

Abstract
Ubrogepant is a novel, oral calcitonin gene‐related peptide (CGRP) receptor antagonist intended for the acute treatment of migraine attacks. Ubrogepant has a chemical structure distinct from previous small‐molecule CGRP receptor antagonists that were associated with elevated serum alanine aminotransferase (ALT) in clinical trials. Here, we report overall and hepatic safety data from two placebo‐controlled phase I trials of ubrogepant, spray‐dried oral compressed tablet (SD‐OCT) in healthy male volunteers. Trial A was a pharmacokinetic (PK) trial of single (100–400 mg) and multiple (40–400 mg) ascending doses. Trial B was a dedicated hepatic safety trial assessing daily use of ubrogepant 150 mg for 28 days. Serum ALT (as hepatotoxicity biomarker) and PK data are reported. Ubrogepant was well‐tolerated in both trials, with a low incidence of adverse events that did not differ greatly from placebo. Changes in mean ALT levels were minimal and similar to placebo. Over 28 days of treatment, the mean percentage change in ALT from baseline was < 5% at all time points. No participant in either trial demonstrated ALT ≥ 3× upper limit of normal at any time. Ubrogepant SD‐OCT demonstrated linear PK appropriate for acute treatment of migraine, with rapid uptake (time of maximum plasma concentration (tmax): 2–3 hours) and no accumulation with daily use. Overall, there was no evidence of ubrogepant‐associated hepatotoxicity with daily doses up to 400 mg for 10 days or with daily ubrogepant 150 mg for 28 days. Supratherapeutic dosing is a useful strategy for characterizing hepatic safety in early drug development.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Ubrogepant is an orally delivered, potent, and specific calcitonin gene‐related peptide (CGRP) receptor antagonist that is anticipated to be a first‐in‐class CGRP‐targeted acute treatment for migraine attacks. The impact of ubrogepant on changes in alanine aminotransferase (ALT) has not yet been evaluated in a dedicated clinical trial.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ What, if any, effect does oral ubrogepant have on ALT levels in healthy adult men?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Single and multiple daily doses of ubrogepant up to the supratherapeutic dose of 400 mg were found to be safe and well‐tolerated, with no evidence of drug‐related ALT elevations in healthy men.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ These findings will provide clinicians with the knowledge that ubrogepant, a small‐molecule CGRP antagonist, is unlikely to be associated with hepatotoxicity.
Migraine is a highly prevalent neurological disease characterized by recurrent attacks of unilateral and pulsating headaches, which are often accompanied by other debilitating symptoms, including nausea, photophobia, and/or phonophobia.1, 2, 3 Migraine is associated with a high personal, familial, and societal burden.4, 5, 6, 7, 8
During migraine attacks, activation of the trigeminovascular system leads to increased release of vasoactive neuropeptides, including calcitonin gene‐related peptide (CGRP) and substance P, as well as nitric oxide, resulting in meningeal blood vessel vasodilation.3 The principal goals of acute treatment for migraine attacks are to provide rapid and durable relief of headache pain and associated symptoms while preventing recurring attacks and restoring the ability to function.2, 9 However, despite the widespread availability of acute treatment options for migraine, treatment optimization remains difficult or impossible for many individuals due to limited effectiveness, poor tolerability, or contraindications to existing therapies.9, 10, 11, 12 As a result, migraine remains one of the most undertreated neurological diseases and continues to be a leading cause of disability worldwide.7, 13
Several lines of evidence have implicated CGRP in the pathophysiology of migraine.14, 15, 16 Elevated levels of CGRP have been found in the external jugular outflow, but not in the cubital fossa, during migraine attacks.17, 18 In addition, a sustained elevation in serum CGRP levels has been reported between attacks in some people with chronic migraine.19, 20 Furthermore, infusion of CGRP can elicit moderate to severe headache pain in people with migraine and in healthy individuals.21, 22, 23
Recognition of the role of CGRP in migraine pathogenesis has led to the evaluation of CGRP antagonists (targeting either CGRP itself or the CGRP receptor) for the treatment of migraine over the past 2 decades.24, 25, 26 Telcagepant, the first oral CGRP receptor antagonist to reach clinical evaluation, demonstrated efficacy for the acute treatment of migraine attacks in several clinical trials.27, 28, 29 Single doses of telcagepant were well‐tolerated, with dry mouth, nausea, dizziness, and somnolence as the most commonly reported adverse events (AEs).27, 28, 29 However, some participants experienced elevated alanine aminotransferase (ALT) levels following treatment with telcagepant once daily for 7 days or twice daily for 12 weeks.30, 31 Concerns regarding hepatotoxicity led to the discontinuation of telcagepant development, despite its clinical efficacy in the treatment of migraine.30 Another oral small‐molecule CGRP receptor antagonist, MK‐3207, was associated with delayed liver test abnormalities in phase I trials. ALT elevations were observed following the discontinuation of MK‐3207 administration, leading to the discontinuation of the MK‐3207 development program.31, 32, 33
The mechanism of the observed liver toxicity associated with telcagepant and MK‐3207 is unclear. It does not appear to be a CGRP receptor antagonist class effect, given that liver problems were not observed in CGRP knockout mice, and there is no evidence of liver toxicity with CGRP neutralizing antibodies.34, 35 Available mechanistic data suggested that hepatic risk with MK‐3207 and telcagepant was related to a bioactivation‐mediated mechanism attributable to reactive metabolites. Subsequent drug development research focused on identifying a small‐molecule CGRP receptor antagonist with characteristics hypothesized to be important for reducing the potential for hepatotoxicity, including greater drug potency, lower dosing for clinical efficacy, and reduced potential to form reactive metabolites.35
Ubrogepant is a small‐molecule CGRP receptor antagonist that is chemically distinct from MK‐3207 and telcagepant. Ubrogepant was developed for the acute treatment of migraine attacks with the goal of providing maximal efficacy with minimal impact on safety, including an absence of vasoconstrictive effects and a greatly reduced hepatic risk. The results of preclinical studies showed that ubrogepant had higher potency than telcagepant (E. Moore, M.E. Fraley, I.M. Bell, C.S. Burgey, R.B. White, C.‐C. Li et al., unpublished data), suggesting that therapeutic efficacy could be attained at lower doses and a smaller chance of exposure to reactive metabolites (i.e., body burden).
