

Abstract
Hemophilia A (HA) is a bleeding disorder characterized by spontaneous and prolonged hemorrhage. The disease is caused by mutations in the coagulation factor 8 gene (F8) leading to factor VIII (FVIII) deficiency. Since FVIII is primarily produced in endothelial cells (ECs) in a non‐diseased human being, ECs hold great potential for development as a cell therapy for HA. We showed that HA patient‐specific induced pluripotent stem cells (HA‐iPSCs) could provide a renewable supply of ECs. The HA‐iPSC‐derived ECs were transduced with lentiviral vectors to stably express the functional B domain deleted F8 gene, the luciferase gene, and the enhanced green fluorescent protein gene (GFP). When transplanted intramuscularly into neonatal and adult immune deficient mice, the HA‐iPSC‐derived ECs were retained in the animals for at least 10‐16 weeks and maintained their expression of FVIII, GFP, and the endothelial marker CD31, as demonstrated by bioluminescence imaging and immunostaining, respectively. When transplanted into HA mice, these transduced HA‐iPSC‐derived ECs significantly reduced blood loss in a tail‐clip bleeding test and produced therapeutic plasma levels (11.2%‐369.2%) of FVIII. Thus, our studies provide proof‐of‐concept that HA‐iPSC‐derived ECs can serve as a factory to deliver FVIII for the treatment of HA not only in adults but also in newborns.
Keywords: cell therapy, endothelial cells, FVIII, gene therapy, hemophilia, induced pluripotent stem cells (iPSCs)
Schematic diagram of the experimental approach.
Significance statement.
The present study demonstrated that induced pluripotent stem cells (iPSCs) derived from hemophilia A (HA) patients can provide an ample supply of endothelial cells (ECs). The HA‐iPSC‐derived ECs can be genetically modified to produce functional factor VIII. The relatively stable engraftment of these ECs, in both neonatal and adult animals, and the functional correction or alleviation of hemophilia by these ECs in animal models, as shown in the study, provide the basis for potential therapeutic development of HA‐iPSC‐derived ECs for treating HA. The current study is a significant step forward in development of autologous gene‐modified cell therapy for HA.
1. INTRODUCTION
Hemophilia A (HA) is an X‐linked recessive bleeding disorder that can lead to disability and even death due to spontaneous or injury‐caused prolonged bleeding if untreated. The incidence of HA is 1 in 5000 male births. The cause of the disease is mutations in the F8 gene leading to deficiency of clotting factor VIII (FVIII). Current standard treatment for HA is factor replacement by repeated infusions of recombinant FVIII proteins. However, this treatment has multiple challenges. Infusion of the FVIII protein can treat but not cure the disease. The frequent (2‐3 times/week) and long‐term intravenous infusion of FVIII can be inconvenient and risky for infections. In addition, this treatment is extremely costly: the median cost of treatment is $98 334 a year and is a lifelong expense.1 Furthermore, bleeding episodes are still common even with factor replacement therapy due to the fluctuation of the infused FVIII levels. Currently, no alternative therapy for HA is clinically available.
Gene and cell therapies have great potential to treat HA because if these therapies can increase plasma FVIII levels only to above 1% to 5% of normal FVIII levels, spontaneous bleeding episodes can be markedly reduced. A recent gene therapy clinical trial for HA showed successfully that a single high dose of an adeno‐associated virus serotype 5 (AAV5) vector encoding a functional B‐domain‐deleted human F8 (bddF8) normalized the plasma FVIII levels in six of seven patients over a period of 1 year.2 However, about 28.8% to 35.6% of HA patients carry AAV neutralizing antibody due to previous exposure to AAV and thus are not suitable for AAV‐based gene therapy.3 Furthermore, new born babies and young children are not suitable for the AAV‐based gene therapy because AAV do not integrate into the host genome and thus AAV‐mediated gene expression is greatly diluted when new born babies and young children grow into adults. Repeated application of AAV therapy is prohibited because administration again could induce a deleterious immune response to the viral capsids.4, 5
Cell therapy is actively being studied as a potential alternative for treatment of HA. Multiple cell types have been tested for the delivery of FVIII in vivo with promising results. These include liver sinusoidal endothelial cells (LSECs),6, 7 blood outgrowth endothelial cells (BOECs),8, 9, 10 skin fibroblasts,11 mesenchymal stem cells,12, 13, 14, 15, 16 and hematopoietic stem cells.17, 18, 19, 20, 21, 22, 23 Moreover, cell‐based therapy could be administered repeatedly if needed without causing significant side effects. However, major challenges remain for the development of a successful HA cell therapy: (a) sufficient supply of the therapeutic cells and (b) sustained engraftment of the therapeutic cells.
Since FVIII is primarily expressed and posttranslationally modified in endothelial cells (ECs) in a non‐diseased human being,24, 25, 26 ECs could be preferable FVIII delivery vehicles to other cell types. LSECs and BOECs are excellent natural EC sources, but their limited availability hinders their clinical applications. Induced pluripotent stem cells (iPSCs) can be generated from any person and extensively expanded. A number of groups including us have efficiently derived ECs from human embryonic stem cells and iPSCs.27, 28, 29, 30 Therefore, iPSCs can produce an ample source of autologous ECs.
Here, we investigated whether the ECs derived from HA patients' iPSCs (HA‐iPSCs) can be used as FVIII delivery vehicles for the treatment of HA in animal models. In order to express FVIII in the HA‐iPSC‐derived ECs (HA‐iPSC‐ECs), we transduced these ECs with lentiviral vectors encoding a functional bddF8 gene. Since HA is a genetic disease, a child born with the disease needs to be treated early in his life. Therefore, we assessed the engraftment of the HA‐iPSC‐ECs at the neonatal stage in comparison to the adult stage, an analysis not previously studied. Finally, we assessed the functionality of the human HA‐iPSC‐ECs in attenuating hemophilia symptoms in mouse models of HA.
2. MATERIALS AND METHODS
2.1. Cell culture
Two independent HA‐iPSC lines, HA‐iPSC1 and HA‐iPSC2, derived from independent HA patients were previously reported by a co‐author, Dr. Pan's group.31, 32 The efficiency of reprogramming was from 0.0006% to 0.0024%.32 These HA‐iPSCs were maintained on Matrigel (Corning, Corning, New York) coated 6‐well plates in mTeSR1 medium (STEMCELL Technologies, Cambridge, Massachusetts) with daily change of the medium. Colonies were passaged every 4‐6 days either by manual picking with a sterile 1 mL pipette tip or ReLeSR (STEMCELL Technologies). The iPSC line derived from a healthy human being, iPS(IMR90)‐4,33 was purchased from WiCell Research Institute (Madison, Wisconsin) and was maintained as previously described.30 The karyotypes of the healthy iPSC line and the HA iPSC lines were confirmed normal. Human LSECs freshly isolated and cryopreserved were purchased from ScienCell Research Laboratories and were used at passage 1 (Carlsbad, California), whereas human coronary artery EC (HCAEC), human cardiac microvascular endothelial cell (HMVECs), and human umbilical vein EC (HUVEC) were purchased from Lonza (Walkersville, Maryland). These primary ECs were cultured in EC growth medium ECGM‐MV2 (Promocell, Heidelberg, Germany).
