the severity of OA in obese mice; instead, it induced inflammation and led to a massive infiltration of CD3+ T cells and neutrophils, but not B cells, into the injured joints. Furthermore, ablation of synovial macrophages by liposomal clodronate led to oxidized low-density lipoproteins (ox-LDL)-induced catabolic processes, including increased pro-inflammatory mediators as well as increased infiltration of monocytes and neutrophils into the joint synovium [89, 90]. Taken together, these findings indicate that macrophages modulate the homeostasis of immune cells with diet-induced obesity in part by regulating levels of ox-LDL in the joint synovial fluid.
While it is well-recognized that the CSF1/CSFR1 signaling axis is required for maintenance of the bone marrow-derived macrophage populations in adult mice [91, 92], recent studies have demonstrated that CSF1R+Kit−CD45+ cells can be detected in the yolk sac at 20-25 somite pairs, and later in the head regions of the embryo from E9.5, and the fetal liver from E10.5 onwards [7, 93]. These findings suggest that a subset of CSF1R+ macrophages is developed during embryonic stage and independent of bone marrow. In addition, using Csf1rcreERR26-tdTomato and Csf1rGFP mice, Culemann et al. observed that tissue-resident interstitial macrophages in embryonic joint synovium express CSF1R and Ki67 (proliferation marker). These results suggest that in a MaFIA mouse model, both tissue-resident and bone marrow-derived macrophages can be targeted by AP20187 as long as these macrophages express CSF1R. Nevertheless, there is still the possibility that certain subpopulations of macrophages may lose their ability to express CSF1R at later stage of development or in adult [94], in which case the treatment of AP20187 would not eliminate CSF1R− macrophages in MaFIA mice.
Despite the fact that several animal models of broad macrophage depletion, including MaFIA and diphtheria toxin receptor (DTR) transgenic mice, have greatly expanded our knowledge on macrophage function in OA, these techniques cannot precisely target specific phenotypes of macrophages without affecting other myeloid lineages such as dendritic cells and neutrophils. For example, CD11c, a conventional marker for pro-inflammatory macrophages, is also expressed by dendritic cells, and some subsets of neutrophils. Furthermore, depletion of CD11c+ macrophages has been shown to lead to distinct neutrophilia responses in three commonly used DTR-based depletion mouse models [95]. Thus, the choice of transgenic mouse lines for the purpose of macrophage depletion must be considered in experimental settings where other myeloid cells may be involved, and caution should be exercised in interpreting the results of these models.
Biomaterial-based and cell engineering-based modulation of macrophages
Biomaterial scaffolds are widely employed to promote cartilage repair. However, scaffold-induced foreign body reaction can lead to innate immune responses from host myeloid cells including neutrophils and macrophages. While the prolonged presence of pro-inflammatory M1-like macrophages is detrimental for tissue repair, it is generally accepted that anti-inflammatory M2-like macrophages facilitate tissue regeneration. Thus, several studies aim to harness the healing capacities of M2-like macrophages by designing scaffolds that can modulate macrophages toward a reparative phenotype. For example, synthetic scaffolds (such as those made of polyethylene glycol, PEG) can elicit a chronic inflammatory response including neutrophil infiltration and the loss of M2-like macrophage markers, while biological scaffolds (such as those derived from extracellular matrix, ECM) induce high expression of the surface marker CD206 on macrophages [96]. This study, although conducted in a mouse model of volumetric muscle loss injury, may provide important insights into the choice of scaffold for cartilage regeneration. While biological scaffolds can promote anti-inflammatory responses, they are known to lack mechanical strength as compared to synthetic ones. One commonly used strategy to improve mechanical properties of a biological scaffold is through cross-linking; however, it is reported that a biological scaffold cross-linked by carbodiimide switches from an M2-like macrophage dominant profile to an M1-like macrophage dominant [97]. Indeed, a recent study shows that macrophage polarization in response to the stiffness of biological scaffolds is dependent on the cross-linking agent, suggesting that both chemical and physical properties of scaffolds are essential in fine-tuning biomaterials to promote a pro-regenerative macrophage phenotype [98].
