

Abstract
Developing a novel drug, including discovery, nonclinical toxicology studies, initial clinical trials, and thorough pivotal studies, may take many years. Once an applicant has generated this comprehensive body of data, the final step prior to regulatory approval is Health Authority review of the marketing authorization application. Review by regulatory authorities to evaluate whether the data support a positive benefit/risk profile takes many months, adding additional time before patients may access therapy. In this paper, we discuss the various opportunities the US Food and Drug Administration and the European Medicines Agency offer to expedite the drug development and regulatory approval timelines for drugs intended to treat serious diseases and unmet medical needs.
In our previous tutorial, we discussed the data required to initiate first‐in‐human clinical trials. Once clinical trials have been initiated, generating the breadth and depth of data required to appropriately assess the benefit/risk of a new drug takes years of effort across multiple disciplines. In this tutorial, we discuss a range of programs implemented by global Health Authorities to expedite both drug development and Health Authority review of marketing applications.
In the United States, the data package submitted to the US Food and Drug Administration (FDA) to support a marketing approval is called either a New Drug Application (NDA) for small‐molecule drugs, or a Biologics License Application (BLA) for large‐molecule drugs (biologics).1 In the European Union, the data package submitted to the European Medicines Agency (EMA) to support a marketing approval is called a Marketing Authorisation Application (MAA). Nonclinical data supporting the pharmacology and toxicology of the drug, data on the drug's chemistry, manufacturing, and controls (CMC), and clinical safety and efficacy data from phase I through phase III programs are synthesized into one cohesive application describing the safety and efficacy profile of the drug in a given patient population. Once these data are generated, Health Authorities require time to evaluate whether the data provided support a marketing approval.
For a new drug, the FDA commits to reviewing most NDAs/BLAs within a total of 12 months. Once a drug is initially approved to treat a specific population or indication, applicants may conduct additional clinical studies to support subsequent FDA approvals in other settings (e.g., in another line of therapy), in combination with other treatments, or in other diseases. For a subsequent marketing application for additional use of an approved drug, the appropriate nonclinical and CMC data may have already been reviewed by the Agency in the initial application; as a result, supplemental marketing applications typically contain less data. Accordingly, the FDA aims to review supplemental applications within a total of 10 months.
Under the centralized procedure, the EMA commits to reviewing both initial and subsequent applications for new indications, known as type II variations, within 210 days, which refers to the number of active review days at the EMA; this review clock stops while the applicant is generating responses to the EMA questions, so the actual review time may be much longer.
In the drug development world, for patients suffering from serious diseases and unmet medical needs waiting anxiously for new therapy options, the process of investigational therapy development and Health Authority application review time can feel exceptionally long. Global Health Authorities, including the FDA2 and EMA,3 have developed multiple mechanisms to expedite both the drug development process and marketing application review timelines for promising drugs intended to treat serious disease and unmet medical needs (see Figure 1).
Figure 1.
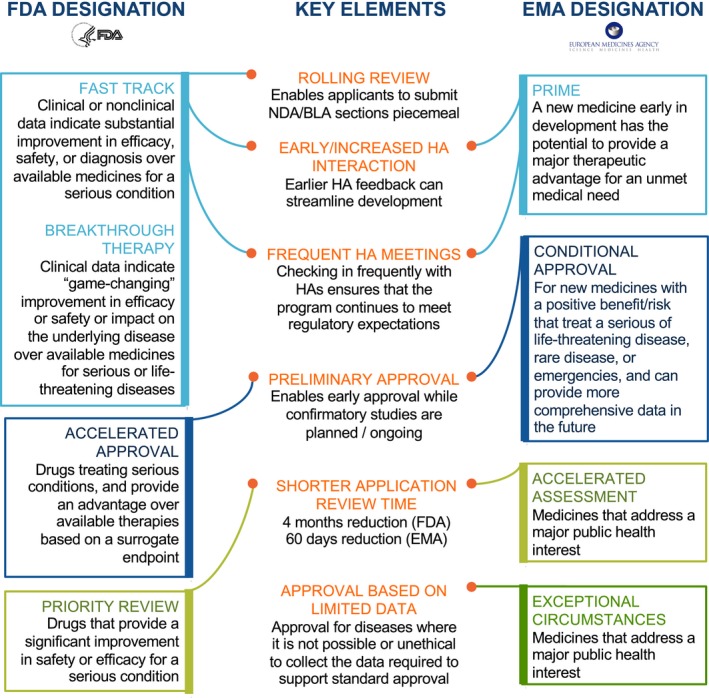
US Food and Drug Administration (FDA) and European Medicines Agency (EMA) expedited programs. Note: Drugs may qualify for more than one expedited program. For US programs, drugs may be eligible for all of these programs, provided they meet the criteria. For EU programs, medicines may be eligible for most of these programs, if criteria are met. The only exception is that drugs pursuing approval under exceptional circumstances are not eligible for conditional approval.1, 2, 3 HA, Health Authority; PRIME, PRIority MEdicines.
Decreasing drug development timelines
Health authorities offer programs that enable more detailed feedback and closer collaboration with the agency, taking some of the guesswork out of submitting a marketing application. Fast Track Designation (United States), Breakthrough Therapy Designation (BTD; United States), and PRIority MEdicines (PRIME) Designation (European Union) are three such programs.
Fast track (FDA)
The FDA’s Fast Track program was initially introduced in 1997 as part of the Food and Drug Administration Modernization Act (FDAMA), and later amended in the Food and Drug Administration Safety and Innovation Act of 2012 (FDASIA). Fast Track is designed to facilitate and expedite the development of drugs to treat serious conditions and fill an unmet medical need.2
Determining whether a condition is serious is a matter of judgment, but is generally based on whether the drug will have an impact on such factors as survival, day‐to‐day functioning, or the likelihood that the condition, if left untreated, will progress to a more serious one. AIDS, Alzheimer’s disease, heart failure, and cancer are obvious examples of serious conditions. Epilepsy, depression, and diabetes are also considered to be serious conditions.
Filling an unmet medical need is defined as providing a therapy where none exists or providing a therapy that may be potentially better than available therapy.
Any drug being developed to treat or prevent a condition with no current therapy is clearly directed at an unmet need. However, in cases in which available therapies exist, a drug must demonstrate an advantage over existing therapies to be eligible for Fast Track designation, such as:
Superior efficacy or effect/improved effect on serious outcomes.
Superior safety or avoiding serious side effects of an existing therapy.
Improved diagnosis of a serious condition, where early diagnosis may result in an improved outcome.
Decreasing a clinically significant toxicity of an available therapy that is common and causes discontinuation of treatment.
Ability to address emerging or anticipated public health need.
Unlike BTD, Fast Track requests may use nonclinical data as evidence to demonstrate the above.
A drug that receives Fast Track designation is eligible for some or all of the following4:
More frequent meetings with the FDA to discuss the drug’s development plan and ensure collection of appropriate data needed to support drug approval.
More frequent written communication from the FDA about issues such as the design of the proposed clinical trials and use of biomarkers.