2.2. EC differentiation and transduction
ECs were differentiated from HA‐iPSCs as previously described by our laboratory.30 The cells on day 4 of differentiation were dissociated from the culture plates with Accutase (Innovative Cell Technologies Inc). These cells were transduced with lentiviral vector pMNDU3‐LUC‐PGK‐eGFP‐WPRE encoding luciferase (LUC) and enhanced green fluorescent protein (GFP) and pMNDU3‐F8‐PGK‐NEO‐WPRE34 encoding the B domain deleted F8 (bddF8) and neomycin resistance gene (NEO) at multiplicity of infection (MOI) range from 1:4 to 1:8 for each vector. The ECs were incubated in a small volume (0.5‐1 mL) of ECGM‐MV2 medium supplemented with 5 μg/mL polybrene (EMD Millipore, Burlington, Massachusetts) for 20 minutes and then plated onto p100 culture dishes in 6 mL of the same medium. Media was changed to ECGM‐MV2 plus additional 50 ng/mL of vascular endothelial growth factor (VEGF) the following day, and every 2 days after that. Cells were harvested at days 7‐9.
2.3. Flow cytometry
The transduced cells were dissociated from the culture plates with Accutase (Innovative Cell Technologies Inc) and washed with phosphate‐buffered saline (PBS). Following filtration through a 70‐μm nylon mesh (ThermoFisher Scientific, Waltham, Massachusetts), these cells were subjected to flow cytometry analysis using a Beckman Coulter Cytomics FC 500 (Beckman Coulter, Indianapolis, Indiana).
2.4. Animal models and cell transplantation
The Institutional Animal Care and Use Committee at the University of California, Davis, approved all animal experiments in the current study. Immune‐deficient NOD/SCID/IL2Rγ−/− (NSG) mice were purchased from the vivarium of University of California, Davis. The HA‐iPSC‐derived ECs that had been transduced to expressed bddF8, LUC, and EGFP (1 × 106cells/mouse) were suspended in 40 μL of ECGM‐MV2 medium and 10 μL of Matrigel and intramuscularly injected into the left hind limb of adult NSG mice at 8‐12 weeks old (mouse number n = 6). Neonatal NSG mice at 4‐7 days old were injected intramuscularly with the transduced ECs (3 × 105cells/mouse) derived from HA‐iPSC1 (mouse number n = 7) or HA‐iPSC2 (mouse number n = 6) in 20 μL of ECGM‐MV2 medium and 5 μL of Matrigel into their left hind limbs.
C57BL/6 mice and HA mouse line B6;129S‐F8tm1Kaz\J (B6F8) carrying a F8 null mutation were purchased from The Jackson Laboratory in Sacramento, California. These hemophilia B6F8 mice were immune‐competent. To repress their immune system, adult B6F8 mice at 8‐ to 16‐week‐old were mated and cyclosporine A was given to the dam and sir in drinking water at 210 mg/L from the time that mating pairs were set up to the pups were sacrificed. The transduced HA‐iPSC‐EC/F8 (2‐3 × 106cells/mouse) were transplanted into the neonatal HA mice at 10 days old (mouse number n = 5) as described above.
To generate an immune‐deficient HA mouse strain to facilitate human cell engraftment, we bred a female B6;129S‐F8 tm1Kaz/J mouse to a male immune‐deficient mouse, B6;129S‐RAG2tm1Fwa CD47tm1Fpl IL2rgtm1Wjl/J (BRG) (The Jackson Laboratory). The F1 progeny were bred back to BRG mice. The progeny was crossed to each other for three more generations. Both female and male mice with the RAG2 IL2rg F8 null (F8RG) were obtained. CD47 was either wild‐type (WT) or heterozygous in these mice. The transduced HA‐iPSC‐EC/F8 (1 × 107cells/mouse) in 300 μL of culture medium supplemented with 30% Matrigel were injected subcutaneously into the adult F8RG mice (mouse number n = 7) as described above.
2.5. Bioluminescence imaging
Luciferase substrate D‐luciferin (Gold Biotechnology, St. Louis, Missouri) was injected intraperitoneally or subcutaneously into the animals at 150‐200 mg/kg weight. Five minutes later, the mice were anesthetized using 2%‐3% isoflurane (Piramal Critical Care, Bethlehem, Pennsylvania) for 5 minutes and then imaged with the IVIS Spectrum (PerkinElmer, Richmond, California) at the Genomic and Molecular Imaging Center (Davis, California) under anesthesia up to 5 minutes. Animals were imaged at the day of cell transplantation and weekly thereafter. The bioluminescence signal was presented as total photons of the region of interest in 5 minutes of acquisition.
2.6. Immunostaining
The mouse limb muscles with bioluminescence signal were dissected and frozen in O.C.T compound (Sakura Finetek USA, Torrance, California) on dry ice and then placed into the −80°C freezer for a minimum of 24 hours. The tissue was then sectioned using a Cryotome FSE (Thermo Scientific, Waltham, Massachusetts) set to 10–15 μm. Immunostaining was performed as we previously described.35 The goat anti‐GFP antibodies or rabbit anti‐GFP antibodies (Novus Biologicals, Littleton, Colorado), rabbit anti‐human CD31 antibodies (Bethyl Laboratories, Montgomery, Texas), and sheep anti‐FVIII antibodies (Affinity Biologicals, Ancaster, Canada) were diluted in PBS with 1% bovine serum albumin (BSA) to 100, 200, and 50 folds, respectively. Secondary antibodies including donkey anti‐goat IgG conjugated with AlexaFluor 488 (ThermoFisher Scientific), donkey anti‐rabbit IgG conjugated with AlexaFluor 594 (ThermoFisher Scientific), and donkey anti‐sheep IgG conjugated with AlexaFluor 647 (EMD Millipore, Burlington, Massachusetts) were diluted to 500 folds in PBS with 1% BSA, respectively. Fluorescence images were captured using a Nikon Eclipse Ti‐U Inverted or Nikon C2 microscope (Nikon Instruments Inc, Melville, New York).
2.7. Quantitative PCR and RT‐PCR
Gene expression was measured by quantitative reverse transcription polymerase chain reaction (qRT‐PCR) as we previously described.30 The primers for VEGFR2, VE‐cadnerin, and NOS3 have been previously described.30 The primers for F8 are the following: 5′‐TCGAGCAATAATTGGAGGGT‐3′ and 5′‐GTGGCAGACTTATCGAGGAAA‐3′. The PCR products of human F8 from animal tissues were analyzed by electrophoresis in a 2.5% agarose gel. For each condition, four to six independent samples were assessed. The genomic DNA was isolated from the transduced cells as previously described.35 The viral copy number in the transduced cells was assessed by quantitative PCR as previously described.36 Standard curve for viral sequence WPRE and human VEGF was generated separately using a VEGF‐coding lentiviral vector as previously described.36 The copy number of WPRE and VEGF in a DNA sample was calculated according to the respective standard curve. The average copy number of viral inserts per cell was calculated by dividing the number of WPRE by the number of VEGF and then multiplying by 2, considering that each cell carries two copies of VEGF.
2.8. Tail clip assay for bleeding and blood collection
A tail‐clip assay was performed to determine the blood loss of the transplanted HA mice compared with non‐transplanted HA mice. Mice were anesthetized using 2%‐3% isoflurane. The mouse tail was clipped at 1.5 cm from the tip. Blood was allowed to flow freely for 5 minutes before applying pressure on the cutting site for 1 minute using a hemostat. Blood was collected into EDTA‐coated tubes for a total of 45 minutes after the cut and weighed. Animals were sacrificed, and tissue at the injection site of the hind limb was collected for RNA isolation. Blood was also collected directly from animal hearts into tubes with citric acid. Mouse plasma was obtained after centrifuging the blood for 5‐10 minutes at 3300 rcf.
