

Abstract
Traditional methods to assess microbial cells during suspension culture require laborious and frequent manual sampling. Approaches to automate sampling and assessment utilize dedicated, sophisticated equipment and suffer from a lack of temporal resolution and sampling efficiency. In this study we describe a simple microfluidic device that allows microbial cells to be sampled from suspension culture and rapidly slowed and concentrated for single-cell imaging on a standard laboratory microscope. We demonstrate a device that:
-
•
slows and concentrates microbial cells, specifically budding yeast, sampled from suspension culture and improves imaging of individual cells by concentrating them in a single focal plane
-
•
provides imaging quality and temporal resolution that is capable of monitoring dynamic spatiotemporal processes, such as nuclear localization of a protein
-
•
is inexpensive and simple enough to be fabricated and used in laboratories equipped for standard molecular and cellular biology
Keywords: Microfluidic, Single-cell microscopy, Microbial cell culture, Bioreactor
Graphical abstract
Specifications table
| Subject Area | Immunology and Microbiology |
| More specific subject area | Single-cell imaging, bioreactors and continuous culture, microfluidic sampling |
| Method name | A Microfluidic Sampling and Imaging Device for Microbial Cultures |
| Name and reference of original method |
Stewart and McClean (2017) Design and Implementation of an Automated Illuminating, Culturing, and Sampling System for Microbial Optogenetic Applications Journal of Visualized Experiments (120) e54894 doi:10.3791/54894 Melendez, et al (2014) Real-time optogenetic control of intracellular protein concentration in microbial cell cultures Integrative Biology 6(3) 366-72 doi:10.1039/c3ib40102b |
| Resource availability | github.com/McCleanResearch |
Method details
Suspension culture techniques are the most widely-used form of culture for microbial cells. When suspension culture is combined with continuous-culture techniques, microbial cells can be maintained in precisely controlled environments for extended periods of time [1,2]. The ability to sample and monitor cells from these suspension cultures is fundamental to the design and implementation of experiments that examine cellular properties such as growth, metabolism, stress-response, and gene expression. Information gained from sampling can even be used in real-time feedback schemes to control culture conditions or cellular processes such as gene expression [3], [4], [5].
Traditionally, samples from suspension culture have been taken manually. Abundance measurements, for example, are often made by manually sampling cells from the culture followed by plating or hemocytometry. Manual sampling and imaging can be combined with more sophisticated staining and imaging to provide information on cell morphology and protein localization. While simple to implement, manual sampling methods are labor-intensive and subject to poor temporal resolution.
Several methods for automated, real-time sampling from microbial suspension cultures exist [5], [6], [7], [8]. Automated optical density measurements are cost effective and rapid, but these measurements lack single-cell resolution and dynamic range. Integration of sampling with flow-cytometry can acquire single-cell information and benefits from the ability to acquire accurate data at high throughput [9]. Appropriate analysis techniques, or integration with an image-based flow cytometer, can allow for single-cell properties such as size, morphology, and protein-localization to be assessed using flow cytometry [10,11]. However, flow cytometry is difficult to automate and requires expensive equipment. It also provides only limited information about cell size, shape, and specific protein localization [12,13]. This is particularly problematic for microbes, where light scattering correlates poorly with size, making it difficult to interpret data even for high-abundance proteins [14].
Recent studies have integrated sampling with microfluidic devices to control and trap cells for more conventional imaging on standard optical microscopes [3,6,15,16]. These methods are advantageous as they allow automated sampling from suspension culture to be integrated with a lab's existing imaging and analysis techniques. Furthermore, the ability to interface with a microscope allows additional aspects of cellular physiology, such as morphology and protein localization to be assayed [3,[15], [16], [17]. However, these methods suffer from poor image quality due to localization of cells at multiple focal planes within a microfluidic device [3,6]. Trapping can improve images, but trapping presents its own challenges. Trapping techniques based on constriction often require complex devices, are low throughput, and cell removal is difficult [18]. Mechanical trapping protocols can slow sampling acquisition, require additional hardware and software, and can have an adverse effect on cellular physiology [15,16].
