

Abstract
The long duration of animal models represents a clear limitation to quickly evaluate the efficacy of drugs targeting nonalcoholic steatohepatitis (NASH). We, therefore, developed a rapid mouse model of liver inflammation (i.e., the mouse fed a high‐fat/high‐cholesterol diet, where cyclodextrin is co‐administered to favor hepatic cholesterol loading, liver inflammation, and NASH within 3 weeks), and evaluated the effects of the dual peroxisome proliferator‐activated receptor alpha/delta agonist elafibranor (ELA). C57BL6/J mice were fed a 60% high‐fat, 1.25% cholesterol, and 0.5% cholic acid diet with 2% cyclodextrin in drinking water (HFCC/CDX diet) for 3 weeks. After 1 week of the diet, mice were treated orally with vehicle or ELA 20 mg/kg q.d. for 2 weeks. Compared with vehicle, ELA markedly reduced liver lipids and nonalcoholic fatty liver disease activity scoring, through steatosis, inflammation, and fibrosis (all P < 0.01 vs. vehicle). Flow cytometry analysis showed that ELA significantly improved the HFCC/CDX diet‐induced liver inflammation by preventing the increase in total number of immune cells (CD45+), Kupffer cells, dendritic cells, and monocytes population, as well as the reduction in natural killer and natural killer T cells, and by blocking conversion of T cells in regulatory T cells. ELA did not alter pyroptosis (Gasdermin D), but significantly reduced necroptosis (cleaved RIP3) and apoptosis (cleaved caspase 3) in the liver. In conclusion, ELA showed strong benefits on NASH, including improvement in hepatic inflammation, necroptosis, and apoptosis in the 3‐week NASH mouse. This preclinical model will be useful to rapidly detect the effects of novel drugs targeting NASH.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ A large effort is put in developing novel therapies targeting nonalcoholic steatohepatitis (NASH), as this comorbidity related to obesity and type 2 diabetes is growing worldwide. However, the long duration of animal models of NASH represents a clear limitation to quickly evaluate the efficacy of drugs before their translation into clinical trials.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study addressed whether a nutritional 3‐week mouse model could enable the rapid evaluation of drugs targeting NASH.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The study demonstrates the possibility of a rapid drug efficacy evaluation in a 3‐week NASH mouse model, as highlighted with the effects of elafibranor showing similar benefits as observed in humans.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ The 3‐week NASH mouse model will favor a faster translation of novel therapies into the clinical setting.
As obesity and diabetes continue to be a growing epidemic worldwide, the prevalence of nonalcoholic fatty liver diseases (NAFLD), is increasing in parallel.1 NAFLD includes simple steatosis, which can turn out to be nonalcoholic steatohepatitis (NASH), characterized by hepatic steatosis, necro‐inflammation, and various stages of fibrosis, that may lead to cirrhosis and ultimately to hepatocellular carcinoma.2 Indeed, NASH is the most rapidly growing indication for liver transplantation in the United States.3
Therefore, a tremendous effort is put in developing novel therapies targeting NASH, which requires the use of animal models that may mimic the human disease. Many animal models are currently available and are based on diet intervention (amino acids deficient diet and high‐fat/cholesterol/fructose diet), chemical intoxication (streptozotocin, carbon tetrachloride, or thioacetamide intoxication), or genetic alteration (Lepob/ob and foz/foz mice).4
Although the genetic and chemical models show clear limitations to represent the human disease, a translational NASH animal model should be ideally set up on a nutritional basis and should present the progressive hepatic metabolic disturbances leading to NASH (i.e., liver fat accumulation, hepatic steatosis, cell death, and inflammation, and ultimately fibrosis). An important component in the pathogenesis of NASH (i.e., transition from simple steatosis to cell death and inflammation), is cholesterol.5 In fact, a 1–2% cholesterol diet in mice is required for a diet‐induced liver inflammation and fibrosis in mice, which cannot be achieved with a regular high‐fat diet.4 Despite this very high percentage of dietary cholesterol, a 2536‐week period of high‐fat/cholesterol diet is needed to reach similar levels of liver lesions observed in human patients with NASH, likely because raising dietary cholesterol substantially reduces intestinal cholesterol absorption.6 This long duration represents a major limitation in drug development, whereas no therapies are indicated for NASH yet.7 Liver inflammation is another important contributor in the transition from simple steatosis to NASH. Hepatocytes injured by the accumulation of lipids (lipotoxicity) play a central role in the induction of innate immunity through Toll‐like receptors and the activation and recruitment of Kupffer cells, lymphocytes, and neutrophils, as well as the inflammasome.8 Therefore, nutritional mouse models presenting this inflammation component are clearly needed to better investigate and target inflammatory processes in NASH.
In this context, we have set up a novel nutritional mouse model, which develops NASH within only 3 weeks under a high‐fat diet supplemented with cholesterol and cholic acid, two components known to favor intestinal cholesterol absorption, hepatic steatosis, and lipotoxicity,9, 10 and 2‐hydroxypropyl‐β‐cyclodextrin (CDX), a cyclic oligosaccharide showing high affinity for sterols,11 in drinking water. To demonstrate its relevance, our aim was to characterize NASH and the hepatic inflammation profile in this 3‐week mouse model, and to perform a pharmacological validation with Elafibranor (ELA), a dual peroxisome‐proliferator activated receptor (PPAR) α/δ agonist, currently evaluated in the clinic for the treatment of NASH.12
METHODS
Animals and experimental design
All animal protocols were reviewed and approved by the local (Comité régional d'éthique de Midi‐Pyrénées) and national (Ministère de l'Enseignement Supérieur et de la Recherche) ethics committees (protocol number CEEA‐122–2014‐15). C57BL6/J mice, male, 8‐week old at delivery, were housed in groups of four to five mice in enriched and ventilated mouse cages (Tecniplast GM500, 500 cm2 surface, 12.7 cm height) in a room with 22 ± 2°C, and 50 ± 10% relative humidity, and a 12‐hour day/night cycle.
The experimental design is shown in Figure S1 . After a 5‐day acclimation period and during the experimental period, mice had free access to a chow diet (reference RM1 (E) 801492; Special Diets Services, Essex, UK) with normal tap water or a high‐fat (60 kcal%), cholesterol (1.25%), and cholic acid (0.5%) diet (reference D11061901, from Research Diets, New Brunswick, NJ) with 2‐hydroxypropyl‐β‐cyclodextrin (reference 10175600; Fisher Scientific, Cedex, France) at 2% in drinking water (HFCC/CDX diet) for up to 3 weeks.