While many researchers focus on modulating macrophage-induced inflammation through the use of biomaterial scaffolds, some seek to use macrophages themselves as a means of drug delivery or therapy. Macrophages possess an intrinsic homing ability, allowing them to migrate to injury or inflammatory sites including arthritic joints. Visser et al. developed a therapy using the transport of nanoparticle-encapsulated drugs inside autologous M1-like macrophages to induce transient phagosome maturation arrest [99]. Recently, by using CRISPR-Cas9 genome editing, our group created a cell-autonomous system (i.e. “SMART” cells) in which mouse induced pluripotent stem cell (miPSC)-derived chondrocytes can modulate inflammation in an auto-regulated manner both in vitro and in an inflammatory arthritis mouse model. Specifically, once these “SMART” chondrocytes sense specifically targeted inflammatory cytokines (e.g., IL-1 or TNF-α), they release a corresponding biologic drug (e.g., IL-1Ra and or soluble TNFR1) to attenuate inflammatory signals [100, 101]. Similarly, self-regulating “SMART” macrophages can be engineered in a manner that not only homes these cells to inflammatory/injury sites, but also enables cytokine-activated feedback-controlled capabilities for effectively targeted therapeutic drug delivery for joint diseases [102].
Conclusions and future perspectives
Accumulating evidence suggests that macrophages, do not have only two polarized states (i.e., M1-like vs. M2-like), but rather should be categorized as a large family of cells with broad and potentially plastic phenotypes that are capable of defending hosts against pathogens, maintaining tissue homeostasis, and/or providing pro- or anti-inflammatory signals that are origin- and tissue microenvironment-dependent. However, whether tissue-resident macrophages in different joint compartments arise from the same or distinct waves during embryonic development remains to be elucidated. Furthermore, although many studies have implied that tissue resident macrophages appear to be pro-healing while circulating bone marrow-derived monocytes/macrophages tend to be pro-inflammatory in tissue injury, whether such an immune activation holds true in OA pathogenesis requires further investigation.
Although the origin and development of tissue resident and bone marrow-derived macrophages have been investigated in the synovium, macrophage lineage development in other joint tissues remains poorly understood, a significant gap we wish to address as elucidating the heterogeneity of macrophage populations in these tissues may provide key insights into OA pathogenesis. Moreover, we believe that the exploration of approaches for modulating macrophages, either via biomaterials or engineered cells, could be an important future research direction for developing novel therapeutics for OA.
Supplementary Material
Acknowledgments
The authors thank Sara Oswald for providing technical writing support for the manuscript. This work was supported by the Nancy Taylor Foundation, the Arthritis Foundation, NIH (AG46927, AG15768, AR67467, AR65956, AR075899, T32 DK108742, T32 EB018266, P30 AR74992, P30 AR073752), and a Taiwan GSSA Scholarship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
All authors declare no conflict of interest. No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
References
- 1.Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum 2008; 58: 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis and cartilage 2013; 21: 16–21. [DOI] [PubMed] [Google Scholar]
- 3.Xie J, Huang Z, Yu X, Zhou L, Pei F. Clinical implications of macrophage dysfunction in the development of osteoarthritis of the knee. Cytokine Growth Factor Rev 2019; 46: 36–44. [DOI] [PubMed] [Google Scholar]
- 4.Chou C-H, Gibson J, Attarian D, Haraden C, Yohn C, Laberge R-M, et al. Profiling human chondrocytes and synoviocytes using single cell RNA sequencing identifies cell diversity in the pathogenesis of osteoarthritis in the joint organ. Osteoarthritis and Cartilage 2019; 27: S27. [Google Scholar]