Rolling review, which means that a drug company can submit completed sections of its BLA/NDA for review by the FDA, rather than waiting until all sections are completed before the entire application can be reviewed.
Fast Track designation requests are usually submitted to the Investigational New Drug (IND), and can be initiated at any time during the drug development process. The FDA will review the request and make a decision within 60 days of the request. All submissions to an IND remain confidential; the FDA does not disclose Fast Track submissions or decisions,5 unless the submission has been publicly disclosed or acknowledged by the applicant.
Once a drug receives Fast Track designation, early and frequent communication between the FDA and applicant is encouraged throughout the entire drug development and review process.
BTD (FDA)
BTD, initially introduced in the FDASIA, is an expedited pathway to facilitate drug development in the United States. An investigational drug can qualify for BTD “…if the drug is intended, alone or in combination with 1 or more other drugs, to treat a serious or life‐threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on 1 or more clinically significant end points, such as substantial treatment effects observed early in clinical development.”2 Unlike with Fast Track, investigational drugs will need preliminary clinical evidence to obtain BTD. To qualify for BTD, the drug should be intended to treat a serious condition and should demonstrate the potential for substantial improvement over existing therapies. It is important to note that the BTD designation is also available for new indications for already approved drugs.
The following are examples of clinical evidence that could support BTD6:
Direct comparison of the investigational drug to available therapy demonstrates a substantial benefit on a clinical end point.
If no existing therapy exists, comparison of the investigational drug to placebo/historical control shows in a substantial effect on a clinically meaningful end point.
The investigational drug in combination with available therapy demonstrates a much greater clinical response than available therapy.
The investigational drug has a substantial effect on the underlying cause of disease in instances where available therapy is perceived to be a symptomatic treatment.
The investigational drug reverses or inhibits disease progression in instances where available therapy only provides symptomatic benefits.
The investigational drug has a better safety profile than available therapy with a similar efficacy profile.
The FDA can rescind BTD later in development if the drug no longer meets the above criteria. For example, the FDA rescinded BTD for Tonix Pharmaceuticals’ Tonmya (cyclobenzaprine HCl) and Trevana’s oliceridine due to the lack of appropriate clinical or safety data needed to support continuation of the designation.7, 8, 9
Benefits of obtaining a BTD include increased interaction and guidance from the FDA during drug development and review. Specifically, senior FDA managers are involved in discussions and reviews, along with an assigned cross‐disciplinary project lead, to provide thorough guidance to ensure efficient drug development. In addition, the applicant can submit parts of the marketing application for a drug granted BTD on a rolling basis, potentially expediting time to approval.5
The applicant can submit the request for BTD, which must include appropriate supportive preliminary clinical evidence, at the time of the IND submission or any time before marketing approval, ideally before the End‐of‐Phase 2 meeting; however, the FDA has made it clear since the initiation of the BTD program that it expects to see potentially “game changing” clinical data to support a BTD application. The FDA response is expected within 60 calendar days of receipt of the request. As with Fast Track designation, the FDA does not publicly disclose any information about BTD requests or status.5
PRIME (EMA)
PRIME is the EMA’s version of an expedited drug development pathway, launched in 2016. To qualify for PRIME designation, the application has to meet the following eligibility criteria:
The investigational drug targets conditions where there is an unmet medical need, and
The investigational drug illustrates the potential to address the unmet medical need.
Critical to obtaining PRIME is the ability to demonstrate that the drug will bring a major therapeutic advantage to patients through improved efficacy or improved morbidity or mortality of the disease. It is important to note that PRIME authorization will only be considered for drugs under development that have not been previously authorized for an indication in the European Union. The Committee for Medicinal Products for Human Use (CHMP) can withdraw PRIME designation in cases that no longer meet the eligibility criteria.10
Some of the key benefits for applicants granted access to the PRIME scheme include the following:
Early appointment of a rapporteur, or lead reviewer, from the EMA’s CHMP or the Committee for Advanced Therapies (CAT) to guide the applicant. In a standard MAA submission, rapporteurs are assigned a few months prior to MAA submission.11
An opportunity to meet with the rapporteur and a team of experts to discuss the development plan and to seek any other advice on the application.
Overall intensive guidance, including scientific advice from the EMA and necessary stakeholders at major development milestones on key issues.
The EMA recommends that the applicant apply for PRIME while in the exploratory clinical trial phase of development and the applicant is able to demonstrate proof of concept that the investigational drug works. Special support is provided to micro‐size, small‐size, and medium‐size enterprises and academic research, and these groups may be eligible for PRIME designation at an earlier stage of development based on strong nonclinical data. Those new drugs that are granted access to the PRIME scheme are included in a cumulative listed updated monthly and available on the EMA website.12
The CHMP’s decision to grant or not grant PRIME designation is expected within 40 days of the start of the assessment of the PRIME application.
Summary
Although timelines and data requirements to support Fast Track, BTD, and PRIME designations are different, all offer additional support from Health Authorities during the development of drugs expected to treat a serious unmet medical need. Once pivotal studies are completed, additional Health Authority support is also available for review of marketing applications, as outlined below.
Decreasing application review time
Review times of marketing applications in the United States and the European Union can be reduced via two pathways: Priority Review in the United States, and Accelerated Assessment in the European Union.
Priority review (FDA)
In the United States, the standard review time for an NDA or BLA is 12 months for an initial application and 10 months for a supplemental application (both timelines include a 60‐day filing review period). Priority Review, initially introduced as part of the Prescription Drug User Fee Act of 1992, is a mechanism for decreasing the review time to 8 and 6 months, respectively. Between 2014 and 2016, 64 of 108 novel drug approvals were approved under priority review.13, 14, 15 To qualify for Priority Review, the application must be for a drug that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. Per the FDA Guidance, “significant improvement” may include:
Evidence of increased effectiveness in treatment, prevention, or diagnosis of a condition.
Elimination or substantial reduction of a treatment‐limiting adverse reaction.
Documented enhancement of patient compliance that is expected to lead to an improvement in serious outcomes.
Evidence of safety and effectiveness in a new subpopulation.
If a marketed product is already approved for the same indication, data to support a significant improvement should come from a clinical trial to demonstrate superiority in either safety or effectiveness. However, significant improvement can also be demonstrated by treating patients who cannot tolerate or do not respond to the approved therapy, normally demonstrated by a randomized trial, although, in some cases, historical controls may be acceptable.
After receiving a request for priority review in an NDA or BLA application, the FDA will inform the applicant of its decision to grant or deny the request within 14 days of the initial 60‐day review of the application. In addition to the standard pathway for obtaining this designation, other mechanisms allow for priority review. These include:
Any supplement that proposes a labeling change pursuant to a report on a pediatric study under section 505A of the Food, Drug, and Cosmetic Act (FD&C) Act (i.e., in response to a request from the FDA under the Best Pharmaceuticals for Children Act).
An application for a drug that has been designated as a qualified infectious disease product.