2.9. Enzyme linked immunosorbent assay for FVIII
A FVIII antigen enzyme linked immunosorbent assay (ELISA) kit (Affinity Biologicals, Ancaster, Canada) was used to quantify the level of human FVIII in the culture media and mouse plasma according to the manufacturer's instructions with minor modifications. The capture antibody specific for human FVIII was first coated onto the wells of the provided microplate strips for 1 hour at room temperature or overnight at 4°C. Human pooled reference plasma standards, collected culture media, and diluted mouse plasma samples were pipetted into the coated wells and incubated for 1 hour at room temperature or overnight at 4°C. Wells were washed with the wash buffer, and a detection antibody was added and incubated for 1‐2 hours. Following wash with the wash buffer, a substrate was added and the color intensity was read at 450 nm with a SpectraMax i3x plate reader (Molecular Devices, San Jose, California). Unknown samples were compared with the standard curve and concentrations estimated by extrapolation.
2.10. Data representation and statistical analysis
Data is presented as average ± standard deviation. Each group had four to six samples. Student's t test or Mann‐Whitney U test was performed to detect statistical difference between two testing groups. One‐way analysis of variance was performed to detect statistical difference between more than two testing groups.
3. RESULTS
3.1. Differentiation of ECs from HA‐iPSCs
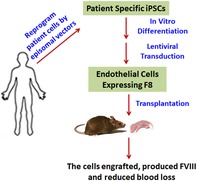
HA‐iPSC1 and HA‐iPSC2 were derived independently from different HA patients and were previously characterized.31, 32 We further verified the presence of pluripotency marker, octamer‐binding transcription factor 4, and the absence of EC marker, cluster of differentiation 31 (CD31), in these HA‐iPSC lines using immunostaining (Figure 1A and data not shown). We differentiated these two lines of HA‐iPSCs into ECs using our previously established protocol.30 Immunostaining of the differentiated cells showed that CD31 was expressed on their cell surfaces (Figure 1B). Quantitative RT‐PCR assessment of the ECs derived from HA‐iPSC1 (HA‐iPSC1‐EC and HA‐iPSC2 (HA‐iPSC2‐ECs) for their expression of endothelial markers revealed that the ECs derived from both HA‐iPSC lines expressed VEGFR2 at the levels comparable to the ECs derived from non‐diseased iPSCs (iPSC‐ECs) as well as primary liver sinusoidal ECs (LSECs) from a person not suffering from HA (Figure 1C). The ECs derived from both HA‐iPSC lines expressed VE‐cadherin and NOS3 at the level around 50% to 80% of those in the iPSC‐ECs, respectively (Figure 1C). The cause of the different expression level of VE‐cadherin and NOS3 in the ECs derived from different iPSC lines is not known. LSECs and HMVECs have been previously shown to produce FVIII,26, 37 whereas HCAEC and HUVEC have not been shown to produce a detectable level of FVIII. Next, we compared the level of the F8 mRNA in the ECs derived from non‐diseased iPSCs or HA‐iPSCs with that in different primary ECs. We found that the iPSC‐ECs did express F8 mRNA at a level slightly higher than that in HMVECs and markedly higher than that in HCAECs or HUVECs (Figure 1D). However, the level of F8 mRNA in LSECs was about 80‐fold higher than that in the iPSC‐ECs and even higher than all other primary ECs tested (Figure 1D). Our data further prove the previous observation that the level of FVIII expression varies markedly among ECs from different tissues, and LSECs have the highest capacity of FVIII production among these ECs. As expected, the ECs derived from both HA‐iPSC lines did not express F8 (Figure 1D).
Figure 1.
Characterization of HA‐iPSCs and the ECs derived from these HA‐iPSCs. A, Immunostaining image of HA‐iPSC1 and HA‐iPSC2 for OCT4 (red) and DAPI (blue). Scale bar = 50 μm. B, Phase (upper) and immunostaining image (lower) of HA‐iPSCs and ECs derived from two HA‐iPSC lines as indicated for CD31 (red) and DAPI (blue). Scale bar=50 µm. C and D, qRT‐PCR analysis of the expression of endothelial cell markers (C) and F8 (D) as indicated in the ECs derived from iPSC, HA‐iPSC1, HA‐iPSC2, and human primary ECs including LSECs, HCAEC, HMVEC, and HUVEC. **P < .01, ****P < .0001 vs iPSC‐EC. ECs, endothelial cells; HA, hemophilia A; HCAEC, human coronary artery EC; HMVEC, human cardiac microvascular endothelial cell; HUVEC, human umbilical vein EC; iPSC, induced pluripotent stem cell; LSECs, liver sinusoidal endothelial cells; qRT‐PCR, quantitative reverse transcription polymerase chain reaction
3.2. Ectopic expression of a functional B‐domain deleted F8 (bddF8), luciferase, and GFP in HA‐iPSC‐derived ECs
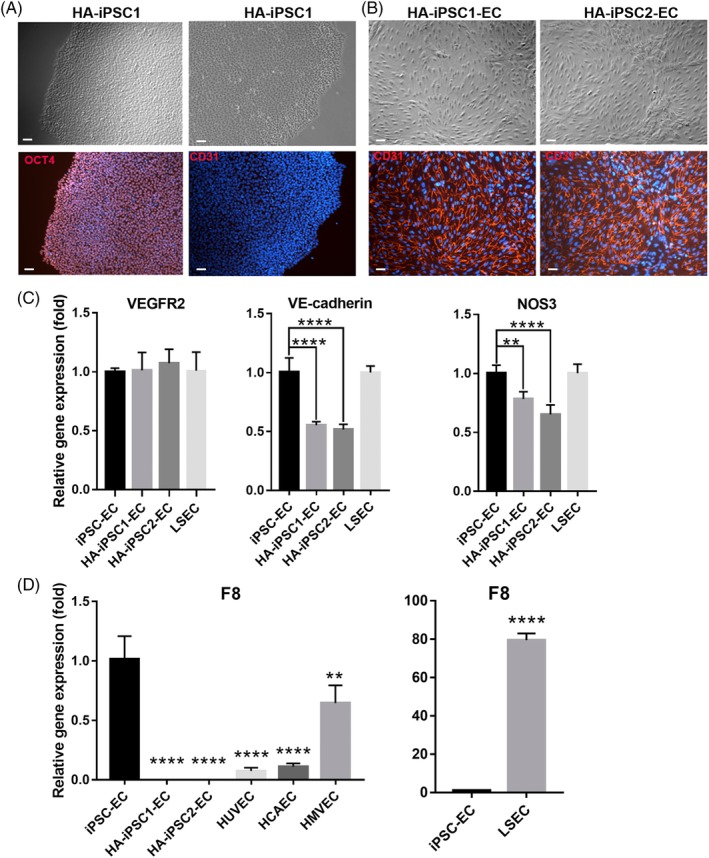
To express a functional F8 gene in the HA‐iPSC‐ECs and to facilitate quantitative assessment of the engraftment of these cells over time, we transduced HA‐iPSC‐ECs with a lentiviral vector pMNDU3‐F8‐PGK‐NEO‐WPRE encoding bddF8 and NEO and a lentiviral vector pMNDU3‐LUC‐PGK‐eGFP‐WPRE encoding the luciferase gene and enhanced GFP.34 The transduced cells expressed GFP as detected under a fluorescent microscope, and flow cytometry analysis of the transduced cells showed that 91.3% of the cells were GFP‐positive (Figure 2A, B). The average copy number of the viral integration per cell was 4.7 when the cells were transduced at MOI 8. The expression of F8 mRNA in these ECs was detected at the level of approximately threefold higher than that in LSECs by qRT‐PCR (Figure 2C). Furthermore, the transduced ECs were capable of secreting FVIII as detected by ELISA (Figure 2D). Apparently, the higher level of the F8 mRNA in HA‐iPSC‐ECs in comparison to those in LSECs did not result in a significant higher level of the secreted FVIII from HA‐iPSC‐ECs compared with that from LSECs. The reason for this apparent inconsistency could be due to differences in translation efficiency, protein stability, or efficiency in secretion of FVIII between the HA‐iPSC‐ECs and LSECs. Our data show that HA‐iPSC‐ECs can be transduced at a high efficiency to produce functional FVIII proteins at a level no less than that in LSECs. These transduced ECs are referred as HA‐iPSC‐ECs/F8 in the following sections.