Ubrogepant has demonstrated significant efficacy in the acute treatment of migraine attacks in two phase III placebo‐controlled clinical trials (ACHIEVE I and ACHIEVE II) that enrolled over 2,500 adults with migraine. Participants treated a single migraine attack of moderate/severe pain intensity with ubrogepant (25 mg (ACHIEVE II), 50 mg (both trials), or 100 mg (ACHIEVE I)) or placebo.36, 37 The 50 mg and 100 mg doses of ubrogepant were significantly more effective than placebo on both coprimary end points of pain freedom and absence of most bothersome migraine‐associated symptom (e.g., photophobia, phonophobia, and nausea) at 2 hours postdose. Overall, ubrogepant was well‐tolerated, with no safety concerns identified.36, 37
The potential hepatotoxicity observed with the early small‐molecule CGRP receptor antagonists30, 31, 32 warranted vigilant monitoring of hepatic safety signals in the clinical trial program for ubrogepant. Two early phase I trials conducted in healthy volunteers included hepatic safety assessments, focusing on ALT elevations as a biomarker for liver toxicity. Trial A was a single‐ascending and multiple‐ascending dose trial that evaluated the pharmacokinetic (PK) profile, safety, and tolerability of increasing doses (40–400 mg) of ubrogepant spray‐dried oral compressed tablet (SD‐OCT). Trial B was a dedicated trial to assess changes in ALT over 28 days of daily dosing with ubrogepant SD‐OCT at a dose of 150 mg, which is 1.5‐fold greater than the highest anticipated clinical dose. The primary objective of this report is to present the overall and hepatic safety data from these two phase I trials. To characterize drug exposure, key PK data from both trials are also reported.
Methods
Trial objectives
The primary objective of this study was to evaluate the safety and tolerability of rising single and multiple oral doses of ubrogepant (trial A) and the safety, tolerability, and effects on ALT levels of multiple oral doses of ubrogepant (trial B) in healthy male volunteers. The secondary objective of both trials was to estimate the serum PK parameters following single‐dose and/or multiple‐dose administration of ubrogepant.
Trial designs and ethical conduct
Trial A (MSD Protocol MK‐1602‐002) was a two‐part, randomized, double‐blind, placebo‐controlled trial. Ubrogepant was administered as an SD‐OCT. Two additional formulations of ubrogepant were also tested, but the results are not included here. Participants were randomized 3:1 to ubrogepant (n = 6) or placebo (n = 2). During part 1, the first panel of participants (panel A) received single once‐daily rising oral doses of ubrogepant (100–400 mg) or placebo under fasting conditions, and 100 mg with a high‐fat meal in five treatment periods, with a minimum 7‐day washout between treatment periods. In part 2, the next four serial panels of participants (panels B, C, D, and F) were administered once‐daily oral doses of ubrogepant (40–400 mg) or placebo, under fasting conditions, for 10 consecutive days.
Trial B (MSD Protocol MK‐1602‐004) was a randomized, double‐blind, placebo‐controlled, single‐center, multiple‐dose trial; participants were randomized 2:1 to once‐daily ubrogepant 150 mg (1.5 times greater than the planned clinical dose) or matching placebo, administered as an SD‐OCT, orally once daily for 28 consecutive days. The 150 mg dose was selected for evaluation in trial B because it was expected to be higher than the anticipated efficacious dose for acute treatment of migraine, which was estimated using an empirical maximal effect (Emax) population PK/pharmacodynamic (PD) model for capsaicin‐induced dermal vasodilation (CIDV) (E. Moore, M.E. Fraley, I.M. Bell, C.S. Burgey, R.B. White, C.‐C. Li et al., unpublished data). In this assay, inhibition of CIDV was measured by laser Doppler scan at 1 and 5 hours after single oral doses of ubrogepant (0.5, 5, and 40 mg) that were selected to capture the expected dynamic range of the exposure‐response curve based on the estimated half‐maximal effective concentration of 3.2 nM from CIDV experiments in rhesus macaques. The CIDV PK/PD modeling results indicated that the threshold concentration of ubrogepant associated with efficacy (effective concentration 90%) is 23 nM.
Each trial was performed in accordance with the Declaration of Helsinki and the principles of Good Clinical Practice. The conduct of each trial was approved by the relevant institutional review boards and/or ethics committees, and all participants provided written informed consent prior to enrollment.
Trial population
Both trials enrolled healthy nonsmoking men aged 18 to 50 years. Additional inclusion criteria were general good health with body mass index ≤ 30 kg/m2 (trial A) or between 18 and 32 kg/m2 (trial B); no clinically significant electrocardiogram (ECG) abnormalities; ALT and aspartate aminotransferase (AST) below the upper limit of normal (ULN); total bilirubin less than twofold ULN and (if total bilirubin is greater than the ULN) direct bilirubin within normal limits at screening and prior to first trial drug dose. Potential participants were excluded if they were mentally or legally incapacitated or had significant emotional problems or a history of mental illness that may have confounded trial results; had an estimated creatinine clearance (Cockroft‐Gault equation) of ≤ 80 mL/minute; had a history of hepatic disease; had a history of stroke, chronic seizures, or major neurological disorder (including migraine); or had a history of excessive intake of alcohol or caffeinated beverages or inability to refrain from prohibited medications.
Assessments
Blood samples for PK determination were collected before dosing and at predetermined times after dosing. Following centrifugation, plasma samples were analyzed for ubrogepant concentration at Merck (West Point, PA) using liquid‐liquid extraction for analyte isolation followed by liquid chromatographic‐tandem mass spectrometric detection for quantitation. For trial B, the lower limit of quantitation was 1 ng/mL (1.82 nM), with a linear calibration range from 1–1,000 ng/mL (1.82–1,820 nM), whereas a more sensitive assay with a lower limit of quantitation of 0.1 ng/mL (0.182 nM), with a linear calibration range from 0.1–1,000 ng/mL (0.182–1,820 nM), was used in trial A.
Safety assessments included tabulation of AEs and serious AEs (SAEs), as reported; and physical examination, vital sign determination, ECG, and laboratory assessments conducted at predetermined time points during the trial. Laboratory assessments included hematology, blood chemistry, and urinalysis; analyte quantitation was performed using standard established methods. Post‐study safety assessments (including ALT evaluation) were conducted 2 weeks and 1 and 2 months after the final dose of study medication.