- 5.Sommerfeld SD, Cherry C, Schwab RM, Chung L, Maestas DR, Laffont P, et al. Single cell RNA-seq in regenerative and fibrotic biomaterial environments defines new macrophage subsets. bioRxiv 2019: 642389. [Google Scholar]
- 6.van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med 1968; 128: 415–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stremmel C, Schuchert R, Wagner F, Thaler R, Weinberger T, Pick R, et al. Yolk sac macrophage progenitors traffic to the embryo during defined stages of development. Nat Commun 2018; 9: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 2016; 44: 439–449. [DOI] [PubMed] [Google Scholar]
- 9.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010; 330: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheng J, Ruedl C, Karjalainen K. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 2015; 43: 382–393. [DOI] [PubMed] [Google Scholar]
- 11.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012; 336: 86–90. [DOI] [PubMed] [Google Scholar]
- 12.Bertrand JY, Jalil A, Klaine M, Jung S, Cumano A, Godin I. Three pathways to mature macrophages in the early mouse yolk sac. Blood 2005; 106: 3004–3011. [DOI] [PubMed] [Google Scholar]
- 13.Davies LC, Rosas M, Smith PJ, Fraser DJ, Jones SA, Taylor PR. A quantifiable proliferative burst of tissue macrophages restores homeostatic macrophage populations after acute inflammation. Eur J Immunol 2011; 41: 2155–2164. [DOI] [PubMed] [Google Scholar]
- 14.Perdiguero EG, Klapproth K, Schulz C, Busch K, de Bruijn M, Rodewald HR, et al. The Origin of Tissue-Resident Macrophages: When an Erythro-myeloid Progenitor Is an Erythro-myeloid Progenitor. Immunity 2015; 43: 1023–1024. [DOI] [PubMed] [Google Scholar]
- 15.Tu J, Hong W, Guo Y, Zhang P, Fang Y, Wang X, et al. Ontogeny of Synovial Macrophages and the Roles of Synovial Macrophages From Different Origins in Arthritis. Front Immunol 2019; 10: 1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurowska-Stolarska M, Alivernini S. Synovial tissue macrophages: friend or foe? RMD Open 2017; 3: e000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein-Wieringa IR, de Lange-Brokaar BJ, Yusuf E, Andersen SN, Kwekkeboom JC, Kroon HM, et al. Inflammatory Cells in Patients with Endstage Knee Osteoarthritis: A Comparison between the Synovium and the Infrapatellar Fat Pad. J Rheumatol 2016; 43: 771–778. [DOI] [PubMed] [Google Scholar]
- 18.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013; 38: 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagemeyer N, Kierdorf K, Frenzel K, Xue J, Ringelhan M, Abdullah Z, et al. Transcriptome-based profiling of yolk sac-derived macrophages reveals a role for Irf8 in macrophage maturation. The EMBO journal 2016; 35: 1730–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Culemann S, Gruneboom A, Nicolas-Avila JA, Weidner D, Lammle KF, Rothe T, et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 2019; 572: 670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Udalova IA, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol 2016; 12: 472–485. [DOI] [PubMed] [Google Scholar]
- 22.Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, et al. Nonclassical Ly6C(−) monocytes drive the development of inflammatory arthritis in mice. Cell Rep 2014; 9: 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wood MJ, Leckenby A, Reynolds G, Spiering R, Pratt AG, Rankin KS, et al. Macrophage proliferation distinguishes 2 subgroups of knee osteoarthritis patients. JCI Insight 2019; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemburg A, Meyer-Lindenberg A, Hewicker-Trautwein M. Immunohistochemical characterization of inflammatory cell populations and adhesion molecule expression in synovial membranes from dogs with spontaneous cranial cruciate ligament rupture. Veterinary immunology and immunopathology 2004; 97: 231–240. [DOI] [PubMed] [Google Scholar]
- 25.van den Bosch MH, Blom AB, Schelbergen RF, Koenders MI, van de Loo FA, van den Berg WB, et al. Alarmin S100A9 Induces Proinflammatory and Catabolic Effects Predominantly in the M1 Macrophages of Human Osteoarthritic Synovium. J Rheumatol 2016; 43: 1874–1884. [DOI] [PubMed] [Google Scholar]
