

Abstract
Background
While multiple studies have assessed molecular changes in chronic atopic dermatitis (AD) lesions, little is known about the transition from acute to chronic disease stages, and the factors and mechanisms that shape chronic inflammatory activity.
Objectives
We sought to assess the global transcriptome changes that characterize the progression from acute to chronic stages of AD.
Methods
We analyzed transcriptome changes in paired non-lesional skin, acute and chronic AD lesions from 11 patients and 38 healthy controls by RNA-seq, and conducted in vivo and histological assays to evaluate findings.
Results
Our data demonstrate that ~74% of the genes dysregulated in acute lesions remain or are further dysregulated in chronic lesions, whereas only 34% of genes dysregulated in chronic lesions are altered already in the acute stage. Non-lesional AD skin exhibited enrichment of TNF, Th1, Th2, and Th17 response genes. Acute lesions showed marked dendritic cell signatures and a prominent enrichment of Th1, Th2 and Th17 responses, along with increased IL-36 and TSLP expression, which were further heightened in chronic lesions. In addition, genes involved in skin barrier repair, keratinocyte proliferation, wound healing and negative regulation of T cell activation showed a significant dysregulation in the chronic versus acute comparison. Furthermore, our data show progressive changes in vasculature and maturation of dendritic cell subsets with chronicity, with FOXK1 acting as immune regulator.
Conclusions
Our results show that the changes accompanying the transition from non-lesional to acute to chronic inflammation in AD are quantitative rather than qualitative, with chronic AD having heightened Th2, Th1, Th17, and IL36 responses and skin barrier repair mechanisms. These findings provide novel insights and highlight underappreciated pathways in AD pathogenesis that may be amenable to therapeutic targeting.
Keywords: Atopic dermatitis, RNA-seq, non-lesional, chronic AD, acute AD
Capsule summary
Our study provides a comprehensive view of the pathologic processes that take place in acute to chronic progression for AD. Changes from acute to chronic AD are quantitative rather than qualitative in terms of shifts in Th2, Th22, Th1 and Th17 responses.
I. Introduction
Atopic dermatitis (AD) is one of the most common inflammatory skin disorders affecting up to 20% of children and 10% of adults in high-income countries, with a worldwide prevalence of approximately 8% 1. While its peak incidence is in childhood, adult AD is common, comprising both chronic persistent and/or relapsing-remitting as well as new-onset disease 2, 3. The pathophysiology of AD is still not completely understood, but it is believed to be driven by epidermal barrier disruption, activation of specific T-cell subsets, and dysbiosis of the commensal skin microbiome 4. The disease’s hallmark clinical sign is eczematous lesions, which may present (sub-)acutely (diffuse scaly erythematous patches and oozing papulovesicles) or chronically (scaly patches and plaques with excoriation and lichenification). Histologically, acute lesions are characterized by spongiosis/edema between epidermal keratinocytes, mild to absent thickening of the epidermis, and infiltration with inflammatory cells along with degranulated mast cells; while chronic lesions show a marked thickening of the epidermis, less pronounced epidermal spongiosis, and a heavier mononuclear inflammatory infiltrate 5. Acute and chronic histopathologic changes exist on a continuum. Patients with moderate-to-severe AD can experience acute and chronic lesions simultaneously, and the distinction between acute and chronic stages is not trivial, since clinical and histopathological features often overlap in lesions 6. It is yet unclear whether acute and chronic stages are driven by different molecular mechanisms or alternatively represent a different magnitude of inflammatory response. One study that investigated the differences between “bona fide” acute and chronic lesions, in a cohort of ten patients using microarray gene expression profiling and quantitative RTPCR, found that acute lesions displayed an increase in IL22, IL32 and S100A7–9 along with type 2 cytokines including IL4 and IL31; all of these, other than IL4, showed progressively greater expression in chronic lesions 7. In contrast, an upregulation of Th1 response genes including MX1 and CXCL9–11, were only seen in chronic lesions, suggesting that acute inflammation in AD is driven by type 2 cytokines, while chronic stages immunological mechanisms also involve Th1 responses. So far, these observations have not been replicated. Our group has previously conducted a large scale and deep molecular transcriptomic profiling for AD using RNA-seq 8. Here, we analyzed acute versus chronic lesions in a subset of 11 AD patients, and by combining RNA-seq technology and paired design, we were able to provide novel insights with enhanced resolution on the mechanisms involved in acute to chronic AD progression. Further, we unraveled FOXK1 as a potential regulatory factor, responsible for differentiating the inflammatory responses in acute versus chronic AD.