Our research group recently described a microfluidic device to sample budding yeast (Saccharomyces cerevisiae) cells from liquid culture and measure protein concentration using fluorescence [3,17]. Continuous sampling of cells and imaging for fluorescence allowed us to develop a real-time control scheme for controlling protein levels using optogenetics [3]. Our microfluidic device consisted of a straight microfluidic channel. Cells were imaged after being allowed to settle to the bottom of the channel. While this technique was effective, and simple to implement, our device suffered limitations including (1) small sample sizes due to the inability to concentrate cells and (2) coarse temporal resolution due to the time needed for cell settling.
In this article, we design and test microfluidic devices that improve on our previous design by using expansion-based trapping to slow and concentrate budding yeast, Saccharomyces cerevisiae, for imaging on an inverted epifluorescence microscope. Using this device, we can continuously sample and image yeast cells for total fluorescence, morphology, and protein localization. Our optimal device improves cell concentration and sampling speed several fold over previous protocols [3,17] but is simple enough to be implemented by most labs currently employing suspension culture and image-based cell assessment.
Design of Microfluidic Channels
In our original microfluidic sampling device, we employed a straight microfluidic channel with dimensions 50 mm X 500 µm X 100 µm [3,17]. Cells were imaged by pumping them into the device positioned on a standard inverted epifluorescence microscope, stopping flow, and waiting for cells to settle before images were taken. Cell settling took several minutes and few cells were captured per image due to low cell density and small volumes in the microfluidic channel. To improve cell settling time and concentration, we decided to test an expansion-based trapping technique [19,20]. In expansion-based trapping, a rapid expansion in the cross-sectional area of a microfluidic channel leads to a decrease in flow velocity as a consequence of the continuity equation for incompressible fluid flow [21]
| (1) |
which states that the flow rate at one point in the channel ( be equal to the flow rate at any other point in the channel (. Here v is the fluid's velocity and A is the area through a cross-section of the channel perpendicular to the flow direction.
Previous cell-trapping microfluidic devices based on expansion have utilized microfluidic devices where the height of the channel rapidly increases to slow fluid flow [19]. However, these devices are constructed using multilevel masks which require more sophisticated photolithography techniques and alignment. We decided to test three microfluidic channels where rapid expansion is achieved by increasing the width of the microfluidic channel. The schematics for these devices are shown in Fig. 1. These designs are modifications of our original straight channel design [3]. Halfway between the fluid inlets and outlets we designed a circular 750µm diameter vestibule. The three variations of this design vary the orientation between the channels entering and leaving the vestibule.
Fig. 1.

Schematics of the 90°, 120° and 180° designs. All designs have a central vestibule 750 µm in diameter for trapping cells. The designs differ in the orientation between the channel entering and the channel leaving the vestibule.
Simulations to Verify Device Design
The motivation for our design is to introduce an area with significantly lower flow velocity than the rest of the channel due to rapid expansion in the cross-sectional area and a concomitant reduction in flow velocity. In order to verify our assumptions before building the devices, Comsol Multiphysics 4.2 [22] was used to model the velocity profile of fluid flow within the channel prototypes (Fig. 1) in comparison to the original straight channel design. The Navier-Stokes equation for incompressible fluid steady-state flow was employed to define the physics of the model:
| (2) |
where F is the body force, u is the fluid velocity, ρ is the fluid density, and µ is the fluid viscosity.
The Comsol microfluidics module used a single-phase flow for testing. Velocity profiles were calculated under the assumption that fluid density ρ = 1000 kg/m3 and fluid viscosity µ= 1 cP. These are the values for water, the primary component of the cell culture media. Body forces were assumed to be zero, since the effect of gravity is negligible on the microscale and no electromagnetic forces are applied during the validation experiments. The volumetric flow was simulated at 4 ml/hr in keeping with our experimental design. Creeping flow, in which the Reynolds number is much less than one, was assumed for the purpose of these models. This is justified because the Reynolds number is small for microfluidic applications due to the small hydraulic diameter of the channel. For illustration, Reynolds number is where u is the fluid velocity, ρ is the fluid density, D is the hydraulic diameter of the microfluidic channel, and µ is the dynamic viscosity of the fluid. If ρ = 1000 kg/m3, µ = 1 cP, and D =100 μm, then the Reynolds number for the flow regime will be less than 1 provided that fluid velocity is less than 10 cm/s. This is a reasonable assumption for most microfluidic flow and greater than our experimental flow rate of ~3.7 cm/s
The flow characteristics of the channel designs simulated in Comsol Multiphysics Modeling Software are show in Fig. 2. Simulations show that introduction of the circular vestibule slows flow, as expected due to the sudden change in channel cross-sectional area. Simulations also show that the bent geometries create significantly larger regions of slowed flow which is preferable for creating larger regions of trapped cells.