After 1 week of diet, mice were either euthanized for fluorescence‐activated cell sorting, biochemical and histological analysis, as described below, or randomized according to their alanine aminotransferase (ALT) and aspartate aminotransferase (AST) plasma levels and body weight into homogenous treatment groups. In this mouse model fed the HFCC/CDX diet, ~ 10–20% of total individuals show extremely high ALT and AST levels (i.e., > 500–1000 U/L) and/or a > 20% body weight loss after the 1‐week diet. These individuals were, therefore, excluded from the study and before the start of the 2‐week treatment period.
Vehicle (0.1% Tween 80 and 1% carboxymethyl cellulose in 98.9% distilled water) or ELA (reference BE163306; Carbosynth, Compton, UK) at 20 mg/kg q.d. were then administrated by oral gavage once a day in the morning for 2 weeks.
At the end of the 1‐week diet period or 2‐week treatment period, 4‐hour fasted mice were anesthetized with isoflurane, blood was taken on EDTA from the retro‐orbital plexus to isolate plasma, and animals were euthanized and exsanguinated by intracardiac perfusion with a sterile PBS1X solution. Liver was collected and processed for fluorescence‐activated cell sorting analysis or weighted before liver sample dissections from the left lateral lobe for biochemical and histology analysis.
Biochemical analysis
Plasma biochemistry was performed by the Genotoul Anexplo platform in Toulouse, France. Plasma ALT and AST, total cholesterol, and triglycerides were assayed, using a Horiba Pentra 400 machine and related Pentra assay kits (Horiba France SAS, Longjumeau, France). Plasma cytokines were assayed using a multiplex assay kit (reference M6000007, NYBio‐Plex Pro Mouse Cytokine Th17 Panel A 6‐Plex; BioRad, Hercules, CA).
Hepatic lipids levels were determined from liver homogenate after lipid solubilization with deoxycholate, as described previously.13
Hepatic gene expression and Western blot analysis
Hepatic gene expression of IL‐1β, MCP‐1, collagen 1α1, and α‐smooth muscle actin was performed by quantitative polymerase chain reaction, as described previously.13 List of primers used, including the house keeping gene 18S, are provided in Table S1 .
Western blot analysis was performed by the We‐Met platform, Toulouse, France. Commercial primary antibodies for Gasdermin D (reference ab209845; Abcam, Cambridge, UK), cleaved caspase 3 (reference 9664; Cell Signaling Technology, Danvers, MA), cleaved receptor interacting protein 3 (RIP3; reference ab56164; Abcam) and actin (reference 4970; Cell Signaling Technology) for control, were used on the WES automated western blot system (Proteinsimple, San Jose, CA). Original noncropped/cut Western blots with molecular weight marker and the full get/membranes are shown in Figure S3 .
Fluorescence‐activated cell sorting analysis
Analysis of immune cells was performed by the I2MC Institute, Toulouse, France, using flow cytometry, as previously described.14 Fixable Viability Dye was used for dead cells’ exclusion. Data were collected on a BD LSRII/Fortessa (BD Biosciences, Franklin Lakes, NJ) and analyzed using FlowJo software (Tree Star).
Histology analysis and NAFLD activity scoring
Histology analysis and blinded NAFLD activity scoring (NAS) was performed by Sciempath Labo, Larcay, France, based on an adapted scoring system of Kleiner et al.15 Hepatocellular steatosis was scored from hematoxylin and eosin staining as 0 (< 5% of liver parenchyma), 1 (5–33%), 2 (33–66%), and 3 (>66%). Lobular inflammation was also scored with hematoxylin and eosin staining as 0 (no inflammatory foci), 1 (< 2 foci per 20× field), 2 (foci per 20× field), and 3 (> 4 foci per 20× field). Fibrosis was scored from Sirius red staining as 0 (none), 1 (perisinusoidal or periportal), 2 (perisinusoidal and periportal), 3 (bridging fibrosis), and 4 (cirrhosis). Total NAS was then calculated for each animal by summing up the scores.
Statistical analysis
Data are presented as mean ± SEM, with n = 6–10 mice per experimental group, as indicated in the legends. Two‐way analysis of variance with Bonferroni post‐test, Mann–Whitney U‐test, or unpaired two‐tailed Student’s t‐test were used for statistical analysis using GraphPad Prism software (GraphPad Software, La Jolla, CA). A P < 0.05 was considered significant.
RESULTS
HFCC/CDX diet rapidly promotes hepatic steatosis and inflammation within 1 week
The 3‐week HFCC/CDX mouse model was set up based on preliminary experiments indicating that the HFCC diet combined with CDX was better at promoting plasma transaminases elevation, liver lipids accumulation, and expression of genes involved in inflammation and fibrosis, than the HFCC diet alone (Figure S2 ). As our objective was to use this 3‐week model to rapidly evaluate the curative effects of drugs targeting NASH over a 2‐week period, we first evaluated the effects of the HFCC/CDX diet for 1 week.
After 1 week of diet, mice fed the HFCC/CDX diet showed a lower body weight (8%; P < 0.05), higher plasma ALT and AST levels (369% and 123%; both P < 0.01), and total cholesterol (80%; P < 0.001), as compared with control chow‐fed mice (Table 1). In contrast, plasma triglycerides levels were significantly lower (−38%; P < 0.001 vs. chow), whereas plasma free fatty acids levels remained unchanged. Compared with chow‐fed mice, plasma cytokines IFN‐γ and IL‐10 levels were significantly increased by 85% and 56% (Table 1). Plasma IL‐1β, IL‐6, IL‐17, and TNF‐α were either undetectable or not different vs. control chow‐fed mice (data not shown).
Table 1.
Body weight, plasma parameters, and liver histopathology scoring in mice fed a chow or HFCC/CDX diet for 1 week
| Parameters | Chow | HFCC/CDX |
|---|---|---|
| Body weight, g | 24 ± 1 | 22 ± 2* |
| Plasma ALT (U/L) | 46 ± 2 | 216 ± 27** |
| Plasma AST (U/L) | 90 ± 14 | 201 ± 29** |
| Plasma total cholesterol, mmol/L | 3.05 ± 0.04 | 5.50 ± 0.36*** |
| Plasma triglycerides, mmol/L | 1.77 ± 0.11 | 1.09 ± 0.09*** |
| Plasma free fatty acids, mmol/L | 0.79 ± 0.06 | 1.13 ± 0.23 |
| Plasma IFN‐γ, pg/mL | 10.6 ± 1.0 | 19.6 ± 1.8*** |
| Plasma IL‐10, pg/mL | 38.6 ± 3.7 | 60.2 ± 8.9* |
| Hepatic steatosis score, 0–3 | 0 | 1.3 ± 0.3 |
| Hepatic inflammation score, 0–3 | 0 | 1.9 ± 0.2 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; HFCC/CDX, 60% high‐fat, 1.25% cholesterol, and 0.5% cholic acid diet with 2% cyclodextrin in drinking water.