II. Methods
Skin biopsies and RNA-seq
Intrapersonal acute, chronic (>72 hrs duration) and non-lesional (≥ 10 cm from active lesions) skin biopsies were collected from 11 consented Caucasian patients with moderate-to-severe AD, along with biopsies from 38 healthy volunteers included in a previous skin transcriptome study 8 under an Institutional Review Board-approved protocol (A110/12) (Suppl Table 1). In all patients, biopsies were taken from the upper arms (flexural side). No systemic or topical treatments were allowed for ≥4 weeks prior to biopsies. All patients used in this study met the following criteria for acute skin lesions: a) new lesions of <72 hours duration, as previously defined7; b) lack of skin lichenification; c) lack of histopathological signs of chronic eczema such as acanthosis, parakeratosis, and regenerative hyperplasia, as defined by epidermal thickness ≤150μm (H&E), and basal or confluent supra-basal Keratin 16 (K16) positivity. Patients were instructed to present at our department within 72 hours when noticing new lesions on previously unaffected skin but not in case of flares of preexisting chronic lesions. All clinical assessments were done by the same investigator. A total of 5 patients were excluded after histopathological examination of biopsies due to signs of chronic eczema (marked acanthosis, parakeratosis, hyperplasia)
After sampling skin tissue specimen were immediately stored in PAXgene® Tissue Containers (PreAnalytiX, Hombrechtikon, Switzerland) at −80°C until isolation of total RNA with the AllPrep DNA/RNA Mini Kit (Qiagen, Hilden, Germany) following the manufacturers protocols. Preceding RNA isolation skin specimens were disrupted using innuSPEED Lysis Tubes W (1.4 – 1.6 mm steel beads & 3.5 mm ceramic beads) (Analytik Jena, Jena, Germany) in a SpeedMill Plus (3× 1min intervals) (Analytik Jena) together with 600μl of RLT-Plus-Buffer (Qiagen) and QIAshredder spin-columns (Qiagen). Quality control on concentration and integrity of the isolated RNA was performed with the Qubit 2.0 Fluorometer (Qubit® RNA HS Assay) (LifeTechnologies, Carlsbad, CA) and the 2200 Tape Station (R6K ScreenTape Assay) (Agilent, Santa Clara, CA) following the manufacturer’s instructions. Only RNA samples with a concentration of >50ng/μl, an OD260/280 ≥1.8 and a RNA integrity number (RIN) >7 were included in subsequent library preparation and sequencing.
RNA samples were prepared for sequencing using the Illumina Truseq® Stranded total RNA Protocol in combination with the RiboZero rRNA removal Kit, and sequenced on the HiSeq2500 in pools of 10 samples with 2×125bp, producing paired-end reads according to the manufacturer’s protocol (Illumina, San Diego, CA).
Filaggrin genotyping
In all patients, the 4 filaggrin loss-of-function variants that are most common in the German population (R501X, 2282del4, R2447X, and S3247X) were typed as described previously8.
RNA-seq processing
All the data processing and analyzes were conducted under the Linux environment, and R was used for all the statistical analyzes. RNA-seq reads were first mapped to the human reference genome (b37) using STAR 9, and the number of uniquely mapped reads for each gene was counted using HTSeq 10. Only genes with an average ≥1 read/sample were used in our analysis. The trimmed mean of M-values normalization method (TMM) was used to normalize the read count across samples 11, and we applied voom transformation to model the mean-variance relationship of the expression data 12. We conducted differential expression analysis between different conditions using an empirical Bayes linear model as implemented in the limma package 13, controlling for individual specific effect (for the non-lesional vs. chronic AD, the non-lesional vs. acute AD, and the acute vs. chronic AD skin comparisons) and gender effect (for the other comparisons); False discovery rate (FDR) ≤10% and |log2 Fold Change (FC)| ≥1 were used to declare significance.
Cell culture, cytokines stimulations, RNA interference
N/TERTs, an immortalized keratinocytes cell line, was grown in KC-SFM medium (ThermoFisher #17005–042) with 30μg/ml bovine pituitary extract, 0.2ng/ml epidermal growth factor, and 0.3mM calcium chloride in 48-well plates and treated with small interfering RNAs (siRNAs) targeting FOXK1(Accell #E-032790–00-0010) and Non-target control siRNAs (Accell #D-001910–01-20) according to Dharmacon Accell siRNA protocol. Cell were either unstimulated or stimulated with the cytokines IL-4 (10ng/ml, R&D #204-IL-010), IL-10(10ng/ml, R&D #217-IL-005), IL-13(10ng/ml, R&D #213-ILB005) for 24h respectively.
RNA isolation and qRT-PCR
RNA was isolated using Qiagen RNeasy plus kit (Cat#74136). Reversed transcription was performed using High Capacity cDNA Transcription kit (ThermoFisher #4368813). qRT-PCR was performed using a 7900HT Fast Real-time PCR system (Thermor Fisher) with Taqman Universal PCR Master Mix (Thermo Fisher #4304437) and Taqman primers (Thermor Fisher: FOXK1 Hs01595620_m1, RPLP0 Hs00420895_gH, CCL5 Hs99999048_m1, IL32 Hs00992441_m1, IL4R Hs00965056_m1.