Any application or supplement for a drug submitted with a priority review voucher. Priority review vouchers are obtained at a previous BLA or NDA approval for a drug whose indication has been designated as a rare pediatric disease. The voucher may be used for any subsequent initial or supplemental NDA or BLA review to obtain priority review. Vouchers are also transferrable between companies and, thus, can be purchased from a different Sponsor.
Priority review does not guarantee approval. Additionally, if major issues arise during review of the application, the timelines may be extended to allow the FDA time to review new information requested of the applicant.
Accelerated assessment (EMA)
In the European Union, accelerated assessment is a similar pathway to reduce the review time of an MAA. Accelerated assessment was first introduced in 2004,16 as Article 14(9) of Regulation (EC) No 726/2004.17 A standard procedure consists of a 210‐day review time, with time added for clock stops. The accelerated procedure reduces this to a 150‐day review time, with time added for clock stops, although there are fewer with accelerated assessment.3 This accelerated review timeline may revert to a standard 210‐day procedure, if major issues arise that cannot be resolved within the accelerated timelines.3
To qualify for accelerated assessment, the product must represent a major public health interest, in particular, those incorporating therapeutic innovation. There is no definition of “major public health interest”; this must be justified by the applicant. The justification should include a description of the unmet medical need and the available methods of prevention, diagnosis, or treatment, including the effect of available therapies and how the unmet medical need is not fulfilled by them; the extent to which the medicinal product is expected to fulfill the unmet medical need; and the strength of evidence to support justifying major public health interest.
The request for accelerated assessment should be submitted 2–3 months prior to the MAA submission; however, the EMA should be informed as early as possible of the intent to request this assessment. A letter of intent is submitted 6–7 months prior to the MAA submission, and the option is discussed during presubmission meetings.
Summary
Accelerated assessment in the European Union takes more upfront planning, discussion, and logistical considerations than priority review in the United States, but the end result is the same—a faster review time. The amount of data required for initial approval, however, is the same as for standard review, although options do exist to enable limited approval on limited data sets. These are described below.
Preliminary approval pending additional data
Accelerated Approval (United States) and Conditional Approval (European Union) are options for applicants to provide preliminary data to Health Authorities in order to support a more limited approval. These pathways are generally reserved for diseases that despite having a significant impact on morbidity and/or mortality are sorely lacking in adequate treatment options, and may be proposed by either the Sponsor or the Health Authority. Additional data are then required to convert these approvals to “full approval.” If additional data do not in fact confirm the earlier promise of benefit, approval may be withdrawn.
Accelerated approval (FDA)
To qualify for accelerated approval, the drug must treat a serious condition and generally provide a meaningful advantage over available therapies and demonstrate an effect on a surrogate end point that is reasonably likely to predict clinical benefit or on an intermediate clinical end point that can be measured earlier than irreversible morbidity or mortality that is reasonably likely to predict clinical benefit. It is important to note that drugs granted accelerated approval must meet the same statutory standards for safety and effectiveness as those granted traditional approval.1 The accelerated approval pathway was introduced in 1992, largely in recognition of the need to bring experimental therapies to patients with AIDS desperate for treatment options. This pathway has subsequently been utilized across therapeutic areas, predominantly oncology.18, 19 As of June 2019, the FDA has granted 198 accelerated approvals.
For drugs granted accelerated approval, postmarketing confirmatory trials have been required to verify and describe the anticipated clinical benefit. These trials must be conducted with due diligence and promptly to facilitate determination of whether clinical benefit has been verified. The confirmatory trial population would ordinarily be the same disease population that was studied to support accelerated approval. In some cases, it is possible to use a later effect in the same trial to verify and describe clinical benefit and the effect seen earlier in the trial that supported accelerated approval. Sometimes, the commercial availability of a drug following accelerated approval may make it difficult to enroll patients in the same disease population. In these cases, a confirmatory trial may be conducted in a different but related population that is capable of verifying the predicted clinical benefit. This is often the case in oncology, where accelerated approval of a drug is granted for late‐stage disease, and the confirmatory trial is conducted in an earlier stage of the same cancer.
The applicant should ordinarily discuss the possibility of accelerated approval with the review division during development, including the use of the planned end point that is reasonably likely to predict the intended benefit of the drug as a basis for approval. The applicant should also discuss the confirmatory trial(s), and there should be agreement between the FDA and the applicant on the design and conduct to support confirmation of clinical benefit. Typically, the confirmatory trial(s) should already be underway at the time of the application for accelerated approval.
The FDA may withdraw approval of a drug or indication approved under the accelerated approval pathway if, for example:
A trial required to verify the predicted clinical benefit of the product fails to verify such benefit.
Other evidence demonstrates that the product is not shown to be safe or effective under the conditions of use.
The applicant fails to conduct any required post‐approval trial of the drug with due diligence.
The applicant disseminates false or misleading promotional materials relating to the product.
If the FDA determines there are grounds for withdrawal, the Agency may ask the applicant to request withdrawal of approval or notify the applicant of the FDA’s proposal to withdraw approval in a notice of opportunity for a hearing (NOOH). Upon receipt of an NOOH, the applicant has 15 days to file a written request for a hearing, otherwise this opportunity is waived. To date, there has been only one hearing to discuss withdrawing an accelerated approval (see Box 1: Case Study). An applicant may also request the Agency to withdraw approval.
Box 1. Case study – bevacizumab (Avastin) metastatic breast cancer indication20 .
August 23, 2007: Supplemental BLA submission of E2100 results intended to support an indication for bevacizumab use in combination with taxane‐based chemotherapy for first‐line treatment of metastatic breast cancer (MBC).
December 5, 2007: Oncologic Drugs Advisory Committee (ODAC) voted 5 to 4 against recommending approval of bevacizumab for treatment of MBC.
February 22, 2008: Against ODAC’s recommendation, the FDA granted accelerated approval for first‐line treatment of MBC based on an improvement in progression‐free survival (PFS) but no increase in overall survival.21, 22
November 16, 2009: Supplemental BLA submission of AVADO and RIBBON1 results intended to confirm/convert the accelerated approval to regular approval.
July 20, 2010: ODAC voted 12 to 1 in favor of removing the MBC indication from the Avastin label, as the two studies did not demonstrate a difference in overall survival, showed smaller improvement in PFS than E2100, and showed an increased risk of serious adverse events.23
December 16, 2010: The FDA proposed to withdraw approval and issued an NOOH.24
January 16, 2011: Genentech rejected the FDA’s proposal to withdraw the indication, requested a hearing, and submitted materials for the FDA and ODAC to review in support of retaining the approval.25
June 28–29, 2011: Public hearing – ODAC voted 6 to 0 in favor of removing the indication.26, 27
November 18, 2011: The FDA Commissioner issued final decision withdrawing approval.28
December 20, 2011: The FDA approved label revisions that removed the MBC indication and information relating to the MBC studies.29
If a drug is granted accelerated approval based on a surrogate end point, the Indications and Usage section of the label must include a “succinct description of the limitations of usefulness of the drug and any uncertainty about anticipated clinical benefits, with reference to the ‘Clinical Studies’ section for a discussion of the available evidence.”30, 31 This section generally also acknowledges that continued approval for the drug (or indication) may be contingent upon verification and description of clinical benefit in confirmatory trial(s). The indication and other sections of the label should be revised, as appropriate, following successful postmarketing studies to reflect the new data, population, condition, clinical studies, etc. If the accelerated approval is withdrawn, the label must be revised to remove information on the withdrawn indication (if the drug remains approved for other indications). In some cases, it may be appropriate to add language to the label by means of a limitation of use and/or safety information on the withdrawn indication.