Figure 2.
The transduced HA‐iPSC‐ECs expressed GFP and F8. HA‐iPSC‐ECs were transduced with the lentiviral vectors encoding bddF8, LUC and the enhance GFP. Two‐three days after the transduction, the cells were assessed by microscopy for GFP fluorescence (A) (scale bar = 50 μm), and cytometry for GFP‐positive cells (B). C, qRT‐PCR analysis of the expression of F8 in the transduced HA‐iPSC‐ECs/F8 and LSECs. *P < .05. D, ELISA analysis of FVIII level in the media collected from HA‐iPSC‐ECs or LSECs in 72 hours using pooled human reference plasma as standards. No statistically significant difference was detected. ECs, endothelial cells; ELISA, enzyme linked immunosorbent assay; FVIII, factor VIII; GFP, green fluorescent protein; HA, hemophilia A; iPSC, induced pluripotent stem cell; qRT‐PCR, quantitative reverse transcription polymerase chain reaction
3.3. Sustained engraftment of HA‐iPSC‐ECs/F8 in neonatal and adult immune‐deficient mice
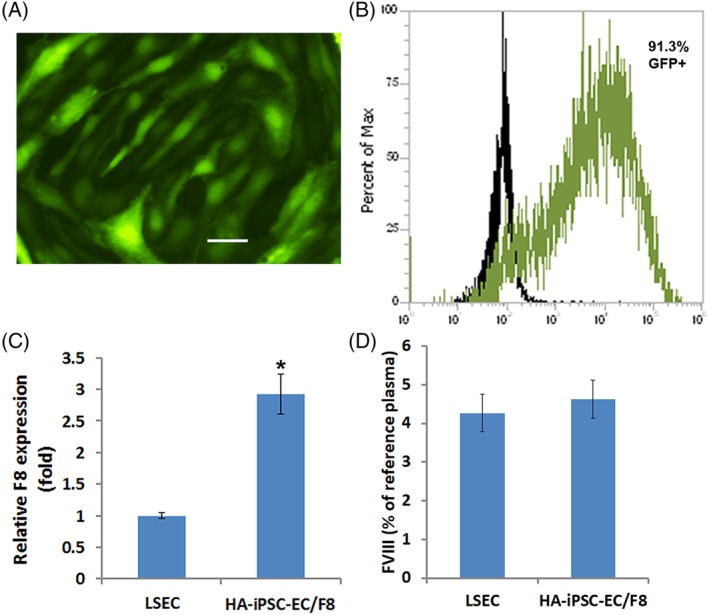
To evaluate whether HA‐iPSC‐ECs/F8 can engraft stably in vivo, we first transplanted these cells intramuscularly into adult immune‐deficient NSG mice. Each mouse received 1 × 106 cells. We assessed the engraftment of HA‐iPSC1‐ECs/F8 in these animals over time using an IVIS spectrum to detect the bioluminescence signal generated by the luciferase in the HA‐iPSC1‐ECs/F8. Bioluminescence imaging demonstrated that the HA‐iPSC1‐ECs/F8 were retained in these animal for 10 weeks after cell transplantation (Figure 3A, B), when this experiment ended. Quantitation of the bioluminescence signals in these mice revealed that there was a dramatic reduction of the bioluminescence signals in the first 2 weeks following cell transplantation and the remaining bioluminescence signals was stably retained thereafter (Figure 3B).
Figure 3.
Evaluation of the engraftment of HA‐iPSC‐EC/F8 in NSG mice. A, Bioluminescent images of the adult NSG mice transplanted with HA‐iPSC1‐EC/F8 at the indicated weeks after the transplantation. B, Bioluminescent signal intensity for the mice in (A) over time (n = 6). C, Bioluminescent images of the NSG mice transplanted with HA‐iPSC1‐EC/F8 at the neonatal stage. Images were taken at the indicated weeks after the transplantation. D, Bioluminescent signal intensity for the mice transplanted with HA‐iPSC1‐EC/F8 (n = 7) or HA‐iPSC2‐EC/F8 (n = 4) at neonatal stage over time. No statistically significant difference was detected between these two mouse groups transplanted with the ECs derived from different HA‐iPSC lines. ECs, endothelial cells; HA, hemophilia A; iPSC, induced pluripotent stem cell; NSG, NOD/SCID/IL2Rγ−/−
Since HA is a genetic disease, the earlier a treatment is given, the greater benefit a patient will receive. The rapid growth of a newborn to an adult could promote either expansion or turnover of the transplanted cells. To assess whether HA‐iPSC‐ECs/F8 can be retained when transplanted at neonatal stage, we next subcutaneously transplanted HA‐iPSC‐ECs/F8 into neonatal NSG mice. Each neonatal mouse received 3 × 105 ECs derived from either HA‐iPSC1 or HA‐iPSC2 line. Bioluminescence imaging of these mice over time demonstrated that bioluminescence signals were retained for at least 16 weeks (Figure 3C, D), when the experiment ended. Again, the most drastic reduction of the bioluminescence signals occurred in the first 2 weeks after cell transplantation. The cells survived the first 2 weeks after being transplanted were able to persist to adult hood (Figure 3C, D). The ECs derived from HA‐iPSC1 line showed a slightly higher level of engraftment compared with the ECs derived from HA‐iPSC2 line (Figure 3D).
To assess whether the HA‐iPSC‐ECs/F8 maintained their expression of F8 as well as the endothelial marker CD31 in vivo, we dissected the limb tissue with bioluminescence signal from the mice 16 weeks after being transplanted with HA‐iPSC1‐ECs/F8 at the neonatal stage. The tissue sections were immunostained with GFP, human CD31, and human FVIII antibodies. As a result, multiple clusters of GFP‐positive cells were detected in the tissue (Figure 4). Importantly, these GFP‐positive cells also expressed human FVIII and human CD31 (Figure 4). Thus, our data suggest that HA‐iPSC‐derived ECs can engraft in vivo for a relatively long time when transplanted not only in adult but also in neonatal mice, and these ECs can maintain their capability of producing FVIII in vivo.
Figure 4.