Statistical analysis
For each trial, all participants who received at least one dose of study medication were included in the analysis of safety. In both trials, hepatic safety was assessed via evaluation of percentage changes from baseline in ALT measurements at various doses and time points.
In trial A, the time to reach steady‐state was assessed by using the effective rate of drug accumulation for each participant, obtained from his accumulation ratio of area under the curve from 0 through 24 hours (AUC0–24hr). The effective rate of drug accumulation (ηi) for each individual (i) was calculated using the following relationship:
where τ is the length of the dosing interval in hours, X is the total number of dosing intervals, and Y = 1. In addition, an exploratory dose proportionality analysis of trial A was conducted using the estimates and 95% confidence intervals (CIs) for the slope from the power law model, with ln(AUC) as the dependent variable and ln(dose) as the explanatory variable. PK parameters (maximum plasma drug concentration (Cmax), mean plasma concentration at 2 hours postdose (C2hr), time to Cmax (tmax), and AUC) were calculated using Phoenix WinNonlin Professional, version 6.3 (Certara USA, Inc, Princeton, NJ).
In trial B, the posterior probability that the true mean percent treatment difference in ALT (ubrogepant/placebo) exceeded 50% was calculated at the time point with the largest mean percent treatment difference. A posterior probability < 70% would provide sufficient evidence to conclude that ubrogepant does not have a clinically meaningful effect on ALT. The principal motivation for the Bayesian methods and use of posterior probability rather than classic, frequentist null hypothesis significance testing is that the latter can obscure the assessment of the target region by using null hypothesis values that are not relevant (e.g., assume 0 as the null value of the parameter) and by using habitual type I error rates (e.g., 0.05) that circumvent an appropriate assessment of risk matched to the current stage of development. Using the clinical target region (50% mean increase) and corresponding posterior probability threshold (70%), a decision rule was set to assess the probability that a clinically meaningful effect exists. The posterior probability provides the level of conditional probability based on randomly observed data. Because it uses a random variable, it is important to summarize its amount of uncertainty. In this case, the hypothesis for this study included the interval of uncertainty (70%) that the observed effect is the true effect that would be seen in a large population.
Results
Participant disposition and baseline demographics
Forty healthy participants (eight per panel) were enrolled in trial A; all 40 completed the trial. Mean age of trial A participants was 39 years (range 21–51 years), and all were non‐Hispanic white men. Thirty‐two healthy male participants were enrolled in trial B; all 32 completed the trial. Participants in trial B had a mean age of 38 years (range 25–49 years); the majority (69%) were African—American. Participant demographics are summarized in Table 1.
Table 1.
Participant characteristics
| Characteristics | Trial A | Trial B | |||||
|---|---|---|---|---|---|---|---|
| Panel A | Panel B | Panel C | Panel D | Panel F | Placebo | 150 mg | |
| No. of participants | 8 | 8 | 8 | 8 | 8 | 10 | 22 |
| Men, % | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Age (years) | |||||||
| Mean | 37 | 42 | 40 | 35 | 43 | 39 | 38 |
| Median | 35 | 45 | 45 | 38 | 46 | 40 | 38 |
| Range | 24–49 | 22–51a | 22–50 | 21–48 | 26–50 | 30–48 | 25–49 |
| Race, % | |||||||
| White | 100 | 100 | 100 | 100 | 100 | 20 | 14 |
| Black or African American | 0 | 0 | 0 | 0 | 0 | 70 | 68 |
| Multiple | 0 | 0 | 0 | 0 | 0 | 10 | 18 |
| Ethnicity, % | |||||||
| Hispanic or Latino | 0 | 0 | 0 | 0 | 0 | 0 | 9 |
| Not Hispanic or Latino | 100 | 100 | 100 | 100 | 100 | 100 | 91 |
Panel E was not conducted.
One participant turned 51 after screening.
Pharmacokinetics
Administered as an SD‐OCT, ubrogepant was rapidly absorbed, with minimal accumulation, following once‐daily dosing. Table 2 summarizes key PK parameters for oral ubrogepant SD‐OCT across both trials in fasted healthy male participants. In trial A, median tmax for ubrogepant SD‐OCT ranged from 2–3 hours, terminal half‐life was ~ 3 to 4 hours after single doses, and ~ 7 to 11 hours following multiple once‐daily doses. Steady‐state was reached within 2 days. There was no evidence for significant accumulation following multiple daily doses. The AUC0–24hr accumulation ratio (90% CI) at day 10 of daily ubrogepant dosing was 0.97 (0.82, 1.15), 1.02 (0.86, 1.20), 0.88 (0.74, 1.04), and 1.07 (0.90, 1.26) for the 40, 100, 200, and 400 mg dose groups, respectively. Mean ubrogepant plasma concentrations vs. time following single oral doses of 100–400 mg ubrogepant in fasted healthy male participants are shown in Figure 1 a. Mean plasma ubrogepant concentrations vs. time following once‐daily oral doses of 40–400 mg ubrogepant for 10 days are shown in Figure 1 b.
Table 2.