Immunohistochemistry and immunofluorescence
For immunohistochemistry, FFPE human skin biopsy specimens (independent of the RNA-seq samples) on slides were heated for 30 minutes at 65°C, rehydrated, epitope retrieved, blocked and incubated with primary antibody against FOXK1(ATLAS ANTIBODIES #HPA017998) overnight at 4°C. Slides were washed, incubated with secondary antibody, developed with diaminobenzidine and counterstained with hematoxylin. Secondary antibody used for immunofluorescence was from (Life technologies, Alexa Flour 488).
III. Results
We performed RNA-seq on biopsies from non-lesional and paired acute and chronic lesions, enabling us to conduct robust analysis of differences and potential progression from non-lesional to acute, to chronic stages of inflammation. We were able to profile 31,207 genes with an average of at least 1 read/sample. Notably, the top three principal components were not able to separate acute from chronic AD (Figure 1a). A previous microarray-based study employing less stringent criteria (i.e. |log2Fold Change (FC)| ≥ 0.585 and p-value ≤ 0.01) 7 identified 47 up- and 96- down regulated genes. Using the same criteria, we found 197 up- and 233- down-regulated genes when comparing acute with chronic AD (Suppl Table 2). When corrected for multiple testing, our RNA-seq data identified 42 statistically significant (|log2 FC| ≥ 1 and false discovery rate, FDR ≤ 10%) genes (29 up and 13 down in chronic AD when compared with acute AD; Suppl Table 3), including genes involved in epidermal differentiation (S100A8 and S100A9) and response to epidermal barrier stress (KRT16, KRT6B), as well as members of the CXC chemokine (CXCL1, CXCL6), TNF (TNIP3) and IL-10 cytokine family (IL19, IL20). Significantly, the majority of these genes overlap with the 15 genes that show a progressive expression pattern from non-lesional to acute to chronic AD (i.e. significantly up-regulated in each of the comparisons) (Figure 1b). Indeed, genes that were dysregulated in the acute AD vs. non-lesional comparison (609 up-regulated and 537 down-regulated) show a modest but consistent shift in their expression levels in the acute vs. chronic AD comparison, indicating the progression of the transcriptomic changes with development of chronicity (Figure 2a). Notably, our results reveal that most of the changes in acute AD are also present in chronic AD, as ~74% of the genes dysregulated in acute AD are also differentially expressed in chronic AD when compared with non-lesional skin (Suppl Table 3). On the contrary, only around 34% of the genes dysregulated in chronic AD are also differentially expressed in acute AD. We identified a set of genes that are statistically differentially expressed only between chronic and acute lesions, while showing little changes between acute vs. non-lesional AD (Figure 2b–d). These include genes that participate in skin barrier maintenance and repair (HRNR), IL-19, IL-20 14–16, immunoglobulin production (IGHG1, IGLC2, IGKC), inhibition of CD4 + T cell activation (CLEC3A) 17, modulation of T-cell responses (TNFAIP3) (Schuijs MJ Science 2015; Vroman H J Allergy Clin Immunol. 2018), wound healing (CXCL1, CXCL6) (Griffith JW 2014), and tryptophan metabolism (KYNU) 18. It is noteworthy that the immunoglobulins as a group have been identified as differentially expressed in chronic lesional skin of psoriasis and AD 8; additionally, KYNU, the L-kynureninase, which regulates tryptophan metabolism, can serve as an immune response modulator for chronic inflammatory conditions 18. No major differences were seen between FLG mutation carriers and non-carriers.
Figure 1. Transcriptomic landscape and progressive profiles in AD disease stages.

a) the top three principal components computed using the whole transcriptomic data; b) heatmap illustrating the expression profiles for genes with progressive pattern of up-regulation from non-lesional to acute to chronic AD.
Figure 2. Molecular profiling for AD progression from acute to chronic.

a) effect size in the acute vs chronic AD comparison for genes dysregulated in non-lesional vs. acute AD; b) effect size comparison between non-lesional vs. acute AD (x-axis) against acute AD vs. chronic AD (y-axis). Genes are colored if they are dysregulated in the first (blue), second (red) or both comparisons (purple); c-d) boxplots to illustrate the expression profiles for 2 genes dysregulated in the acute vs. chronic comparison but not in the non-lesional vs. acute comparison.