Fig. 2.

Creeping flow was simulated in the three device designs, as well as the original straight channel design, using Comsol. Fluid density (ρ=1000 kg/m3) and fluid viscosity (µ=1 cP) were assumed to match the values for water, the major component of cell culture media. Flow rates are in mm/s.
Photolithography and Soft Lithography to Fabricate Microfluidic Mold
Based on the promising simulation results, we fabricated the channel designs by creating a photolithographic master mold for repeated casting of polydimethylsiloxane (PDMS) microfluidic chips. The following procedures were used to construct the SU8 master mold and PDMS chips:
Materials
-
•
Appropriate design software (e.g. AutoCad)
-
•
Photomask (10160 DPI, Dark Field Right Read Down Orientation and Polarity, Fine Line Imaging, Colorado Springs, CO)
-
•
3” Silicon Wafers (P(100) 0-100 ohm-cm SSP 500µm Test 100 crystal orientation, one-side polished) (University Wafers, South Boston, MA)
-
•
Negative Photoresist SU8-50 (Microchem Corp, Newton, MA)
-
•
SU Developer Propylene Glycol Methyl Ether Acetate (PGMEA) (Microchem Corp, Newton, MA)
-
•
Spincoater (Spincoater Model P6700, Specialty Coating Systems, Indianapolis, IN)
-
•
4” Petri Dish with lid
-
•
3M Scotch tape, Matte Finish, Magic Tape (3M, St. Paul, MN)
-
•
Disposable plastic dropper
-
•
Aluminum Foil
-
•
Hot plate at 65°C; Hot plate at 95°C
-
•
Wafer tweezers (Unifit)
-
•
Large glass container for developing wafer (at least 4” deep and 6” in diameter)
-
•
Isopropyl alcohol in a squeeze bottle
-
•
Dow Corning SLYGARD 184 Elastomer kit containing Slygard 184 base, Sylgard184 curing agent (Polydimethylsiloxane-PDMS) (Ellsworth Adhesive Systems)
-
•
Cover Glass 23 × 60 mm No. 1.5 Cat. No 12544G (Fisher Scientific)
-
•
Omnicure 365 nm Lumen Dynamics S1500
-
•
Diener Fempto Plasma Cleaner (Diener Electronic GmbH, Ebhausen, DE)
-
•
VWR Gravity Convection Oven Model 1300 U (VWR International Inc)
-
•
1.2 mm inner diameter biopsy punch (Harris Uni-core WHAWB100028)
-
•
190 mm vacuum desiccator (Bel-Art)
Transparency Mask and Master Mold Creation
-
1.
Channel designs were rendered in AutoCad. AutoCad designs were converted to plotter-ready files and printed as Mylar photomasks by Fine Line Imaging (Colorado Springs, Colorado) at 10,160 DPI resolution with Dark Field Right Read Down (DFRRD) orientation and polarity.
-
2.
Master molds for PDMS casting were constructed on 3” silicon wafers. Wafers were placed on a 2” chuck in the P6700 Spincoater. SU8-50 was poured onto the middle of the wafer until approximately 1/3 of the wafer surface was covered, taking care to avoid bubbles. The wafer was coated with SU8 using the following spin program: Step 1) 500 rpm, ramp 100 rpm/s, duration 10 s; Step 2) 1500 rpm, ramp 300 rpm/s, duration 30 s; Step 3) speed 0 rpm, ramp 500 rpm/s, duration 0 s
-
3.
Before UV exposure, coated wafers were baked at 65°C for 8 minutes, followed by 25 minutes at 95°C.