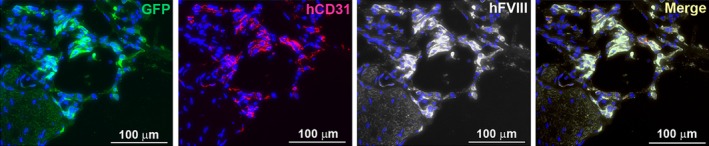
Histological analysis of the transplanted HA‐iPSC1‐EC/F8 in NSG mice. Neonatal NSG mice were transplanted with HA‐iPSC1‐EC/F8 at their left hind limb. Sixteen weeks after the transplantation, a limb tissue with bioluminescence signal was immunostained with antibodies against GFP, human CD31, and human FVIII. Fluorescence images for GFP (green), human CD31 (red), and human FVIII (white) and the merge image of all three were shown. Nuclei were shown in blue (DAPI stain). Scale bar = 100 μm. ECs, endothelial cells; FVIII, factor VIII; GFP, green fluorescent protein; HA, hemophilia A; iPSC, induced pluripotent stem cell; NSG, NOD/SCID/IL2Rγ−/−
3.4. HA‐iPSC‐ECs/F8 reduced blood loss in a mouse model of HA
To assess the functional effect of HA‐iPSC‐ECs/F8 in treating HA, we purchased a F8 knock‐out mouse strain B6;129S‐F8tm1Kaz/J (B6F8) from The Jackson Laboratory. This B6F8 mouse strain is immune‐competent. To allow human cell engraftment, we suppressed the immune system in neonatal hemophilia mice by administration of cyclosporine A at 210 mg/L in drinking water38 to the dam from the day of mating to the end of the experiment. Lactational transfer of cyclosporine to nursing pups has been previously demonstrated.39 We transplanted HA‐iPSC1‐ECs/F8 intramuscularly into the hind limb of the neonatal hemophilia B6F8 mice. Each mouse received 2 to 3 million cells. We detected bioluminescence signals in these mice (Figure 5A) at 1 week after the transplantation, suggesting the retention of the transplanted cells. Next, a tail‐clip bleeding test was performed to evaluate the blood loss in these mice as well as the control age‐matched B6F8 mice and the control WT C57BL/6 mice (WT mice). We found that the blood loss in the HA mice transplanted with HA‐iPSC1‐ECs/F8 was significantly reduced compared with that in the control HA mice (Figure 5B), and only slightly more compared with that in the WT mice (Figure 5B). Although no F8 expression was detected in the control B6F8 mice by qRT‐PCR, the expression of F8 was detected in the mice transplanted with HA‐iPSC1‐ECs/F8 (Figure 5C). Therefore, our data demonstrated that transplantation of HA‐iPSC1‐ECs/F8 is effective in delivering FVIII and attenuating the symptom of HA.
Figure 5.
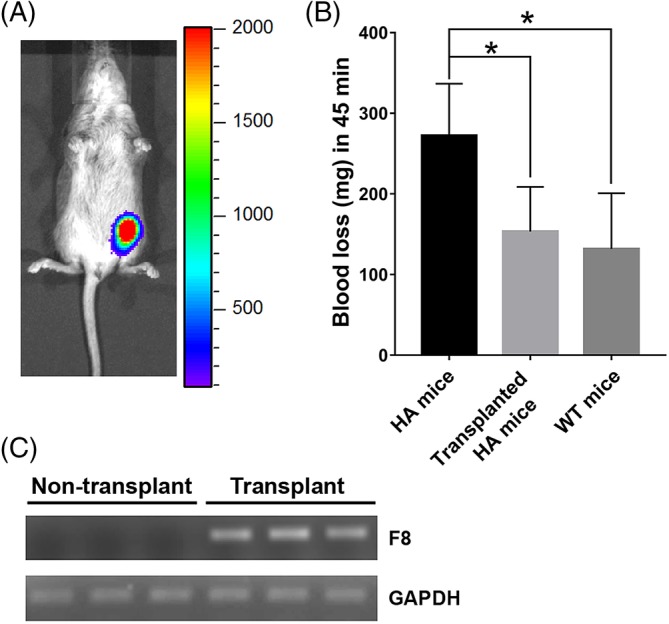
Evaluation of the effect of HA‐iPSC‐ECs/F8 on blood loss in B6F8 HA mice. Neonatal B6F8 HA mice were transplanted with HA‐iPSC1‐EC/F8 and analyzed 1 week after the transplantation. A, A representative bioluminescent image of the mice. B, Blood loss in non‐transplanted B6F8 mice or HA‐iPSC1‐EC/F8‐transplanted B6F8 mice and wild‐type C57BL6 (WT) mice in a tail‐clip assay. (n = 4‐5) *P < .05. C, The F8 mRNA in non‐transplanted and HA‐iPSC1‐EC/F8 transplanted HA mice as detected by qRT‐PCR and agarose gel electrophoresis. ECs, endothelial cells; HA, hemophilia A; iPSC, induced pluripotent stem cell; qRT‐PCR, quantitative reverse transcription polymerase chain reaction
3.5. HA‐iPSC‐ECs/F8 produced therapeutic levels of FVIII in an immune‐deficient HA mouse model
Our preceding study showed that HA‐iPSC1‐ECs/F8 were retained in immune‐competent HA neonatal mice treated with cyclosporine A for 1 week. However, we found that the immune suppression by cyclosporine A through milk might not be sufficient, because we could not detect bioluminescence signal in those mice 2 weeks after the cell transplantation. This is in contrast with our data using immune‐deficient NSG mice, which showed persistent bioluminescence signal from the transplanted cells over 10‐16 weeks (Figure 3). To facilitate human cell engraftment, we generated an immune‐deficient HA mouse strain by breeding a female B6;129S‐F8 tm1Kaz/J mouse to a male immune‐deficient mouse, B6;129S‐RAG2tm1Fwa CD47tm1Fpl IL2rgtm1Wjl/J (BRG) (The Jackson Laboratory) and breeding their progeny. The mice with F8 null RAG2 null IL2rg null genotype were obtained and referred as F8RG mice.
We transplanted HA‐iPSC1‐ECs/F8 subcutaneously into the hind limb of the adult F8RG mice. Each mouse received 10 million cells. We estimated that the weight of an adult mouse is about fourfold to eightfold higher than a neonatal mouse. Therefore, we increased the cell dose for adult mice to 1 × 107/mouse, which was around fivefold higher than that for neonatal mice. These mice were analyzed 2 weeks posttransplantation because we had shown in the preceding section that the HA‐iPSC‐ECs/F8 could persist for much longer time in immune‐deficient mice if they were able to survive the first 2 weeks after transplantation (Figure 3). The transplanted cell‐Matrigel plugs were easily identified 2 weeks after the transplantation (Figure 6A). Some of the cell‐matrigel plugs attracted more blood vessels than others. Immunostaining of the transplanted cell‐Matrigel plugs showed that the GFP‐positive cells also expressed human FVIII and formed a vessel‐like network in vivo (Figure 6B).
Figure 6.