Conditional approval (EMA)
The EMA also offers a pathway enabling approval based on a more limited data set for new medicines that have the potential to address an unmet medical need.32 Conditional Approval (as introduced in 2004 in Regulation (EC) No 726/200417 and further defined in Commission Regulation [EC] No 507/2006)33 is available only for seriously debilitating or life‐threatening diseases, for drugs that may be used in emergency situations (e.g., pandemic flu vaccine), or for rare diseases.
To qualify for Conditional Approval, the applicant must demonstrate all of the following:
The drug has a positive risk/benefit (even in the context of the uncertainties surrounding a more limited clinical data set),
The applicant will likely be able to provide more comprehensive data in the future,
The medicine fulfills an unmet medical need, and
The benefit of providing the drug to patients outweighs the potential risks of the uncertainty inherent in a more limited clinical data package.
The CHMP encourages applicants to seek Scientific Advice to discuss a potential Conditional Approval strategy, and either the applicant or the EMA may suggest pursuing Conditional Approval. Conditional Approval is granted based on less clinical data than would normally support approval. Although conditional approval is predicated on the applicant providing a more limited clinical data package, the CHMP generally expects a comprehensive nonclinical and CMC data package to support the application, except possibly in the assessment of a product to be used in an emergency situation.
A key aspect of Conditional Approval is that the applicant commits to “Specific Obligations,” which are mandatory postmarketing requirements with specified timelines to ensure that the applicant does eventually provide a comprehensive data set to support “full approval.” The additional data could include safety and efficacy from longer duration of treatment or follow‐up, assessment of additional clinical end points, or results on a more meaningful clinical end point, if an intermediate end point that may translate to clinical benefit was used to support the initial conditional approval. In contrast to the Accelerated Approval pathway in the United States, Conditional Approval is not necessarily predicated on a surrogate end point. If the applicant fails to meet these Obligations, Conditional Approval can be withdrawn.34
Conditional Approval is only valid for 1 year at a time, until such time as the Specific Obligations are fulfilled and approval is converted to a standard marketing authorization. Each year, the applicant submits documentation requesting renewal of the Conditional Approval. In a 60‐day procedure, the CHMP assesses whether the benefit/risk remains positive, and reviews the progress on the Specific Obligations. With justification, modification to the timelines for the Specific Obligations may be discussed. Conditional Approval renewals are required annually until the Specific Obligations are met, and the Conditional Approval is converted into a standard marketing authorization not subject to Specific Obligations. According to a report published by the EMA detailing Conditional Approvals from 2006–2016, Conditional Approvals are converted, on average, within 4 years.35 To date, the EMA has only withdrawn one approval: olaratumab (Lartruvo), for the treatment of soft tissue sarcoma, which failed to provide confirmatory evidence of efficacy, and the EMA recommended that the drug be withdrawn from the market (see Box 2: Case Study).
Box 2. Case study – olaratumab (Lartruvo) soft tissue sarcoma indication.
June 8, 2015: Lilly disclosed that the FDA granted olaratumab Breakthrough Therapy Designation. (Note: olaratumab was also granted Fast Track Designation.)49
May 4, 2016: The FDA granted the olaratumab Biologics License Application for the treatment of soft tissue sarcoma (STS) Priority Review.50
July 22, 2016: The EMA disclosed that the olaratumab STS Marketing Authorisation Application is being reviewed under accelerated assessment.51
October 19, 2016: The FDA granted accelerated approval for olaratumab, in combination with doxorubicin, for the treatment of a subset of adult patients with STS based on results from Trial JGDG. Trial JGDG was an open‐label, single arm, US‐only study, which met its primary end point based of PFS. Trial JGDG also demonstrated an improvement in overall survival, although this was not the primary end point. The FDA granted accelerated approval based on the heterogeneous enrolled population (with > 25 different histologic subtypes) and small sample size.52
November 23, 2016: The EMA granted conditional marketing approval for olaratumab in combination with doxorubicin for the treatment of a subset of adult patients with advanced STS.53
January 18, 2019: Eli Lilly released clinical trial results from ANNOUNCE, the randomized double‐blind multinational confirmatory study intended to convert olaratumab to full approval. ANNOUNCE enrolled 509 patients, and did not meet its primary end points of an improvement in overall survival for either the overall STS population or the leiomyosarcoma subpopulation.54, 55, 56
January 24, 2019: The FDA recommended that patients currently on olaratumab consult their physician, and no new patients initiate olaratumab treatment outside a clinical trial.57
February 1, 2019: The EMA’s Committee for Health and Medicinal Products referred olaratumab for review under Article 20 of Regulation (EC) No 726/2004.58
April 25, 2019: Eli Lilly announced plans to withdraw olaratumab from the market.59
April 26, 2019: The EMA recommended withdrawal of olaratumab from the market.60
July 19, 2019: The European Commission adopted the EMA recommendation to withdraw olaratumab and issues a final legally binding decision applicable in all EU Member States, the first time a conditional approval has been withdrawn.61
The Summary of Product Characteristics (Pharmacodynamic Properties) and package leaflet will state that a conditional marketing authorization (CMA) has been granted and further evidence is awaited as the product is subject to certain specific obligations.32, 36 The EMA will review new information annually, and the labeling may be updated as necessary.
Summary
Under specific circumstances, both the US and EU regulatory pathways allow applicants to submit limited data to support a drug approval, with the expectation that complete data will be subsequently provided to support a regular approval. However, the European Union does provide one pathway for approval of drugs without what is usually considered “complete” data: Exceptional Circumstances.
Approval based on a limited data set
The EMA also offers an Exceptional Circumstances approval pathway for those applications where it would not be possible or ethical to collect the standard level of evidence typically required to support approval.
Exceptional circumstances (EMA)
In some cases, specific circumstances preclude obtaining the usual data required for marketing approval (e.g., in the case of some rare diseases). In these cases, approval in the European Union may still be granted under Exceptional Circumstances.37 Exceptional circumstances was introduced in 2004 in Article 14 (8) of Regulation (EC) No 726/2004.17, 38
Exceptional Circumstances designation is a type of marketing authorization in the European Union that is granted on the basis of one of the following three main criteria:
The applicant is unable to gather and provide comprehensive clinical evidence due to the rarity of disease occurrence, or
Comprehensive scientific knowledge is unavailable at the time of application submission, or
It is considered unethical to collect the necessary information for a standard approval.
Once approval under Exceptional Circumstances has been granted, the applicant is required to introduce specific procedures/obligations, usually concerning safety, as part of an annual reassessment to maintain approval.17 Proposals for specific obligations should include a plan for safety procedures and outlines of studies. Along with the proposals, the applicant submits information on prescription of the drug or conditions of its use indicating whether the product may be supplied on medical prescription only or administered only under strict medical supervision.