-
4.
Wafers were placed under the UV light source (Omnicure 365 nm, Lumen Dynamics S1500) and exposed to 1 minute of light for approximately 375 mJ/cm2. Exposure times will vary depending on the output from the available light source.
-
5.
Wafers were post-exposure baked for 1 minute at 65°C and 7 minutes at 95°C
-
6.
Once baked, wafers were developed by submerging them in PGMEA with constant gentle agitation. Roughly 3 minutes of development were required to remove uncured SU8. Development was monitored by eye and using a stereoscope to check the channel edges for complete removal of undeveloped SU8.
-
7.
Developed wafers were washed with isopropyl alcohol and dried gently with a filtered stream of nitrogen.
-
8.
Wafers were again baked at 120°C for 30 minutes and placed in a clean petri dish for subsequent use.
Soft Lithography for Microfluidic Channel Creation
-
1.
PDMS channels were made by first casting PDMS onto the master masks. The Sylgard184 base was weighed in a clean weigh boat up to 30 g. To the base, 3 grams of Slygard184 curing agent were added and the solution was mixed using a plastic fork.
-
2.
Bubbles were removed from the PDMS by degassing in a vacuum desiccator hooked up to a vacuum source. The mixing container was monitored to assure that the PDMS did not bubble over during degassing. If this starts to happen the vacuum can be temporarily released to avoid spillage.
-
3.
A square piece of foil was placed in a petri dish, and the master mold to be cast was placed on top of this.
-
4.
Once degassed, the PDMS was poured over the master wafer and covered with a petri dish lid.
-
5.
The PDMS was baked at 65°C for 8 hours.
-
6.
Once cured, the PDMS was removed from the oven and the PDMS carefully peeled from the wafer. Channels were manually separated using a razor blade and the side of the PDMS containing the channel was covered with tape to avoid contamination with dust and other particulates.
-
7.
Inlet and outlet ports were punched using a 1.2 mm inner diameter biopsy punch.
-
8.
PDMS channels were cleaned three times by covering and removing tape. They were then bonded to clean glass coverslips using a Diener Fempto Plasma Machine. Both the glass coverslip and the PDMS chip were exposed to plasma for 60 seconds after which point the channel side of the PDMS was applied to the glass coverslip.
-
9.
Bonded channels were baked again at 65°C for 8 hours to improve adhesion of the PDMS to the glass.
-
10.
For stability and to protect the fragile coverslips, bonded channels were mounted to a custom aluminum frame with outer dimensions of 7.5 cm X 3 cm, thickness of 0.1 cm, and a central cutout to allow for imaging with dimensions of 2.25 cm X 2.25 cm. The coverslip was mounted by gently taping it to the frame using standard laboratory tape.
-
11.
Tubing (24 AWG PTFE) was manually inserted into the inlets and outlets to prepare the microfluidic device for processing of cell culture.
Cell Enrichment in the Imaging Areas
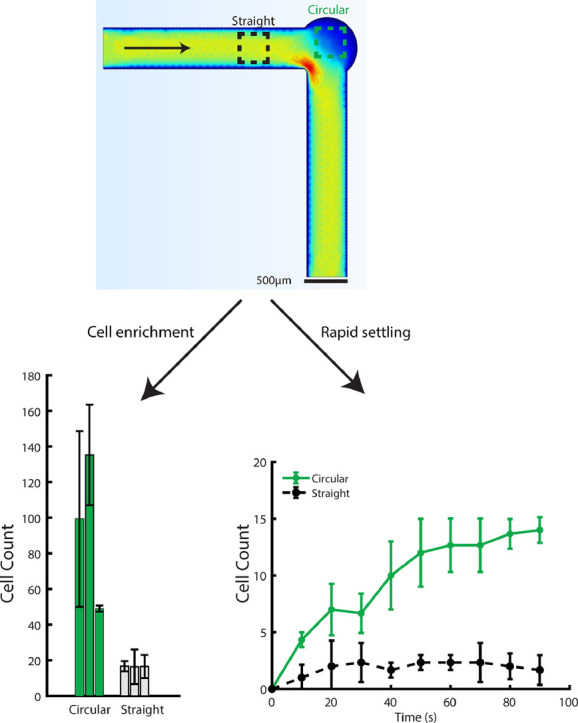
In order to image as many cells as possible from suspension culture, our device should concentrate cells in the imaging vestibules. In order to measure enrichment in each device, we used a syringe pump (Chemyx Fusion 100) to pump 2 ml of constitutively fluorescent yeast cells (TEF1-GFP) in 10 ml syringes (BD syringes) through each of the devices. The cell culture was at a concentration of 1 × 108 cells/ml. The devices were placed on an inverted Nikon microscope (see Supplemental Note 1) as in Melendez, et al and Stewart, et al [3,17]. The microscope is equipped with an automated stage and was programmed to take images at three stage positions within the circular imaging area and three within the straight channel areas (Fig. 3). Cell culture was pumped through the imaging apparatus at a rate of 4 ml/hr for 30 seconds. Flow was stopped and cells were allowed to settle for 60 seconds. After settling, z-stacks were taken in the GFP channel. This procedure was repeated at least three times per stage position. A custom ImageJ script (see Supplemental Note 2) was used to count cells at each stage position.