We then used the molecular profiles to provide a deeper insight into the changes in cellular compositions during disease progression, by comparing the control skin versus the non-lesional, acute, and chronic lesional skin of AD patients. By utilizing an in silico technique to study the expression signatures for different human cell types 19, we identified a significant trend of increasing infiltration of immune cells (i.e. dendritic; Th1, Th2, pro B-cells) and endothelial cells during the acute-to-chronic AD progression (Figure 3). The increase in a type-2 expression signature (Figure 4a) in chronic AD corresponded with the heightened immune response and the more pronounced epidermal hyperplasia/growth signaling in chronic AD. The presence of dendritic cell signatures in acute AD skin is of interest, and could correspond to the crucial role they play in type-2 response induction in this early inflammatory disease stage. Vasculature is known to play an important role in the pathophysiology of AD, as AD lesions are characterized by activated endothelial cells and their interactions with T cells are important for leukocyte trafficking into inflamed AD skin 20. Thus, these findings align with the gradual increase in endothelial cell signature we observed in progression to chronic AD (Figure 3).
Figure 3. Heatmap to indicate the presence of cell type signature comparing control vs different AD disease stages.

The color in the heatmap correlates with the presence of the cell type specific signature. The bars on the left indicate the significance level for the difference in cell type signatures when comparing control with different AD skin types.
Figure 4. Cytokine expression and effect in AD.
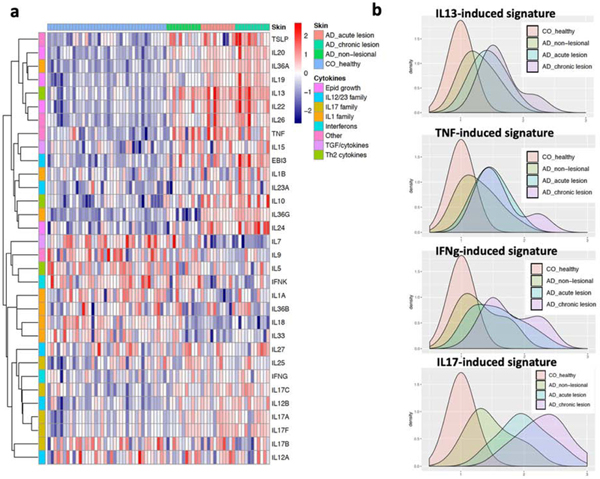
a) heatmap shows the expression profiles for different cytokines in healthy skin, non-lesional, acute and chronic AD subtypes; b) distributions of the cytokine stimulated “burden” in healthy skin, non-lesional, acute and chronic stages of AD. The values are normalized referencing the control samples.
To better understand the cytokine expression and cytokine responses in acute vs. chronic AD, we compared the mRNA expression of the major cytokines across our dataset, as well as how well each skin type aligns with the cytokine response signatures (the down-stream responses) in keratinocytes. Using unbiased hierarchical clustering, we determined the grouping of the expression patterns of different cytokine families across acute and chronic AD, with the type 2 family (in particular, IL13) grouping with IL10 family cytokines (IL19, IL20, IL22), IL1/IL36 family (IL36A), and TSLP (Figure 4a). In concordance with our previous finding, we did not detect expression of IL4 in acute AD skin. In addition to IL19 and IL22, both IL36G and TSLP show progressive expression profiles from acute to chronic AD skin (p=8.6×10−4 and p=2.6×10−3, respectively). Assessing the down-stream cytokine responses 8 using healthy control skin as a reference, we observed that non-lesional AD skin already has enriched TNF, IFN, Th2, and Th17 response gene expression, and these cytokine response “burdens” become more pronounced in lesional AD (Figure 4b). Notably, chronic AD exhibits heightened inflammatory responses for all inflammatory signals that we measured when compared to acute AD (Figure 5). We also observed a large variation in these signatures within the chronic or acute AD stage, consistent with the clinical and molecular heterogeneity of AD. By correlating the genome-wide fold change between chronic vs acute versus the effect size during cytokine stimulation, we further consolidated the above findings, demonstrating that chronic AD is typified by dysregulated gene responses to IL17, IL13, TNF, and IFN, as well as an elevated response to IL36 (Supplementary Figure 1).
Figure 5. Changes in gene expression from control to different types of AD skin.

*FDR ≤0.1; **FDR≤0.01; ***FDR≤1×10−5
To understand the transcriptional regulatory network that differentiates acute from chronic AD we screened for transcription factor (TF) binding motif enrichment within the promoter regions of the genes that are significantly up-regulated in chronic AD (compared to non-lesional skin) but not differentially expressed in acute AD. Only the binding sites of FOXK1, a transcription factor that modulates developmental and cell differentiation processes 21–23, were found to be significantly enriched (p=1.5×10−6; FDR=9.5×10−4). While FOXK1 mRNA expression was neither dysregulated in non-lesional nor lesional AD, its protein expression was increased and showed prominent nuclear localization in epidermal keratinocytes in lesional skin, particularly in chronic lesions (Figure 6a). We next investigated the effect of FOXK1 on differentially expressed genes (in chronic AD) that were predicted to have FOXK1 binding sites (i.e. IL4R, CCL5, IL32). While IL4R was not affected, CCL5, IL32, and FOXK1 itself were significantly induced upon stimulation with different type 2 associated cytokines, including IL-4, IL-10, and IL13, which we have previously outlined as the most prominent Th2 cytokines in AD 8. Notably, under IL-13 induction we observed a significant effect upon FOXK1 knockdown, which led to increased expression of CCL5 and IL32, suggesting that FOXK1 has a role as a negative immune regulator in chronic AD (Figure 6b).