Data are shown as mean ± SEM, n = 8–10 per group.
P < 0.05, **P < 0.01, and ***P < 0.001 vs. chow.
Within only 1 week, microvesicular steatosis and multifocal mixed inflammation were observed in the liver, as shown by hematoxylin and eosin staining (Figure 1), and confirmed by histopathological scoring (Table 1). However, fibrosis (Sirius red staining) was yet not observed (Figure 1).
Figure 1.
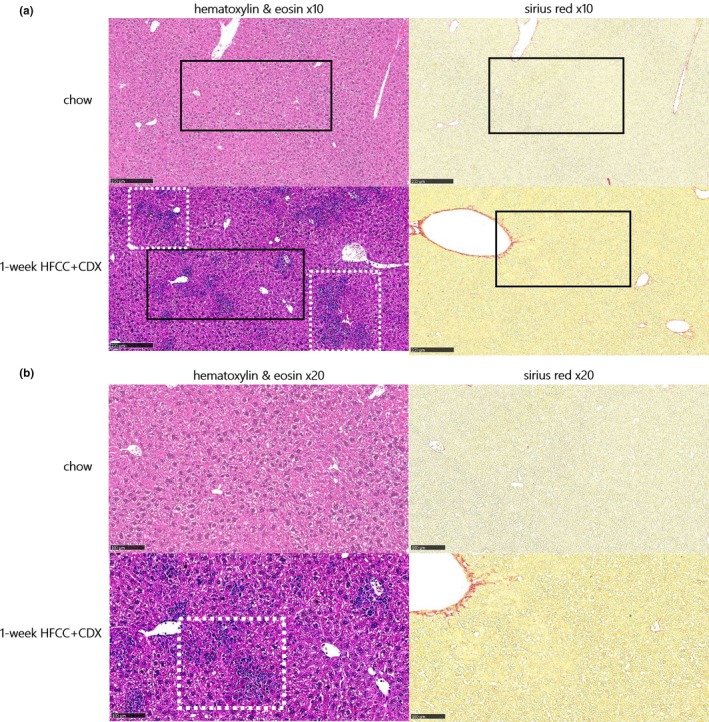
Hematoxylin and eosin and Sirius red stained liver section in mice fed a chow or a 60% high‐fat, 1.25% cholesterol, and 0.5% cholic acid diet with 2% cyclodextrin in drinking water (HFCC/CDX) diet for 1‐week at ×10 (a) and ×20 (b) magnification. Black squares localize the area of the ×20 magnification shown in panel b. Dashed white squares indicate liver steatosis and inflammatory foci.
Hence, a 1‐week HFCC/CDX diet period was sufficient to already induce liver lipids accumulation and advanced liver inflammation at treatment start.
ELA markedly improves liver lesions in HFCC/CDX fed mice
We next evaluated whether the dual PPAR α/δ agonist ELA would be beneficial in this HFCC/CDX diet‐induced NASH mouse model. After 2 weeks of treatment, mice under ELA treatment tended to have lower body weight, although this effect was not found to be significant (Table 2). ELA increased plasma ALT levels (+158%; P < 0.001 vs. vehicle), but not AST. Plasma total cholesterol and high‐density lipoprotein (HDL)‐cholesterol levels were significantly higher by 68% and 94% (both P < 0.001 vs. vehicle) with ELA, wheres it reduced triglyceride levels by 48% (P < 0.01 vs. vehicle). Plasma‐free fatty acids remained unchanged. Compared with vehicle, ELA significantly reduced plasma cytokines IL‐6, IL‐1β, IL‐17, and TNF‐α, INF‐γ, and IL‐10 levels by 66, 55, 48, 52, 48, and 47% (Table 2).
Table 2.
Body weight, plasma parameters, and liver histopathology scoring in HFCC/CDX mice treated p.o. q.d. with vehicle or elafibranor 20 mg/kg for 2 weeks
| Parameters | Vehicle | Elafibranor |
|---|---|---|
| Body weight, g | 23 ± 1 | 22 ± 1 |
| Plasma ALT, (U/L) | 173 ± 23 | 446 ± 23*** |
| Plasma AST, (U/L) | 289 ± 26 | 245 ± 25 |
| Plasma total cholesterol, mmol/L | 4.78 ± 0.17 | 8.03 ± 0.18*** |
| Plasma HDL‐cholesterol, mmol/L | 2.19 ± 0.18 | 4.26 ± 0.10*** |
| Plasma triglycerides, mmol/L | 0.73 ± 0.09 | 0.38 ± 0.07** |
| Plasma free fatty acids, mmol/L | 0.76 ± 0.06 | 0.78 ± 0.08 |
| Plasma IL‐6, pg/mL | 5.8 ± 0.8 | 2.0 ± 0.3*** |
| Plasma IL‐1β, pg/mL | 27.8 ± 3.5 | 12.5 ± 1.8*** |
| Plasma IL‐17A, pg/mL | 61.4 ± 10.3 | 32.2 ± 4.2* |
| Plasma TNF‐α, pg/mL | 59.9 ± 5.5 | 28.7 ± 3.6*** |
| Plasma IFN‐γ, pg/mL | 21.7 ± 3.0 | 11.4 ± 1.5** |
| Plasma IL‐10, pg/mL | 68.5 ± 9.3 | 36.5 ± 3.5** |
| Liver weight, g | 1.51 ± 0.04 | 2.07 ± 0.07*** |
| Hepatic total cholesterol, µg/mg | 74.3 ± 3.3 | 19.3 ± 0.7*** |
| Hepatic triglycerides, µg/mg | 31.3 ± 2.5 | 22.0 ± 2.3* |
| Hepatic fatty acids, nmol/mg | 44.8 ± 1.8 | 16.6 ± 0.6*** |
| Hepatic IL‐1β gene expression (fold change vs. vehicle) | 1.00 ± 0.19 | 0.07 ± 0.01*** |
| Hepatic MCP‐1 gene expression (fold change vs. vehicle) | 1.00 ± 0.25 | 0.13 ± 0.03*** |
| Hepatic α‐smooth muscle actin gene expression (fold change vs. vehicle) | 1.00 ± 0.19 | 0.55 ± 0.11* |
| Hepatic coll1α1 gene expression (fold change vs. vehicle) | 1.00 ± 0.17 | 0.27 ± 0.06*** |
| Hepatic steatosis score, 0–3 | 2.80 ± 0.13 | 0.90 ± 0.10*** |
| Hepatic inflammation score, 0–3 | 3.00 ± 0.00 | 0.40 ± 0.16*** |
| Hepatic fibrosis score, 0–4 | 2.00 ± 0.00 | 0.10 ± 0.10*** |
| Total NAS score, 0–10 | 7.80 ± 0.13 | 1.40 ± 0.27*** |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; HDL, high‐density lipoprotein; HFCC/CDX, 60% high‐fat, 1.25% cholesterol, and 0.5% cholic acid diet with 2% cyclodextrin in drinking water; NAS, nonalcoholic fatty liver disease activity scoring.