Fig. 3.

The ability of each device to concentrate flow was tested by flowing yeast cells at a density of 1 × 108 cells/ml through the device for 30 seconds, stopping flow, and allowing cells to settle for 60 seconds. (A) Counting cells collected at positions P1-P6 shows that cells preferentially collect in the vestibules (green), especially towards the outer edge of the vestibule where flow is slowest. The 90° device performs best, enriching the cell concentration seven-fold over other parts of the same device. By ANOVA followed by Tukey's HSD the circular vestibule positions contained statistically significantly more cells (all p < 0.0059) while the 90° bent design collects more cells than either the straight or angled design (all p < 6.7 × 10−7). In the 90° device the P1-P3 positions collect significantly more cells than positions P4-P6 (p < 0.008). (B) Stage positions where cells were counted. Positions P1-P3 are within the circular viewing areas and positions P4-P6 are within the straight channels. The arrow indicates the direction of flow.
Fig. 3 shows the average cell counts at each stage position for each device. We found that after stopping flow cells preferentially collect at the outer regions of the imaging vestibules, consistent with the expected flow profiles in Fig. 2. Regions in the center of the imaging vestibules yield cell counts comparable to those in the straight channel, consistent with those areas having higher flow rates. The 90° device provided the largest enrichment, especially at position P2, which is predicted from the Comsol simulations to have the slowest flow. This experiment confirms that low flow velocity areas preferentially trap and concentrate cells after flow is stopped. This increase in average cell count per image (seven-fold) allows higher sample sizes of imaged cells to be obtained in fewer imaging cycles, improving this sampling technique from microbial suspension cultures.
Verification of enrichment at the focal plane in the circular vestibules
Due to the differences in flow dynamics between the circular imaging area and the straight channel, we decided to investigate the possibility of image quality differences between these areas. For optimal image quality and imaging speed, all cells should align on the same z-directional focal plane. To determine how cells aligned in the circular vestibules relative to the straight channels, we analyzed z-stacks taken at each stage position. At each stage position, the microscope was programmed to take a z-stack spanning 40 µm with a Δz of 2 µm. The z-stack was centered around a prime focal plane determined by flowing cells through the channel, stopping flow and allowing them to settle for 60 seconds, and then manually determining the appropriate z-plane for optimal focus. We analyzed the fluorescence intensity of each cell over the span of the z-stack at each stage position (Fig. 4). The focal plane of the cell was determined using a custom ImageJ script to find the z-stack with the maximal intensity (see Supplemental Note 2).
Fig. 4.

The percentage of cells at a specific z-position relative to the optimal focal plane (which is at z = 0 µm). While the straight channel device shows the narrowest spread at all viewing positions, the bent device also shows a narrow spread within the vestibule. In the 90° bent design the vestibule traps cells significantly closer to the focal plane than the straight regions of the same device.