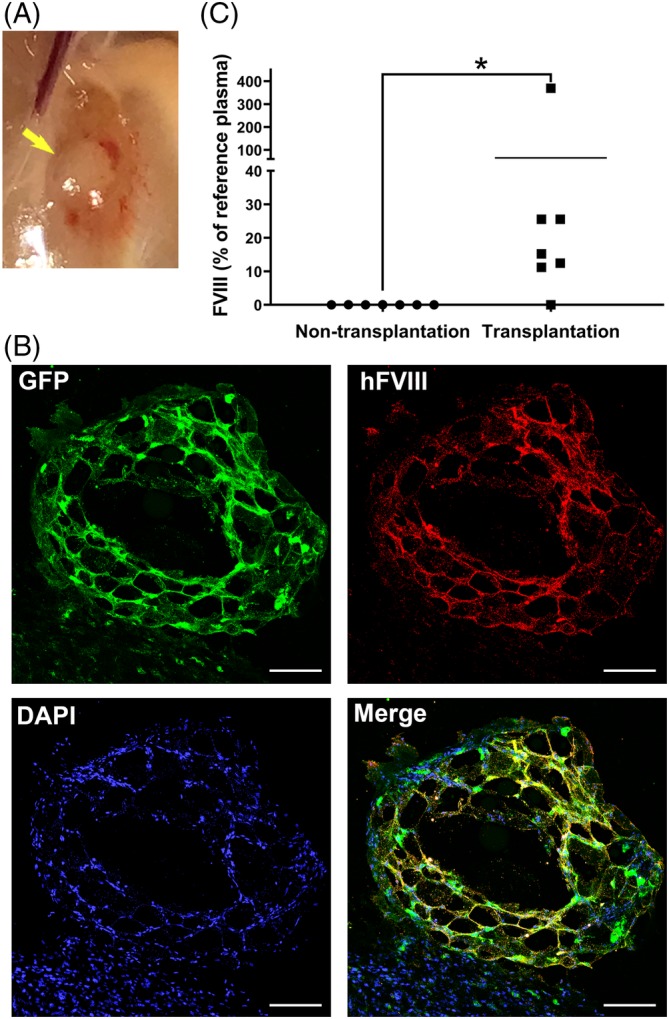
Evaluation of HA‐iPSC‐ECs/F8 in immune deficient F8RG mouse model. Adult F8RG mice were subcutaneously transplanted with HA‐iPSC1‐EC/F8 at their hind limbs and were analyzed 2 weeks after the transplantation. A, A transplanted cell‐Matrigel plug at the transplantation site. B, Immunostaining of a transplanted cell‐Matrigel plug retrieved from the mice for GFP (green), human FVIII (red), and DAPI (blue). Scale bar = 100 μm. C, Plasma FVIII levels in non‐transplanted and transplanted F8RG mice were assayed by FVIII antigen ELISA (n = 7). *P = .015. ECs, endothelial cells; ELISA, enzyme linked immunosorbent assay; FVIII, factor VIII; GFP, green fluorescent protein; HA, hemophilia A; iPSC, induced pluripotent stem cell
We collected plasma samples from these mice and from non‐transplanted control F8RG mice. Human FVIII protein was nearly undetectable (<0.1%) in non‐transplanted mouse plasma by ELISA analysis (Figure 6C). Remarkably, the six of seven mice that received HA‐iPSC1‐ECs/F8 had plasma human FVIII protein ranged from 11.2% to 369.2% relative to human plasma standard (Figure 6C). The severity of HA is clinically classified into severe, moderate and mild, which is defined as <1%, 1%‐5%, and >5%‐49% of FVIII activity, respectively. Thus, our data suggest that the HA‐iPSC1‐ECs/F8 produced therapeutic levels of FVIII in HA mice and at least attenuated severe HA into mild HA.
4. DISCUSSION
HA is one of the best disease candidates suitable for cell therapy because correction of the disease only requires a single gene product that functions outside the cell that had produced it. Therefore, as long as the potential therapeutic cells can produce and secrete functional FVIII, engraft well in vivo and are safe, there is no additional functional requirement for these cells. We showed here that HA patient‐derived iPSCs can be efficiently differentiated into ECs. These ECs can be modified with lentiviral vectors to produce high levels of functional BDD‐FVIII, comparable to primary human LSECs. These ECs can deliver therapeutic levels of FVIII to the circulation for the treatment of HA.
Sustained engraftment of therapeutic cells is crucial for the treatment of HA as well as other genetic diseases. It has been shown that fetal liver endothelial progenitors have much robust engraftment potential in newborn animals compared with adult LSEC.40, 41 We found that, when transplanted at the neonatal stage, the ECs derived from two different HA‐iPSC lines were retained in mice for at least 16 weeks (Figure 3), suggesting consistent engraftment of the ECs. When the ECs derived from the same HA‐iPSC line were tested in both neonatal and adult mice, the ECs were retained in all tested mice but slightly more cells were retained when transplanted at the neonatal stage (Figure 3). Therefore, our data suggest that the HA‐iPSC‐ECs are capable of relatively stable engraftment in mice, and that transplantation of HA‐iPSC‐ECs at the neonatal stage is at least as effective, if not more so, as at the adult stage. Since children born with HA need to be treated early after birth to prevent potential bleeding, neonatal cell therapy could be especially beneficial in the clinical setting.
Based on no increase of the bioluminescence signal upon transplantation, no significant in vivo expansion of the transplanted HA‐iPSC‐ECs was evident in neonatal as well as in adult mice in the study (Figure 3). Consistent with the bioluminescence signal, no teratoma was detected in any of the mice transplanted with the HA‐iPSC‐ECs over 3‐4 months. Therefore, the HA‐iPSC‐ECs were not proliferating extensively in vivo and not likely tumorigenic. However, the current study is an initial proof‐of‐concept study in small animal models. To completely rule out the tumorigenic potential of the HA‐iPSCs and HA‐iPSC‐ECs, whole‐genome sequencing or deep sequencing for genes involved in tumorigenesis and a much longer observation time are necessary in future studies toward clinical applications of these cells, which is beyond the scope of the current study.
We used lentiviral vectors to express the functional bddF8 gene in HA‐iPSC‐ECs at a relatively low MOI of 4 to 8. The transduced HA‐iPSC‐ECs secreted FVIII at a level comparable to those by LSECs in vitro (Figure 2D). We showed previously the activity of this bdd‐FVIII using the same viral vector.42 These transduced HA‐iPSC‐ECs produced FVIII after being transplanted into mice by immunostaining (Figure 4), qRT‐PCR (Figure 5), and ELISA (Figure 6). In situ gene editing of the F8 mutations in HA‐iPSCs using transcription activator‐like effector nucleases or clusters of regularly interspaced palindromic repeats engineered nucleases/Cas‐derived RNA‐guided endonucleases has now been proven feasible to correct the F8 mutations.43, 44, 45 We noticed, however, that the ECs derived from non‐diseased iPSCs expressed around 80‐fold less F8 mRNA compared with liver sinusoid ECs (Figure 1), whereas other tested primary ECs had even lower expression levels of F8 compared with the health iPSC‐ECs (Figure 1). A new method is needed to specifically differentiate HA‐iPSCs into liver sinusoid ECs, if in situ gene editing approach is used. Most recently, strategies to express F8 in HA‐iPSCs by inserting the functional F8 gene into a safe harbor site or rDNA locus have been reported.46, 47 These ectopic gene expression approaches can ensure levels of F8 expression. However, the possibilities of random DNA deletion in the iPSC genome by the gene editing system and the selection of the p53 pathway inactivated cells48, 49, 50, 51 need to be carefully addressed before moving forward to clinical trials.
We used two HA mouse models to assess the therapeutic effects of HA‐iPSC‐ECs/F8. In immune‐competent B6F8 mice that received immunosuppressive agent cyclosporine A, the transduced HA‐iPSC‐ECs significantly reduced the blood loss in a tail‐clip assay, when transplanted at neonatal stage (Figure 5). However, we noticed much lower number of cells and shorter time of cell retention in the B6F8 mice compared with the immunodeficient NSG mice, suggesting incompletely immune suppression in these B6F8 mice. We generated a new immunodeficient mouse stain, F8RG. When HA‐iPSC‐ECs/F8 were transplanted into F8RG mice, they secreted human FVIII to the mouse circulation at levels ranged from 11.2% to 369.2% at 2 weeks post‐transplantation. The marked variation on the FVIII levels among these mice may be caused by different microenvironments that impact cell survival and integration of the transplanted cells into the host vascular system (Figure 6A). Nonetheless, this study provides proof‐of‐concept evidence that HA‐iPSC‐ECs at least attenuate severe HA to mild HA. In future studies, long‐term therapeutic evaluation of HA‐iPSC‐ECs are needed.