Data are shown as mean ± SEM, n = 8–10 per group.
P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle.
ELA induced hepatic hypertrophy with a 38% higher liver mass (P < 0.001 vs. vehicle). ELA significantly reduced hepatic total cholesterol, triglycerides, and fatty acids by 63%, 30%, and 74% (Table 2). Additionally, hepatic gene expression of IL‐1β, MCP‐1, coll1α1, and α‐smooth muscle actin was significantly reduced by 93%, 87%, 45%, and 73% (Table 2).
As shown in Figure 2, mice fed a 3‐week HFCC/CDX diet and treated with vehicle showed aggravated liver lesions, as compared with those observed after 1 week of HFCC/CDX diet: steatosis (as shown by hematoxylin and eosin staining) ranged from score‐2, limited to zone‐2/3, to a score‐3 pan‐lobular steatosis (see Figure 2), which was microvesicular in appearance. Maximal inflammation (score 3) was observed in all individuals treated with vehicle (see hematoxylin and eosin staining). This was characterized by the presence of a large number of variably‐sized interstitial foci composed of lymphocytes and macrophages. Portal fibrosis (score 2) was also observed (see Sirius red staining, Figure 2). In line with the improvement in plasma and hepatic parameters above, mice treated with ELA showed major reduction in steatosis and inflammation, as shown by liver sections stained with hematoxylin and eosin (Figure 2). Hepatic fibrosis was also markedly reduced, as shown by Sirius red staining (Figure 2). Accordingly, significant reduction in hepatic steatosis, inflammation, fibrosis, and total NAS scores was observed with ELA (Table 2).
Figure 2.
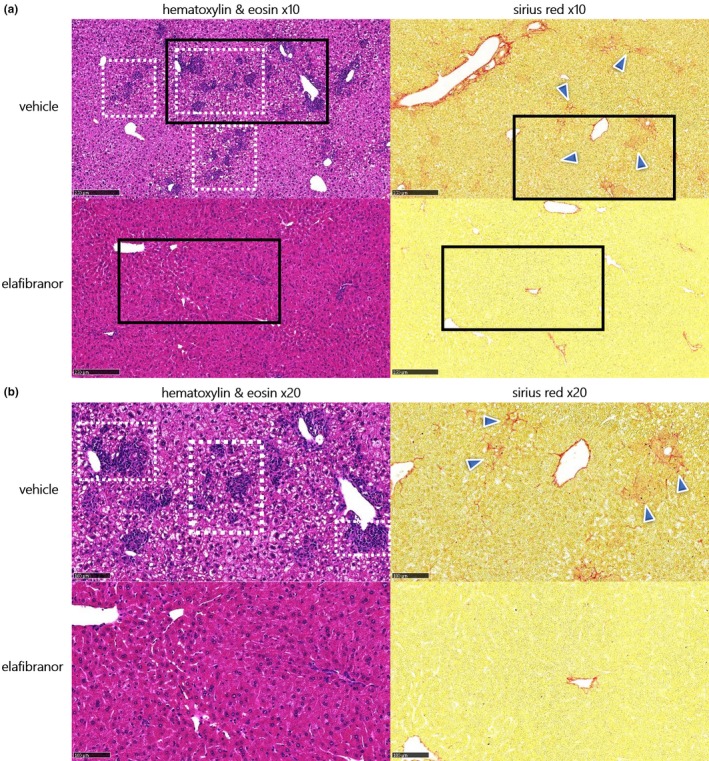
Hematoxylin and eosin and Sirius red stained liver sections at ×10 (a) and ×20 (b) magnification, in 3‐week 60% high‐fat, 1.25% cholesterol, and 0.5% cholic acid diet with 2% cyclodextrin in drinking water diet fed mice treated orally with vehicle or elafibranor 20 mg/kg q.d. for 2 weeks. Black squares localize the area of the ×20 magnification shown in panel (b). Dashed white squares indicate liver steatosis and inflammatory foci, blue arrows indicate portal fibrosis.
Overall, the present data indicate that a 3‐week HFCC/CDX period induces NASH, including major inflammation and portal fibrosis, and that ELA markedly reverses these diet‐induced liver lesions.
ELA improves diet‐induced liver immune cells alteration in HFCC/CDX fed mice
As ELA showed marked improvement in liver inflammation, we further studied the effects on liver immune cells population by flow cytometry. Compared with chow‐fed mice, the total number of immune cells (CD45+) in the liver gradually increased after 1 week‐HFCC/CDX diet (Figure 3 a) and was even higher in vehicle‐treated mice, which corresponds to 3 weeks of HFCC diet (P < 0.05 vs. 1‐week diet). ELA treatment significantly reduced the total number of immune cells (P < 0.05 vs. vehicle).
Figure 3.