- 26.Manferdini C, Paolella F, Gabusi E, Silvestri Y, Gambari L, Cattini L, et al. From osteoarthritic synovium to synovial-derived cells characterization: synovial macrophages are key effector cells. Arthritis Res Ther 2016; 18: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis JS Jr, Furman BD, Zeitler E, Huebner JL, Kraus VB, Guilak F, et al. Genetic and cellular evidence of decreased inflammation associated with reduced incidence of posttraumatic arthritis in MRL/MpJ mice. Arthritis & Rheumatism 2013; 65: 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bondeson J, Wainwright SD, Lauder S, Amos N, Hughes CE. The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis research & therapy 2006; 8: R187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bondeson J, Blom AB, Wainwright S, Hughes C, Caterson B, Van Den Berg WB. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis & Rheumatism 2010; 62: 647–657. [DOI] [PubMed] [Google Scholar]
- 30.Sun AR, Panchal SK, Friis T, Sekar S, Crawford R, Brown L, et al. Obesity-associated metabolic syndrome spontaneously induces infiltration of pro-inflammatory macrophage in synovium and promotes osteoarthritis. PloS one 2017; 12: e0183693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez-Aristizabal A, Gandhi R, Mahomed NN, Marshall KW, Viswanathan S. Synovial fluid monocyte/macrophage subsets and their correlation to patient-reported outcomes in osteoarthritic patients: a cohort study. Arthritis Res Ther 2019; 21: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakurai Y, Fujita M, Kawasaki S, Sanaki T, Yoshioka T, Higashino K, et al. Contribution of synovial macrophages to rat advanced osteoarthritis pain resistant to cyclooxygenase inhibitors. Pain 2019; 160: 895–907. [DOI] [PubMed] [Google Scholar]
- 33.de Visser HM, Mastbergen SC, Kozijn AE, Coeleveld K, Pouran B, van Rijen MH, et al. Metabolic dysregulation accelerates injury-induced joint degeneration, driven by local inflammation; an in vivo rat study. J Orthop Res 2018; 36: 881–890. [DOI] [PubMed] [Google Scholar]
- 34.Larranaga-Vera A, Lamuedra A, Perez-Baos S, Prieto-Potin I, Pena L, Herrero-Beaumont G, et al. Increased synovial lipodystrophy induced by high fat diet aggravates synovitis in experimental osteoarthritis. Arthritis Res Ther 2017; 19: 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu CL, Jain D, McNeill JN, Little D, Anderson JA, Huebner JL, et al. Dietary fatty acid content regulates wound repair and the pathogenesis of osteoarthritis following joint injury. Ann Rheum Dis 2015; 74: 2076–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun AR, Wu X, Liu B, Chen Y, Armitage CW, Kollipara A, et al. Pro-resolving lipid mediator ameliorates obesity induced osteoarthritis by regulating synovial macrophage polarisation. Sci Rep 2019; 9: 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hui W, Litherland GJ, Elias MS, Kitson GI, Cawston TE, Rowan AD, et al. Leptin produced by joint white adipose tissue induces cartilage degradation via upregulation and activation of matrix metalloproteinases. Annals of the rheumatic diseases 2012; 71: 455–462. [DOI] [PubMed] [Google Scholar]
- 38.Clockaerts S, Bastiaansen-Jenniskens YM, Runhaar J, Van Osch GJ, Van Offel JF, Verhaar JA, et al. The infrapatellar fat pad should be considered as an active osteoarthritic joint tissue: a narrative review. Osteoarthritis Cartilage 2010; 18: 876–882. [DOI] [PubMed] [Google Scholar]
- 39.de Jong AJ, Klein-Wieringa IR, Andersen SN, Kwekkeboom JC, Herb-van Toorn L, de Lange-Brokaar BJE, et al. Lack of high BMI-related features in adipocytes and inflammatory cells in the infrapatellar fat pad (IFP). Arthritis Res Ther 2017; 19: 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klein-Wieringa IR, Kloppenburg M, Bastiaansen-Jenniskens YM, Yusuf E, Kwekkeboom JC, El-Bannoudi H, et al. The infrapatellar fat pad of patients with osteoarthritis has an inflammatory phenotype. Ann Rheum Dis 2011; 70: 851–857. [DOI] [PubMed] [Google Scholar]
