

Abstract
The identification of biomarkers for early diagnosis of Parkinson’s disease (PD) is of pivotal importance for improving approaches for clinical intervention. The use of translatable animal models of pre-motor PD therefore offers optimal opportunities for novel biomarker discovery in vivo. Peptidylarginine deiminases (PADs) are a family of calcium-activated enzymes that contribute to protein misfolding through post-translational deimination of arginine to citrulline. Furthermore, PADs are an active regulator of extracellular vesicle (EV) release. Both protein deimination and extracellular vesicles (EVs) are gaining increased attention in relation to neurodegenerative diseases, including in PD, while roles in pre-motor PD have yet to be investigated. The current study aimed at identifying protein candidates of deimination in plasma and plasma-EVs in a rat model of pre-motor PD, to assess putative contributions of such post-translational changes in the early stages of disease. EV-cargo was further assessed for deiminated proteins as well as three key micro-RNAs known to contribute to inflammation and hypoxia (miR21, miR155, and miR210) and also associated with PD. Overall, there was a significant increase in circulating plasma EVs in the PD model compared with sham animals and inflammatory and hypoxia related microRNAs were significantly increased in plasma-EVs of the pre-motor PD model. A significantly higher number of protein candidates were deiminated in the pre-motor PD model plasma and plasma-EVs, compared with those in the sham animals. KEGG (Kyoto encyclopedia of genes and genomes) pathways identified for deiminated proteins in the pre-motor PD model were linked to “Alzheimer’s disease”, “PD”, “Huntington’s disease”, “prion diseases”, as well as for “oxidative phosphorylation”, “thermogenesis”, “metabolic pathways”, “Staphylococcus aureus infection”, gap junction, “platelet activation”, “apelin signalling”, “retrograde endocannabinoid signalling”, “systemic lupus erythematosus”, and “non-alcoholic fatty liver disease”. Furthermore, PD brains showed significantly increased staining for total deiminated proteins in the brain vasculature in cortex and hippocampus, as well as increased immunodetection of deiminated histone H3 in dentate gyrus and cortex. Our findings identify EVs and post-translational protein deimination as novel biomarkers in early pre-motor stages of PD.
Keywords: Peptidylarginine deiminases (PADs); protein deimination; extracellular vesicles (EVs); pre-motor Parkinson’s disease; microRNA (miR21, miR155, miR210)
1. Introduction
Identification of biomarkers for early diagnosis of Parkinson’s disease (PD) is essential for developing early clinical intervention strategies. The use of animal models that present symptoms comparable to those observed in PD patients in the early stages of the disease, prior to the appearance of motor dysfunctions, therefore offers promising avenues for novel biomarker discovery in vivo. Peptidylarginine deiminases (PADs) are a family of calcium-activated enzymes that contribute to protein misfolding, as well as changes in protein function, through post-translational deimination of arginine to citrulline [1,2]. Furthermore, PADs are an active regulator of extracellular vesicle (EVs) release [3,4,5,6,7]. Both protein deimination and EVs are gaining increasing attention in relation to neurodegenerative diseases [8,9], including in PD, while their roles in pre-motor PD have yet to be investigated. Increased PAD-mediated protein deimination is indeed observed in several autoimmune, chronic and neurodegenerative diseases, including in PD [8,9,10,11,12,13,14]. In PD, deiminated proteins have been detected in substantia nigra of post-mortem human brain samples [10]. Furthermore, in PD iPSC neuronal models, derived from fibroblasts of patients carrying α-synuclein triplication [15], an increase and change in deiminated protein patterns has also been reported [9].
EVs are a recognized contributor to neurodegenerative diseases, and both neurotoxic and neuroprotective roles via distribution of EV-mediated cargo, including microRNAs and misfolded proteins, have been implicated [16,17]. In addition, EVs may be usable as a “liquid biopsy” for identification of disease related biomarkers [8,16,17,18,19,20]. EVs have indeed recently been suggested as putative biomarkers in PD [21]. For example, PD patients have been shown to have greater amounts of circulating small EVs [22]. However, less is known about EVs in early and pre-motor stages of PD.
The current study aimed at profiling EVs and identifying protein candidates of deimination in plasma and plasma-EVs in a rat model of pre-motor PD to determine putative contributions of such post-translationally mediated changes in early stages of PD, including further downstream effects that may be related to disease progression. To this end, we used a toxin-induced pre-motor PD model that was shown to display non-motor symptoms in the absence of motor symptoms and pathologies mimicking those observed in patients (See 4.1 in the Methods section). EV-cargo was assessed for deiminated proteins as well as for three key microRNAs known to contribute to inflammation and hypoxia (miR21, miR155, and miR210), all of which have also been associated to neurodegenerative diseases and PD [23,24,25], but have not been related to, or assessed in, pre-motor PD before the current study. Epigenetic mechanisms of PD have indeed received increasing attention, including histone modifications, DNA methylation, as well as microRNA involvement [26,27]. Our findings identify EVs and protein deimination as novel markers in pre-motor stages of PD.
2. Results
2.1. Circulating Plasma Extracellular Vesicles (EVs) are Significantly Increased in the Pre-Motor Parkinson’s Disease (PD) Model Compared with Sham-Treated Rats
Significant changes were observed in the number of circulating plasma-EVs in the pre-motor PD, compared with sham-treated rats, with a 2-fold increase in plasma-EVs (p = 0.031; Figure 1A). Modal plasma-EV size did not show a significant change between the pre-motor PD and shams (Figure 1B).
Figure 1.

Plasma-extracellular vesicles (EVs) are elevated in pre-motor Parkinson’s disease (PD) model rats. (A) The number of circulating plasma-EVs was significantly increased in plasma of the pre-motor PD model rats, compared with that in plasma of sham control rats (p < 0.05; unpaired t-test). (B) Modal size of plasma-EVs did not differ between plasma from the pre-motor PD rat model compared to control shams. Exact p-values are indicated (n = 3 biological replicates for all; ctrl = sham; PD = pre-motor PD models).
Figure 2 shows representative nanoparticle tracking analysis (NTA) profiles of EV size distribution from shams and pre-motor PD animals (Figure 2A,B). Additional EV characterisation was carried out by western blotting (WB) using the EV-specific markers CD63 and flotillin-1 (Flot-1), which showed positive for the rat EVs (Figure 2C), as well as by transmission electron microscopy (TEM), revealing typical EV morphology (Figure 2D,E).
Figure 2.

EV characterisation from rat plasma. (A) Representative Nanosight graphs showing nanoparticle tracking analysis (NTA) analysis of plasma EV profiles from sham-treated rats (Sham; n = 3). (B) Representative Nanosight graphs showing NTA analysis of plasma EV profiles from the pre-motor PD rat models (PD; n = 3). (C) Western blotting (WB) confirms that rat plasma-EVs are positive for the EV-specific markers CD63 and flotillin-1 (Flot-1); the molecular weight standard is indicated in kilo Daltons (kDa). (D,E) Transmission electron microscopy (TEM) images showing EVs isolated from sham (D) and pre-motor PD model (PD; (E)) rat plasma, revealing typical EV morphology; composite images are shown and the scale bar represents 50 nm in all images. In the NTA curves the black line represents the mean of the 5 repetitive readings per individual sample and the red line represents standard error (+/−) between those same 5 readings per sample. Each treatment group was measured in 3 biological replicates (sham; PD = pre-motor PD models).
2.2. Inflammatory and Hypoxia Related microRNA EV-Cargo is Increased in Plasma of Pre-Motor PD Models
When assessing EV-cargo for two inflammatory (miR21, miR155) and one hypoxia related microRNA (miR210), a significant increase in relative expression was found for all three microRNAs in plasma-EVs of the pre-motor PD models, compared with that in sham-treated animals (Figure 3). The pro-inflammatory miR21 was increased by 7.77-fold (p = 0.00014) in pre-motor PD plasma-EVs, compared with shams (Figure 3A); the pro-inflammatory miR155 was 11.34-fold increased (p < 0.0001) in the pre-motor PD plasma-EVs, compared with shams (Figure 3B); and the hypoxia-related miR210 was 6.88-fold increased (p < 0.0001) in the pre-motor PD plasma-EVs, compared with shams (Figure 3C).
Figure 3.

MicroRNA analysis of inflammatory and hypoxia associated microRNAs in circulating plasma-EVs of pre-motor PD and sham animals. (A) Significantly increased relative expression of the inflammatory miR21 was observed in plasma-EVs of the pre-motor PD rats, compared with shams. (B) Significantly increased relative expression of the inflammatory miR155 was observed in plasma-EVs of pre-motor PD rats, compared with shams. (C) The relative expression of the hypoxia related miR210 was significantly increased in plasma-EVs of pre-motor PD rats, compared with those of sham-treated animals. Results are represented as relative microRNA (miR) expression compared to the internal control miRs (2Λ(-DDCT)); exact p-values are indicated, error bars show SD (n = 3 biological and three technical replicates for all; ctrl = sham; PD = pre-motor PD models).
2.3. Deiminated Protein Targets and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways Enriched in Deiminated Proteins Differ between Pre-Motor PD and Sham-Treated Rat Plasma and Plasma-EVs
Deiminated proteins isolated by F95-enrichment were assessed by silver staining, revealing a number of protein bands in the range of 15–200 kDa (Figure 4). There was no obvious difference observed in protein banding patterns of F95-enriched protein eluates (F95_IP) between the sham (ctrl) and pre-motor PD-model; neither in plasma nor plasma-EVs, while concentration of F95-enriched proteins unexpectedly seemed slightly higher in the sham (ctrl) animals, without verifying specific target proteins of deimination at this point. The F95-enriched eluates were further assessed by liquid chromatography mass spectrometry (LC-MS/MS) analysis for identification of differences in deiminated target proteins in sham, compared with pre-motor PD animals, both for plasma and plasma-EVs.
Figure 4.