Statistical summary for plasma pharmacokinetic parameters of oral ubrogepant (SD‐OCT formulation)
| Pharmacokinetic parameters | Trial A, part 1 (single dose of ubrogepant) | Trial A, part 2 (once‐daily ubrogepant for 10 days) | Trial B (once‐daily ubrogepant for 28 days) | |||||
|---|---|---|---|---|---|---|---|---|
|
100 mg (n = 6) |
200 mg (n = 6) |
400 mg (n = 6) |
40 mg (n = 6) |
100 mg (n = 6) |
200 mg (n = 6) |
400 mg (n = 6) |
150 mg (n = 22) |
|
| Day 1 (first dose) | ||||||||
| AUC0–∞ (95% CI), nM·houra | 1,960 (1,450, 2,670) | 4,420 (3,260, 6,000) | 6,850 (5,050, 9,290) | – | – | – | – | – |
| AUC0–24 h (95% CI), nM·houra | 1,950 (1,430, 2,650) | 4,350 (3,200, 5,910) | 6,680 (4,910, 9,080) | 598 (461, 776) | 1,690 (1,300, 2,190) | 3,250 (2,500, 4,220) | 7,050 (5,430, 9,150) | 2,210 (1,870, 2,610) |
| C2hr, (95% CI), nMa | 344 (241, 491) | 846 (590, 1,210) | 1,160 (810, 1,670) | – | – | – | – | 247 (186, 329) |
| Cmax, (95% CI)a nM | 344 (241, 491) | 846 (590, 1,210) | 1,160 (810, 1,670) | 134 (106, 169) | 316 (250, 400) | 591 (467, 746) | 1,140 (900, 1,440) | 411 (341, 496) |
| tmax, hourb | 3 (1, 4) | 3 (2, 4) | 2 (2, 4) | 1.75 (1, 3) | 2.5 (2, 4) | 3 (1, 3) | 3.5 (2, 4) | 2.26 (1, 6) |
| t1/2, hourc | 3.4 (7.9) | 4.2 (18.4) | 4.3 (20.3) | – | – | – | – | – |
| Day 10 (last dose) | ||||||||
| Cmax, nM (95% CI)a | – | – | – | 95 (75.2, 120) | 302 (239, 382) | 543 (430, 686) | 1,340 (1,060, 1,700) | – |
| tmax, hourb | – | – | – | 3 (2, 3) | 3.5 (1, 4) | 2 (1,3) | 2 (1, 4) | – |
| t1/2, hourc | – | – | – | 7.3 (34.7) | 6.7 (62.7) | 11.2 (80.2) | 7.8 (29.1) | – |
| Day 28 (last dose) | ||||||||
| Cmax, nM (95% CI)a | – | – | – | – | – | – | – | 465 (385, 561) |
| tmax, hourb | – | – | – | – | – | – | – | 3 (0.70, 4.07) |
| t1/2, hourc | – | – | – | – | – | – | – | 9.9 (51.6) |
AUC0‐∞, area under the concentration‐time curve from zero to infinity; AUC0–24h, 0–24‐hour area under the concentration‐time curve; C2hr, mean plasma concentration at 2 hours postdose; CI, confidence interval; Cmax, maximum plasma concentration; SD‐OCT, spray‐dried oral compressed tablet; t1/2, half‐life; tmax, time to Cmax.
Back‐transformed least squares mean and 95% CI from linear mixed effects model performed on natural log‐transformed values.
Median (min, max) reported for tmax.
Geometric mean and percentage geometric coefficient of variation reported for apparent terminal t1/2.
Figure 1.

Trial A: Mean (± SD) ubrogepant plasma concentrations in healthy participants. (a) Part 1 following administration of 100–400 mg doses in 24 hours, and (b) part 2 following administration of 40–400 mg doses once daily for 10 days. Top panel: Linear scale, bottom panel: semi‐log scale.
Administration of 100 mg ubrogepant SD‐OCT with a high‐fat breakfast (fed condition) reduced the observed area under the curve from 0 to infinity (AUC0–∞) by 16%, C2hr by 59%, and Cmax by 33% when compared with administration of 100 mg ubrogepant under fasted conditions. Ubrogepant tmax was delayed by 1 hour when administered with food (median (min, max): 4.0 hours (2.0, 6.0)) compared with the fasted state (3.0 hours (1.0, 4.0)). Following multiple oral doses of ubrogepant SD‐OCT for 10 days (trial A), a C2hr of > 23 nM was attained at doses of 40 mg and higher. This value is the threshold PK target predicted to be associated with efficacy for ubrogepant based on a population PK/PD model for CIDV in healthy young men (E. Moore, M.E. Fraley, I.M. Bell, C.S. Burgey, R.B. White, C.‐C. Li et al., unpublished data).
In trial B, AUC0–24hour and Cmax values following once‐daily administration of ubrogepant SD‐OCT 150 mg were consistent with trial A results in the 40–400 mg dose range (Table 2, Figure S1 ). Mean ubrogepant plasma concentrations vs. time in fasted healthy adult men following once‐daily 150 mg ubrogepant SD‐OCT are plotted in Figure 2. In trial B, median tmax for ubrogepant SD‐OCT was 2.26–3 hours and the terminal half‐life was 9.9 hours at day 28. The AUC0–24hr accumulation ratio (90% CI) at day 28 of daily dosing (trial B) was 1.09 (1.00, 1.18). In trial A, ubrogepant PK exposure increased with dose over the investigated dose range (40–400 mg); estimates of slope (95% CI) were 1.08 (0.90, 1.25) for AUC0–24hr and 1.13 (1.00, 1.26) for Cmax, suggesting approximately dose‐proportional exposure. Because ubrogepant has minimal urinary excretion in humans, urine PK was not included in these studies.
Figure 2.

Trial B: Mean (± SD) ubrogepant plasma concentration following administration of once‐daily 150 mg doses for 28 days in fasted healthy participants. Top panel: Linear scale, bottom panel: semi‐log scale.
Safety
In trial A, single (100–400 mg) and multiple (40–400 mg) doses of ubrogepant were well‐tolerated in healthy male participants. There were no SAEs reported, and no participant discontinued due to an AE. No clinically significant abnormalities were noted in routine serum chemistry, hematology, urinalysis, ECG, or vital signs. All reported clinical AEs were mild or moderate in intensity. In trial A, part 1, no AE was experienced in ≥ 1 participant, except for nasopharyngitis (one participant each from the placebo and ubrogepant groups). In trial A, part 2, AEs reported by more than two participants included nausea (ubrogepant n = 2, placebo n = 2), headache (ubrogepant n = 1, placebo n = 2), diarrhea (ubrogepant n = 1, placebo n = 2), and nasopharyngitis (ubrogepant n = 3, placebo n = 0).
In trial B, once‐daily ubrogepant 150 mg for 28 days was well‐tolerated, with no SAEs and no discontinuations due to an AE. There were no clinically significant abnormalities with respect to laboratory assessments, ECGs, or vital signs. All reported AEs were mild or moderate in intensity, with the exception of one participant who reported severe flank pain and nephrolithiasis; this participant continued in the trial after diagnosis of a kidney stone unrelated to trial drug administration. AEs reported by more than two participants included contusion (ubrogepant n = 5, placebo n = 1), excoriation (ubrogepant n = 2, placebo n = 1), scratch (ubrogepant n = 1, placebo n = 2), and dizziness (ubrogepant n = 4, placebo n = 0).