The applicant is responsible for submitting the following items at the time of the marketing application submission:
A claim that the applicant can show that it is unable to provide comprehensive nonclinical or clinical data on the efficacy and safety of the drug under normal conditions of use,
A list of the nonclinical or clinical efficacy or safety data that cannot be comprehensively provided,
Justification on the grounds for approval under exceptional circumstances, and
Proposals for detailed information on the specific procedures/obligations to be conducted postapproval (safety procedures, list of studies to be performed, prescription or administration conditions, product information, etc.).
Approval under Exceptional Circumstances is a type of approval unique to the European Union, and a drug cannot be approved under both Exceptional Circumstances and Conditional Approval. However, accelerated assessment can be requested for products requesting approval under Exceptional Circumstances. An approval of a drug under Exceptional Circumstances is not normally converted to a standard marketing authorization, unlike the expectation for Conditional Approvals.
Similar to Conditional Approval, the summary of product characteristics and package leaflet will state that an exceptional circumstances marketing authorization has been granted and the information on the product is as yet incomplete in certain specified respects, and that the EMA (or another Agency) will review new information submitted with the annual reassessment, and the label may be updated as necessary.32, 36
Discussion
Health Authorities carry the significant responsibility of evaluating the totality of data generated throughout a drug’s development to determine whether the drug is likely to be reasonably safe and effective in the indicated population. Recognizing that patients with serious diseases are willing to trade some certainty regarding clinical benefit for the opportunity to access potentially transformative therapies as soon as possible, many Health Authorities have developed programs to facilitate the development of new drugs, as well as expedite the review of marketing applications.18, 19, 35, 39, 40 How well have these programs accomplished their goal of expediting drug development and marketing application review timelines?
Programs to accelerate drug development: FastTrack, BTD, and PRIME
Between fiscal years 2012 and 2018, a total of 1,052 requests for Fast Track designation were submitted to the FDA’s Center for Drug Evaluation and Research (CDER), which reviews the majority of NDAs. A total of 737 (70%) of these Fast Track applications were granted, 258 (25%) were denied, and 57 (5%) were unable to be granted (e.g., the IND was withdrawn).41 Since the inception of BTD in 2012 through September 2018, CDER received a total of 636 BTD requests.42 Of these 636 requests, 250 (39%) designations were granted, 304 (48%) were denied, and 82 (13%) were withdrawn.
A recent review showed that between January 2012 and December 2016, the clinical development time (from IND application to FDA approval) for drugs under at least one FDA expedited program (i.e., Accelerated Approval, Priority Review, Fast Track, or BTD) was 0.9 years shorter than those that were not under any expedited program.43 Although development timelines for drugs that received Fast Track designation were reduced by ~ 1 year (7.0 years (interquartile range (IQR), 5.2–9.5) vs. 8.0 years (IQR, 6.2–10.3)), drugs that received BTD demonstrated the greatest advantage, reducing development times from 8.0 years (IQR, 6.2–10.3) to 4.8 years (IQR, 3.6–7.7).43
These data suggest that BTD has a higher bar for approval than Fast Track designation. This is also reflected in the different eligibility requirements: whereas both programs are available for drugs that treat serious or life‐threatening condition and unmet needs, BTD requires at least preliminary clinical evidence, but a Fast Track designation may be granted on the basis of nonclinical or clinical data. In addition, drugs that qualify for BTD proceed through drug development more quickly than Fast Track drugs, likely due to their “game‐changing” potential to treat serious or life‐threatening diseases.
As the EMA program PRIME has been in effect only since March 2016, there are limited data on this program in expediting drug development timelines. In the first 2 years of PRIME, the EMA granted 36 of the 177 applications, predominantly in oncology and hematology.39 As of September 2019, MAAs have been granted for three drugs with PRIME designation, seven are under review, and five have been withdrawn at the request of the applicant due to discontinuation of the drug development in the PRIME indication.12 Time will tell whether PRIME reduces drug development timelines without significantly increasing the risk that patients with a serious disease are exposed to a drug that ultimately does not have a positive benefit/risk.
Programs to shorten marketing application review times: Priority review and accelerated assessment
In the United States, Priority Review reduces the targeted marketing application review timeline by 4 months. Of the 28 applications submitted in fiscal year 2018 that were granted priority review, the average review time was 7.3 months.44 In contrast, of the nine marketing applications submitted in fiscal year 2018 that were granted standard review, the average review time was 10 months. These data support that Priority Review effectively reduces the timelines for US marketing application reviews. However, data on US drug approvals between January 2012 and December 2016 showed Priority Review is not linked to faster overall development times.43
In the European Union, ~ 80 requests for accelerated assessment were received between 2012 and 2016, over one‐half of which were accepted.45 As described above, the accelerated procedure reduces the standard 210‐day review time to a 150‐day review time, with time added for clock stops. However, if an applicant is unable to respond adequately to questions during the shortened clock stop periods or there are major objections that are not resolvable under accelerated assessment, the assessment reverts to the standard timeframe. Between 2012 and 2016, approximately one half of the programs with accelerated approval were reverted to standard timelines.3, 45 Thus, although accelerated assessment can effectively reduce EU marketing application timelines, the onus is on the applicant to rapidly and thoroughly respond to questions to retain the accelerated assessment timelines.
Preliminary approval pending additional data: Accelerated approval and conditional approval
As of June 2019, the FDA had granted 198 Accelerated Approvals for new drugs or new indications of an approved drug.18 Of these, 114 (57.5%) have converted to full approval, and 15 (7.5%) applications/indications were withdrawn. The remaining 69 (35%) approvals have not yet converted. In an analysis of 93 accelerated approvals granted in the oncology setting over a 25‐year period (1992–2017), the FDA found that 51 (55%) of these were converted to full approval, in a median of 3.4 years. Only 5% of the drugs described in this review were withdrawn from the market. However, 37 (40%) have not yet provided confirmatory evidence of clinical benefit.46 As described above, Accelerated Approval is predicated on a surrogate end point “reasonably likely” to predict clinical benefit; as a result, there is less certainty of the clinical benefit of these drugs at the time of approval.47 Approximately one‐third of drugs that have been approved under the Accelerated Approval program are still awaiting confirmatory data; as a result, patients are taking drugs that may ultimately not demonstrate a clinically meaningful benefit. However, the majority of these drugs have continued on to demonstrate clinical benefit, supporting the conclusion that overall, Accelerated Approval effectively provides therapeutic options with an appropriate benefit/risk to patients with serious unmet medical needs years before they would have otherwise been available.
In the European Union, as Conditional Approval is only available for new molecular entities,32 there are fewer approvals under this paradigm than under the Accelerated Approval pathway in the United States. Of the 30 CMAs granted in the first 10 years of the pathway’s availability (2006–2016), 11 (37%) remained CMAs, 2 (7%) were withdrawn for commercial reasons, and 14 (47%) converted to full marketing authorization. At the time this assessment was published, conversion was pending on three applications.48 Although the rate of conversion to full approval for CMAs is not as high as for Accelerated Approvals, the rate of withdrawals is similar. The EMA has only withdrawn a single conditional approval, for olaratumab for the treatment of soft tissue sarcoma (see Box 2).