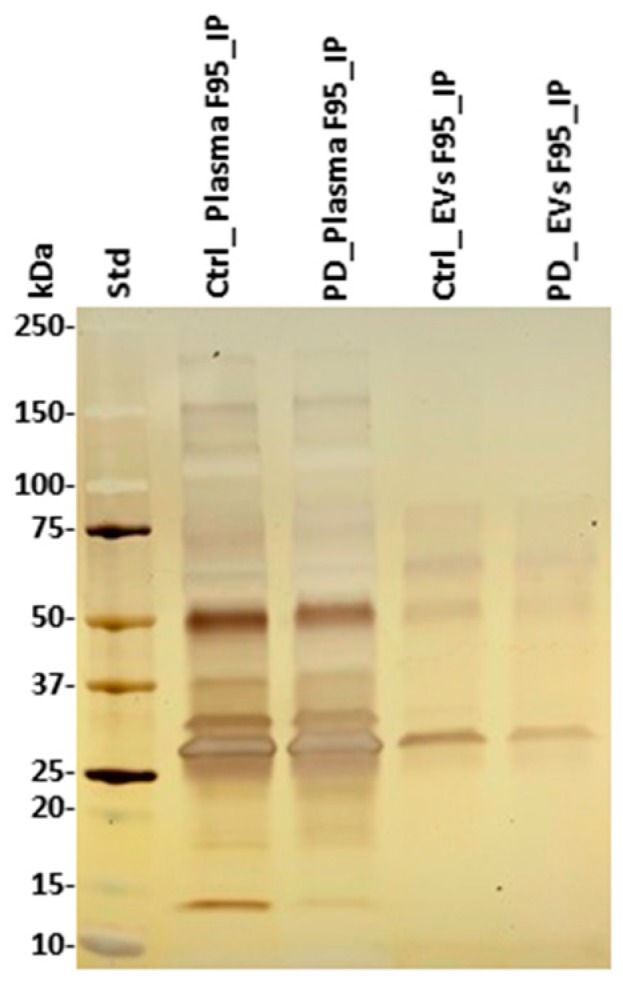
F95-enriched fractions from plasma and plasma-EVs from pre-motor PD model and sham rats. Silver stained sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel (4–20% gradient gel), showing F95-enriched protein bands from whole plasma and plasma-EVs in pre-motor PD and sham (control) rats. The protein standard is indicated on the far left in kilodaltons (kDa) (ctrl = sham; PD = pre-motor PD models).
Following LC-MS/MS analysis, F95-enriched protein candidates identified were analysed for protein–protein interaction networks using STRING (https://string-db.org/). STRING analysis revealed some differences in Kyoto encyclopedia of genes and genomes (KEGG) pathways enriched in deiminated proteins between the pre-motor PD model and the shams, both for plasma-EVs as well as whole plasma. Overall, more protein hits were identified as deiminated in the pre-motor PD models, both in EVs and whole plasma, compared with shams, with some common targets but some specific to either shams or the pre-motor PD models as summarised in the Venn diagram in Figure 5. In summary, 118 protein hits were specific to the pre-motor PD model whole plasma and 9 deiminated protein hits were specific to sham plasma (Figure 5A; Table 1 and Table S1). In plasma-EVs of the pre-motor PD rats, 49 deiminated protein hits were specific, while 33 deimination hits were specific to plasma-EVs from shams (Figure 5B, Table 2, and Table S2).
Figure 5.

Venn diagram showing deiminated protein hits identified in plasma and plasma-EVs of pre-motor PD model and of sham rats. (A) A considerable higher number of deiminated protein hits was identified in plasma of the pre-motor PD model rats, compared with that in shams, with some overlapping protein hits. (B) A higher number of protein hits was identified to be deiminated in circulating plasma-EVs of the pre-motor PD rat models, compared with that in shams, with some hits overlapping.
Table 1.
Deiminated proteins in sham-treated rat plasma (Rattus norvegicus), as identified by F95-enrichment and liquid chromatography mass spectrometry (LC-MS/MS) analysis. Deiminated proteins were isolated from sham-treated rat plasma by immunoprecipitation using the pan-deimination F95 antibody. The resulting F95-enriched eluate was then analysed by LC-MS/MS and peak list files submitted to mascot. Rattus norvegicus species-specific peptide sequence hits are listed, showing number of sequences for protein hits and total score. Blue highlighted rows indicate protein hits identified in whole plasma of sham-treated rats, but not identified as deiminated in plasma of the pre-motor PD model.
| Protein Name | Symbol | Sequences | Total Score (p < 0.05) ⱡ |
|---|---|---|---|
| Serum albumin | A0A0G2JSH5_RAT | 38 | 2280 |
| Serum albumin | ALBU_RAT | 37 | 2215 |
| Alpha-1-macroglobulin | A1M_RAT | 34 | 1890 |
| Alpha-1-inhibitor 3 | A1I3_RAT | 25 | 1615 |
| Complement C3 | M0RBF1_RAT | 30 | 1611 |
| Similar to histidine-rich glycoprotein | A0A0G2K9Y5_RAT | 28 | 1561 |
| Murinoglobulin-1 | D4A6E3_RAT | 22 | 1330 |
| Kininogen-1 | KNG1_RAT | 20 | 1112 |
| Ac1873 | Q7TQ70_RAT | 21 | 1008 |
| Serotransferrin | TRFE_RAT | 18 | 936 |
| Isoform LMW of Kininogen-1 | KNG1_RAT | 15 | 860 |
| Ig gamma-2C chain C region | IGG2C_RAT | 13 | 739 |
| Isoform 2 of Fibrinogen beta chain | FIBB_RAT | 14 | 545 |
| Ig gamma-2A chain C region | IGG2A_RAT | 8 | 520 |
| Isoform Gamma-A of Fibrinogen gamma chain | FIBG_RAT | 10 | 516 |
| Carboxypeptidase N catalytic chain | CBPN_RAT | 8 | 460 |
| Uncharacterized protein | A0A0G2JZW1_RAT | 8 | 446 |
| C-reactive protein | CRP_RAT | 6 | 432 |
| Fibronectin | A0A096P6L8_RAT | 9 | 404 |
| Ig kappa chain C region, A allele | KACA_RAT | 6 | 383 |
| Uncharacterized protein | A0A0G2JVP4_RAT | 7 | 341 |
| Uncharacterized protein | A0A0G2K477_RAT | 6 | 333 |
| Hemoglobin subunit beta-1 | HBB1_RAT | 6 | 319 |
| Apolipoprotein E | A0A0G2K151_RAT | 7 | 316 |
| Uncharacterized protein | D4ACR1_RAT | 4 | 300 |
| Hemoglobin subunit alpha-1/2 | HBB1_RAT | 6 | 295 |
| Uncharacterized protein | D4ACR1_RAT | 4 | 274 |
| Kininogen-1 | A0A0G2JVQ5_RAT | 6 | 251 |
| Carboxypeptidase N subunit 2 | F1LQT4_RAT | 6 | 250 |
| Ig gamma-2B chain C region | IGG2B_RAT | 5 | 237 |
| Hemopexin | HEMO_RAT | 6 | 231 |
| Ceruloplasmin | A0A0G2K9I6_RAT | 6 | 220 |
| Hemoglobin subunit beta-2 | HBB2_RAT | 4 | 216 |
| Uncharacterized protein | M0RA79_RAT | 3 | 213 |
| Keratin, type II cytoskeletal 5 | A0A0G2K509_RAT | 3 | 210 |
| Alpha-1-antiproteinase | A0A0G2JY31_RAT | 3 | 209 |
| Uncharacterized protein | A0A0G2K290_RAT | 3 | 203 |
| Keratin, type II cytoskeletal 6A | K2C6A_RAT | 3 | 198 |
| Ab1-233 | A0A0G2JXK5_RAT | 4 | 197 |
| Uncharacterized protein | F1LVL4_RAT | 3 | 186 |
| Keratin, type I cytoskeletal 14 | K1C14_RAT | 3 | 181 |
| Uncharacterized protein | F1M3 × 3_RAT | 4 | 177 |
| Kallikrein B, plasma 1 | Q5FVS2_RAT | 3 | 175 |
| Complement C2 | Q6MG73_RAT | 4 | 175 |
| Uncharacterized protein | A0A0G2JY98_RAT | 3 | 172 |
| Haptoglobin | A0A0H2UHM3_RAT | 3 | 151 |
| Selenoprotein P | SEPP1_RAT | 4 | 151 |
| Apolipoprotein A-I | APOA1_RAT | 2 | 134 |
| Serine protease inhibitor A3K | SPA3K_RAT | 4 | 130 |
| Uncharacterized protein | F1LXY6_RAT | 2 | 123 |
| Uncharacterized protein | A0A0G2JW41_RAT | 1 | 115 |
| Keratin 16 | Q6IFU9_RAT | 3 | 115 |
| Ig lambda-2 chain C region | LAC2_RAT | 2 | 112 |
| Uncharacterized protein | A0A0G2K4K2_RAT | 3 | 111 |
| Coagulation factor XII | A0A0H2UI19_RAT | 3 | 110 |
| Inter-alpha-trypsin inhibitor heavy chain family, member 4 | D3ZFC6_RAT | 2 | 105 |
| Uncharacterized protein | F1LZH0_RAT | 2 | 93 |
| Uncharacterized protein | F1M0U4_RAT | 2 | 93 |
| RCG21066 | D3ZJW6_RAT | 2 | 86 |
| Uncharacterized protein | M0RAB8_RAT | 1 | 85 |
| Uncharacterized protein | M0R816_RAT | 1 | 83 |
| C-type lectin domain family 11 member A | CLC11_RAT | 2 | 80 |
| Complement factor H | F1M983_RAT | 2 | 79 |
| Beta-globin | Q62669_RAT | 2 | 71 |
| Uncharacterized protein | A0A0G2JV42_RAT | 2 | 62 |
| Uncharacterized protein | F1LM19_RAT | 1 | 53 |
| Inter-alpha-trypsin inhibitor heavy chain 2 | D3ZFH5_RAT | 2 | 53 |
| Uncharacterized protein | F1M5L5_RAT | 2 | 52 |
| Fetub protein | Q6IRS6_RAT | 1 | 45 |
| Uncharacterized protein | A0A0G2JWX0_RAT | 1 | 45 |
| Uncharacterized protein | A0A0G2JTG4_RAT | 1 | 42 |
| Ubiquitin carboxyl-terminal hydrolase isozyme L1 | UCHL1_RAT | 1 | 41 |
| Uncharacterized protein | A0A0G2JUY3_RAT | 1 | 41 |
| Apolipoprotein A-IV | A0A0G2JVX7_RAT | 1 | 39 |
| Extracellular calcium-sensing receptor | CASR_RAT | 1 | 37 |
| Uncharacterized protein | F1LZ11_RAT | 1 | 36 |
| Uncharacterized protein | M0R9U2_RAT | 1 | 34 |
| Golgi apparatus protein 1 | G3V8G5_RAT | 1 | 33 |
ⱡ Ions score is −10*Log(P), where P is the probability that the observed match is a random event. Individual ions scores > 31 indicated identity or extensive homology (p < 0.05). Protein scores were derived from ions scores as a non-probabilistic basis for ranking protein hits.