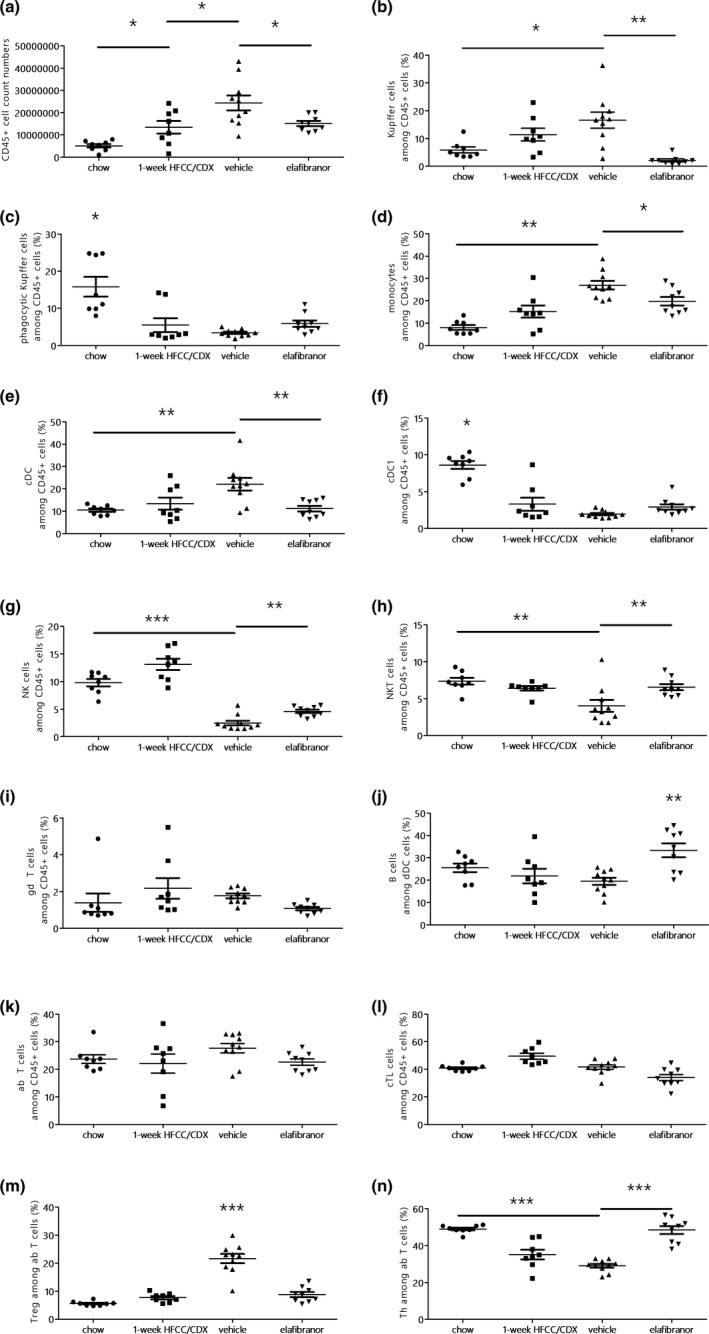
Quantification of liver immune cells by flow cytometry analysis in mice fed with a chow or a 60% high‐fat, 1.25% cholesterol, and 0.5% cholic acid diet (HFCC) diet for 1‐week or HFCC with 2% cyclodextrin in drinking water (CDX) fed mice treated with vehicle or elafibranor for 2 weeks: total CD45+ cell counts (a), F4/80+ Kupffer cells (b), CD68+ phagocytic Kupffer cells (c), CX3CR1+ monocytes (d), CD11c+ conventional dendritic (CD) cells (e), CD8a+ conventional dendritic cells 1 (f), NK1.1+ TCRab‐ natural killer (NK) cells (g), NK1.1+ TCRab+ NK T cells (h), TCRgd+ γδ T cells (i), CD19+ B220+ B cells (j), CD4+/CD8+ T cells (k), CD8+ cytotoxic T lymphocytes (l), FoxP3+ regulatory T lymphocytes (m), and CD4+ helper T lymphocytes (n). Data are shown as mean ± SEM, n = 8–10 per group. *P < 0.05, **P < 0.01, and ***P < 0.001.
The number of Kupffer cells (i.e., liver macrophages), significantly increased during the 3‐week HFCC/CDX diet, as compared with chow‐fed mice, whereas this increase was blunted with ELA (Figure 3 b). Phagocytic Kupffer cells frequency was significantly reduced with HFCC/CDX diet and unchanged with ELA (Figure 3 c).
As shown in Figure 3 d, a gradual increase in monocytes was observed during the 3‐week HFCC/CDX period (P < 0.01 vs. chow‐fed mice), whereas ELA treatment induced a reduction (P < 0.05). HFCC + CDX diet for 3 weeks significantly increased the frequency of conventional dendritic cells (Figure 3 e), whereas ELA treatment induced a decrease of this cell subset (P < 0.001 vs. vehicle). Among those cells, the proportion of conventional dendritic cells 1 was significantly reduced with HFCC/CDX diet, but not altered by ELA (Figure 3 f).
We next tracked lymphoid immune cells as a marker of liver inflammation. The HFCC/CDX diet for 3 weeks significantly reduced the frequency of natural killer (NK) cells, compared with chow‐fed mice (Figure 3 g), wheres ELA had the opposite effect (P < 0.01 vs. vehicle). The HFCC + CDX diet for 3 weeks significantly decreased the frequency of NKT cells (Figure 3 h), which was restored by ELA (P < 0.01 vs. vehicle). The frequencies of unconventional γδ T cells were similar between groups (Figure 3 i).
Although the HFCC/CDX diet showed no effect, ELA significantly increased B cells frequency (Figure 3 j). Frequency of conventional T cells was similar in all groups (Figure 3 k). Frequency of cytotoxic T cells was also similar (Figure 3 l), but the 3‐week HFCC + CDX diet induced a large increase of the Treg population (Figure 3 m), while it decreased the proportion of effector Th cells among conventional T cells (Figure 3 n). ELA treatment blocked this process, with similar frequencies of Treg and effector Th cells compared with chow‐fed mice.
Taken together, these data indicate that the 3‐week HFCC + CDX diet induced liver inflammation with significant alteration in the immune cell’s population, which was markedly improved by ELA treatment.
ELA treatment reduces necroptosis and apoptosis in HFCC/CDX fed mice
We also investigated whether ELA benefits observed at the liver level may alter hepatic cell death pathways (i.e., pyroptosis, necroptosis, and apoptosis) in HFCC/CDX mice. Compared with vehicle‐treated mice, Western blot analysis showed no change in the protein levels of Gasdermin D (a marker of pyroptosis) in ELA‐treated mice (data not shown). However, ELA treatment resulted in a significant increase in cleaved RIP3 levels (P < 0.05 vs. vehicle), indicating lower necroptosis (Figure 4 a,b). Additionally, ELA inhibited the protein expression of cleaved caspase 3, indicating lower apoptosis (Figure 4 a,c). Overall, our data indicate that ELA also has major benefits on necroptosis and apoptosis in the liver, although it does not alter pyroptosis.
Figure 4.