Figure 6. Expression of FOXK1 in AD and its modulating effect upon cytokine stimulation.

a) immunostaining for FOXK1 in normal, acute, and chronic AD lesional skin; b) RNA expression (from RT-PCR) of FOXK1, IL4R, CCL5, and IL32 upon the indicated cytokine stimulation in keratinocytes under control (NC) or FOXK1 knockdown (siFOXK1). *p<0.05; **p<0.01; ***p<0.001.
IV. Discussion
Clinically, AD presents with eczematous lesions in different acuity stages. Acute and chronic lesions are often found in the same individual, often overlap, and clinically are sometimes difficult to distinguish 24. Histologically, acute and chronic AD have fairly distinctive features. For instance, acute AD lesions exhibit spongiosis with mild to moderate acanthosis in addition to a superficial perivascular infiltrate of lymphocytes and macrophages. Mast cells show degranulation 25, and occasionally eosinophils may be present 26. With increased chronicity, usually first noted in clinically subacute lesions, the acanthosis becomes more prominent, which can take on psoriasis-like features. In addition, the number of blood vessels increases 27. However, the processes and mechanisms that are involved in the transition from acute to chronic AD are still not completely understood.
Only a limited number of studies have attempted to address the molecular differences between acute and chronic AD. The most comprehensive study on this subject was a microarray transcriptomic study on 10 patients with paired acute and chronic AD biopsy samples. This study in addition measured selected cytokines and chemokines using quantitative RT-PCR7. Based on the results, the authors suggested that acute AD is primarily triggered through action of IL-22, with smaller contributions from IL-17 and IFN-γ, accompanied by Th2 cytokines including IL-4/IL-13 and IL-31. In chronic lesions expression of Th2 and Th22 cytokines further increased, and higher activation of Th1 and Th17 responses was observed, accompanied by increase in epidermal growth and thickness 7. To enable comparison with previous studies, we used the same clinical and histopathological criteria for differentiating acute from chronic AD. To further reduce variability, all patients were instructed to present at our department within 72 hours when noticing new lesions on previously unaffected skin, but not in case of flares of preexisting chronic lesions. All clinical assessments were done by the same investigator, and all biopsies were taken from the same anatomical region. Using higher resolution RNA-seq, our study highlights a more comprehensive and nuanced picture illustrating the continuum of the AD progression. Specifically, our results confirm that rather than a qualitative shift in the cytokine network and activated immune axes in chronic AD, all the major Th1, Th2, and Th17 responses are progressively heightened from non-lesional AD to acute and then chronic lesions (Figure 4b). This is also evident by our finding that there is not a clear distinction between the cytokine responses in acute AD vs. chronic AD, and frequently the acute lesions show biphasic responses suggesting an ongoing transition towards the chronic phase. Furthermore, it is striking that 74% of genes dysregulated in acute AD are also differentially expressed in chronic AD. In contrast, only 34% of DEGs found in chronic lesions (versus non-lesional skin) were altered in acute lesions. DEGs found only in chronic but not acute lesions have annotated functions in particular in “skin barrier maintenance and repair”, “wound healing”, “modulation of T-cell responses” and “immunoglobulin production”. This suggests that the main distinction between acute and chronic AD in terms of the cytokine network activation is quantitative rather than qualitative in nature, but that there are additional specific features associated with perpetuation and chronicity of inflammation.
IL-22 has been suggested to play a major role particularly in the early phases of AD 7, 28, 29. IL-22 belongs to the IL-20 family of cytokines 30 and was not the only prominent IL-20 family member we observed to be upregulated in both acute and chronic AD lesions. Interestingly, in our study other IL-20 family members including IL-19, IL-20, IL-24, and IL-26 also showed prominent increases in both acute and chronic AD lesions to similar degree. This could provide an explanation for the limited clinical improvement seen with anti-IL-22 treatment in AD 31, as it is likely that in the presence of IL-22 inhibition, other members of this family are able to compensate given their similarities and overlapping receptor usage.