Hepatic safety
In trial A, part 1, the mean percentage changes from baseline in ALT at 24 hours with single‐dose ubrogepant under fasted conditions ranged from − 0.21% to 1.91%, compared with a placebo change of − 1.63% (Table 3). Under fed conditions, the percentage ALT change from baseline was 10.04% and −6.25% for ubrogepant and placebo, respectively. In trial A, part 2, the mean percentage changes from baseline in ALT at day 10 following once‐daily SD‐OCT ubrogepant (40–400 mg) ranged from −33.57% to 3.69%, compared with a − 2.86% change with placebo (Table 3). There was no evidence of a dose relationship with respect to percentage change from baseline in ALT. No participant demonstrated an ALT elevation ≥ 3 × ULN.
Table 3.
Percentage change from baseline in ALT associated with oral ubrogepant in fasted healthy men for 10 days
| Treatment | Trial A, part 1 (single doses of ubrogepant)a | Trial A, part 2, once‐daily ubrogepant for 10 daysb | ||||
|---|---|---|---|---|---|---|
| N | Percentage change from baselinec | SE | N | Percentage change from baselinec | SE | |
| Placebo | 8 | −1.63 | 7.66 | 8 | −2.86 | 4.93 |
| 40 mg | – | – | – | 6 | −11.28 | 7.65 |
| 100 mg | 6 | 1.91 | 7.30 | 6 | 3.69 | 7.75 |
| 200 mg | 6 | 0.51 | 9.15 | 6 | −19.39 | 7.23 |
| 400 mg | 6 | −0.21 | 3.69 | 6 | −33.57 | 3.66 |
ALT, alanine aminotransferase.
Data from 24 hours following single doses of 100–400 mg of oral ubrogepant.
Data from day 10, 24 hours following multiple‐dose administration of 40–400 mg oral ubrogepant.
Day 1, predose measurement serves as baseline.
In trial B, mean serum ALT levels in the ubrogepant 150 mg group were similar to placebo throughout the 28‐day trial duration (Figure 3). The mean percentage change from baseline in ALT 24 hours after the day 28 dose was −4.15% vs. 7.09% in ubrogepant and placebo, respectively (Table 4). During the 28‐day treatment period, the mean percentage change from baseline in ALT ranged from − 15.69% to − 0.83% for ubrogepant and − 2.22% to 9.22% for placebo. No individual participant demonstrated an ALT elevation ≥ 3 × ULN.
Figure 3.
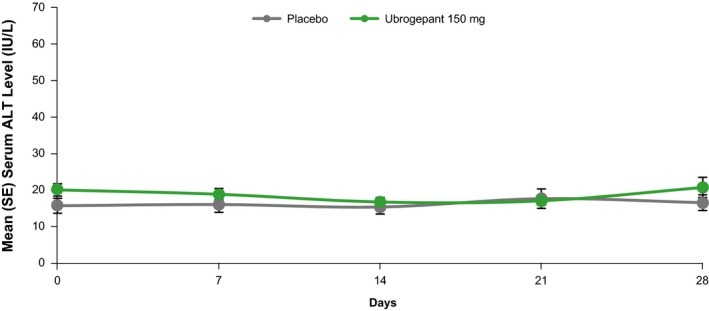
Trial B: Mean (± SE) serum ALT following the administration of ubrogepant 150 mg or placebo for 28 days. ALT, serum alanine aminotransferase.
Table 4.
Statistical summary for serum ALT following multiple‐dose administration of ubrogepant 150 mg or placebo
| Time | Percentage change (95% CI) from baselinea | |
|---|---|---|
| Placebo (n = 10), mean | Ubrogepant 150 mg (n = 22), mean | |
| Day 1, 24 hours | −2.22 (−15.52, 13.18) | −3.59 (−12.64, 6.40) |
| Day 7, predose | 3.05 (−10.97, 19.27) | −5.08 (−13.99, 4.76) |
| Day 14, predose | −1.76 (−15.12, 13.71) | −15.69 (−23.60, −6.95) |
| Day 21, predose | 9.22 (−5.64, 26.42) | −12.65 (−20.85, −3.60) |
| Day 28, predose | 5.63 (−8.74, 22.26) | −0.83 (−10.14, 9.44) |
| Day 28, 24 hours | 7.09 (−7.48, 23.95) | −4.15 (−13.14, 5.78) |
ALT, alanine aminotransferase; CI, confidence interval.
Baseline is the geometric mean of the day − 1 and day 1 predose values.
Mean treatment differences in ALT (ubrogepant/placebo percentage change) and posterior probabilities are summarized in Table S1 . Based on the observed patterns of ALT values, the percentage changes in treatment differences were negative at all time points. Given these values, the corresponding probability that the “true” percentage change exceeded 50% was < 0.1% throughout the trial.
Discussion
Taken together, the results of these two trials provide support for the safety and tolerability of ubrogepant, and, importantly, its hepatic safety profile. Ubrogepant, administered at single doses up to 400 mg and multiple doses up to 400 mg q.d. for 10 days (trial A) and at 150 mg q.d. for 28 days (trial B) to healthy male participants, was not associated with any SAEs or clinically significant abnormalities in ECGs, vital signs, or laboratory safety assessments. In trial A, changes in serum ALT were minimal and similar between ubrogepant and placebo; there was no evidence of a dose effect. In trial B, once‐daily administration of ubrogepant 150 mg (a dose 1.5‐fold greater than the anticipated clinical dose) did not have a clinically meaningful effect on serum ALT levels over 28 days of administration. Percentage changes from baseline in serum ALT following ubrogepant treatment were low (< 5%) and lower than the time‐matched changes from baseline for placebo. In addition, participants were evaluated for 2 months after the final ubrogepant dose to rule out the possibility of delayed ALT elevations as were observed in previous trials of first‐generation CGRP antagonists; no elevations were detected.