Mouse iPSC‐derived ECs had been shown to have therapeutic effect in HA mice.52 While we were preparing this manuscript, Olgasi et al reported that HA patient's iPSC‐derived ECs transduced with F8 expressing lentivirus produced approximately 6% plasma FVIII when transplanted via portal vein or intraperitoneally in a different HA mouse model.53 Hu et al also reported that infusion of the ECs derived from HA patient's iPSCs that had being gene edited to express F8 via orbital vein could partially rescue the bleeding phenotype in HA mice.54 To our knowledge, we are the first group to show the relatively stable engraftment as well as the therapeutic effect of iPSC‐derived ECs at the neonatal stage. Considering the size of a human body, we think that our subcutaneous transplantation approach allows transplanting a large number of cells and easy removal of the transplanted cells if any adverse effects from the transplant are evident. In future studies, improvement in cell engraftment to reduce cell loss after transplantation and assessment of the potential long‐term therapeutic cells in animals must be undertaken.
5. CONCLUSION
ECs can be efficiently generated from HA patients' iPSCs and genetically modified to express high levels of a functional F8. These HA‐iPSC‐ECs are capable of durable engraftment in both neonatal and adult animals, and they either attenuated or functionally corrected the symptoms of hemophilia in animal models. Our studies provide proof‐of‐concept that HA‐iPSC‐derived ECs can serve as an autologous cell factory to deliver FVIII for the treatment of HA in not only adults but also in newborns.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
M.R., K.G.: collection and/or assembly of data, data analysis and interpretation, manuscript writing; E.C., E.A., A.H.: collection and/or assembly of data; G.P.: provision of study material, data analysis and interpretation; J.N.: data analysis and interpretation, financial support, manuscript writing; A.W., P.Z.: conception and design, data analysis and interpretation, financial support, manuscript writing, final approval of manuscript.
ACKNOWLEDGMENTS
This work is supported by the Milstein Medical Asian American Partnership Foundation (P.Z.), an Innovative Development Award from University of California, Davis (P.Z.), an Interdepartmental Seed Grant from UC Davis (A.W. and P.Z.), NIH R01 R01GM099688 (J.A.N.), the Shriners Hospitals for Children Postdoctoral Fellowship (84705‐NCA‐19 to K.G.), the University of California, Davis, School of Medicine Dean's Fellowship (to A.W), the California Institute for Regenerative Medicine training grant TB1‐01184 to California State University Sacramento (M.R., E.C, and A.H.), and the University of California Davis Impact Program (J.N.).
Rose M, Gao K, Cortez‐Toledo E, et al. Endothelial cells derived from patients' induced pluripotent stem cells for sustained factor VIII delivery and the treatment of hemophilia A. STEM CELLS Transl Med. 2020;9:686–696. 10.1002/sctm.19-0261
Melanie Rose and Kewa Gao contributed equally to the study.
Funding information California State University; California Institute for Regenerative Medicine, Grant/Award Number: TB1‐01184; University of California, Davis, School of Medicine; Shriners Hospitals for Children, Grant/Award Number: 84705‐NCA‐19; NIH, Grant/Award Number: R01GM099688; University of California, Davis; Milstein Medical Asian American Partnership Foundation
Contributor Information
Aijun Wang, Email: aawang@ucdavis.edu.
Ping Zhou, Email: pizhou@ucdavis.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author.
REFERENCES
- 1. Armstrong EP, Malone DC, Krishnan S, Wessler MJ. Costs and utilization of hemophilia a and B patients with and without inhibitors. J Med Econ. 2014;17:798‐802. [DOI] [PubMed] [Google Scholar]
- 2. Rangarajan S, Walsh L, Lester W, et al. AAV5‐factor VIII gene transfer in severe Hemophilia a. N Engl J Med. 2017;377:2519‐2530. [DOI] [PubMed] [Google Scholar]
- 3. Mimuro J, Mizukami H, Shima M, et al. The prevalence of neutralizing antibodies against adeno‐associated virus capsids is reduced in young Japanese individuals. J Med Virol. 2014;86:1990‐1997. [DOI] [PubMed] [Google Scholar]
- 4. Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV‐factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342‐347. [DOI] [PubMed] [Google Scholar]
- 5. Mingozzi F, Maus MV, Hui DJ, et al. CD8(+) T‐cell responses to adeno‐associated virus capsid in humans. Nat Med. 2007;13:419‐422. [DOI] [PubMed] [Google Scholar]
- 6. Follenzi A, Benten D, Novikoff P, Faulkner L, Raut S, Gupta S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia a mice. J Clin Invest. 2008;118:935‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fomin ME, Zhou Y, Beyer AI, Publicover J, Baron JL, Muench MO. Production of factor VIII by human liver sinusoidal endothelial cells transplanted in immunodeficient uPA mice. PLoS One. 2013;8:e77255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matsui H, Shibata M, Brown B, et al. Ex vivo gene therapy for hemophilia a that enhances safe delivery and sustained in vivo factor VIII expression from lentivirally engineered endothelial progenitors. Stem Cells. 2007;25:2660‐2669. [DOI] [PubMed] [Google Scholar]
- 9. Tatsumi K, Sugimoto M, Lillicrap D, et al. A novel cell‐sheet technology that achieves durable factor VIII delivery in a mouse model of hemophilia a. PLoS One. 2013;8:e83280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin Y, Chang L, Solovey A, Healey JF, Lollar P, Hebbel RP. Use of blood outgrowth endothelial cells for gene therapy for hemophilia a. Blood. 2002;99:457‐462. [DOI] [PubMed] [Google Scholar]
- 11. Roth DA, Tawa NE Jr, O'Brien JM, et al. Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia a. N Engl J Med. 2001;344:1735‐1742. [DOI] [PubMed] [Google Scholar]
- 12. Porada CD, Sanada C, Kuo CJ, et al. Phenotypic correction of hemophilia a in sheep by postnatal intraperitoneal transplantation of FVIII‐expressing MSC. Exp Hematol. 2011;39:1124‐1135. e1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Q, Gong X, Gong Z, et al. The mesenchymal stem cells derived from transgenic mice carrying human coagulation factor VIII can correct phenotype in hemophilia a mice. J Genet Genomics. 2013;40:617‐628. [DOI] [PubMed] [Google Scholar]
- 14. Sokal EM, Lombard CA, Roelants V, et al. Biodistribution of liver‐derived mesenchymal stem cells after peripheral injection in a hemophilia a patient. Transplantation. 2017;101:1845‐1851. [DOI] [PubMed] [Google Scholar]
- 15. Kashiwakura Y, Ohmori T, Mimuro J, et al. Intra‐articular injection of mesenchymal stem cells expressing coagulation factor ameliorates hemophilic arthropathy in factor VIII‐deficient mice. J Thromb Haemost. 2012;10:1802‐1813. [DOI] [PubMed] [Google Scholar]
- 16. Ravanbod R, Torkaman G, Mophid M, Mohammadali F. Experimental study on the role of intra‐articular injection of MSCs on cartilage regeneration in haemophilia. Haemophilia. 2015;21:693‐701. [DOI] [PubMed] [Google Scholar]
- 17. Moayeri M, Ramezani A, Morgan RA, Hawley TS, Hawley RG. Sustained phenotypic correction of hemophilia a mice following oncoretroviral‐mediated expression of a bioengineered human factor VIII gene in long‐term hematopoietic repopulating cells. Mol Ther. 2004;10:892‐902. [DOI] [PubMed] [Google Scholar]