Both TSLP and the IL-36 family of cytokines are expressed predominantly by epithelial cells 32, 33, and act on a number of cells including epithelial cells and immune cells 34. TSLP induces dendritic cells (DCs) to express OX40 ligand (OX40L), which binds to the OX40L receptor on T cells to stimulate the production of Th2 cytokines 35. IL-36 cytokines do not act directly on T cells but instead can stimulate maturation and function of DCs and through them drive T cell proliferation 36, thereby propagating and amplifying immune responses in skin. The shift to chronic AD is accompanied by an increase in TSLP and IL-36 activity, in accordance with their established role in amplifying and sustaining inflammatory reactions in the skin, suggesting that TSLP, its inducers, and the IL-36 cytokine axis might be potential therapeutic targets in AD. The potential for success in this strategy is evident, as therapeutic targeting of TSLP in 113 patients with AD showed promising responses in a phase II clinical trial 37. Current clinical studies are ongoing, focusing on the OX-40 inhibition in atopic dermatitis. While no data yet exists on anti-IL-36 treatment in AD, agents targeting this inflammatory axis have already been tested with promising results in pustular psoriasis 38. Therefore, the contribution of IL-36 family cytokines in the pathogenesis of chronic AD and their therapeutic relevance should be assessed in the near future.
The histologic features of acute and chronic AD, as outlined above, are well established. Our findings are consistent with acute to chronic AD histologic changes. They highlight a progressive enrichment in gene signatures, attributed to endothelial cells, Th1 and Th2 cells being more prominent in chronic AD compared to acute AD (Figure 3). By contrast, with increased chronicity we observed decreased signatures for myeloid, lymphoid progenitor, and hematopoietic stem cells, likely reflecting the increased activation and maturation of resident immune cells during chronic AD. We also observed an increased pro B-cell signature in chronic lesions. Whereas regulatory B cells have been shown to be decreased in patients with AD 39, B cell counts are increased in the skin and blood of patients with AD; this may correlate with increased IgE levels 40, which is a prominent feature of AD 4. Thus, our inflammatory cell signatures align closely with what has been described clinically for AD.
The mechanisms involved in regulating chronic inflammation in AD are not well established. Our data provide some novel insights into these mechanisms. We observed a prominent activation of signatures related to skin barrier repair and wound healing, and we identified two transcription factors that may have relevance for the regulation of inflammatory responses. These factors include FOXE1, which was one of a limited group of genes showing progressive changes from inflamed acute to chronic AD (Suppl Table 2), and FOXK1, which did not show changes in its mRNA expression but had enriched binding sites amongst genes having differential expression in chronic AD lesions. FOXK1 is a transcription factor that modulates developmental and cell differentiation process 21–23. Not much is known about the role of FOXK1 in inflammatory responses, but it has been shown to regulate CCL2 expression and recruitment of tumor associated macrophages 41. Our data suggest that in chronic AD it has an immune-regulatory role that is fairly specific to IL-13 responses, which we have previously shown to be the dominant inflammatory response in AD 8. Little is known about the role of FOXE1 in epidermal biology or inflammatory responses, with most of the data on this transcription factor being from cancer biology, where it has been shown to have anti-proliferative effects 42.
Taken together, our study provides a high-resolution view of the pathologic processes that take place during the acute to chronic conversion that occurs in AD. Our findings suggest that the changes from acute to chronic AD are quantitative rather than qualitative in terms of shifts in Th2, Th22, Th1 and Th17 responses, with additional features developing only in chronic inflammation. We also illustrate the elevated IL-36 responses in the chronic phase of AD. These findings provide novel insights into the pathogenesis of atopic dermatitis and highlight previously understudied pathways in AD pathogenesis that may be amenable to future therapeutic targeting. They need to be corroborated, however, by investigation in larger well assembled cohorts.
Supplementary Material
{kind=link}
Key messages.
Our study highlights the continuum of AD progression from acute to chronic stages with progressively heightened inflammatory responses for all major immune axes.
Our study identified 42 significantly dysregulated genes in chronic AD when compared with acute AD, including genes involved in epidermal differentiation (and response to barrier stress (IL-20, KRT16, KRT6B, S100A8 and S100A9), antimicrobial and immunomodulatory chemokines (CXCL1, CXCL6), and negative T cell regulation (TNIP3, CLEC3A) and Th2 cell differentiation (IL-19).
FOXK1 may have a role as a negative immune regulator in chronic AD
Acknowledgments
Funding support
This work was supported by BIOMAP (Biomarkers in Atopic Dermatitis and Psoriasis), a project funded by the Innovative Medicines Initiative 2 Joint Undertaking under Grant Agreement No. 821511 and in-kind contributions of the participating pharma companies. The Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA. It further received support by the Babcock Endowment Fund (LCT, MKS, JT, JEG), by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health under Award Number R01-AR060802 (JEG), P30-AR075043 (JEG), and K01-AR072129 (LCT), and National Institute of Allergy and Infectious Diseases (NIAID) under Award Number R01-AR069071 (JEG), the A. Alfred Taubman Medical Research Institute (JEG, JMK), and the Kenneth and Frances Eisenberg Emerging Scholar Award (JEG). LCT is supported by the Dermatology Foundation, Arthritis National Research Foundation, and National Psoriasis Foundation. Infrastructure support was provided through the DFG Cluster of Excellence “Precision Medicine in Inflammation” (grant EXC2167).