Table 2.
Deiminated proteins in pre-motor PD rat plasma (Rattus norvegicus), as identified by F95-enrichment and LC-MS/MS analysis. Deiminated proteins from pre-motor PD rat plasma were isolated by immunoprecipitation using the pan-deimination F95 antibody. The resulting F95-enriched eluate was then analysed by LC-MS/MS and peak list files submitted to mascot. Rattus norvegicus species-specific peptide sequence hits are listed, showing number of sequences for protein hits and total score. Pink highlighted rows indicate protein hits identified in pre-motor PD plasma but not sham-treated rat plasma.
| Protein Name | Symbol | Sequences | Total Score (p < 0.05) ⱡ |
|---|---|---|---|
| Complement C3 | M0RBF1_RAT | 89 | 5530 |
| Alpha-1-macroglobulin | A1M_RAT | 74 | 4911 |
| Alpha-1-inhibitor 3 | A1I3_RAT | 59 | 3845 |
| Fibronectin | A0A096P6L8_RAT | 64 | 3722 |
| Murinoglobulin-1 | MUG1_RAT | 57 | 3618 |
| Serum albumin | ALBU_RAT | 56 | 3564 |
| Serum albumin | A0A0G2JSH5_RAT | 56 | 3550 |
| Fibrinogen beta chain | FIBB_RAT | 53 | 3399 |
| Serotransferrin | TRFE_RAT | 53 | 3384 |
| Ac1873 | Q7TQ70_RAT | 56 | 3317 |
| Isoform Gamma-A of Fibrinogen gamma chain | FIBG_RAT | 31 | 2185 |
| Similar to histidine-rich glycoprotein | A0A0G2K9Y5_RAT | 38 | 2137 |
| Kininogen-1 | KNG1_RAT | 33 | 1981 |
| Complement C2 | Q6MG73_RAT | 30 | 1875 |
| Ceruloplasmin | A0A0G2K9I6_RAT | 32 | 1816 |
| Complement factor H | G3V9R2_RAT | 28 | 1570 |
| Kallikrein B, plasma 1 | Q5FVS2_RAT | 27 | 1552 |
| Ig gamma-2C chain C region | IGG2C_RAT | 21 | 1447 |
| Complement factor B | A0A0U1RRP9_RAT | 24 | 1405 |
| Hemopexin | HEMO_RAT | 25 | 1388 |
| Keratin, type II cytoskeletal 5 | K2C5_RAT | 24 | 1284 |
| Uncharacterized protein | G3V9J1_RAT | 22 | 1197 |
| Inter alpha-trypsin inhibitor, heavy chain 4 | Q5EBC0_RAT | 22 | 1192 |
| Carboxypeptidase N catalytic chain | CBPN_RAT | 18 | 1172 |
| Uncharacterized protein | A0A0G2JVP4_RAT | 18 | 1133 |
| Ig gamma-2A chain C region | IGG2A_RAT | 16 | 1108 |
| Complement C4 | CO4_RAT | 20 | 1070 |
| Carboxypeptidase N subunit 2 | F1LQT4_RAT | 19 | 1042 |
| Plasminogen | PLMN_RAT | 20 | 1001 |
| Apolipoprotein A-IV | APOA4_RAT | 17 | 989 |
| Uncharacterized protein | A0A0G2JZW1_RAT | 13 | 980 |
| Inter-alpha trypsin inhibitor, heavy chain 1 | B2RYM3_RAT | 18 | 960 |
| Complement C5 | A0A096P6L9_RAT | 21 | 959 |
| Apolipoprotein E | A0A0G2K151_RAT | 13 | 813 |
| Keratin, type I cytoskeletal 14 | K1C14_RAT | 14 | 779 |
| Complement C4B (Chido blood group) | Q6MG90_RAT | 13 | 769 |
| Ab1-233 | A0A0G2JYK0_RAT | 14 | 760 |
| Inter-alpha-trypsin inhibitor heavy chain 2 | D3ZFH5_RAT | 15 | 755 |
| Uncharacterized protein | F1LTJ5_RAT | 14 | 735 |
| Serine protease inhibitor A3L | SPA3L_RAT | 14 | 734 |
| Serine protease inhibitor A3K | SPA3K_RAT | 12 | 685 |
| Keratin, type I cytoskeletal 42 | K1C42_RAT | 12 | 660 |
| Uncharacterized protein | A0A0G2K477_RAT | 11 | 656 |
| Haptoglobin | A0A0H2UHM3_RAT | 14 | 650 |
| Keratin, type I cytoskeletal 17 | A0A0G2K9Q9_RAT | 14 | 634 |
| Alpha-1-antiproteinase | A0A0G2JY31_RAT | 11 | 613 |
| Inter-alpha-trypsin inhibitor heavy chain H3 | D3ZBS2_RAT | 13 | 610 |
| Selenoprotein P | SEPP1_RAT | 9 | 590 |
| C-type lectin domain family 11 member | CLC11_RAT | 12 | 587 |
| Uncharacterized protein | M0RA79_RAT | 10 | 571 |
| Apolipoprotein A-I | APOA1_RAT | 10 | 558 |
| C-reactive protein | CRP_RAT | 7 | 554 |
| Coagulation factor XII | A0A0H2UI19_RAT | 11 | 550 |
| Ig kappa chain C region, A allele | KACA_RAT | 7 | 536 |
| Uncharacterized protein | F1M3X3_RAT | 9 | 536 |
| Insulin-like growth factor binding protein, acid labile subunit, isoform CRA_b | F1LRE2_RAT | 9 | 529 |
| Kininogen-1 | A0A0G2JVQ5_RAT | 9 | 524 |
| Uncharacterized protein | F1LXY6_RAT | 7 | 507 |
| Keratin 16 | Q6IFU9_RAT | 9 | 504 |
| Uncharacterized protein | A0A0G2K828_RAT | 7 | 500 |
| Keratin, type II cytoskeletal 75 | A0A0H2UHH5_RAT | 9 | 496 |
| Uncharacterized protein | D4ACR1_RAT | 8 | 471 |
| Apolipoprotein B-100 | F1M6Z1_RAT | 12 | 457 |
| Keratin, type II cytoskeletal 6A | K2C6A_RAT | 9 | 446 |
| Keratin, type II cytoskeletal 8 | K2C8_RAT | 8 | 435 |
| Apolipoprotein H | Q5I0M1_RAT | 9 | 435 |
| Uncharacterized protein | D3ZZ08_RAT | 7 | 433 |
| Ig gamma-2B chain C region | IGG2B_RAT | 8 | 433 |
| Carboxylesterase 1C | EST1C_RAT | 8 | 431 |
| Hemoglobin subunit alpha-1/2 | HBA_RAT | 8 | 411 |
| Angiopoietin-like 6 | B2RYM1_RAT | 8 | 407 |
| Alpha-2-HS-glycoprotein | FETUA_RAT | 8 | 403 |
| Uncharacterized protein | M0R9U2_RAT | 6 | 403 |
| Hemoglobin subunit beta-1 | HBB1_RAT | 6 | 380 |
| Uncharacterized protein | A0A0G2K290_RAT | 6 | 378 |
| Uncharacterized protein | F1LZ11_RAT | 7 | 373 |
| Ig lambda-2 chain C region | LAC2_RAT | 6 | 340 |
| Uncharacterized protein | M0RAB8_RAT | 5 | 330 |
| Fetub protein | Q6IRS6_RAT | 7 | 314 |
| Uncharacterized protein | D4A3D1_RAT | 5 | 312 |
| Afamin | G3V9R9_RAT | 7 | 312 |
| Keratin, type I cytoskeletal 19 | K1C19_RAT | 7 | 308 |
| Uncharacterized protein | A0A0G2JUY4_RAT | 5 | 307 |
| Uncharacterized protein | M0RDF2_RAT | 5 | 281 |
| Uncharacterized protein | M0RBK4_RAT | 3 | 272 |
| Uncharacterized protein | A0A0G2JY98_RAT | 4 | 268 |
| Uncharacterized protein | A0A0G2JXP0_RAT | 4 | 257 |
| Extracellular matrix protein 1 | ECM1_RAT | 5 | 255 |
| Group specific component | Q68FY4_RAT | 6 | 247 |
| Desmoplakin | F1LMV6_RAT | 6 | 244 |
| Vitronectin | Q3KR94_RAT | 6 | 233 |
| Uncharacterized protein | M0RAV0_RAT | 3 | 225 |
| Peptidoglycan recognition protein 2 | M0R485_RAT | 5 | 220 |
| Uncharacterized protein | A0A0G2K7I1_RAT | 2 | 211 |
| Uncharacterized protein | A0A0G2JW41_RAT | 2 | 210 |
| Uncharacterized protein | A0A0G2K7P6_RAT | 3 | 204 |
| Glutathione peroxidase | A0A0G2K531_RAT | 4 | 201 |
| Uncharacterized protein | A0A0G2JTG4_RAT | 3 | 200 |
| Uncharacterized protein | F1LVL4_RAT | 3 | 199 |
| C4b-binding protein alpha chain | C4BPA_RAT | 6 | 196 |
| RCG21066 | D3ZJW6_RAT | 4 | 194 |
| Alpha-2-macroglobulin | A2MG_RAT | 4 | 194 |
| Uncharacterized protein | A0A0G2K304_RAT | 4 | 193 |
| Uncharacterized protein | A0A0G2K4I8_RAT | 4 | 188 |
| Uncharacterized protein | M0R693_RAT | 4 | 187 |
| Complement C1q subcomponent subunit B | G3V7N9_RAT | 3 | 186 |
| T-kininogen 1 | KNT1_RAT | 4 | 174 |
| Uncharacterized protein | A0A0G2K332_RAT | 3 | 169 |
| Protein AMBP | AMBP_RAT | 2 | 169 |
| Uncharacterized protein | A0A0G2JZV7_RAT | 3 | 164 |
| Uncharacterized protein | A0A0G2JXB7_RAT | 4 | 161 |
| Uncharacterized protein | M0RD98_RAT | 2 | 161 |
| Junction plakoglobin | PLAK_RAT | 2 | 154 |
| Actin, cytoplasmic 1 | A0A0G2K3K2_RAT | 3 | 153 |
| Uncharacterized protein | A0A0G2JV42_RAT | 2 | 153 |
| Uncharacterized protein | F1M7B3_RAT | 2 | 151 |
| Uncharacterized protein | A0A0G2K245_RAT | 2 | 150 |
| Uncharacterized protein | M0R4Z4_RAT | 3 | 142 |
| Uncharacterized protein | F1M1R0_RAT | 2 | 138 |
| Uncharacterized protein | F1LYF1_RAT | 2 | 134 |