The PK profile of ubrogepant SD‐OCT as observed in trials A and B, achieving maximal plasma levels in 1.75 to 3.5 hours and demonstrating a half‐life of ~ 7 to 11 hours, is consistent with its suitability for use as an acute treatment for migraine attacks. The SD‐OCT formulation differs slightly from the final formulation of ubrogepant used in phase III clinical trials.36, 37 The final ubrogepant formulation is a hot‐melt extrusion tablet, which exhibited faster absorption (ka increased by approximately eightfold) and higher bioavailability (F1 increased by 14%) and, thus, modestly higher C2hr than the SD‐OCT formulation.38 In trial A, C2hr following 10 days of dosing with the SD‐OCT formulation ranged from 77.51 to 110 nM, well above the human effective concentration 90% target of 23 nM estimated using the CIDV model of CGRP target engagement shown to predict clinical efficacy with previously studied CGRP antagonists (E. Moore, M.E. Fraley, I.M. Bell, C.S. Burgey, R.B. White, C.‐C. Li et al., unpublished data).39, 40 The 2‐hour time point is an important point of reference, given that pain freedom at 2 hours is a recommended efficacy measure in clinical trials for treatment of migraine attacks.
The upper range of the doses evaluated in trial A resulted in peak concentrations after 10 days of daily dosing that were more than fourfold greater (Cmax = 1,340 nM with ubrogepant 400 mg) than observed with the highest phase III clinical dose (100 mg; Cmax = 302 nM). The 150 mg dose evaluated in trial B resulted in maximum plasma drug concentrations that were 54% higher on the 28th day of daily dosing (Cmax = 465 nM) than those observed with the 100 mg dose after 10 days of daily dosing in trial A. Thus, both trials evaluated the safety profile of doses of ubrogepant that resulted in plasma concentrations that were substantially greater than therapeutically relevant levels (i.e., supratherapeutic). Assessing safety outcomes with supratherapeutic doses is recommended for the evaluation of the cardiovascular safety of new drugs41 and is an important strategy for characterizing hepatic safety in early drug development.
Ubrogepant demonstrated significant efficacy in the acute treatment of migraine attacks in two phase III clinical trials, ACHIEVE I and ACHIEVE II. In both trials, participants were administered one dose of trial medication (placebo or ubrogepant 25, 50, or 100 mg) to treat a single migraine attack of moderate/severe pain intensity, with an optional second dose of study medication after 2 hours.36, 37 Thus, the dosage and administration frequency of ubrogepant in the current trials were higher than in the phase III trials or what would be expected in real‐world use. All cases of ALT or AST elevation ≥ 3 × ULN in the ACHIEVE trials were evaluated by a panel of liver experts blinded to treatment allocation. Among the total safety population of the two ACHIEVE trials (N = 2,901), there were six cases of ALT or AST elevation ≥ 3 × ULN in ACHIEVE I and four cases in ACHIEVE II. In ACHIEVE I, four of the six cases (all ubrogepant) were adjudicated as unlikely to be related, two of the six (one ubrogepant and one placebo) were adjudicated as possibly related, and no cases were adjudicated as probably related to study medication.36 In ACHIEVE II, three of the four cases (all ubrogepant) were adjudicated as unlikely to be related, one of four cases (placebo) was adjudicated as possibly related, and no cases were adjudicated as probably related to study medication.37
The long‐term safety of the 50 and 100 mg doses of ubrogepant was also studied in a 52‐week trial.42 In this trial, ALT or AST ≥ 3 × ULN were found in 1.3% (5/399) and 2.7% (11/406) of participants receiving ubrogepant 50 and 100 mg, respectively. In this trial, three cases were considered possibly or probably related to ubrogepant; however, these three ubrogepant participants with ALT or AST elevations ≥ 3 × ULN had possible confounding factors, including alcohol use, urinary tract infection, and psoriasis flare, that may have caused the abnormalities. All three cases were asymptomatic, with no concurrent bilirubin elevation, and resolved in participants who continued treatment. The findings from this trial suggested little to no sign of ubrogepant‐related hepatotoxicity with long‐term ubrogepant use.
In a dedicated hepatic safety trial of ubrogepant, 518 healthy adults were randomized to placebo or ubrogepant 100 mg administered via high‐frequency intermediate dosing (2 days of ubrogepant followed by 2 days of placebo).43 A total of seven cases of ALT/AST ≥ 3 × ULN were reported during the 8 weeks of treatment, with five cases occurring in placebo participants and two cases reported in the ubrogepant group. Within the ubrogepant group, one case was judged possibly related to trial medication and the other case was judged probably related to trial medication. Both cases were asymptomatic and resolved with continued dosing.
Limitations of the current trials include a relatively small sample size and the inclusion of healthy men only. However, results from recently completed clinical trials in men and women with migraine have demonstrated a hepatic safety profile consistent with the results reported in the current study, suggesting these results are generalizable to the larger migraine population. Trial strengths include the use of varying doses (40–400 mg in trial A; 150 mg in trial B) and duration of treatment (single dose, 10 or 28 days of once‐daily treatment). These regimens involve dosing and an administration frequency well above those expected in either clinical trials or real‐world clinical practice. The absence of hepatotoxicity in these trials, along with the lack of serious hepatic enzyme elevations in larger randomized clinical trials, strongly supports the absence of ubrogepant‐associated hepatotoxicity.
In conclusion, the PK and safety results from these two studies, particularly the hepatic safety profile, support the continued development of ubrogepant for treatment of migraine. In addition to its PK profile, which is suitable for the acute treatment of migraine attacks, single and multiple daily ubrogepant doses up to 400 mg were safe and well‐tolerated. There was no evidence of clinically meaningful drug‐related ALT elevations in healthy men who received daily dosing of ubrogepant up to 400 mg over 10 days or 150 mg over 28 days, supporting its lack of hepatotoxicity. More generally, although these studies were relatively small and short, results from subsequent larger and longer safety studies support the use of short‐duration, supratherapeutic dosing as an appropriate derisking assessment for hepatic safety in early drug development.
Funding
These studies were sponsored by Merck & Co, Inc., Kenilworth, New Jersey. Writing and editorial assistance was provided to the authors by Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, USA, and was funded by Allergan plc. The opinions expressed in this paper are those of the authors. The authors received no honorarium/fee or other form of financial support related to the development of this paper.