JEG received research grants from AbbVie, AnaptysBio, Pfizer, Novartis, Celgene, and Eli Lilly, and serves as advisory board in Novartis, AbbVie, Eli Lilly, MiRagen, and Almirall. SW has received institutional research grants from Novartis, Pfizer and L’Oreal, has performed consultancies for Sanofi-Genzyme, Regeneron, LEO Pharma, Incyte, Lilly, Abbvie and Novartis, has lectured at educational events sponsored by Sanofi-Genzyme, Regeneron, LEO Pharma, Abbvie and Galderma, and is involved in performing clinical trials with pharmaceutical industries that manufacture drugs used for the treatment of atopic dermatitis. JMK serves as advisory boards and received consulting fees from AstraZeneca, BMI, BMS, Eli Lilly, and also received grant funding from Celgene/BMS.
Abbreviations
- AD
atopic dermatitis
- FDR
False discovery rate
- FC
Fold Change
Footnotes
Conflict of interest
All other authors declare no relevant conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lloyd-Lavery A, Solman L, Grindlay DJC, Rogers NK, Thomas KS, Harman KE. What’s new in atopic eczema? An analysis of systematic reviews published in 2016. Part 2: Epidemiology, aetiology and risk factors. Clin Exp Dermatol 2019; 44:370–5. [DOI] [PubMed] [Google Scholar]
- 2.Abuabara K, Ye M, McCulloch CE, Sullivan A, Margolis DJ, Strachan DP, et al. Clinical onset of atopic eczema: Results from 2 nationally representative British birth cohorts followed through midlife. J Allergy Clin Immunol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abuabara K, Yu AM, Okhovat JP, Allen IE, Langan SM. The prevalence of atopic dermatitis beyond childhood: A systematic review and meta-analysis of longitudinal studies. Allergy 2018; 73:696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers 2018; 4:1. [DOI] [PubMed] [Google Scholar]
- 5.Oyoshi MK, He R, Kumar L, Yoon J, Geha RS. Cellular and molecular mechanisms in atopic dermatitis. Adv Immunol 2009; 102:135–226. [DOI] [PubMed] [Google Scholar]
- 6.Leung DY, Boguniewicz M, Howell MD, Nomura I, Hamid QA. New insights into atopic dermatitis. J Clin Invest 2004; 113:651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gittler JK, Shemer A, Suarez-Farinas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQ, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol 2012; 130:1344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsoi LC, Rodriguez E, Degenhardt F, Baurecht H, Wehkamp U, Volks N, et al. Atopic dermatitis is an IL-13 dominant disease with greater molecular heterogeneity compared to psoriasis. J Invest Dermatol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015; 31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 2010; 11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014; 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Koning HD, van den Bogaard EH, Bergboer JG, Kamsteeg M, van Vlijmen-Willems IM, Hitomi K, et al. Expression profile of cornified envelope structural proteins and keratinocyte differentiation-regulating proteins during skin barrier repair. Br J Dermatol 2012; 166:1245–54. [DOI] [PubMed] [Google Scholar]
- 15.Rahrig S, Dettmann JM, Brauns B, Lorenz VN, Buhl T, Kezic S, et al. Transient epidermal barrier deficiency and lowered allergic threshold in filaggrin-hornerin (FlgHrnr(−/−) ) double-deficient mice. Allergy 2019; 74:1327–39. [DOI] [PubMed] [Google Scholar]
- 16.Takaishi M, Makino T, Morohashi M, Huh NH. Identification of human hornerin and its expression in regenerating and psoriatic skin. J Biol Chem 2005; 280:4696–703. [DOI] [PubMed] [Google Scholar]
- 17.Lau D, Elezagic D, Hermes G, Morgelin M, Wohl AP, Koch M, et al. The cartilage-specific lectin C-type lectin domain family 3 member A (CLEC3A) enhances tissue plasminogen activator-mediated plasminogen activation. J Biol Chem 2018; 293:203–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harden JL, Lewis SM, Lish SR, Suarez-Farinas M, Gareau D, Lentini T, et al. The tryptophan metabolism enzyme L-kynureninase is a novel inflammatory factor in psoriasis and other inflammatory diseases. J Allergy Clin Immunol 2016; 137:1830–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017; 18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steinhoff M, Steinhoff A, Homey B, Luger TA, Schneider SW. Role of vasculature in atopic dermatitis. J Allergy Clin Immunol 2006; 118:190–7. [DOI] [PubMed] [Google Scholar]
- 21.Hannenhalli S, Kaestner KH. The evolution of Fox genes and their role in development and disease. Nat Rev Genet 2009; 10:233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer 2007; 7:847–59. [DOI] [PubMed] [Google Scholar]