| Complement C8 gamma chain | D3ZPI8_RAT | 2 | 134 |
| Uncharacterized protein | D3ZC54_RAT | 2 | 133 |
| Uncharacterized protein | F1M3E9_RAT | 3 | 132 |
| CD5 antigen-like | Q4KM75_RAT | 2 | 129 |
| Collagen type XVIII alpha 1 chain | F1LR02_RAT | 2 | 128 |
| Serpin family F member 2 | F7FHF3_RAT | 2 | 126 |
| Uncharacterized protein | M0R628_RAT | 2 | 124 |
| Uncharacterized protein | G3V8Z5_RAT | 3 | 118 |
| Uncharacterized protein | F1LZH0_RAT | 2 | 117 |
| Ab1-233 | A0A0H2UHI5_RAT | 2 | 105 |
| Papilin, proteoglycan-like sulfated glycoprotein | D3ZD40_RAT | 3 | 104 |
| Uncharacterized protein | A0A0G2K3K8_RAT | 2 | 104 |
| Uncharacterized protein | F1LWS4_RAT | 3 | 103 |
| Uncharacterized protein | D3ZPL2_RAT | 1 | 101 |
| Uncharacterized protein | D4A4L6_RAT | 2 | 101 |
| Uncharacterized protein | M0R816_RAT | 1 | 99 |
| Glutamine synthetase | GLNA_RAT | 3 | 99 |
| Uncharacterized protein | A0A0G2K5D2_RAT | 2 | 98 |
| Uncharacterized protein | F1M663_RAT | 1 | 96 |
| Uncharacterized protein | M0R8G6_RAT | 1 | 93 |
| Uncharacterized protein | M0R8Q4_RAT | 1 | 91 |
| Uncharacterized protein | F1LWD0_RAT | 2 | 91 |
| Similar to RIKEN cDNA | A0A0G2K896_RAT | 2 | 86 |
| Uncharacterized protein | M0R5A0_RAT | 2 | 83 |
| Uncharacterized protein | F1M5L5_RAT | 2 | 77 |
| Uncharacterized protein | D3ZFF8_RAT | 2 | 75 |
| Uncharacterized protein | A0A0G2K4K2_RAT | 2 | 73 |
| Prothrombin | G3V843_RAT | 2 | 73 |
| Uncharacterized protein | M0R7Q2_RAT | 2 | 69 |
| Uncharacterized protein | A0A096P6M7_RAT | 1 | 65 |
| Tripartite motif-containing 33 | D3ZUK4_RAT | 2 | 65 |
| Uncharacterized protein | M0R8B5_RAT | 1 | 61 |
| Uncharacterized protein | M0R4C5_RAT | 1 | 60 |
| Ig kappa chain V region S211 | KVX01_RAT | 1 | 59 |
| Uncharacterized protein | F1LTY5_RAT | 1 | 57 |
| Uncharacterized protein | M0R4G1_RAT | 1 | 55 |
| Suprabasin | F7FEM5_RAT | 1 | 53 |
| NADH:ubiquinone oxidoreductase subunit B10 | D4A0T0_RAT | 2 | 52 |
| Complement C1q subcomponent subunit A | C1QA_RAT | 1 | 52 |
| Ciliary rootlet coiled-coil, rootletin | F1M6Q2_RAT | 2 | 52 |
| E3 ubiquitin-protein ligase HUWE1 | A0A0G2JVW5_RAT | 2 | 52 |
| Complement factor properdin | B0BNN4_RAT | 1 | 52 |
| Uncharacterized protein | A0A0G2JX36_RAT | 1 | 51 |
| Tubulin beta-2B chain | TBB2B_RAT | 1 | 51 |
| Uncharacterized protein | A0A0G2K0N6_RAT | 1 | 50 |
| Cadherin-1 | CADH1_RAT | 1 | 46 |
| Caspase 8-associated protein 2 | D4A7V6_RAT | 2 | 46 |
| Phospholipase A2 | A0A0G2JZ44_RAT | 1 | 46 |
| PEX5-related protein | F1LMT5_RAT | 2 | 44 |
| Collectin sub-family member 11 | F1LSS7_RAT | 1 | 44 |
| Oxysterol-binding protein | A0A0G2K0D5_RAT | 2 | 43 |
| Uncharacterized protein | F1M6N0_RAT | 1 | 42 |
| Obscurin-like protein 1 | OBSL1_RAT | 2 | 42 |
| Serum amyloid A protein | Q5M878_RAT | 1 | 41 |
| Proline-rich protein 5 | A0A0H2UHJ6_RAT | 1 | 41 |
| Complement component C9 | F7F389_RAT | 1 | 39 |
| Methyltransferase-like 3 | Q4V8G6_RAT | 1 | 36 |
| Complement C8 alpha chain | D3ZWD6_RAT | 1 | 35 |
| Serine (Or cysteine) peptidase inhibitor, clade C (Antithrombin), member 1 | Q5M7T5_RAT | 1 | 33 |
| Cell cycle checkpoint control protein RAD9B | RAD9B_RAT | 1 | 33 |
| Heparin cofactor 2 | A0A0G2K8K3_RAT | 1 | 33 |
| Histone H4 | H4_RAT | 1 | 33 |
| Uncharacterized protein (Fragment) | A0A096MK45_RAT | 1 | 32 |
| Phosphatidylinositol glycan anchor biosynthesis, class Q | Q642B8_RAT | 1 | 32 |
ⱡ Ions score is −10*Log(P), where P is the probability that the observed match is a random event. Individual ions scores > 31 indicated identity or extensive homology (p < 0.05). Protein scores were derived from ions scores as a non-probabilistic basis for ranking protein hits.
STRING analysis of the KEGG pathways identified for deiminated protein hits in total plasma of the pre-motor PD model and sham-treated rat plasma respectively, is shown in Figure 6 (Figure 6A,B; see also Table 1 and Table 2 as well as Supplementary Tables S1 and S2 for full LC-MS/MS data analysis of all protein hits). In whole plasma, KEGG pathways enriched in deimination and found to be common between shams and pre-motor PD models whole plasma were “complement and coagulation cascades” and “oestrogen signalling pathway” (Figure 6A,B). KEGG pathways identified to be enriched in deiminated proteins in the pre-motor PD models compared to plasma from sham-treated rats were KEGG pathways for “Parkinson’s disease”, “Alzheimer’s disease”, “Huntington’s disease” and “prion disease”, “retrograde endocannabinoid signalling”, “systemic lupus erythematosus (SLE)”, and “non-alcoholic fatty liver disease (NAFLD)” (Figure 6B). Pathways identified as enriched in deiminated proteins in sham plasma only belonged to “cholesterol metabolism”, “fat digestion and absorption”, “vitamin digestion and absorption”, “platelet activation”, “PPAR signalling pathway”, “African trypanosomiasis”, “S. aureus infection”, and “malaria” (Figure 6A).
Figure 6.

Kyoto encyclopedia of genes and genomes (KEGG) pathways for deiminated proteins identified in plasma of sham and pre-motor PD model rats. (A) KEGG pathways identified in circulating EVs of sham-treated (control) rat plasma are highlighted. Colour codes for nodes included in the figure. (B) KEGG pathways identified for deiminated proteins in circulating EVs of pre-motor PD model rat plasma are highlighted; notably pathways for PD and other neurodegenerative disease are identified for deiminated proteins in the pre-motor PD plasma-EVs. See colour codes for nodes and for connecting lines in the figure.
The KEGG pathways identified for deiminated protein hits in plasma-EVs of the pre-motor PD models and sham-treated rats are shown in Figure 7 (Figure 7A,B; see also Table 3 and Table 4 as well as Supplementary Tables S3 and S4 for full LC-MS/MS data analysis of all protein hits). Deiminated proteins in plasma-EVs of the pre-motor PD models were enriched in KEGG pathways for “Parkinson’s disease”, “Alzheimer’s disease”, and “Huntington’s disease”, while none of these KEGG pathways were identified for deiminated protein hits in sham-treated rat plasma-EVs (Figure 7A,B). Furthermore, pathways for “oxidative phosphorylation”, “complement coagulation cascades”, “thermogenesis”, “metabolic pathways”, “S. aureus infection”, “gap junction”, “platelet activation”, and “apelin signalling” were only identified in plasma-EVs from the pre-motor PD models (Figure 7B). “Oestrogen signalling pathway” was common to EVs of both PD and sham-treated rat plasma. In addition, pathways enriched in deiminated proteins in sham-treated plasma-EVs and not identified in the PD models belonged to “cell cycle”, “progesterone mediated oocyte maturation”, “Epstein Barr virus infection”, “antigen processing and presentation”, “viral carcinogenesis”, “HTLV-1 infection”, “ubiquitin mediated proteolysis”, “protein processing in ER”, “cellular senescence”, “longevity regulating pathway”, “glycolysis”, and “p53 signalling pathway” (Figure 7A).
Figure 7.