- 41.Bastiaansen-Jenniskens YM, Clockaerts S, Feijt C, Zuurmond AM, Stojanovic-Susulic V, Bridts C, et al. Infrapatellar fat pad of patients with end-stage osteoarthritis inhibits catabolic mediators in cartilage. Ann Rheum Dis 2012; 71: 288–294. [DOI] [PubMed] [Google Scholar]
- 42.Schmidli MR, Fuhrer B, Kurt N, Senn D, Drogemuller M, Rytz U, et al. Inflammatory pattern of the infrapatellar fat pad in dogs with canine cruciate ligament disease. BMC Vet Res 2018; 14: 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harasymowicz NS, Clement ND, Azfer A, Burnett R, Salter DM, Simpson AHW. Regional differences between perisynovial and infrapatellar adipose tissue depots and their response to class II and class III obesity in patients with osteoarthritis. Arthritis & Rheumatology 2017; 69: 1396–1406. [DOI] [PubMed] [Google Scholar]
- 44.Barboza E, Hudson J, Chang WP, Kovats S, Towner RA, Silasi-Mansat R, et al. Profibrotic Infrapatellar Fat Pad Remodeling Without M1 Macrophage Polarization Precedes Knee Osteoarthritis in Mice With Diet-Induced Obesity. Arthritis Rheumatol 2017; 69: 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei W, Rudjito E, Fahy N, Verhaar JA, Clockaerts S, Bastiaansen-Jenniskens YM, et al. The infrapatellar fat pad from diseased joints inhibits chondrogenesis of mesenchymal stem cells. Eur Cell Mater 2015; 30: 303–314. [DOI] [PubMed] [Google Scholar]
- 46.Wickham MQ, Erickson GR, Gimble JM, Vail TP, Guilak F. Multipotent stromal cells derived from the infrapatellar fat pad of the knee. Clin Orthop Relat Res 2003: 196–212. [DOI] [PubMed] [Google Scholar]
- 47.Chuckpaiwong B, Charles HC, Kraus VB, Guilak F, Nunley JA. Age-associated increases in the size of the infrapatellar fat pad in knee osteoarthritis as measured by 3T MRI. Journal of Orthopaedic Research 2010; 28: 1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li G, Yin J, Gao J, Cheng TS, Pavlos NJ, Zhang C, et al. Subchondral bone in osteoarthritis: insight into risk factors and microstructural changes. Arthritis Res Ther 2013; 15: 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geurts J, Patel A, Hirschmann MT, Pagenstert GI, Muller-Gerbl M, Valderrabano V, et al. Elevated marrow inflammatory cells and osteoclasts in subchondral osteosclerosis in human knee osteoarthritis. J Orthop Res 2016; 34: 262–269. [DOI] [PubMed] [Google Scholar]
- 50.Van Lent P, Blom A, Van Der Kraan P, Holthuysen A, Vitters E, Van Rooijen N, et al. Crucial role of synovial lining macrophages in the promotion of transforming growth factor β-mediated osteophyte formation. Arthritis & Rheumatism: Official Journal of the American College of Rheumatology 2004; 50: 103–111. [DOI] [PubMed] [Google Scholar]
- 51.Bakker AC, van de Loo FA, van Beuningen HM, Sime P, van Lent PL, van der Kraan PM, et al. Overexpression of active TGF-beta-1 in the murine knee joint: evidence for synovial-layer-dependent chondro-osteophyte formation. Osteoarthritis Cartilage 2001; 9: 128–136. [DOI] [PubMed] [Google Scholar]
- 52.Kaur S, Raggatt LJ, Batoon L, Hume DA, Levesque JP, Pettit AR. Role of bone marrow macrophages in controlling homeostasis and repair in bone and bone marrow niches. Semin Cell Dev Biol 2017; 61: 12–21. [DOI] [PubMed] [Google Scholar]
- 53.Batoon L, Millard SM, Wullschleger ME, Preda C, Wu AC, Kaur S, et al. CD169(+) macrophages are critical for osteoblast maintenance and promote intramembranous and endochondral ossification during bone repair. Biomaterials 2019; 196: 51–66. [DOI] [PubMed] [Google Scholar]
- 54.Lohmander LS, Englund PM, Dahl LL, Roos EM. The long-term consequence of anterior cruciate ligament and meniscus injuries: osteoarthritis. The American journal of sports medicine 2007; 35: 1756–1769. [DOI] [PubMed] [Google Scholar]
- 55.Muir P, Schamberger GM, Manley PA, Hao Z. Localization of cathepsin K and tartrate-resistant acid phosphatase in synovium and cranial cruciate ligament in dogs with cruciate disease. Veterinary surgery 2005; 34: 239–246. [DOI] [PubMed] [Google Scholar]