- 23.Yang XF, Fang P, Meng S, Jan M, Xiong X, Yin Y, et al. The FOX transcription factors regulate vascular pathology, diabetes and Tregs. Front Biosci (Schol Ed) 2009; 1:420–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weidinger S, Novak N. Atopic dermatitis. Lancet 2016; 387:1109–22. [DOI] [PubMed] [Google Scholar]
- 25.Fischer M, Harvima IT, Carvalho RF, Moller C, Naukkarinen A, Enblad G, et al. Mast cell CD30 ligand is upregulated in cutaneous inflammation and mediates degranulation-independent chemokine secretion. J Clin Invest 2006; 116:2748–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gahr N, Folster-Holst R, Weichenthal M, Christophers E, Schroder JM, Bartels J. Dermal fibroblasts from acute inflamed atopic dermatitis lesions display increased eotaxin/CCL11 responsiveness to interleukin-4 stimulation. Br J Dermatol 2011; 164:58692. [DOI] [PubMed] [Google Scholar]
- 27.Soter NA, Mihm MC. Morphology of atopic eczema. Acta Derm Venereol 1980; Suppl:115. [Google Scholar]
- 28.Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, et al. IL-22producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol 2009; 123:1244–52 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brunner PM, Pavel AB, Khattri S, Leonard A, Malik K, Rose S, et al. Baseline IL-22 expression in patients with atopic dermatitis stratifies tissue responses to fezakinumab. J Allergy Clin Immunol 2019; 143:142–54. [DOI] [PubMed] [Google Scholar]
- 30.Rutz S, Wang X, Ouyang W. The IL-20 subfamily of cytokines--from host defence to tissue homeostasis. Nat Rev Immunol 2014; 14:783–95. [DOI] [PubMed] [Google Scholar]
- 31.Guttman-Yassky E, Brunner PM, Neumann AU, Khattri S, Pavel AB, Malik K, et al. Efficacy and safety of fezakinumab (an anti-IL-22 monoclonal antibody) in adults with moderateto-severe atopic dermatitis inadequately controlled by conventional treatments - A randomized, double-blind, phase 2a trial. J Am Acad Dermatol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ebner S, Nguyen VA, Forstner M, Wang YH, Wolfram D, Liu YJ, et al. Thymic stromal lymphopoietin converts human epidermal Langerhans cells into antigen-presenting cells that induce proallergic T cells. J Allergy Clin Immunol 2007; 119:982–90. [DOI] [PubMed] [Google Scholar]
- 33.Johnston A, Xing X, Guzman AM, Riblett M, Loyd CM, Ward NL, et al. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J Immunol 2011; 186:2613–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bassoy EY, Towne JE, Gabay C. Regulation and function of interleukin-36 cytokines. Immunol Rev 2018; 281:169–78. [DOI] [PubMed] [Google Scholar]
- 35.Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med 2005; 202:1213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foster AM, Baliwag J, Chen CS, Guzman AM, Stoll SW, Gudjonsson JE, et al. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J Immunol 2014; 192:6053–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simpson EL, Parnes JR, She D, Crouch S, Rees W, Mo M, et al. Tezepelumab, an antithymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: A randomized phase 2a clinical trial. J Am Acad Dermatol 2019; 80:1013–21. [DOI] [PubMed] [Google Scholar]
- 38.Bachelez H, Choon SE, Marrakchi S, Burden AD, Tsai TF, Morita A, et al. Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N Engl J Med 2019; 380:981–3. [DOI] [PubMed] [Google Scholar]
- 39.Yoshihara Y, Ishiuji Y, Yoshizaki A, Kurita M, Hayashi M, Ishiji T, et al. IL-10-Producing Regulatory B Cells Are Decreased in Patients with Atopic Dermatitis. J Invest Dermatol 2019; 139:475–8. [DOI] [PubMed] [Google Scholar]
- 40.Czarnowicki T, Gonzalez J, Bonifacio KM, Shemer A, Xiangyu P, Kunjravia N, et al. Diverse activation and differentiation of multiple B-cell subsets in patients with atopic dermatitis but not in patients with psoriasis. J Allergy Clin Immunol 2016; 137:118–29 e5. [DOI] [PubMed] [Google Scholar]
- 41.Nakatsumi H, Matsumoto M, Nakayama KI. Noncanonical Pathway for Regulation of CCL2 Expression by an mTORC1-FOXK1 Axis Promotes Recruitment of Tumor-Associated Macrophages. Cell Rep 2017; 21:2471–86. [DOI] [PubMed] [Google Scholar]
- 42.Ding Z, Ke R, Zhang Y, Fan Y, Fan J. FOXE1 inhibits cell proliferation, migration and invasion of papillary thyroid cancer by regulating PDGFA. Mol Cell Endocrinol 2019; 493:110420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.