- 18. Kuether EL, Schroeder JA, Fahs SA, et al. Lentivirus‐mediated platelet gene therapy of murine hemophilia a with pre‐existing anti‐factor VIII immunity. J Thromb Haemost. 2012;10:1570‐1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Du LM, Nurden P, Nurden AT, et al. Platelet‐targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat Commun. 2013;4:2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shi Q, Kuether EL, Chen Y, Schroeder JA, Fahs SA, Montgomery RR. Platelet gene therapy corrects the hemophilic phenotype in immunocompromised hemophilia a mice transplanted with genetically manipulated human cord blood stem cells. Blood. 2014;123:395‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ohmori T, Mimuro J, Takano K, et al. Efficient expression of a transgene in platelets using simian immunodeficiency virus‐based vector harboring glycoprotein Ibalpha promoter: in vivo model for platelet‐targeting gene therapy. FASEB J. 2006;20:1522‐1524. [DOI] [PubMed] [Google Scholar]
- 22. Ramezani A, Zweier‐Renn LA, Hawley RG. Factor VIII delivered by haematopoietic stem cell‐derived B cells corrects the phenotype of haemophilia a mice. Thromb Haemost. 2011;105:676‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gangadharan B, Parker ET, Ide LM, Spencer HT, Doering CB. High‐level expression of porcine factor VIII from genetically modified bone marrow‐derived stem cells. Blood. 2006;107:3859‐3864. [DOI] [PubMed] [Google Scholar]
- 24. Everett LA, Cleuren AC, Khoriaty RN, et al. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. 2014;123:3697‐3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fahs SA, Hille MT, Shi Q, Weiler H, Montgomery RR. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood. 2014;123:3706‐3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shahani T, Covens K, Lavend'homme R, et al. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J Thromb Haemost. 2014;12:36‐42. [DOI] [PubMed] [Google Scholar]
- 27. Tan JY, Sriram G, Rufaihah AJ, Neoh KG, Cao T. Efficient derivation of lateral plate and paraxial mesoderm subtypes from human embryonic stem cells through GSKi‐mediated differentiation. Stem Cells Dev. 2013;22:1893‐1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lian X, Bao X, Al‐Ahmad A, et al. Efficient differentiation of human pluripotent stem cells to endothelial progenitors via small‐molecule activation of WNT signaling. Stem Cell Rep. 2014;3:804‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patsch C, Challet‐Meylan L, Thoma EC, et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol. 2015;17:994‐1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harding A, Cortez‐Toledo E, Magner NL, et al. Highly efficient differentiation of endothelial cells from pluripotent stem cells requires the MAPK and the PI3K pathways. Stem Cells. 2017;35:909‐919. [DOI] [PubMed] [Google Scholar]
- 31. Jia B, Chen S, Zhao Z, et al. Modeling of hemophilia a using patient‐specific induced pluripotent stem cells derived from urine cells. Life Sci. 2014;108:22‐29. [DOI] [PubMed] [Google Scholar]
- 32. Xue Y, Cai X, Wang L, et al. Generating a non‐integrating human induced pluripotent stem cell bank from urine‐derived cells. PLoS One. 2013;8:e70573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu J, Vodyanik MA, Smuga‐Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917‐1920. [DOI] [PubMed] [Google Scholar]
- 34. Kumar P, Gao K, Wang C, et al. In utero transplantation of placenta‐derived mesenchymal stromal cells for potential fetal treatment of hemophilia A. Cell Transplant. 2018;27:130‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou P, Hohm S, Olusanya Y, Hess DA, Nolta J. Human progenitor cells with high aldehyde dehydrogenase activity efficiently engraft into damaged liver in a novel model. Hepatology. 2009;49:1992‐2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beegle JR, Magner NL, Kalomoiris S, et al. Preclinical evaluation of mesenchymal stem cells overexpressing VEGF to treat critical limb ischemia. Mol Ther Methods Clin Dev. 2016;3:16053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shahani T, Lavend'homme R, Luttun A, et al. Activation of human endothelial cells from specific vascular beds induces the release of a FVIII storage pool. Blood. 2010;115:4902‐4909. [DOI] [PubMed] [Google Scholar]
- 38. Huang R, Baranov P, Lai K, Zhang X, Ge J, Young MJ. Functional and morphological analysis of the subretinal injection of human retinal progenitor cells under Cyclosporin a treatment. Mol Vis. 2014;20:1271‐1280. [PMC free article] [PubMed] [Google Scholar]
- 39. Padgett EL, Seelig LL Jr. Effects on T‐cell maturation and proliferation induced by lactational transfer of cyclosporine to nursing pups. Transplantation. 2002;73:867‐874. [DOI] [PubMed] [Google Scholar]
- 40. Canete A, Comaills V, Prados I, et al. Characterization of a fetal liver cell population endowed with long‐term multiorgan endothelial reconstitution potential. Stem Cells. 2017;35:507‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Serrano LJ, Canete A, Garcia‐Leal T, et al. Searching for a cell‐based therapeutic tool for haemophilia a within the embryonic/foetal liver and the aorta‐gonads‐Mesonephros region. Thromb Haemost. 2018;118:1370‐1381. [DOI] [PubMed] [Google Scholar]
- 42. Gao K, Kumar P, Cortez‐Toledo E, et al. Potential long‐term treatment of hemophilia a by neonatal co‐transplantation of cord blood‐derived endothelial colony‐forming cells and placental mesenchymal stromal cells. Stem Cell Res Ther. 2019;10:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Park CY, Kim DH, Son JS, et al. Functional correction of large factor VIII gene chromosomal inversions in hemophilia A patient‐derived iPSCs using CRISPR‐Cas9. Cell Stem Cell. 2015;17:213‐220. [DOI] [PubMed] [Google Scholar]
- 44. Park CY, Kim J, Kweon J, et al. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc Natl Acad Sci U S A. 2014;111:9253‐9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu Y, Hu Z, Li Z, et al. In situ genetic correction of F8 intron 22 inversion in hemophilia a patient‐specific iPSCs. Sci Rep. 2016;6:18865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Park CY, Sung JJ, Cho SR, Kim J, Kim DW. Universal correction of blood coagulation factor VIII in patient‐derived induced pluripotent stem cells using CRISPR/Cas9. Stem Cell Rep. 2019;12:1242‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pang J, Wu Y, Li Z, et al. Targeting of the human F8 at the multicopy rDNA locus in Hemophilia a patient‐derived iPSCs using TALENickases. Biochem Biophys Res Commun. 2016;472:144‐149. [DOI] [PubMed] [Google Scholar]
- 48. Kosicki M, Tomberg K, Bradley A. Repair of double‐strand breaks induced by CRISPR‐Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36:765‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shin HY, Wang C, Lee HK, et al. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat Commun. 2017;8:15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR‐Cas9 genome editing induces a p53‐mediated DNA damage response. Nat Med. 2018;24:927‐930. [DOI] [PubMed] [Google Scholar]
- 51. Ihry RJ, Worringer KA, Salick MR, et al. p53 inhibits CRISPR‐Cas9 engineering in human pluripotent stem cells. Nat Med. 2018;24:939‐946. [DOI] [PubMed] [Google Scholar]
- 52. Xu D, Alipio Z, Fink LM, et al. Phenotypic correction of murine hemophilia a using an iPS cell‐based therapy. Proc Natl Acad Sci U S A. 2009;106:808‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Olgasi C, Talmon M, Merlin S, et al. Patient‐specific iPSC‐derived endothelial cells provide long‐term phenotypic correction of Hemophilia a. Stem Cell Rep. 2018;11:1391‐1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hu Z, Zhou M, Wu Y, et al. ssODN‐mediated in‐frame deletion with CRISPR/Cas9 restores FVIII function in Hemophilia A‐patient‐derived iPSCs and ECs. Mol Ther Nucleic Acids. 2019;17:198‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.