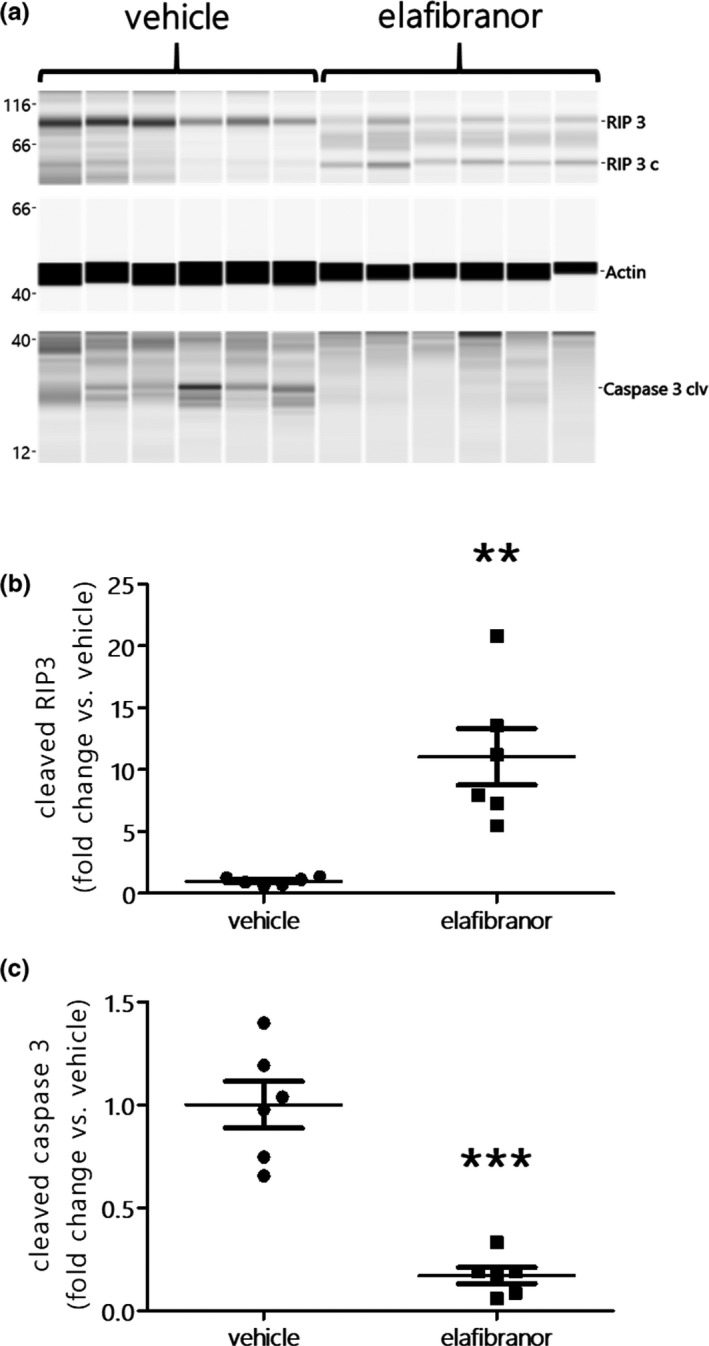
Western blots (a) and protein levels of hepatic cleaved receptor interacting protein 3 (RIP‐3), cleaved caspase 3, and actin from 3‐week 60% high‐fat, 1.25% cholesterol, and 0.5% cholic acid diet with 2% cyclodextrin in drinking water (HFCC/CDX) diet fed mice treated orally with vehicle or elafibranor 20 mg/kg q.d. for 2 weeks. Data are shown as mean ± SEM, n = 6 per group. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle.
DISCUSSION
As effective therapies for the treatment of NASH are urgently needed, we have developed and pharmacologically validated a rapid NASH mouse model for drug efficacy studies. Although other available animal models require chemical, genetic, or long dietary interventions, the HFCC/CDX diet‐fed mouse develops NASH in 3 weeks, with cholesterol and liver inflammation as major drivers.
Our flow cytometry analysis indicated an increase in the number of Kupffer cells, monocytes, and dendritic cells. Notably, this increase was concomitant with a raise in INF‐γ and IL‐10 plasma levels, two cytokines secreted by Kupffer cells, whereas hepatic dendritic cells mostly secrete IL‐10.8, 16 Increased activation of hepatic Kupffer cells and recruitment of monocytes, both representing above 80% of the body’s macrophages,17 has been shown to play a role in the transition from NAFLD to NASH.16 Additionally, phagocytic Kupffer cells, which are the main players in defending the organism in a healthy liver,16 were reduced with the HFCC/CDX diet. We also observed a reduced frequency of NK cells, which play an important role in inhibiting the development of fibrosis, by directly killing newly activated and senescent hepatic stellate cells.18 As well, NKT cells’ frequency was reduced after 3 weeks of diet, which would be in line with human studies indicating decreased frequencies of NKT cells in C57BL6/J mice and patients with NASH.19 Meanwhile, other studies have shown opposite results with NKT cell accumulation in NAFLD and patients with NASH.20 Indeed, the liver disease stage together with the kinetics of NKT cell infiltration in the liver should be considered carefully to compare and interpret the present results. In the same line, the HFCC/CDX diet induced a marked increase in Treg cells frequency in the liver, whereas the opposite effect is observed in other animal or human studies investigating the role of Treg cells in NASH.21 As the frequency of conventional T cells remained unchanged, we also observed a reduction in Th cells after 3 weeks of the HFCC/CDX diet. Although this would require further investigations, it is possible that the maximal levels of liver inflammation in our 3‐week NASH mouse model, as shown by the histopathological inflammation scoring, may actually favor the plasticity of effector Th cells to become induced Treg cells.
By activating PPARα, ELA modulates fatty acids transport and β‐oxidation, reduces plasma triglycerides, and increases HDL‐cholesterol, and inhibits inflammatory genes.22 ELA also activates PPARδ, resulting in improved fatty acid transport and oxidation, increased HDL levels, and anti‐inflammatory effects in macrophages and Kupffer cells.23, 24 Our data suggest that all these metabolic benefits occurred in our 3‐week NASH mouse model, resulting in reduced steatosis and inflammation, thereby preventing cell death and fibrosis. Interestingly, ELA also resolved NASH, without fibrosis worsening, and demonstrated clear reduction in systemic inflammatory markers in the GOLDEN‐505 human clinical trial.12 Although it does reduce plasma ALT in human patients with NASH, ELA induced a significant increase in the 3‐week HFCC/CDX diet fed mice. However, this effect is actually expected in rodents treated with PPARα and PPARδ agonist,25, 26 as well as the hepatic hypertrophy,27 observed in the present study. An increase in ALT levels can occur without any liver damage,28 as demonstrated in rats and humans showing elevated serum ALT activity when treated with fibrates,26, 29 which are PPARα agonists.