Across these 30 CMAs, applicants committed to a total of 107 Specific Obligations, ~ 70% of which were submitted to the EMA by the agreed upon timelines. In addition, as previously noted, conditional approvals are valid for 1 year and can be renewed annually. Thus, although earlier approval under the conditional approvals pathway has demonstrated ability to bring medicines intended to treat an unmet medical need to patients sooner, there is substantial burden upon the applicant to renew the approval and meet Specific Obligations on the road to converting to a full marketing authorization.
Approval based on a limited data set: Exceptional circumstances
Finally, also in the European Union, the exceptional circumstances pathway is used infrequently for those indications in which it may not be possible to collect the same level of evidence as in standard indications. This pathway has enabled approval of seven drugs, largely in endocrinology, from 2016 to 2018.62, 63, 64
Conclusion
Here, we have described expedited drug development and marketing application review programs in the United States and the European Union, provided data to support how effective these programs are at reducing the overall timelines for drug development and/or marketing application reviews, and described some of the considerations in pursuing these approaches. Although not described herein, other Health Authorities (e.g., Japan65, 66 and Brazil67) also offer opportunities to expedite drug development and approval for drugs intended to treat rare or serious conditions and/or fill unmet medical needs.
Applicants may pursue more than one of these expedited pathways in parallel. In the United States, a drug that obtains either BTD or Fast Track designation may also be approved under Accelerated Approval if the approval is based on a surrogate end point reasonably likely to predict clinical benefit. Whether the approval pathway is accelerated or standard, marketing applications for BTD or Fast Track drugs may be reviewed on a standard or a priority timeline, depending on whether they meet priority criteria. Similarly, in the European Union, a drug accepted into the PRIME scheme may also be eligible for accelerated assessment, and may be approved under conditional or exceptional circumstances. As the criteria for these designations are largely overlapping in both the United States and the European Union, many drugs are granted more than one of these designations.
It is important to note that these expedited pathways are not intended to lower the bar for approval; instead, they enable Health Authorities to partner with applicants to generate the most relevant data and allow Health Authorities to prioritize review of applications for drugs that may be transformative for patients with serious conditions. That said, drugs approved via an expedited pathway may have less data available at the time of initial approval than a drug pursuing a more standard development program. As these programs are intended to provide access to therapy for serious and life‐threatening diseases that lack adequate therapeutic options, expedited programs trade some level of increased confidence in the drug’s benefit/risk profile in favor of earlier patient access, predicated on robust enough data to justify the trade‐off.47
Finally, a drug development program under an expedited pathway does not guarantee drug approval. As noted above, some companies whose drugs have been granted one or more of these designations ultimately withdraw their marketing application or discontinue drug development.
Overall, for patients with serious or life‐threatening conditions that lack effective therapies, these pathways have proven useful in supporting applicants in expediting both development of promising new drugs, and the Health Authority review and approval process.
Funding
No funding was received for this work.
Conflict of Interest
The authors declared no competing interests for this work.
Erica M. Cox, Anita V. Edmund, Erica Kratz, Sarah H. Lockwood, and Aishwarya Shankar contributed equally to this work.
References
- 1. US Food and Drug Administration (FDA) . Code of Federal Regulations Title 21 (21 CFR). Part 601, Subpart A–General Provisions and Subpart E–Accelerated approval of biological products for serious or life‐threatening illnesses and Part 314, Subpart B–Applications and Subpart H–Accelerated approval of new drugs for serious or life‐threatening illnesses. <https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm> (2018). Accessed June 25, 2019.
- 2. US Food and Drug Administration (FDA) . Guidance for industry. Expedited programs for serious conditions – drugs and biologics. <https://www.fda.gov/files/drugs/published/Expedited-Programs-for-Serious-Conditions-Drugs-and-Biologics.pdf> (2014). Accessed June 25, 2019.
- 3. European Medicines Agency (EMA) . Guideline on the scientific application and the practical arrangements necessary to implement the procedure for accelerated assessment pursuant to Article 14(9) of Regulation (EC) No 726/2004. <https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-scientific-application-practical-arrangements-necessary-implement-procedure-accelerated/2004_en.pdf> (2016). Accessed June 25, 2019.
- 4. US Food and Drug Administration (FDA) . Fast track. <https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/fast-track> (2018). Accessed September 30, 2019.
- 5. US Food and Drug Administration (FDA) . Frequently asked questions: breakthrough therapies. <https://www.fda.gov/regulatory-information/food-and-drug-administration-safety-and-innovation-act-fdasia/frequently-asked-questions-breakthrough-therapies> (2019). Accessed September 30, 2019.
- 6. US Food and Drug Administration (FDA) . Breakthrough therapy. <https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/breakthrough-therapy> (2018). Accessed September 30, 2019.
- 7. US Food and Drug Administration (FDA) . CDER Breakthrough Therapy Designation withdrawn after granting (WAG) and rescinded. <https://www.fda.gov/regulatory-information/food-and-drug-administration-safety-and-innovation-act-fdasia/cder-breakthrough-therapy-designation-withdrawn-after-granting-wag-and-rescinded> (2019). Accessed July 15, 2019.
- 8. Regulatory Affairs Professionals Society (RAPS) . FDA rescinds two Breakthrough Therapy Designations. <https://www.raps.org/news-and-articles/news-articles/2019/3/fda-rescinds-two-breakthrough-therapy-designations> (2019). Accessed July 15, 2019.
- 9. Global News Wire . Tonix Pharmaceuticals announces that breakthrough therapy designation remains in effect for Tonmya® for the treatment of posttraumatic stress disorder. <https://www.globenewswire.com/news-release/2019/04/22/1807231/0/en/Tonix-Pharmaceuticals-Announces-that-Breakthrough-Therapy-Designation-Remains-in-Effect-for-Tonmya-for-the-Treatment-of-Posttraumatic-Stress-Disorder.html> (2019). Accessed July 15, 2019.
- 10. European Medicines Agency (EMA) . European Medicines Agency guidance for applicants seeking access to PRIME scheme. <https://www.ema.europa.eu/en/documents/other/european-medicines-agency-guidance-applicants-seeking-access-prime-scheme_en.pdf> (2018). Accessed September 30, 2019.
- 11. European Medicines Agency (EMA) . Procedural Advice on CHMP/CAT/PRAC Rapporteur/CoRapporteur appointment principles, objective criteria and methodology in accordance with Article 62 (1) of Regulation (EC) No 726/2004. <https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/procedural-advice-chmp/cat/prac-rapporteur/co-rapporteur-appointment-principles-objective-criteria-methodology-accordance-article-62-1/2004_en.pdf> (2016). Accessed June 25, 2019.
- 12. European Medicines Agency (EMA) . PRIME: priority medicines. <https://www.ema.europa.eu/en/human-regulatory/research-development/prime-priority-medicines> (2019). Accessed October 4, 2019.