We found that within the imaging vestibules of all devices most cells fell within a 6 µm spread centered at the optimal focal plane (these cells are ~5µm in diameter). In the straight and 90° devices a significant number of cells settled off plane in the straight areas of the channels. Cells in the straight channel portion fall within a 12 µm spread. This indicates that cells settle closer to the focal plane within the vestibule. This result makes sense; some slow media flow remains in the straight portion after the cells settle, displacing them from the focal plane, but this effect is attenuated in the vestibule. These results indicate that images taken within the vestibule will be more in-focus than those in the straight channel, facilitating automated analysis by software such as ImageJ.
Verification of Cell Settling in the Microfluidic Device
We further characterized the settling time within the 90° device as it provided the best cell trapping capabilities (Fig. 3). One of the motivating factors for creating this device was to improve cell settling time, and therefore sampling temporal resolution. In order to verify that our expansion-based trapping scheme rapidly trapped cells we performed validation experiments with Saccharomyces cerevisiae cells using the same imaging setup described above. Cell culture was pumped through the bent channel apparatus as before. Flow was stopped and phase contrast images were taken at the predetermined positions every ten seconds for a period of 90 seconds. Cell counts were manually conducted on the images obtained at each XY position. Fig. 5 shows cell counts in two representative stage positions within the circular viewing area and within the straight channel. Cells within the circular viewing area took approximately 60 seconds to settle, a substantial improvement over the several minutes reported for Melendez, et al and Stewart, et al [3,17]. Cells in the straight channel in fact never stopped moving during the course of the experiment, as seen in the poorly focused representative images.
Fig. 5.

The number of counted cells over time at position 1 and position 2 (same as in Fig. 3) in the 90° device. In the straight channel area (position 7 and position 8) cells can be counted at each timepoint, but they do not concentrate or settle (as shown in the representative images in the bottom panel).
Application to Measurement of Dynamic Yeast Stress Response (Hog1 localization)
One powerful application of an automatic sampling and imaging device is the ability to monitor dynamic processes happening in cell culture. In order to evaluate the capability of our device to monitor dynamic changes in cellular physiology we followed localization of Hog1-GFP in response to osmotic stress. In response to osmotic shock, the HOG (high-osmolarity glycerol) mitogen-activated kinase pathway is activated in yeast culminating in the phosphorylation and nuclear localization of the Hog1 kinase. We utilized a GFP-tagged version (Hog1-GFP) and monitored its localization in response to 0.5M NaCl in images taken in our sampling device (90° design) every two minutes. We used a culturing and sampling setup similar to that used in [17], illustrated in Supplemental Figure 1.
As shown in Fig. 6, Hog1 localization is clearly visible in images taken in the 90° device, and follows the characteristic localization timing and magnitude expected in response to 0.5M NaCl as seen in previous experiments utilizing single-cell microfluidic devices [23], [24], [25]. We quantified Hog1-GFP localization using the common metric of nuclear/cytoplasmic fluorescence but an alternative metric (maximum Hog1-GFP intensity) also showed the expected dynamics (Supplemental Figure 2). Fig. 6 shows images taken in the circular trapping region. While images from the straight channel also show localization, out-of-focus images lead to noisiness and loss of fidelity in following the Hog1-GFP localization signal (Supplemental Figure 2).
Fig. 6.

(A) Hog1-GFP nuclear localization in response to a 0.5 NaCl osmotic shock (dashed red line). The magnitude of Hog1 localization was quantified using an automated image analysis script as described in the Supplementary Material. Briefly, a nuclear marker (Htb2-mCherry) was used to identify the nuclear and cytoplasmic fractions of Hog1-GFP and Hog1 localization is quantified as the nuclear to cytoplasmic ratio. (B) Representative images throughout the time course taken in the circular vestibule showing clear Hog1-GFP localization in response to the 0.5M NaCl shock and subsequent delocalization following adaptation.
Our ability to follow Hog1-GFP localization with a resolution of minutes indicates that dynamic cellular processes with a clear fluorescence or morphological signature could be automatically tracked utilizing our sampling device.