KEGG pathways for deiminated proteins in plasma-EVs of sham and pre-motor PD model rats. (A) KEGG pathways identified in circulating plasma-EVs of sham (control) rats are highlighted. See colour code for nodes included in the figure. (B) KEGG pathways identified for deiminated proteins in circulating plasma-EVs of the pre-motor PD model rats are highlighted. See colour codes for nodes and connecting lines in the figure.
Table 3.
Deiminated proteins in plasma-EVs of sham-treated rats (Rattus norvegucus), as identified by Figure 95. and LC-MS/MS analysis. Deiminated proteins in sham-treated rat plasma were isolated by immunoprecipitation using the pan-deimination F95 antibody. The resulting F95-enriched eluate was then analysed by LC-MS/MS and peak list files submitted to mascot. Rattus norvegicus species-specific peptide sequence hits are listed, showing number of sequences for protein hits and total score. Green highlighted rows indicate protein hits identified in sham-treated (control) rat plasma only.
| Protein Name | Symbol | Sequences | Total Score (p < 0.05) ⱡ |
|---|---|---|---|
| Serum albumin | A0A0G2JSH5_RAT | 13 | 829 |
| RCG36700 | A0A0G2K3G0_RAT | 13 | 654 |
| Keratin, type II cytoskeletal 5 | K2C5_RAT | 14 | 624 |
| Keratin, type I cytoskeletal 17 | A0A0G2K9Q9_RAT | 12 | 571 |
| Keratin, type I cytoskeletal 14 | K1C14_RAT | 8 | 500 |
| Actin, cytoplasmic 1 | A0A0G2K3K2_RAT | 8 | 461 |
| Keratin, type II cytoskeletal 6A | K2C6A_RAT | 8 | 401 |
| Keratin, type I cytoskeletal 19 | K1C19_RAT | 9 | 391 |
| Keratin 16 | Q6IFU9_RAT | 6 | 368 |
| Heat shock protein HSP 90-beta | A0A0G2K793_RAT | 9 | 337 |
| Keratin, type II cytoskeletal 8 | K2C8_RAT | 6 | 330 |
| Keratin, type II cytoskeletal 75 | A0A0H2UHH5_RAT | 7 | 320 |
| Heat shock 70 kDa protein 1A | HS71A_RAT | 5 | 312 |
| Heat shock protein HSP 90-alpha | HS90A_RAT | 7 | 298 |
| Uncharacterized protein | A0A0G2K828_RAT | 4 | 284 |
| Ig gamma-2A chain C region | IGG2A_RAT | 5 | 281 |
| Isoform LMW of Kininogen-1 | KNG1_RAT | 6 | 272 |
| Alpha-1-macroglobulin | A1M_RAT | 8 | 267 |
| Keratin, type I cytoskeletal 42 | K1C42_RAT | 5 | 265 |
| Heat shock 70 kDa protein 1-like | HS71L_RAT | 4 | 257 |
| Complement C3 | M0RBF1_RAT | 6 | 238 |
| Ig gamma-2C chain C region | IGG2C_RAT | 5 | 224 |
| Serotransferrin | A0A0G2QC06_RAT | 5 | 222 |
| Uncharacterized protein | D4ACR1_RAT | 3 | 179 |
| Murinoglobulin-1 | A0A0G2JUP5_RAT | 3 | 148 |
| Ig kappa chain C region, A allele | KACA_RAT | 2 | 142 |
| Desmoplakin | F1LMV6_RAT | 3 | 131 |
| C-reactive protein | A0A0G2K8V5_RAT | 2 | 119 |
| Histone H4 | H4_RAT | 2 | 109 |
| 14-3-3 protein theta | 1433T_RAT | 2 | 103 |
| 40S ribosomal protein | RSSA_RAT | 1 | 93 |
| Elongation factor 1-alpha | M0R757_RAT | 2 | 87 |
| Ubiquitin-like modifier-activating enzyme 1 | UBA1_RAT | 2 | 87 |
| Ig gamma-2B chain C region | IGG2B_RAT | 2 | 82 |
| Uncharacterized protein | A0A0G2JUY4_RAT | 1 | 82 |
| L-lactate dehydrogenase A chain | LDHA_RAT | 2 | 69 |
| Uncharacterized protein | M0RA79_RAT | 1 | 66 |
| Apolipoprotein E | A0A0G2K151_RAT | 1 | 64 |
| Globin a4 | A0A0G2JSW3_RAT | 1 | 63 |
| Uncharacterized protein | A0A0G2K980_RAT | 1 | 60 |
| Uncharacterized protein | D3ZE63_RAT | 1 | 57 |
| Histone H2A.Z | H2AZ_RAT | 2 | 55 |
| Peptidyl-prolyl cis-trans isomerase FKBP4 | FKBP4_RAT | 1 | 55 |
| Peroxiredoxin-2 | A0A0G2JSH9_RAT | 1 | 55 |
| L-lactate dehydrogenase B chain | LDHB_RAT | 2 | 54 |
| Eukaryotic initiation factor 4A-II | A0A0G2K8B7_RAT | 2 | 52 |
| Clathrin heavy chain | F1M779_RAT | 1 | 49 |
| Tubulin alpha chain | A0A0H2UHM7_RAT | 1 | 49 |
| Uncharacterized protein | A0A096P6M7_RAT | 1 | 46 |
| cAMP responsive element binding protein 3, isoform CRA | A0A0G2K331_RAT | 2 | 45 |
| Transportin 1 | F1LQP9_RAT | 1 | 44 |
| Unconventional myosin-Va | A0A0G2K4Y7_RAT | 2 | 44 |
| 6-phosphogluconate dehydrogenase, decarboxylating | A0A0G2K7Q8_RAT | 1 | 43 |
| Uncharacterized protein | A0A0G2K8S2_RAT | 1 | 41 |
| Phospholipase A2 | A0A0G2JZ44_RAT | 1 | 41 |
| Histone H2B | A0A0G2JXE0_RAT | 1 | 40 |
| Ventricular zone-expressed PH domain-containing protein homolog 1 | A0A0G2K6S2_RAT | 1 | 40 |
| Uncharacterized protein | A0A0G2JVP4_RAT | 1 | 37 |
| Pyruvate kinase | A0A0G2JVG3_RAT | 1 | 37 |
| Centromere protein E | D3ZV60_RAT | 1 | 36 |
| Methyltransferase-like 3 | Q4V8G6_RAT | 1 | 35 |
| Transketolase | G3V826_RAT | 1 | 35 |
| Uncharacterized protein | A0A0G2KB28_RAT | 1 | 35 |
| Enhancer of mRNA-decapping protein 4 | EDC4_RAT | 1 | 35 |
| T-complex protein 1 subunit eta | D4AC23_RAT | 1 | 34 |
| Signal-induced proliferation-associated 1 | E9PSX8_RAT | 1 | 34 |
| Stress-70 protein, mitochondrial | F1M953_RAT | 1 | 32 |
| Filamin-C | A0A0H2UHR7_RAT | 1 | 32 |
ⱡ Ions score is −10*Log(P), where P is the probability that the observed match is a random event. Individual ions scores > 31 indicated identity or extensive homology (p < 0.05). Protein scores were derived from ions scores as a non-probabilistic basis for ranking protein hits.
Table 4.
Deiminated proteins in plasma-EVs of pre-motor PD rats (Rattus norvegicus), as identified by F95-enrichment and LC-MS/MS analysis. Deiminated proteins in pre-motor PD rat plasma were isolated by immunoprecipitation using the pan-deimination F95 antibody. The resulting F95-enriched eluate was then analysed by LC-MS/MS and peak list files submitted to mascot. Rattus norvegicus species-specific peptide sequence hits are listed, showing number of sequences for protein hits and total score. Orange highlighted rows indicate protein hits identified in plasma-EVs of pre-motor PD rats, but not in plasma-EVs of sham-treated rats.