Conflict of Interest
W.A., P.B., J.P., W.L., M.F.D., C.M., and D.P. are employees of Merck Sharp & Dohme, a subsidiary of Merck Kenilworth, NJ, USA, and owns/holds stock/stock options of Merck, Kenilworth, NJ, USA. E.E.M., C.‐C.L., and J.A.W. worked for Merck at the time of the study and owned Merck stock. T.R. is an employee of MSD Belgium, with stock in the company. A.J. is an employee and shareholder of Allergan plc. As Editor‐in‐Chief for Clinical and Translational Science, J.A.W. was not involved in the review or decision process for this paper. S.M. has no conflicts to report. W.K.K. has no conflicts to report.
Author Contributions
All authors wrote the manuscript. J.P., D.P., J.A.W., E.E.M., C.C.L., and T.R. designed the research. W.K.K. and S.M. performed the research. J.P., D.P., E.E.M., C.C.L., and W.A. analyzed the data.
Supporting information
Figure S1. Comparison of mean AUC0–24hr and Cmax of ubrogepant vs. dose following administration of multiple oral doses in healthy male participants. AUC0–24hr, area under the curve from 0 through 24 hours; Cmax, maximum plasma concentration.
Table S1. Summary of percentage change in treatment difference and posterior probabilities.
Table S2. Adverse events reported by ≥ 1 participant in trial A (SD‐OCT formulation).
Table S3. Adverse events reported by ≥ 1 participant in trial B.
Data Availability Statement
Data reported in this paper are available within the article and its Supplementary Material s. Allergan may share de‐identified patient‐level data and/or study‐level data, including protocols and clinical study reports, for phase I trials completed after 2008 that are registered on http://ClinicalTrials.gov or EudraCT. The indication studied in the trial must have regulatory approval in the United States and/or the European Union and the primary manuscript from the trial must be published prior to data sharing. To request access to the data, the researcher must sign a data use agreement. All shared data are to be used for noncommercial purposes only. More information can be found on http://www.allerganclinicaltrials.com/.
References
- 1. Headache Classification Committee of the International Headache Society . The international classification of headache disorders. Cephalalgia 38, 1–211 (2018). [DOI] [PubMed] [Google Scholar]
- 2. Chowdhury, D. Acute management of migraine. J. Assoc. Physicians India 58 (suppl.), 21–25 (2010). [PubMed] [Google Scholar]
- 3. Pietrobon, D. Migraine: new molecular mechanisms. Neuroscientist 11, 373–386 (2005). [DOI] [PubMed] [Google Scholar]
- 4. Vos, T. et al Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2163–2196 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lipton, R.B. , Bigal, M.E. , Diamond, M. , Freitag, F. , Reed, M.L. & Stewart, W.F. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology 68, 343–349 (2007). [DOI] [PubMed] [Google Scholar]
- 6. Buse, D.C. et al Life with migraine, effect on relationships, career and finances from the chronic migraine epidemiology and outcomes (CAMEO) study. Headache 59, 1286–1299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. World Health Organization . Headache disorders. World Health Organization, 2016. <http://www.who.int/en/news-room/fact-sheets/detail/headache-disorders>. Accessed July 18, 2018. [Google Scholar]
- 8. Buse, D.C. et al Chronic migraine prevalence, disability, and sociodemographic factors: results from the American Migraine Prevalence and Prevention study. Headache 52, 1456–1470 (2012). [DOI] [PubMed] [Google Scholar]
- 9. Buse, D.C. , Pearlman, S.H. , Reed, M.L. , Serrano, D. , Ng‐Mak, D.S. & Lipton, R.B. Opioid use and dependence among persons with migraine: results of the AMPP study. Headache 52, 18–36 (2012). [DOI] [PubMed] [Google Scholar]
- 10. Alwhaibi, M. , Deb, A. & Sambamoorthi, U. Triptans use for migraine headache among nonelderly adults with cardiovascular risk. Pain Res. Treat. 2016, 8538101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schwedt, T.J. et al Factors associated with acute medication overuse in people with migraine: results from the 2017 migraine in America symptoms and treatment (MAST) study. J. Headache Pain 19, 38 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goadsby, P.J. , Lipton, R.B. & Ferrari, M.D. Migraine–current understanding and treatment. N. Engl. J. Med. 346, 257–270 (2002). [DOI] [PubMed] [Google Scholar]
- 13. GBD 2016 Disease and Injury Incidence and Prevalence Collaborators . Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1211–1259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosenfeld, M.G. et al Production of a novel neuropeptide encoded by the calcitonin gene via tissue‐specific RNA processing. Nature 304, 129–135 (1983). [DOI] [PubMed] [Google Scholar]
- 15. O'Connor, T.P. & van der Kooy, D. Enrichment of a vasoactive neuropeptide (calcitonin gene related peptide) in the trigeminal sensory projection to the intracranial arteries. J. Neurosci. 8, 2468–2476 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iyengar, S. , Ossipov, M.H. & Johnson, K.W. The role of calcitonin gene‐related peptide in peripheral and central pain mechanisms including migraine. Pain 158, 543–559 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goadsby, P.J. , Edvinsson, L. & Ekman, R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann. Neurol. 28, 183–187 (1990). [DOI] [PubMed] [Google Scholar]
- 18. Goadsby, P.J. , Edvinsson, L. & Ekman, R. Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann. Neurol. 23, 193–196 (1988). [DOI] [PubMed] [Google Scholar]
- 19. Ashina, M. , Bendtsen, L. , Jensen, R. , Schifter, S. & Olesen, J. Evidence for increased plasma levels of calcitonin gene‐related peptide in migraine outside of attacks. Pain 86, 133–138 (2000). [DOI] [PubMed] [Google Scholar]
- 20. Cernuda‐Morollon, E. , Larrosa, D. , Ramon, C. , Vega, J. , Martinez‐Camblor, P. & Pascual, J. Interictal increase of CGRP levels in peripheral blood as a biomarker for chronic migraine. Neurology 81, 1191–1196 (2013). [DOI] [PubMed] [Google Scholar]
- 21. Lassen, L.H. , Haderslev, P.A. , Jacobsen, V.B. , Iversen, H.K. , Sperling, B. & Olesen, J. CGRP may play a causative role in migraine. Cephalalgia 22, 54–61 (2002). [DOI] [PubMed] [Google Scholar]