The pronounced hepatoprotective effects of ELA in the 3‐week HFCC/CDX fed mice also resulted in reduced necroptosis and apoptosis in the liver, as tracked with cleaved RIP3 and cleaved caspase 3 protein levels. Although this obviously results from a drastic improvement in NASH, a direct effect of ELA on necroptosis and apoptosis may not be excluded, as other mouse studies have indicated reduction in apoptosis in adipose tissue and kidneys in models of alcoholic liver disease and NASH.30, 31 Studies in mice have suggested a direct link between IL‐1β secretion and pyroptosis through Gasdermin D, which would play an important role in NASH‐related cell death.32 Despite the major improvement in liver lesions, the significant reduction in IL‐1β hepatic gene expression and plasma IL‐1β levels with ELA, hepatic Gasdermin D protein levels still remained unaffected. Hence, the contribution of pyroptosis in liver cell death and its eventual alteration by ELA treatment remain to be clarified in other NASH models.
Although other reference drugs targeting NASH will have to be evaluated in our 3‐week HFCC/CDX fed mice, the impaired inflammatory profile and the positive effects of ELA suggest that this model will be useful to rapidly detect the efficacy of novel therapies targeting NASH prior to clinical trials. To our knowledge, there are few models developing advanced liver lesions in such a short period of time, and they have limitation for translating data to human NASH, because they require chemical intoxication (e.g., carbon tetrachloride)33 or amino acids depleted diets, inducing severe body weight loss (e.g., methionine choline deficient diet).34 However, the amino acids depleted diet‐induced weight loss effect may be compensated with the use of genetically modified obese mice. In the db/db mouse model fed a methionine choline deficient diet and simultaneously treated with ELA for 7 weeks, significant reduction in steatosis and inflammation, but not fibrosis, was observed Staels et al.35 The diet‐induced obese NASH models have also been used to demonstrate the benefits of ELA. When administered for 8 weeks, ELA induced significant reduction in steatosis, inflammation, and fibrosis scores in both C57BL6/J and Lepob/ob mice fed a high‐fat/cholesterol/fructose diet for 30 and 21 weeks, respectively, Tolbol et al.36 Thus, other models also demonstrated similar benefits of ELA, but a substantial duration of diet and treatment were required. In contrast, the sensitivity of the HFCC/CDX diet fed mouse model, which rapidly develops NASH, enabled to detect the benefits of ELA within 3 weeks.
One major limitation of this very short HFCC/CDX diet period is the lack of weight gain, obesity, and diabetes, which are important factors in favoring NAFLD and NASH. Although the high fat content of the diet compensates for the weight loss, the diet supplementation with cholic acid certainly prevents any weight gain induction in mice, likely through higher energy expenditure.37
In conclusion, the present study indicates that our 3‐week NASH mouse model allows a rapid evaluation of the efficacy of drugs targeting NASH and will also be helpful to dissect the inflammatory mechanisms shifting simple steatosis into more complicated liver disease states, including liver cell death and fibrosis.
Funding
This study was funded by Physiogenex.
Conflict of Interest
F.B., L.B., E.B., M.Q., and T.S. are employees of Physiogenex. F.B., R.B., and T.S. declare shares in Physiogenex. All other authors declared no competing interests for this work.
Authors Contribution
F.B. wrote the manuscript. F.B., R.B., and T.S. designed the research. C.H., L.B., T.A., J.C., M.B., E.B., and M.Q. performed the research. F.B. and N.F. analyzed the data.
Supporting information
Figure S1. Experimental design.
Figure S2. Plasma ALT/AST levels (A), liver total cholesterol, triglycerides and fatty acids (B) and hepatic gene expression of α‐smooth muscle actin (α‐SMA), collagen 1α1 (coll1α1), interleukin 1β (IL1β) and monocyte chemoattractant protein 1 (MCP‐1) in mice fed a control chow diet, HFCC or HFCC/CDX diet for 3 weeks. Data are presented as mean ± SEM, n = 7–10, *P < 0.05, **P < 0.01 and ***P < 0.001 vs. control chow.
Figure S3. Original non‐cropped/cut western blots corresponding to Figure 4, with molecular weight marker and the full get/membrane.
Acknowledgments
The authors thank colleagues and collaborators from Physiogenex (Dominique Lopes, Paolo Demoor, Cindy Moriceau, and Hélène Marty for animal care, Clément Costard, Isabelle Urbain, Noémie Burr, and Mylène Bernes for technical assistance, and Sura Setau for audits and quality control of experiments and data), Inserm 1048 (Laurent Monbrun and Alexandre Lucas (We‐Met platform) for technical assistance) and Sciempath Labo (Audrey Chabrat and Virgile Richard for histology expertise).