Summary
The results presented here demonstrate that the updated channel design outperforms the original straight channel [3,17] in all categories of interest. The observed seven-fold increase in average cell count per image will allow for the collection of sufficient data to accurately quantify protein expression in cultured cells. Furthermore, the decrease in settling time from three minutes to one minute ensures that the data acquired will more closely reflect conditions within the cell culture, since metabolic changes in protein expression will have less time to take effect. Finally, the 50% reduction in cell spread from the focal plane demonstrated in the updated design will allow for the collection of higher-quality images with an increased percentage of in-focus cells. This facilitates automated analysis of the images and more accurate quantification of protein expression.
We anticipate that this design will be useful for microbiologists and other researchers using suspension culture. While we have optimized the device for Saccharomyces cerevisiae cells, we do not anticipate that significant modifications would be needed to trap other commonly used cell types such as Escherichia coli or CHO cells. Recent studies have taken advantage of chemical and optogenetic inducers to dynamically control cellular processes in suspension culture [5,26,27]. We anticipate that sampling devices such as the one presented here will be useful for interfacing with these induction experiments, to measure cellular responses in real-time and potentially develop real-time interventions or feedback control [28,29].
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors thank Mitchell Tyler for his guidance and feedback on study design. We would also like to thank members of the McClean laboratory for access to resources, assistance with training, and feedback on the manuscript. This research was supported by funding from the Wisconsin Alumni Research Foundation and the National Institutes of Health R21AI135166 to MNM. Megan Nicole McClean, Ph.D. holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.mex.2020.100891.
Appendix. Supplementary materials
References
- 1.Novic A., Szilard L. Description of the chemostat. Science. 1950;112(2920):715–716. doi: 10.1126/science.112.2920.715. [DOI] [PubMed] [Google Scholar]
- 2.Monod J. La technique de culture continue: theorie et applications. Annales de l'institute Pasteur. 1950;79:390–410. [Google Scholar]
- 3.Melendez J., Patel M., Oakes B., Xu P., Morton P., McClean M. Real-time optogenetic control of intracellular protein concentration in microbial cell cultures. Integrative Biology. 2014;6(3):366–372. doi: 10.1039/c3ib40102b. [DOI] [PubMed] [Google Scholar]
- 4.Milias-Argeitis A., Summers S., Stewart-Ornstein J., Zuleta I., Pincus D., El-Samad H., Khammash M., Lygeros J. In silico feedback control for in vivo regulation of a gene expression circuit. Nature Biotechnology. 2011;29(12):1114–1116. doi: 10.1038/nbt.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milias-Argeitis A., Rullan M., Aoki S., Buchmann P., Khammash M. Automated optogenetic feedback control for precise and robust regulation of gene expression and cell growth. Nature Communications. 2016;7(12546) doi: 10.1038/ncomms12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merritt J., Kuehn S. Quantitative high-throughput population dynamics in continuous-culture by automated microscopy. Scientific Reports. 2016;6(33173) doi: 10.1038/srep33173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zuleta I., Aranda-Diaz A., Li H., El-Samad H. Dynamic characterization of growth and gene expression using high-throughput automated flow cytometry. Nature Methods. 2014;11(4):443–448. doi: 10.1038/nmeth.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toprak E., Veres A., Michel J., Chait R., Hartl D., Kishony R. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nature Genetics. 2011;44(1):101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Givan A. Wiley-Liss; New York: 2001. Flow cytometry: First principles. [Google Scholar]
- 10.Ramdzan Y., Polling S., Chia C., Ng I., Ormsby A., Croft N., Purcell A., Bogoyevitch M., Ng D., Gleeson P. Tracking protein aggregation and mislocalization in cells with flow cytometry. Nature methods. 2012;9(5):467–470. doi: 10.1038/nmeth.1930. [DOI] [PubMed] [Google Scholar]