| Protein Name | Symbol | Sequences | Total Score (p < 0.05) ⱡ |
|---|---|---|---|
| Serum albumin | ALBU_RAT | 41 | 2238 |
| Alpha-1-macroglobulin | A1M_RAT | 33 | 1650 |
| Keratin, type II cytoskeletal 5 | K2C5_RAT | 29 | 1395 |
| Similar to histidine-rich glycoprotein | A0A0G2K9Y5_RAT | 28 | 1305 |
| Serotransferrin | TRFE_RAT | 28 | 1238 |
| Alpha-1-inhibitor | A1I3_RAT | 21 | 976 |
| Kininogen-1 | KNG1_RAT | 17 | 898 |
| Complement C3 | M0RBF1_RAT | 16 | 815 |
| Keratin, type II cytoskeletal 75 | A0A0H2UHH5_RAT | 13 | 714 |
| Ig gamma-2C chain C region | IGG2C_RAT | 14 | 699 |
| Murinoglobulin-1 | A0A0G2JUW7_RAT | 15 | 683 |
| Ig gamma-2A chain C region | IGG2A_RAT | 12 | 670 |
| Keratin, type I cytoskeletal 17 | A0A0G2K9Q9_RAT | 14 | 663 |
| Keratin, type II cytoskeletal 6A | K2C6A_RAT | 12 | 649 |
| Keratin, type II cytoskeletal 1b | A0A0G2JZQ9_RAT | 12 | 595 |
| Keratin, type I cytoskeletal 14 | K1C14_RAT | 12 | 592 |
| Actin, cytoplasmic 1 | A0A0G2K3K2_RAT | 11 | 539 |
| Desmoplakin | F1LMV6_RAT | 14 | 523 |
| Keratin, type II cytoskeletal 8 | K2C8_RAT | 10 | 503 |
| Junction plakoglobin | PLAK_RAT | 10 | 481 |
| Keratin, type I cytoskeletal 42 | K1C42_RAT | 11 | 472 |
| Uncharacterized protein | A0A0G2JVP4_RAT | 9 | 447 |
| Uncharacterized protein | A0A0G2JVP4_RAT | 7 | 417 |
| Keratin 16 | Q6IFU9_RAT | 9 | 413 |
| C-reactive protein | CRP_RAT | 6 | 325 |
| Uncharacterized protein | M0RA79_RAT | 5 | 303 |
| Tubulin alpha-1B chain | TBA1B_RAT | 7 | 296 |
| Uncharacterized protein | A0A0G2K9Y0_RAT | 6 | 293 |
| Ig kappa chain C region, A allele | KACA_RAT | 6 | 280 |
| Ac1873 | Q7TQ70_RAT | 7 | 256 |
| Tubulin beta-5 chain | TBB5_RAT | 5 | 246 |
| Tubulin beta-2B chain | TBB2B_RAT | 5 | 244 |
| Tubulin beta-4B chain | TBB4B_RAT | 5 | 229 |
| Isoform Long of Annexin A2 | ANXA2_RAT | 3 | 228 |
| Histone H4 | H4_RAT | 4 | 221 |
| Uncharacterized protein | A0A0G2K290_RAT | 4 | 218 |
| Isoform Gamma-A of Fibrinogen gamma chain | FIBG_RAT | 4 | 215 |
| Carboxypeptidase N catalytic chain | CBPN_RAT | 6 | 202 |
| Uncharacterized protein | A0A0G2K828_RAT | 3 | 176 |
| Histone H2B | A0A0G2JXE0_RAT | 3 | 172 |
| Neurofilament heavy polypeptide | F1LRZ7_RAT | 3 | 164 |
| Keratin 83 | A0A0G2JUU5_RAT | 4 | 150 |
| Uncharacterized protein | A0A0G2K3S3_RAT | 2 | 137 |
| Uncharacterized protein | M0RD98_RAT | 2 | 132 |
| Hemopexin | HEMO_RAT | 4 | 130 |
| Ig gamma-2B chain C region | IGG2B_RAT | 3 | 129 |
| Uncharacterized protein | D4ACR1_RAT | 2 | 126 |
| Carboxypeptidase N subunit 2 | F1LQT4_RAT | 3 | 123 |
| Keratinocyte proline-rich protein | G3V9A5_RAT | 3 | 122 |
| Uncharacterized protein | F1LXY6_RAT | 2 | 116 |
| Plakophilin 1 | D3ZY51_RAT | 2 | 115 |
| Selenoprotein P | SEPP1_RAT | 3 | 106 |
| Uncharacterized protein | F1M3X3_RAT | 2 | 102 |
| Apolipoprotein E | A0A0G2K151_RAT | 3 | 97 |
| Epsilon 1 globin | O88752_RAT | 1 | 91 |
| Globin c2 | A0A0G2JSV6_RAT | 2 | 82 |
| Keratin 78 (Fragment) | A0A0G2JUR6_RAT | 2 | 81 |
| Isoform 2 of Fibrinogen beta chain | FIBB_RAT | 2 | 81 |
| Keratin, type I cytoskeletal 40 | K1C40_RAT | 2 | 80 |
| Uncharacterized protein | A0A096P6M7_RAT | 1 | 80 |
| Neurofilament medium polypeptide | G3V7S2_RAT | 2 | 73 |
| Vitronectin | Q3KR94_RAT | 2 | 72 |
| Heat shock 70 kDa protein 1A | HS71A_RAT | 2 | 72 |
| Uncharacterized protein | A0A0G2K6T8_RAT | 2 | 69 |
| Uncharacterized protein | F1M6N0_RAT | 2 | 66 |
| Ras homolog family member C | A0A0G2K6E0_RAT | 1 | 64 |
| Chymotrypsinogen B | F1MA56_RAT | 2 | 61 |
| Uncharacterized protein | A0A0G2JZV7_RAT | 1 | 59 |
| Lactadherin | A0A0G2K506_RAT | 1 | 57 |
| Uncharacterized protein | A0A0G2JTG4_RAT | 1 | 56 |
| Uncharacterized protein | A0A0G2K099_RAT | 1 | 51 |
| Uncharacterized protein | A0A0G2JV42_RAT | 1 | 51 |
| Dystrophin | DMD_RAT | 2 | 50 |
| Cell division control protein 42 homolog | A0A0G2JSM8_RAT | 1 | 50 |
| Ig lambda-2 chain C region | LAC2_RAT | 2 | 49 |
| Uncharacterized protein | M0RBK4_RAT | 1 | 43 |
| Desmoglein 1 | D3ZM39_RAT | 1 | 42 |
| Elongation factor 1-alpha | M0R757_RAT | 1 | 42 |
| Uncharacterized protein | A0A0G2K4K2_RAT | 2 | 41 |
| Cationic trypsinogen | G3V7Q8_RAT | 1 | 41 |
| Histidine ammonia-lyase | HUTH_RAT | 1 | 40 |
| Trypsin V-A | TRYA_RAT | 1 | 39 |
| Cingulin | D4A4X4_RAT | 1 | 39 |
| Uncharacterized protein | A0A0G2K8S2_RAT | 1 | 39 |
| Centrosomal protein 290 | A0A0G2K715_RAT | 1 | 39 |
| Proline-rich protein 5 | A0A0H2UHJ6_RAT | 1 | 38 |
| Uncharacterized protein | A0A0G2JUY3_RAT | 1 | 38 |
| Uncharacterized protein | M0RAB8_RAT | 1 | 37 |
| Spliceosome RNA helicase Ddx39b | A0A0G2KAT4_RAT | 1 | 37 |
| Uncharacterized protein | A0A0G2K0N8_RAT | 1 | 37 |
| Complement factor B (Fragment) | A0A096MKF9_RAT | 1 | 36 |
| Methyltransferase-like 3 | Q4V8G6_RAT | 1 | 35 |
| Golgi apparatus protein 1 | G3V8G5_RAT | 1 | 35 |
| Ceruloplasmin | A0A0G2K9I6_RAT | 1 | 35 |
| Histone H2A.Z | H2AZ_RAT | 1 | 35 |
| DNA-(apurinic or apyrimidinic site) lyase | D3ZPZ3_RAT | 1 | 34 |
| Keratin, type II cytoskeletal 80 | K2C80_RAT | 1 | 33 |
| Uncharacterized protein (Fragment) | A0A096MK45_RAT | 1 | 32 |
ⱡ Ions score is −10*Log(P), where P is the probability that the observed match is a random event. Individual ions scores > 31 indicated identity or extensive homology (p < 0.05). Protein scores were derived from ions scores as a non-probabilistic basis for ranking protein hits.
2.4. Deiminated Proteins are Increased in Brain Tissue of Pre-Motor PD Rats
The pre-motor PD model brains presented an increased level of F95 immuno-positive staining compared with that of sham-treated brains. A strong positive staining for pan-deiminated proteins, by F95 detection, was observed in the cortex and hippocampus, as well as in white matter, with a highly notable positive staining in the brain vasculature (Figure 8A–C). F95 immuno-positive protein detection, assessed by thresholding intensity, showed an increase by 1.7-fold (p ***; p < 0.001) in the cortex and by 1.4-fold (p ***; p < 0.001) in the hippocampal CA1 region (Figure 8D,E), compared with shams.
Figure 8.


Total deiminated proteins are increased in brain tissue of pre-motor PD rats. Total deiminated protein expression was assessed by immunohistochemistry using the pan-deimination F95 antibody. (A) Increased protein deimination is observed in the brain vasculature in the cortex (ctx) of the pre-motor PD rat brains compared with that in the sham control brains. (B) Increased protein deimination is observed in white matter (wm) of the pre-motor PD rat brains compared with sham brains. (C) Increased protein deimination (F95 positive) is observed in the hippocampus (hip) of the pre-motor PD rat brains, compared with sham brains; the scale bars indicate 100 µm in all figures (ctx = cortex, hip = hippocampus, wm = white matter). (D,E) Thresholding intensity analysis of F95 positive staining showed a significant increase in the cortex (D), and the CA1 region of the hippocampus (E) of pre-motor PD models, compared with that in control sham brains (unpaired t-test, *** p < 0.001).
Immunohistochemical staining for deiminated histone H3 (citH3) also revealed increased detection in brains of pre-motor PD models, compared with shams. CitH3 positive staining was increased in the dentate gyrus (Figure 9A), and cortex (Figure 9B) of the pre-motor PD brains, compared with shams.
Figure 9.

Deiminated histone H3 staining is increased in the cortex and dentate gyrus of pre-motor PD models. Immunohistochemical staining showed increased protein detection of deiminated histone H3 (citH3) in: (A)) the dentate gyrus; (B) the cortex of the pre-motor PD models, compared with those in the control sham animals. The scale bars indicate 100 µm in all figures.
Immunoreactivity for PAD2, PAD3, and PAD4 was also confirmed in both sham-treated and pre-motor PD model brains, with some elevated protein levels for PAD4 observed in the pre-motor PD model, while PAD3 showed a slight elevation and PAD2 levels were not markedly changed in the pre-motor PD models (Figure 10A–C).
Figure 10.

PAD2, PAD3, and PAD4 protein detection in pre-motor PD model and sham brain tissue. (A) PAD2 protein detection in cortex (ctx), white matter (wm) and hippocampus (hip) of sham and pre-motor PD models. (B) PAD3 protein detection in cortex, white matter, and hippocampus of shams and pre-motor PD rats. (C) PAD4 protein detection in cortex, white matter and hippocampus of shams and pre-motor PD rats. The scale bars indicate 100 µm in all figures (ctx = cortex, hip = hippocampus, wm = white matter).
3. Discussion
The regulation of EV release is critical for cellular communication, and particularly for the modulation of the cellular microenvironment in a range of pathologies. Mounting evidence has linked EVs to neurodegenerative diseases, indicating critical roles for EV-mediated transport of pathogenic protein and genetic cargo, including in PD [21,22]. However, roles for such EV-mediated communication in early stages of PD have received limited attention. To this end, a rat pre-motor PD model was used in this study. While this model may not display all the signs observed in PD patients, it has been shown to display some non-motor symptoms (hyposmia and cognitive impairment) in the absence of motor dysfunction. This model allows therefore a valuable insight into the time window prior to the appearance of typical PD motor symptoms and highlight potential markers of early stages of the disease. The small sample sizes in each experimental group are justified by the clear statistical differences observed between the sham animals and pre-motor models indicating good compliance of this study to the 3Rs principles.