References
- 1. Yonoussi, Z.M. Non‐alcoholic fatty liver disease – a global public health perspective. J. Hepatol. 70, 531–544 (2019). [DOI] [PubMed] [Google Scholar]
- 2. Kutlu, O. , Kaleli, H.N. & Ozer, E. Molecular pathogenesis of nonalcoholic steatohepatitis‐ (NASH‐) related hepatocellular carcinoma. Can. J. Gastroenterol. Hepatol. 29, 8543763 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Samji, N.S. et al Liver transplantation for nonalcoholic steatohepatitis: pathophysiology of recurrence and clinical challenges. Dig. Dis. Sci. 64, 3413–3430 (2019). [DOI] [PubMed] [Google Scholar]
- 4. Friedman, S.L. , Neuschwander‐Tetri, B.A. , Rinella, M. & Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–922 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arguello, G. , Balboa, E. , Arrese, M. & Zanlungo, S. Recent insights on the role of cholesterol in non‐alcoholic fatty liver disease. Biochim. Biophys. Acta 1852, 1765–1778 (2015). [DOI] [PubMed] [Google Scholar]
- 6. Sehayek, E. , Shefer, S. , Nguyen, L.B. , Ono, J.G. , Merkel, M. & Breslow, J.L. Apolipoprotein E regulates dietary cholesterol absorption and biliary cholesterol excretion: studies in C57BL/6 apolipoprotein E knockout mice. Proc. Natl. Acad. Sci. U S A 97, 3433–3437 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Povsic, M. , Oliver, L. , Jiandani, N.R. , Perry, R. & Bottomley, J. A structured literature review of interventions used in the management of nonalcoholic steatohepatitis (NASH). Pharmacol. Res. Perspect. 7, e00485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Farrell, G.C. , van Rooyen, D. , Gan, L. & Chitturi, S. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut Liver 6, 149–171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murphy, C. , Parini, P. , Wang, J. , Björkhem, I. , Eggertsen, G. & Gåfvels, M. Cholic acid as key regulator of cholesterol synthesis, intestinal absorption and hepatic storage in mice. Biochim. Biophys. Acta 1735, 167–175 (2005). [DOI] [PubMed] [Google Scholar]
- 10. Vergnes, L. , Phan, J. , Strauss, M. , Tafuri, S. & Reue, K. Cholesterol and cholate components of an atherogenic diet induce distinct stages of hepatic inflammatory gene expression. J. Biol. Chem. 278, 42774–42784 (2003). [DOI] [PubMed] [Google Scholar]
- 11. Christian, A.E. , Haynes, M.P. , Phillips, M.C. & Rothblat, G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 38, 2264–2272 (1997). [PubMed] [Google Scholar]
- 12. Ratziu, V. et al Elafibranor, an agonist of the peroxisome proliferator‐activated receptor‐α and ‐δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150, 1147–1159.e5 (2016). [DOI] [PubMed] [Google Scholar]
- 13. Briand, F. , Thiéblemont, Q. , Muzotte, E. & Sulpice, T. High‐fat and fructose intake induces insulin resistance, dyslipidemia, and liver steatosis and alters in vivo macrophage‐to‐feces reverse cholesterol transport in hamsters. J. Nutr. 142, 704–709 (2012). [DOI] [PubMed] [Google Scholar]
- 14. Lolmède, K. et al Interrelationship between lymphocytes and leptin in fat depots of obese mice revealed by changes in nutritional status. J. Physiol. Biochem. 71, 497–507 (2015). [DOI] [PubMed] [Google Scholar]
- 15. Kleiner, D.E. et al Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005). [DOI] [PubMed] [Google Scholar]
- 16. Cha, J.Y. , Kim, D.H. & Chun, K.H. The role of hepatic macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Lab. Anim. Res. 34, 133–139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Narayanan, S. , Surette, F.A. & Hahn, Y.S. The immune landscape in nonalcoholic steatohepatitis. Immune Netw. 16, 147–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krizhanovsky, V. et al Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kremer, M. et al Kupffer cell and interleukin‐12‐dependent loss of natural killer T cells in hepatosteatosis. Hepatology 51, 130–141 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Syn, W.K. et al Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 51, 1998–2007 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paquissi, F.C. Immune imbalances in non‐alcoholic fatty liver disease: from general biomarkers and neutrophils to interleukin‐17 axis activation and new therapeutic targets. Front. Immunol. 7, 490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pawlak, M. , Lefebvre, P. & Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non‐alcoholic fatty liver disease. J. Hepatol. 62, 720–733 (2015). [DOI] [PubMed] [Google Scholar]
- 23. Bojic, L.A. & Huff, M.W. Peroxisome proliferator‐activated receptor δ: a multifaceted metabolic player. Curr. Opin. Lipidol. 24, 171–177 (2013). [DOI] [PubMed] [Google Scholar]
- 24. Odegaard, J.I. et al Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity‐induced insulin resistance. Cell Metab. 7, 496–507 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kobayashi, A. , Suzuki, Y. , Kuno, H. , Sugai, S. , Sakakibara, H. & Shimoi, K. Effects of fenofibrate on plasma and hepatic transaminase activities and hepatic transaminase gene expression in rats. J. Toxicol. Sci. 34, 377–387 (2009). [DOI] [PubMed] [Google Scholar]
- 26. Faiola, B. et al PPAR alpha, more than PPAR delta, mediates the hepatic and skeletal muscle alterations induced by the PPAR agonist GW0742. Toxicol. Sci. 105, 384–394 (2008). [DOI] [PubMed] [Google Scholar]
- 27. Gonzalez, F.J. Recent update on the PPAR alpha‐null mouse. Biochimie 79, 139–144 (1997). [DOI] [PubMed] [Google Scholar]
- 28. Thulin, P. et al PPARalpha regulates the hepatotoxic biomarker alanine aminotransferase (ALT1) gene expression in human hepatocytes. Toxicol. Appl. Pharmacol. 231, 1–9 (2008). [DOI] [PubMed] [Google Scholar]
- 29. Blane, G.F. Comparative toxicity and safety profile of fenofibrate and other fibric acid derivatives. Am. J. Med. 83, 26–36 (1987). [DOI] [PubMed] [Google Scholar]
- 30. Li, T.H. et al Elafibranor interrupts adipose dysfunction‐mediated gut and liver injury in mice with alcoholic steatohepatitis. Clin. Sci. (Lond.) 133, 531–544 (2019). [DOI] [PubMed] [Google Scholar]
- 31. Tsai, H.C. et al Elafibranor inhibits chronic kidney disease progression in NASH mice. Biomed. Res. Int. 2019, 6740616 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu, B. et al Gasdermin D plays a key role as a pyroptosis executor of non‐alcoholic steatohepatitis in humans and mice. J. Hepatol. 68, 773–782 (2018). [DOI] [PubMed] [Google Scholar]
- 33. Kim, Y.O. , Popov, Y. & Schuppan, D. Optimized mouse models for liver fibrosis. Methods Mol. Biol. 1559, 279–296 (2017). [DOI] [PubMed] [Google Scholar]
- 34. Zhang, S. , Wang, J. , Liu, Q. & Harnish, D.C. Farnesoid X receptor agonist WAY‐362450 attenuates liver inflammation and fibrosis in murine model of non‐alcoholic steatohepatitis. J. Hepatol. 51, 380–388 (2009). [DOI] [PubMed] [Google Scholar]
- 35. Staels, B. et al Hepatoprotective effects of the dual peroxisome proliferator‐activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 58, 1941–1952 (2013). [DOI] [PubMed] [Google Scholar]
- 36. Tølbøl, K. et al Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet‐induced obese mouse models of biopsy‐confirmed nonalcoholic steatohepatitis. World J. Gastroenterol. 24, 179–194 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Watanabe, M. et al Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439, 484–489 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Experimental design.
Figure S2. Plasma ALT/AST levels (A), liver total cholesterol, triglycerides and fatty acids (B) and hepatic gene expression of α‐smooth muscle actin (α‐SMA), collagen 1α1 (coll1α1), interleukin 1β (IL1β) and monocyte chemoattractant protein 1 (MCP‐1) in mice fed a control chow diet, HFCC or HFCC/CDX diet for 3 weeks. Data are presented as mean ± SEM, n = 7–10, *P < 0.05, **P < 0.01 and ***P < 0.001 vs. control chow.
Figure S3. Original non‐cropped/cut western blots corresponding to Figure 4, with molecular weight marker and the full get/membrane.