- 22. Hansen, J.M. , Hauge, A.W. , Olesen, J. & Ashina, M. Calcitonin gene‐related peptide triggers migraine‐like attacks in patients with migraine with aura. Cephalalgia 30, 1179–1186 (2010). [DOI] [PubMed] [Google Scholar]
- 23. Asghar, M.S. et al Evidence for a vascular factor in migraine. Ann. Neurol. 69, 635–645 (2011). [DOI] [PubMed] [Google Scholar]
- 24. Olesen, J. et al Calcitonin gene‐related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N. Engl. J. Med. 350, 1104–1110 (2004). [DOI] [PubMed] [Google Scholar]
- 25. Luo, G. et al Discovery of BMS‐846372, a potent and orally active human CGRP receptor antagonist for the treatment of migraine. ACS Med. Chem. Lett. 3, 337–341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deen, M. et al Blocking CGRP in migraine patients – a review of pros and cons. J. Headache Pain 18, 96 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ho, T.W. et al Randomized controlled trial of an oral CGRP receptor antagonist, MK‐0974, in acute treatment of migraine. Neurology 70, 1304–1312 (2008). [DOI] [PubMed] [Google Scholar]
- 28. Ho, T.W. et al Efficacy and tolerability of MK‐0974 (telcagepant), a new oral antagonist of calcitonin gene‐related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo‐controlled, parallel‐treatment trial. Lancet 372, 2115–2123 (2008). [DOI] [PubMed] [Google Scholar]
- 29. Connor, K.M. et al Randomized, controlled trial of telcagepant for the acute treatment of migraine. Neurology 73, 970–977 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ho, T.W. et al Randomized controlled trial of the CGRP receptor antagonist telcagepant for prevention of headache in women with perimenstrual migraine. Cephalalgia 36, 148–161 (2016). [DOI] [PubMed] [Google Scholar]
- 31. Ho, T.W. et al Randomized controlled trial of the CGRP receptor antagonist telcagepant for migraine prevention. Neurology 83, 958–966 (2014). [DOI] [PubMed] [Google Scholar]
- 32. Hewitt, D.J. et al Randomized controlled trial of the CGRP receptor antagonist MK‐3207 in the acute treatment of migraine. Cephalalgia 31, 712–722 (2011). [DOI] [PubMed] [Google Scholar]
- 33. Merck updates status of clinical development programs for investigational CGRP receptor antagonist treatments for acute migraine; MK‐3207 clinical development discontinued [press release]. BusinessWire, 2009. <https://www.businesswire.com/news/home/20090910005709/en/Merck-Updates-Status-Clinical-Development-Programs-Investigational>. Accessed December 10, 2018.
- 34. Walker, C.S. et al Mice lacking the neuropeptide alpha‐calcitonin gene‐related peptide are protected against diet‐induced obesity. Endocrinology 151, 4257–4269 (2010). [DOI] [PubMed] [Google Scholar]
- 35. Hargreaves, R. & Olesen, J. Calcitonin gene‐related reptide modulators – the history and renaissance of a new migraine drug class. Headache 59, 951–970 (2019). [DOI] [PubMed] [Google Scholar]
- 36. Dodick, D.W. et al Ubrogepant for the treatment of migraine. N Engl J Med 381, 2230–2241 (2019). [DOI] [PubMed] [Google Scholar]
- 37. Lipton, R.B. et al Effect of ubrogepant vs placebo on pain and the most bothersome associated symptom in the acute treatment of migraine: the ACHIEVE II randomized clinical trial. JAMA 322, 1887–1898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li, C.C. et al Population PK analyses of ubrogepant (MK‐1602), a CGRP receptor antagonist: enriching in‐clinic plasma PK sampling with outpatient dried blood spot sampling. J. Clin. Pharmacol. 58, 294–303 (2018). [DOI] [PubMed] [Google Scholar]
- 39. Li, C.C. et al Characterizing the PK/PD relationship for inhibition of capsaicin‐induced dermal vasodilatation by MK‐3207, an oral calcitonin gene related peptide receptor antagonist. Br. J. Clin. Pharmacol. 79, 831–837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Salvatore, C.A. et al Pharmacological characterization of MK‐0974 [N‐[(3R,6S)‐6‐(2,3‐difluorophenyl)‐2‐oxo‐1‐(2,2,2‐trifluoroethyl)azepan‐3‐yl]‐4‐(2‐oxo‐2,3‐dihydro‐1H‐imidazo[4,5‐b]pyridin‐1‐yl)piperidine‐1‐carboxamide], a potent and orally active calcitonin gene‐related peptide receptor antagonist for the treatment of migraine. J. Pharmacol. Exp. Ther. 324, 416–421 (2008). [DOI] [PubMed] [Google Scholar]
- 41. Guidance for industry E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs. U.S. Department of Health and Human Services. Food and Drug Administration, 2005. <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073153.pdf>. Accessed October 26, 2018. [Google Scholar]
- 42. Ailani, J. et al Long‐term safety evaluation of ubrogepant for the acute treatment of migraine: phase 3, randomized, 52-week extension trial. Headache. in press 60, 141–152 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goadsby, P.J. et al Safety and tolerability of ubrogepant following intermittent, high‐frequency dosing: randomized, placebo‐controlled trial in healthy adults. Cephalalgia 39, 1753–1761 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Comparison of mean AUC0–24hr and Cmax of ubrogepant vs. dose following administration of multiple oral doses in healthy male participants. AUC0–24hr, area under the curve from 0 through 24 hours; Cmax, maximum plasma concentration.
Table S1. Summary of percentage change in treatment difference and posterior probabilities.
Table S2. Adverse events reported by ≥ 1 participant in trial A (SD‐OCT formulation).
Table S3. Adverse events reported by ≥ 1 participant in trial B.
Data Availability Statement
Data reported in this paper are available within the article and its Supplementary Material s. Allergan may share de‐identified patient‐level data and/or study‐level data, including protocols and clinical study reports, for phase I trials completed after 2008 that are registered on http://ClinicalTrials.gov or EudraCT. The indication studied in the trial must have regulatory approval in the United States and/or the European Union and the primary manuscript from the trial must be published prior to data sharing. To request access to the data, the researcher must sign a data use agreement. All shared data are to be used for noncommercial purposes only. More information can be found on http://www.allerganclinicaltrials.com/.