- 56.Muir P, Hayashi K, Manley PA, Colopy SA. Evaluation of tartrate-resistant acid phosphatase and cathepsin K in ruptured cranial cruciate ligaments in dogs. American journal of veterinary research 2002; 63: 1279–1284. [DOI] [PubMed] [Google Scholar]
- 57.Barrett JG, Hao Z, Graf BK, Kaplan LD, Heiner JP, Muir P. Inflammatory changes in ruptured canine cranial and human anterior cruciate ligaments. American journal of veterinary research 2005; 66: 2073–2080. [DOI] [PubMed] [Google Scholar]
- 58.Madsen DH, Ingvarsen S, Jürgensen HJ, Melander MC, Kjøller L, Moyer A, et al. The non-phagocytic route of collagen uptake a distinct degradation pathway. Journal of Biological Chemistry 2011; 286: 26996–27010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saito I, Koshino T, Nakashima K, Uesugi M, Saito T. Increased cellular infiltrate in inflammatory synovia of osteoarthritic knees. Osteoarthritis and cartilage 2002; 10: 156–162. [DOI] [PubMed] [Google Scholar]
- 60.Xie J, Zhang D, Lin Y, Yuan Q, Zhou X. Anterior Cruciate Ligament Transection–Induced Cellular and Extracellular Events in Menisci: Implications for Osteoarthritis. The American journal of sports medicine 2018; 46: 1185–1198. [DOI] [PubMed] [Google Scholar]
- 61.Fuhrmann IK, Steinhagen J, Rüther W, Schumacher U. Comparative immunohistochemical evaluation of the zonal distribution of extracellular matrix and inflammation markers in human meniscus in osteoarthritis and rheumatoid arthritis. Acta histochemica 2015; 117: 243–254. [DOI] [PubMed] [Google Scholar]
- 62.Blomgran P, Blomgran R, Ernerudh J, Aspenberg P. A possible link between loading, inflammation and healing: Immune cell populations during tendon healing in the rat. Scientific reports 2016; 6: 29824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abraham AC, Shah SA, Golman M, Song L, Li X, Kurtaliaj I, et al. Targeting the NF-kappaB signaling pathway in chronic tendon disease. Sci Transl Med 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chamberlain CS, Clements AEB, Kink JA, Choi U, Baer GS, Halanski MA, et al. Extracellular Vesicle-Educated Macrophages Promote Early Achilles Tendon Healing. Stem Cells 2019; 37: 652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Manning CN, Martel C, Sakiyama-Elbert SE, Silva MJ, Shah S, Gelberman RH, et al. Adipose-derived mesenchymal stromal cells modulate tendon fibroblast responses to macrophage-induced inflammation in vitro. Stem Cell Res Ther 2015; 6: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ackerman JE, Geary MB, Orner CA, Bawany F, Loiselle AE. Obesity/Type II diabetes alters macrophage polarization resulting in a fibrotic tendon healing response. PLoS One 2017; 12: e0181127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karalaki M, Fili S, Philippou A, Koutsilieris M. Muscle regeneration: cellular and molecular events. In Vivo 2009; 23: 779–796. [PubMed] [Google Scholar]
- 68.Dumont N, Frenette J. Macrophages protect against muscle atrophy and promote muscle recovery in vivo and in vitro: a mechanism partly dependent on the insulin-like growth factor-1 signaling molecule. Am J Pathol 2010; 176: 2228–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brigitte M, Schilte C, Plonquet A, Baba-Amer Y, Henri A, Charlier C, et al. Muscle resident macrophages control the immune cell reaction in a mouse model of notexin-induced myoinjury. Arthritis Rheum 2010; 62: 268–279. [DOI] [PubMed] [Google Scholar]
- 70.Wang H, Melton DW, Porter L, Sarwar ZU, McManus LM, Shireman PK. Altered macrophage phenotype transition impairs skeletal muscle regeneration. Am J Pathol 2014; 184: 1167–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akhmedov D, Berdeaux R. The effects of obesity on skeletal muscle regeneration. Front Physiol 2013; 4: 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Godziuk K, Prado CM, Woodhouse LJ, Forhan M. The impact of sarcopenic obesity on knee and hip osteoarthritis: a scoping review. BMC Musculoskelet Disord 2018; 19: 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Misra D, Fielding RA, Felson DT, Niu J, Brown C, Nevitt M, et al. Risk of Knee Osteoarthritis With Obesity, Sarcopenic Obesity, and Sarcopenia. Arthritis Rheumatol 2019; 71: 232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. The Journal of Clinical Endocrinology & Metabolism 2001; 86: 5755–5761. [DOI] [PubMed] [Google Scholar]