- 13. US Food and Drug Administration (FDA) . 2014 Novel drugs summary report (charts). <https://wayback.archive-it.org/7993/20170112023851/http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm429873.htm> (2016). Accessed October 4, 2019.
- 14. US Food and Drug Administration (FDA) . Novel drugs summary 2015. <https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drugs-summary-2015> (2016). Accessed October 4, 2019.
- 15. US Food and Drug Administration (FDA) . 2016 Novel drugs summary. <https://www.fda.gov/media/102618/download> (2017). Accessed October 4, 2019.
- 16. European Medicines Agency (EMA) . Guideline on the procedure for accelerated assessment pursuant to Article 14 (9) of Regulation (EC) No 726/2004. <https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/guideline-procedure-accelerated-assessment-pursuant-article-149-regulation-ec-no-726/2004_en.pdf> (2006). Accessed October 4, 2019.
- 17. European Medicines Agency (EMA) . Commission Regulation (EC) No 726/2004 of the European parliament and of the council laying down community procedures for the authorisation and supervision of medicinal products for human and veterinary use. <https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2004_726/reg_2004_726_en.pdf> (2004). Accessed June 25, 2019.
- 18. US Food and Drug Administration . CDER drug and biologic accelerated approvals based on a surrogate endpoint as of June 30, 2019. <https://www.fda.gov/media/88907/download> (2019). Accessed October 4, 2019.
- 19. Johnson, J.R. , Ning, Y.M. , Farrell, A. , Justice, R. , Keegan, P. & Pazdur, R. Accelerated approval of oncology products: the Food and Drug Administration experience. J. Natl. Cancer Inst. 103, 636–644 (2011). [DOI] [PubMed] [Google Scholar]
- 20. Vitry, A. , Nguyen, T. , Entwistle, V. & Roughead, E. Regulatory withdrawal of medicines marketed with uncertain benefits: the bevacizumab case study. J. Pharma. Pol. Pract. 8, 25 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. US Food and Drug Administration (FDA) . Letter to applicant. <https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2008/125085s091ltr.pdf> (2008). Accessed July 15, 2019.
- 22. US Food and Drug Administration (FDA) . CDER. Approval package for Application BLA 125085/S‐091. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2008/125085_s091_avastin.pdf> (2008). Accessed July 15, 2019.
- 23. US Food and Drug Administration (FDA) . Summary minutes of the Oncologic Drugs Advisory Committee July 20, 2010. <http://wayback.archive-it.org/7993/20170113054917/http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM224753.pdf> (2010). Accessed June 13, 2019.
- 24. US Food and Drug Administration (FDA) . Proposal to withdraw approval for the breast cancer indication for bevacizumab; hearing. <https://www.regulations.gov/document?D=FDA-2010-N-0621-0143> (2011). Accessed June 13, 2019.
- 25. Genentech Inc .Press release: FDA grants Genentech a hearing on Avastin's use for metastatic breast cancer in the United States. <https://www.gene.com/media/press-releases/13267/2011-02-24/fda-grants-genentech-a-hearing-on-avastin> (2011). Accessed June 13, 2019.
- 26. US Food and Drug Administration (FDA) . Proposal to withdraw approval for the breast cancer indication for bevacizumab (Avastin) public hearing, June 28, 2011 – Transcript. <https://www.regulations.gov/document?D=FDA-2010-N-0621-0537> (2011b). Accessed June 13, 2019.
- 27. US Food and Drug Administration (FDA) . Proposal to withdraw approval for the breast cancer indication for bevacizumab (Avastin) public hearing, June 28, 2011 – Transcript. <https://www.regulations.gov/document?D=FDA-2010-N-0621-0538> (2011c). Accessed June 13, 2019.
- 28. US Food and Drug Administration (FDA) . Proposal to withdraw approval for the breast cancer indication for bevacizumab – commissioner's decision. Docket No. FDA–2010–N–0621. <https://www.fda.gov/media/82467/download> (2011d). Accessed June 13, 2019.
- 29. US Food and Drug Administration (FDA) . Letter to applicant. <https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2011/125085s0241ltr.pdf> (2011e). Accessed June 13, 2019.
- 30. US Food and Drug Administration (FDA) . CFR – Code of Federal Regulations Title 21. <https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=201.57> (2019). Accessed October 4, 2019.
- 31. US Food and Drug Administration (FDA) . Guidance for industry: labeling for human prescription drug and biological products approved under the accelerated approval regulatory pathway. <https://www.fda.gov/media/119755/download> (2019). Accessed October 4, 2019.
- 32. European Medicines Agency (EMA) . Guideline on the scientific application and the practical arrangements necessary to implement commission regulation (EC) No 507/2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of regulation (EC) No 726/2004. <https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-scientific-application-practical-arrangements-necessary-implement-commission/2006-conditional-marketing-authorisation-medicinal-products-human-use-f_en.pdf> (2006). Accessed October 4, 2019.
- 33. European Medicines Agency (EMA) . Commission Regulation (EC) No 507/2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of Regulation (EC) No 726/2004 of the European Parliament and of the Council. <https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2006_507/reg_2006_507_en.pdf> (2006). Accessed June 25, 2019.
- 34. European Medicines Agency (EMA) . Guideline on the scientific application and the practical arrangements necessary to implement Commission Regulation (EC) No 507/2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of Regulation (EC) No 726/2004. <https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐scientific‐application‐practical‐arrangements‐necessary‐implement‐commission‐regulation‐ec/2006‐conditional‐marketing‐authorisation‐medicinal‐products‐human‐use‐falling_en.pdf > (2016). Accessed June 25, 2019.
- 35. European Medicines Agency (EMA) . Conditional marketing authorization: Report on ten years of experience at the European Medicines Agency. <https://www.ema.europa.eu/en/documents/report/conditional-marketing-authorisation-report-ten-years-experience-european-medicines-agency_en.pdf> (2017). Accessed June 25, 2019.
- 36. European Commission Enterprise and Industry Directorate‐General . A guideline on summary of product characteristics (SmPC). <https://ec.europa.eu/health//sites/health/files/files/eudralex/vol-2/c/smpc_guideline_rev2_en.pdf> (2009). Accessed October 18, 2019.
- 37. European Medicines Agency (EMA) . Marketing authorisation. <https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation> Accessed June 25, 2019.
- 38. European Medicines Agency (EMA) . Guideline on procedures for the granting of a marketing authorisation under exceptional circumstances, pursuant to Article 14 (8) of Regulation (EC) No 726/2004. <https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/guideline-procedures-granting-marketing-authorisation-under-exceptional-circumstances-pursuant/2004_en.pdf> (2005). Accessed October 18, 2019.
- 39. European Medicines Agency (EMA) . PRIME: a two‐year overview. <https://www.ema.europa.eu/en/documents/report/prime-two-year-overview_en.pdf> (2018). Accessed October 18, 2019.
- 40. US Food and Drug Administration (FDA) . Fast track approvals. <https://www.fda.gov/drugs/nda-and-bla-approvals/fast-track-approvals> (2019). Accessed October 18, 2019.