Roles for peptidylarginine deiminases (PADs) and post-translational deimination in various neurodegenerative diseases are an area of increasing interest. Data from a human RNA-Seq transcriptome and splicing database of glia, neurones, and vascular cells of the cerebral cortex have shown that levels of PAD2 are highest in mature astrocytes, oligodendrocytes, and microglia [28]. In the majority of studies of protein deimination in neurodegenerative diseases, a focus has so far been on histological analysis of postmortem human brain samples with increased pan-protein deimination detected in both PD and Alzheimer’s disease (AD) [10,11,29,30,31]. Deimination has hitherto not been assessed in pre-motor PD and was therefore the subject of our current study as such post-translationally mediated changes may result in early onset pro-inflammatory and neurodegenerative processes and therefore offer an opportunity for early intervention. Interestingly, in the current study using a rat model of pre-motor PD, that has been shown to display non motor symptoms in the absence of motor dysfunction and neuroinflammation [32], an increase in deiminated proteins was observed in the brain vasculature, particularly in the cortex and hippocampus. Such elevated F95 positive detection observed here correlates somewhat with a previous study on human PD post-mortem brain tissue, although that study reported elevation of deiminated proteins in the substantia nigra [10]. In the current rat pre-motor PD model, substantia nigra did neither show significant changes in F95 staining (data not shown) nor in citH3 staining (Supplementary Figure S1). The increase in F95 positivity detected in other brain areas in the current study does correlate with a previous study using cellular iPSC neuronal models derived from fibroblasts from patients carrying α-synuclein triplication [15], in which an increase in total deiminated proteins was observed in vitro [9].
When assessing deiminated proteins in total plasma and plasma-EVs, KEGG pathways for AD, PD, Huntington’s disease (HD), and prion disease were found to be enriched for deiminated proteins in the pre-motor PD model, compared with shams. Protein deimination has previously been linked to AD [11,29,30,31], PD [10,12], and prion disease, including Creutzfeldt–Jakob disease (CJD) [33,34,35]. However, via F95-enrichment and protein–network analysis, the current study reveals for the first time a link between protein deimination and HD, indicative of some common pathways with other dementias, including PD, via protein deimination. Enrichment for deiminated proteins in KEGG pathways for all these neurodegenerative diseases identified in the pre-motor PD model in this current study therefore indicates putative common pathways for these neurodegenerative disorders, regulated by deimination, including some contribution of deiminated protein cargo via circulating plasma-EVs. This is in agreement with previous studies assessing KEGG pathways in AD, PD, and HD, with the aim to identify shared pathways for distinct but related diseases that may share common underlying pathogenic mechanisms [36]. Such identification may be of importance to further current understanding of these neurodegenerative disorders and for the design of new treatment strategies, especially in the early stage of the disease.
Furthermore, in the current study, KEGG pathways for “oxidative phosphorylation”, “complement coagulation cascades”, “thermogenesis”, “metabolic pathways”, “S. aureus infection”, “gap junction”, “platelet activation”, and “apelin signalling pathway” were only identified for F95-enriched proteins in plasma-EVs of the pre-motor PD model, and were therefore specific to the circulating plasma-EVs in the pre-motor PD model. The relevance of these pathways in neurodegeneration, and specifically in relation to what is reported in the literature for PD, is further discussed below:
Oxidative phosphorylation is a vital part of metabolism, takes place in the mitochondria, and contributes to the major source of ATP. This process also leads to the generation of reactive oxygen species (ROS) and resulting oxidative stress and bioenergetics defects, which are linked to a range of neurodegenerative disorders, including PD [37,38,39]. While oxidative stress has been shown to play significant roles both in the onset and progression of PD [40], it still remains open to debate whether the mitochondrial respiratory deficiencies observed in a range of neurodegenerative disorders are initiators or consequences of prior insult [41]. Mitochondrial dysfunction has been found to be severe in cortex and in advanced stages of PD patients, based on algorithm analysis [42]. Identification of post-translational deimination in KEGG pathways of oxidative phosphorylation that was here identified for the first time in the pre-motor PD rat model, may provide novel insights into EV-mediated transport of deiminated proteins and protein deimination mediated effects on mitochondrial dysfunction via oxidative phosphorylation pathways in the pathogenesis of neurodegenerative diseases.
The complement system is an increasingly recognised factor in neurodegenerative diseases, also with links to PD [43,44,45]. Neuroinflammation can also result in the disruption of the blood–brain barrier and therefore lead to the direct participation of the adaptive immune system [46]. While complement mediated responses are involved in chronic inflammation, the complement system forms also part of the innate and humoral responses and has been recently linked to non-motor PD [47]. Interestingly, levels of the key complement components of the alternative and classical pathways, C3 and C4, were previously reported to be lower in non-motor PD compared with healthy controls [47], while severity of disease has been related to C3 and factor H levels in PD and AD patients [48]. However, no studies have discussed putative effects of post-translational modifications of these complement components in neurodegeneration, which may be a contributing factor to altered immune and inflammatory responses in early disease stages. The post-translational deimination of several complement components, including C3, C4, and factor H, alongside a number of other complement components, has been recently identified in a range of taxa [49,50,51,52,53,54]. Indeed, in the current study, C2, C3, as well as factor H and C-type lectin were found to be deimination candidates in rat plasma, both in shams and the pre-motor PD model, while C1q, factor B, C4, C8, C9, C4-binding protein, properdin, and collectin were only identified as deiminated in the pre-motor PD model. This indicates differences in complement pathway regulation via post-translational deimination in early stages of PD. As a number of arginines may undergo post-translational deimination in all these proteins, and therefore contribute to their structural and consequently downstream functional changes, the role for deimination of complement components in early onset neurodegeneration will need to be further evaluated. Furthermore, the involvement of inflammatory factors and modified inflammatory responses and their involvement with the brain–gut axis of PD also needs consideration [55]; particularly if such factors can be identified at early stages of disease.
Thermogenesis was identified as a deimination enriched KEGG pathway. Loss of body weight and fat mass is one of the non-motor symptoms of Parkinson’s disease and has, amongst others, been related to brown fat tissue meditated thermogenesis [56]. Thermogenesis is related to mitochondrial function [57,58] and also linked to synaptic transmission and neurodegeneration [59]. Alteration in mitochondrial complexes, restricting energy output, are related to sepsis as well as PD [57] and implicated in other neurodegenerative disease such as AD, as well as being associated with diabetes [60]. Interestingly, thermogenesis is also linked to CNS activity in hibernating animals [61,62]. Deimination in thermogenesis pathways has not been identified before and provides a novel insight into putative regulation via such post-translational modifications.
Metabolic KEGG pathways were identified in this study as deiminated in plasma-EVs of pre-motor PD rats. Mitochondria are the key regulators of cellular metabolism and their dysfunction is a hallmark of PD pathogenesis [63]. Furthermore, PD has been linked to a range of inherited metabolic disorders [64]. The metabolic landscape of neurodegenerative diseases is therefore receiving increased attention [65] and alterations in metabolic pathways in PD, including in early pre-motor stages in PD, have been discussed [66]. KEGG pathways for metabolic pathways have been linked to PD development based on microarray analysis of blood samples from PD patients and matched controls [67]. The identification here of enrichment of deiminated proteins in KEGG metabolic pathways in EVs provides a novel angle of post-translational regulation of such processes.
S. aureus infection KEGG pathway was identified to be enriched in deiminated proteins in plasma-EVs of the pre-motor PD rats. S. aureus has been found to be significantly increased in the conjunctival flora in PD patients [68]. Furthermore, α-synuclein upregulation and over-expression has been correlated with elevated innate immune responses and been verified to display antimicrobial peptide-like properties and antibacterial activity against S. aureus, alongside other bacteria and fungi [69]. Interestingly, phagocytic activity and bactericidal activity of neutrophils have been shown to be affected in early AD [70]. However, the precise involvement of protein deimination in bacterial infection pathways and in the regulation of immune responses in PD remains to be investigated. Deimination has previously been associated with bacterial immune evasion [71], as well as with bacterial membrane vesicle release and antibiotic resistance, highlighting roles for PAD-mediated mechanisms in host–pathogen interactions [6]. This may be of some interest as the brain–gut axis relationship between the gut microflora and PD has been a topic of investigation due to gastrointestinal symptoms being one of the earliest features of PD [72]. Roles for the gut microbiota in neuroinflammation has also been hypothesised and assessed for other neurodegenerative diseases, including AD and amyotrophic lateral sclerosis (ALS) [73]. Deimination of KEGG bacterial infection pathways, or in dysbacteriosis, has not been revealed in PD or other neurodegenerative diseases in previous studies, while KEGG pathways for bacterial infection were recently identified to be enriched in deiminated proteins in glioblastoma [7].
Gap junction and platelet activation were identified in this study as KEGG pathways enriched in deiminated proteins in pre-motor PD plasma EVs. Reduced platelet activation has been described in PD [74] and furthermore a recent study has identified gap junctions and platelet activation as KEGG pathways in early PD [26]. Gap junctions are formed by pannexins and connexins and allow for exchange of ions, second messengers, and small metabolites between adjacent cells [75]. Gap junctions have critical roles in homeostasis and roles in neurodegenerative disease, including PD, have been suggested [76,77,78]. Deimination in these pathways has not been reported before and may provide novel insights into such post-translationally mediated regulation in disease and disease progression.
Apelin signalling KEGG pathway was identified to be enriched in deiminated proteins in plasma-EVs of pre-motor PD rats only. Apelin signalling is involved in a broad range of physiological functions and furthermore associated with pathologies that result from decreased vascularisation—for example ischaemia, or neovascularisations events such as tumours and retinopathes [79]. Apelin is a neuropeptide with regulatory roles of many cellular functions and involvement in a range of physiological processes including metabolic, cardiovascular function, and regulation of body fluid homeostasis [80,81]. Apelin expression has been described in endothelial cells and the CNS [82] and has also been found to be involved in neuronal dysfunction related to inflammation during ageing [83]. Modulation of apelin signalling has been discussed in a range of pathologies [84] and has recently also been associated with neurodegenerative diseases [85], including AD [86,87,88,89] and PD [90,91,92]. However, specific roles in PD remain to be further investigated. Apelin signalling has been linked to autophagy in PD, both in cellular in vitro and in vivo mouse models [90,92]. PD mouse models have shown roles for apelin in MPTP-induced damage affecting the substantia nigra, behavioral dysfunction and dopaminergic neurodegeneration [90]. Roles for apelin signalling in endoplasmic reticulum (ER) stress have also been identified [91] and neuroprotective roles of apelin signalling pathways have recently been identified in PD mouse models [93]. The current study is the first to identify apelin signalling pathways to be associated with protein deimination, and such post-translational regulation of apelin via deimination, as identified here in pre-motor PD model plasma-EVs, may play important roles in the multifaceted roles of apelin signalling.