- 75.Fink LN, Costford SR, Lee YS, Jensen TE, Bilan PJ, Oberbach A, et al. Pro-inflammatory macrophages increase in skeletal muscle of high fat-fed mice and correlate with metabolic risk markers in humans. Obesity (Silver Spring) 2014; 22: 747–757. [DOI] [PubMed] [Google Scholar]
- 76.Collins KH, Hart DA, Reimer RA, Seerattan RA, Waters-Banker C, Sibole SC, et al. High-fat high-sucrose diet leads to dynamic structural and inflammatory alterations in the rat vastus lateralis muscle. J Orthop Res 2016; 34: 2069–2078. [DOI] [PubMed] [Google Scholar]
- 77.Collins KH, Hart DA, Smith IC, Issler AM, Reimer RA, Seerattan RA, et al. Acute and chronic changes in rat soleus muscle after high-fat high-sucrose diet. Physiol Rep 2017; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Khan IM, Perrard XY, Brunner G, Lui H, Sparks LM, Smith SR, et al. Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int J Obes (Lond) 2015; 39: 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest 2017; 127: 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lara-Castro C, Garvey WT. Intracellular lipid accumulation in liver and muscle and the insulin resistance syndrome. Endocrinology and metabolism clinics of North America 2008; 37: 841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pellegrinelli V, Rouault C, Rodriguez-Cuenca S, Albert V, Edom-Vovard F, Vidal-Puig A, et al. Human Adipocytes Induce Inflammation and Atrophy in Muscle Cells During Obesity. Diabetes 2015; 64: 3121–3134. [DOI] [PubMed] [Google Scholar]
- 82.Tang R, Harasymowicz NS, Wu C-L, Collins KH, Choi Y-R, Oswald SJ, et al. Gene Therapy for Follistatin Mitigates Systemic Metabolic Inflammation and Post-Traumatic Osteoarthritis in High-Fat Diet-Induced Obese Mice. bioRxiv 2019: 619239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blom AB, van Lent PL, Holthuysen AE, van der Kraan PM, Roth J, van Rooijen N, et al. Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthritis and cartilage 2004; 12: 627–635. [DOI] [PubMed] [Google Scholar]
- 84.Blom AB, van Lent PL, Libregts S, Holthuysen AE, van der Kraan PM, van Rooijen N, et al. Crucial role of macrophages in matrix metalloproteinase–mediated cartilage destruction during experimental osteoarthritis: involvement of matrix metalloproteinase 3. Arthritis & Rheumatism 2007; 56: 147–157. [DOI] [PubMed] [Google Scholar]
- 85.Chamberlain CS, Leiferman EM, Frisch KE, Wang S, Yang X, Van Rooijen N, et al. The influence of macrophage depletion on ligament healing. Connective tissue research 2011; 52: 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.de la Durantaye M, Piette AB, van Rooijen N, Frenette J. Macrophage depletion reduces cell proliferation and extracellular matrix accumulation but increases the ultimate tensile strength of injured Achilles tendons. Journal of Orthopaedic Research 2014; 32: 279–285. [DOI] [PubMed] [Google Scholar]
- 87.Hays PL, Kawamura S, Deng X-H, Dagher E, Mithoefer K, Ying L, et al. The role of macrophages in early healing of a tendon graft in a bone tunnel. JBJS 2008; 90: 565–579. [DOI] [PubMed] [Google Scholar]
- 88.Wu CL, McNeill J, Goon K, Little D, Kimmerling K, Huebner J, et al. Conditional Macrophage Depletion Increases Inflammation and Does Not Inhibit the Development of Osteoarthritis in Obese Macrophage Fas-Induced Apoptosis–Transgenic Mice. Arthritis & Rheumatology 2017; 69: 1772–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Munter W, van den Bosch M, Slöetjes A, Croce K, Vogl T, Roth J, et al. High LDL levels lead to increased synovial inflammation and accelerated ectopic bone formation during experimental osteoarthritis. Osteoarthritis and cartilage 2016; 24: 844–855. [DOI] [PubMed] [Google Scholar]