- 11.Hui H., Fuller K., Erber W., Linden M. Imaging flow cytometry in the assessment of leukocyte-platelet aggregates. Methods. 2017;112:46–54. doi: 10.1016/j.ymeth.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Tracy B., Gaida S., Papoutsakis E. Flow cytometry for bacteria: enabling metabolic engineering, synthetic biology and the elucidation of complex phenotypes. Current opinion in biotechnology. 2010;21(1):85–99. doi: 10.1016/j.copbio.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 13.Bongaerts R., Hautefort I., Sidebothm J., Hinton J. Green fluorescent protein as a marker for conditional gene expression in bacterial cells. Methods in Enzymology. 2002;358:43–66. doi: 10.1016/s0076-6879(02)58080-0. [DOI] [PubMed] [Google Scholar]
- 14.Felip M., Andreatta S., Sommaruga R., Sraskrabova V., Catalan J. Suitability of flow cytometry for estimating bacterial biovolume in natural plankton samples: comparison with microscopy data. Applied and environmental microbiology. 2007;73(14):4508–4515. doi: 10.1128/AEM.00733-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okumus B., Landgraf D., Lai G., Bakshi S., Arias-Castro J., Yildiz S., Huh D., Fernandez-Lopez R., Peterson C., Toprak E., El Karoui M., Paulsson J. Mechanical slowing-down of cytoplasmic diffusion allows in vivo counting of proteins in individual cells. Nature Communications. 2016;7(11641) doi: 10.1038/ncomms11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okumus B., Baker C., Arias-Castro J., Lai G., Leoncini E., Bakshi S., Luro S., Landgraf D., Paulsson J. Single-cell microscopy of suspension cultures using a microfluidics-assisted cell screening platform. Nature Protocols. 2018;13(1):170. doi: 10.1038/nprot.2017.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stewart C., McClean M. Design and Implementation of an Automated Illuminating, Culturing, and Sampling System for Microbial Optogenetic Applications. Journal of Visualized Experiments. 2017;120:e54894. doi: 10.3791/54894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan W., Takeuchi S. A trap-and-release integrated microfluidic system for dynamic microarray applications. Proceedings of the National Academy of Sciences. 2007;104(4):1146–1151. doi: 10.1073/pnas.0606625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park Y., Choi Y., Mitra D., Kang T., Lee L. Study of microscale hydraulic jump phenomenon for hydrodynamic trap-and-release of microparticles. Applied Physics Letters. 2010;97 doi: 10.1063/1.3479052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitra D., Park Y., Choi Y., Patel H., Pham B., Waldeisen J., Kang T., Lee L. Cell 'Trap and Release' using novel microfluidic hydraulic jump trap. 14th International Conference on Miniaturized Systems for Chemistry and Life Sciences; Groningen; 2010. [Google Scholar]
- 21.Truskey G., Yuan F., Katz D. Pearson; 2009. Transport phenomena in biological systems. [Google Scholar]
- 22."COMSOL Multiphysics Reference Manual, Version 4.2," [Online]. Available:www.comsol.com.
- 23.Hersen P., McClean M., Mahadevan L., Ramanathan S. Signal processing by the HOG MAP kinase pathway. Proceedings of the National Academy of Sciences. 2008;105(20):7165–7170. doi: 10.1073/pnas.0710770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchell A., Wei P., Wendell L. Oscillatory stress stimulation uncovers an Achilles’ heel of the yeast MAPK signaling network. Science. 2015;350(6266):1379–1383. doi: 10.1126/science.aab0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mettetal J., Muzzey D., Gomez-Uribe C., van Oudenaarden A. The Frequency Dependence of Osmo-Adaptation in Saccharomyces cerevisiae. Science. 2008;319(4862):482–484. doi: 10.1126/science.1151582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao E., Zhang Y., Mehl J., Park H., Lalwani M., Toettcher J., Avalos J. Optogenetic regulation of engineered cellular metabolism for microbial chemical production. Nature. 2018;555(7698):683–687. doi: 10.1038/nature26141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benzinger D., Khammash M. Pulsatile inputs achieve tunable attenuation of gene expression variability and graded multi-gene regulation. Nature communications. 2018;9(1):3521. doi: 10.1038/s41467-018-05882-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lugagne J., Dunlop M. Cell-machine interfaces for characterizing gene regulatory network dynamics. Current opinions in systems biology. 2019;14:1–8. doi: 10.1016/j.coisb.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott T., Sweeney K., McClean M. Biological signal generators: integrating synthetic biology tools and in silico control. Current opinion in systems biology. 2019;14:58–65. doi: 10.1016/j.coisb.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.