- 41. US Food and Drug Administration (FDA) . CDER fast track designation requests received, fiscal year 1998 – fiscal year 2018. <https://www.fda.gov/media/97830/download> (2019). Accessed October 18, 2019.
- 42. US Food and Drug Administration (FDA) . CDER breakthrough therapy designation requests received by fiscal year. Cohort: July 9, 2012‐September 30, 2018. <https://www.fda.gov/media/95292/download> (2019). Accessed October 18, 2019.
- 43. Hwang, T.J. , Darrow, J.J. & Kesselheim, A.S. The FDA’s expedited programs and clinical development times for novel therapeutics, 2012–2016. JAMA 318, 2137–2138 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. US Food and Drug Administration (FDA) . FY 2018 performance report to Congress for the Prescription Drug User Fee Act. <https://www.fda.gov/media/130665/download> (2018). Accessed October 18, 2019.
- 45. European Medicines Agency (EMA) . Accelerated assessment (AA): review of 10 months experience with the new AA process. <https://www.ema.europa.eu/en/documents/presentation/presentation-accelerated-assessment-aa-review-10-months-experience-new-aa-process-victoria-palmi_en.pdf> (2017). Accessed October 18, 2019.
- 46. Beaver, J.A. et al A 25‐year experience of US Food and Drug Administration accelerated approval of malignant hematology and oncology drugs and biologics: a review. JAMA Oncol. 4, 849–856 (2018). [DOI] [PubMed] [Google Scholar]
- 47. Woodcock, J. Expediting drug development for serious illness: trade‐offs between patient access and certainty. Clin. Trials 15, 230–234 (2018). [DOI] [PubMed] [Google Scholar]
- 48. European Medicines Agency (EMA) . Conditional Marketing Authorisation. How early access to medicines has helped patients from 2006 to 2016. <https://www.ema.europa.eu/en/documents/other/conditional-marketing-authorisation-how-early-access-medicines-has-helped-patients-2006-2016_en.pdf> (2016). Accessed October 18, 2019.
- 49. Friends of Cancer Research . Breakthrough therapies. <https://www.focr.org/breakthrough-therapies?title=olara%26field_sponsor_value=> (2017). Accessed October 18, 2019.
- 50. Eli Lilly and Company . FDA grants priority review for Lilly’s olaratumab, an investigational medicine for advanced soft tissue sarcoma. <https://investor.lilly.com/news‐releases/news‐release‐details/fda‐grants‐priority‐review‐lillys‐olaratumab‐investigational > (2016). Accessed October 4, 2019.
- 51. European Medicines Agency (EMA) . Committee for Medicinal Products for Human Use (CHMP). Minutes for the meeting on 20–23 June 2016. <https://www.ema.europa.eu/en/documents/minutes/minutes-chmp-meeting-20-23-june-2016_en.pdf> (2016). Accessed October 18, 2019.
- 52. US Food and Drug Administration (FDA) . Center for Drug Evaluation and Research. Application Number: 761038Orig1s000. Multi‐disciplinary review. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761038Orig1s000MultiDisciplineR.pdf> (2016). Accessed October 18, 2019.
- 53. European Medicines Agency (EMA) . Lartruvo. <https://www.ema.europa.eu/en/medicines/human/EPAR/lartruvo> (2019). Accessed October 18, 2019.
- 54. Eli Lilly and Company . Lilly reports results of Phase 3 soft tissue sarcoma study of LARTRUVO®. <https://investor.lilly.com/news-releases/news-release-details/lilly-reports-results-phase-3-soft-tissue-sarcoma-study> (2019). Accessed October 18, 2019.
- 55. National Institutes of Health . A study of doxorubicin plus olaratumab (LY3012207) in participants with advanced or metastatic soft tissue sarcoma. <https://clinicaltrials.gov/ct2/show/NCT02451943> (2019). Accessed October 18, 2019.
- 56. European Medicines Agency (EMA) . Assessment report. Lartruvo. <https://www.ema.europa.eu/en/documents/referral/lartruvo-article-20-referral-chmp-assessment-report_en.pdf> (2019). Accessed October 18, 2019.
- 57. US Food and Drug Administration (FDA) . FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. <https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-new-treatment-advanced-soft-tissue-sarcoma> (2016). Accessed October 18, 2019.
- 58. European Medicines Agency (EMA) . Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 28–31 January 2019. <https://www.ema.europa.eu/en/news/meeting-highlights-committee-medicinal-products-human-use-chmp-28-31-january-2019> (2019). Accessed October 18, 2019.
- 59. Eli Lilly and Company . Lilly to establish an access program for patients as it prepares to withdraw Lartruvo from the global market. <https://investor.lilly.com/news-releases/news-release-details/lilly-establish-access-program-patients-it-prepares-withdraw> (2019). Accessed October 18, 2019.
- 60. European Medicines Agency (EMA) . EMA recommends withdrawal of marketing authorisation for cancer medicine Lartruvo. <https://www.ema.europa.eu/en/documents/referral/lartruvo-article-20-referral-ema-recommends-withdrawal-marketing-authorisation-cancer-medicine_en.pdf> (2019). Accessed October 18, 2019.
- 61. European Medicines Agency (EMA) . EMA recommends withdrawal of marketing authorisation for cancer medicine Lartruvo. <https://www.ema.europa.eu/en/documents/referral/lartruvo-article-20-referral-ema-recommends-withdrawal-marketing-authorisation-cancer-medicine_en-0.pdf> (2019). Accessed October 18, 2019.
- 62. European Medicines Agency (EMA) . Human medicines highlights 2016. <https://www.ema.europa.eu/en/documents/leaflet/human-medicines-highlights-2016_en.pdf> (2016). Accessed October 18, 2019.
- 63. European Medicines Agency (EMA) . Human medicines highlights 2017. <https://www.ema.europa.eu/en/documents/report/human-medicines-highlights-2017_en.pdf> (2017). Accessed October 18, 2019.
- 64. European Medicines Agency (EMA) . Human medicines highlights 2018. <https://www.ema.europa.eu/en/documents/report/human-medicines-highlights-2018_en.pdf> (2018). Accessed October 18, 2019.
- 65. Ministry of Health, Labour and Welfare (MHLW) . Strategy of SAKIGAKE. <http://www.mhlw.go.jp/english/policy/health-medical/pharmaceuticals/140729-01.html> (2014). Accessed June 25, 2019.
- 66. Nagai, S. Flexible and expedited regulatory review processes for innovative medicines and regenerative medical products in the US, the EU, and Japan. Int. J. Mol. Sci. 20, 3801 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fagundes, P. , Dresel, P. & Miler, E. Brazil’s regulatory environment offers positive changes for clinical trials. Regulatory Focus. Regulatory Affairs Professionals Society. <https://www.ppdi.com/-/media/Files/PPDI-Files/news/PPD-In-The-News/2018-June-Regulatory-Focus-Brazil-Fagundes-Dresel-Miller.ashx> (2018). Accessed June 25, 2019.