- 90.de Munter W, Geven E, Blom A, Walgreen B, Helsen M, Joosten L, et al. Synovial macrophages promote TGF-β signaling and protect against influx of S100A8/S100A9-producing cells after intra-articular injections of oxidized low-density lipoproteins. Osteoarthritis and cartilage 2017; 25: 118–127. [DOI] [PubMed] [Google Scholar]
- 91.MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood 2010; 116: 3955–3963. [DOI] [PubMed] [Google Scholar]
- 92.Hume DA, Pavli P, Donahue RE, Fidler IJ. The effect of human recombinant macrophage colony-stimulating factor (CSF-1) on the murine mononuclear phagocyte system in vivo. J Immunol 1988; 141: 3405–3409. [PubMed] [Google Scholar]
- 93.Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015; 518: 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Burnett SH, Kershen EJ, Zhang J, Zeng L, Straley SC, Kaplan AM, et al. Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J Leukoc Biol 2004; 75: 612–623. [DOI] [PubMed] [Google Scholar]
- 95.Tittel AP, Heuser C, Ohliger C, Llanto C, Yona S, Hämmerling GJ, et al. Functionally relevant neutrophilia in CD11c diphtheria toxin receptor transgenic mice. Nature methods 2012; 9: 385. [DOI] [PubMed] [Google Scholar]
- 96.Sadtler K, Wolf MT, Ganguly S, Moad CA, Chung L, Majumdar S, et al. Divergent immune responses to synthetic and biological scaffolds. Biomaterials 2019; 192: 405–415. [DOI] [PubMed] [Google Scholar]
- 97.Badylak SF, Valentin JE, Ravindra AK, McCabe GP, Stewart-Akers AM. Macrophage phenotype as a determinant of biologic scaffold remodeling. Tissue Engineering Part A 2008; 14: 1835–1842. [DOI] [PubMed] [Google Scholar]
- 98.Sridharan R, Ryan EJ, Kearney CJ, Kelly DJ, O’Brien FJ. Macrophage Polarization in Response to Collagen Scaffold Stiffness Is Dependent on Cross-Linking Agent Used To Modulate the Stiffness. ACS Biomaterials Science & Engineering 2018; 5: 544–552. [DOI] [PubMed] [Google Scholar]
- 99.Visser JG, Van Staden ADP, Smith C. Harnessing Macrophages for Controlled-Release Drug Delivery: Lessons From Microbes. Front Pharmacol 2019; 10: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pferdehirt L, Ross AK, Brunger JM, Guilak F. A Synthetic Gene Circuit for Self-Regulating Delivery of Biologic Drugs in Engineered Tissues. Tissue Eng Part A 2019; 25: 809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brunger JM, Zutshi A, Willard VP, Gersbach CA, Guilak F. Genome engineering of stem cells for autonomously regulated, closed-loop delivery of biologic drugs. Stem cell reports 2017; 8: 1202–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Adkar SS, Brunger JM, Willard VP, Wu C-L, Gersbach CA, Guilak F. Genome engineering for personalized arthritis therapeutics. Trends in molecular medicine 2017; 23: 917–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Distel E, Cadoudal T, Durant S, Poignard A, Chevalier X, Benelli C. The infrapatellar fat pad in knee osteoarthritis: An important source of interleukin-6 and its soluble receptor. Arthritis & Rheumatism: Official Journal of the American College of Rheumatology 2009; 60: 3374–3377. [DOI] [PubMed] [Google Scholar]
- 104.Verma P, Dalal K. ADAMTS-4 and ADAMTS-5: key enzymes in osteoarthritis. Journal of cellular biochemistry 2011; 112: 3507–3514. [DOI] [PubMed] [Google Scholar]
- 105.Murata M, Yudoh K, Masuko K. The potential role of vascular endothelial growth factor (VEGF) in cartilage: how the angiogenic factor could be involved in the pathogenesis of osteoarthritis? Osteoarthritis and cartilage 2008; 16: 279–286. [DOI] [PubMed] [Google Scholar]
- 106.Meng X-M, Wang S, Huang X-R, Yang C, Xiao J, Zhang Y, et al. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell death & disease 2016; 7: e2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marcelin G, Ferreira A, Liu Y, Atlan M, Aron-Wisnewsky J, Pelloux V, et al. A PDGFRα-mediated switch toward CD9high adipocyte progenitors controls obesity-induced adipose tissue fibrosis. Cell metabolism 2017; 25: 673–685. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.