In whole plasma, KEGG pathways identified to be enriched in deiminated proteins in the pre-motor PD model compared with plasma from sham animals were KEGG pathways for PD, AD, HD, and prion disease, retrograde endocannabinoid signalling, SLE, and NAFLD. These are further discussed below:
Retrograde endocannabinoid signalling KEGG pathways were identified in this study to be enriched in deiminated proteins in the pre-motor PD plasma only. The retrograde endocannabinoid signalling system is a lipid-based neuromodulatory system with important roles in the CNS including in physiological and neurodegenerative processes [94,95]. It has been associated to pathogenesis of PD as well as AD and ischemia [95,96,97] and suggested as a therapeutic target for treatment of a range of neurodegenerative disorders including AD, PD, HD, multiple sclerosis (MS), and ALS [98,99,100]. Putative roles for its regulation via post-translational deimination have not been previously identified and therefore bring a novel angle of modulation of endocannabinoid signalling pathways in neurodegeneration, including in early disease stages.
Systemic lupus erythematosus (SLE) KEGG pathway was identified to be enriched in deiminated proteins in plasma of the pre-motor PD rat model. SLE is a multisystemic autoimmune disease, with association to cerebrovascular diseases [101,102,103] and also with PD [104]. Furthermore, Parkinsonian syndrome has been implicated in complicating SLE [105,106]. A population-based study assessing association of PD with SLE found an inverse association between the two, with SLE patients displaying a decreased risk of subsequent PD, although this study did not include early stages of PD [107]. The deimination of SLE KEGG pathway in pre-motor PD model may provide novel insights into the previously observed interplay between SLE and PD.
NAFLD KEGG pathways were enriched in deiminated proteins in pre-motor PD model plasma-EVs. Interestingly, a link via mitochondrial dysfunction has been made between metabolic syndrome, diabetes, obesity, and non-alcoholic fatty liver disease in the progression of AD, PD, and other neurodegenerative diseases [108]. Furthermore, lipid metabolism (in particular non-alcoholic fatty liver disease pathway) and mitochondrial dysregulation have been identified as molecular pathways and putative biomarkers linked to PD [109]. Post-translational deimination is revealed for the first time for NAFLD associated KEGG pathways in this study and may be of relevance for the interplay of neurodegeneration and such comorbidities.
Recent work has assessed circulating amino acid signatures in PD patient sera, where free citrulline was found to be lower in PD than controls [110]. Furthermore, assessment of molecular signatures in circulating small EVs have been identified in relationship to mitochondria and systemic inflammation, including CD9, NDUFS3, C-reactive protein (CRP), fibroblast growth factor 21, interleukin 9, macrophage inflammatory protein 1β, and tumour necrosis factor alpha [22]. Such changes have not been assessed in pre-motor PD animal models, and interestingly in this current study, CRP is one of the deimination candidates identified in both sham and pre-motor PD model plasma. Whether the deimination levels of CRP differ between sham and pre-motor PD plasma needs to be further investigated; indeed differences in CRP deimination and between CRP forms have been recently reported in teleost fish immunity [111] but have yet to be assessed in relation to human pathologies.
Overall, deimination signatures in EVs have yet to be assessed in relation to PD or other neurodegenerative diseases. Other proteomic approaches, including Raman profiling of circulating EVs, have been used to elucidate putative links to PD progression and treatment [21]. Furthermore, proteomic analysis of serum EVs in PD patients have revealed the expression levels of seven proteins, including pigmented epithelium-derived factor, afamin, apolipoprotein D and J, are significantly increased in PD patients. Moreover, expression levels of complement C1q and protein immunoglobulins, have been reported to be decreased in PD patients [112]. Some of these proteins including C1q, immunoglobulins, and apolipoprotein A, B, and E (but not D and J) were identified to be deiminated in the pre-motor PD model, bringing a novel aspect of post-translationally mediated regulation and modification of protein function, possibly contributing to disease mechanisms. A recent study using a cellular model of PD, treated with MPP+, a well-established parkinsonian toxicant, reported that microglial EVs, when not stimulated by aggregated α-synuclein, appeared to be protective, possibly involving mitochondrial dynamics and mitochondrial fission [113]. Furthermore, deimination of Nicotinamide- N-methyltransferase (NNMT), which is implicated in several chronic diseases as well as PD, has recently been identified [114]. These previous findings, as well as our present data, further support roles for EVs and circulating deiminated proteins, including in plasma-EVs, in the involvement of PD, including in early pre-motor stages, as identified in the current study.
In the present study, the brain tissue of pre-motor PD rats showed significant increase in total deiminated proteins, as assessed by the pan-deimination F95 antibody and for deiminated histone H3. While PAD2, PAD3, and PAD4 protein expression was confirmed in the rat brains, including in the cortex, hippocampus, and white matter, there was no significant difference in PAD isozyme protein levels, although some elevated levels were noted for PAD4 and to a lesser level for PAD3 protein in the pre-motor PD brains. Their deiminated protein products showed though a significant and marked increase in the pre-motor PD brains, particularly in the cortex, white matter, and hippocampus, while deiminated histone H3 was increased in the dentate gyrus as well as in the cortex. As protein deimination is a result of PAD activation, increased levels of PAD proteins per se would not necessarily be expected at this stage, while an increased production of deiminated protein products would be the result of PAD activation, leading to increased production of deiminated protein products. As dysregulation of calcium homeostasis is a known contributor to a number of neurodegenerative diseases, including in PD [115,116], and PADs are calcium activated enzymes, an increase in protein deimination would be one of such downstream factors. In previous studies on post-mortem PD brain samples, increased levels of total protein deimination and deimination-positive extracellular plaques have indeed been reported [10]. Furthermore, the presence of mutated misfolded α-synuclein protein has also been related to increased protein deimination [12]. Supporting those post-mortem studies, in vitro studies using iPSC derived neuronal models carrying α-synuclein triplication, revealed increased protein deimination levels, although specific target proteins of deimination were not further assessed [9].
A hitherto unrecognised contribution of deiminated proteins to the inflammatory responses, which previously have been shown in the pre-motor PD rat model [32], has to also be considered via the release of deiminated neuronal proteins from necrotic neurons, as well as via the circulation of deiminated proteins in the cerebrospinal fluid, which can contribute to progressive pathology due to generation of autoantibodies [117]. Furthermore, the significant increase observed in F95-positive brain vasculature of the pre-motor PD model, in particular in the cortex and hippocampus, and to some extent in the white matter, may also affect both local and systemic EV release and contribute to the spread and progression of pathology. Deiminated proteins also expose neo-epitopes which, in addition to leakage of deiminated proteins from dying cells, can further contribute to neuro-inflammatory responses. This also includes histone deimination which, in addition to gene regulatory effects, may cause extracellular trap formation which can contribute to local tissue damage [118]. Increased levels of deiminated histone H3 were here observed in the dentate gyrus as well as in cortex of the pre-motor PD brains. Such increase in citH3 positive detection may be of considerable importance in the neuroinflammatory environment; indeed, previous studies of CNS injury, including via hypoxic damage, have shown that pharmacological inhibition of PADs, and associated reduction of citH3, correlates with neuroprotective effects [119,120,121].
Recent work has emphasised the association of selected microRNAs to PD [27,122,123] and roles for microRNAs in neuroinflammation have received increased attention [25,124]. As neuroinflammation is thought to be a facilitator of PD pathogenesis, and was observed in the pre-motor PD model used in this study [32,125], we assessed changes in relative expression levels of three inflammatory and hypoxia associated microRNAs: miR21, miR155, and miR210. These microRNAs have previously been related to neurodegenerative diseases, including PD, although none of these microRNA (miRs) have been assessed, or associated with, pre-motor PD before the current study. All the microRNAs were here found to be significantly increased in circulating plasma-EVs of the pre-motor PD model. Known roles for these microRNAs in relation to neurodegeneration, including PD, are discussed below:
miR21 is a key regulator of inflammation [126], also associated with oxidative stress [127] and involved in neuroinflammatory regulation [128,129,130]. miR21 has been found to be upregulated in multiple sclerosis (MS), although specific functions have not been identified [131]. In PD, miR21 has been associated to autophagy [132]. Moreover, miR21 has been identified as a biomarker in plasma/serum for AD, prion disease, as well as traumatic brain injury [24,124,133,134], where it has also been found to be increased in EVs [135]. The current study is the first one to identify elevated miR21 as a marker of pre-motor PD.
miR155 has previously been identified as a contributor to the induction of neuroinflammation [25,136] and to increase blood–brain barrier (BBB) permeability [137]. In PD, miR155 has been linked to microglial inflammatory response induced by α-synuclein [23]. It has also been associated with various other neuroinflammatory disorders including MS, where it has been identified as a biomarker [138]. mir155 is also found to be elevated in AD and to contribute to neuroninflammation in this disease [139] and identified as a biomarker [124,133]. In PD, miR155 has been implicated in mitochondrial regulation in dopaminergic cell death in later stage [140], however this study presents a link between miR155 and pre-motor PD for the first time.
miR210 is a hypoxia-related microRNA, also linked to inflammation, and is induced under hypoxic conditions. It plays key roles in mitochondrial metabolism, apoptosis, cell proliferation, and the DNA damage response [141,142,143,144]. miR210 has been associated with mitochondrial dysfunction and oxidative stress in relation to neurodegenerative diseases [145,146]. It has previously been linked to PD in cell culture models, assessing environmental neurotoxicant mangane, in which miR210 was identified in EVs [147]. The interplay between oxidative stress and microRNAs has been shown in a number of neurodegenerative diseases including AD, PD, HD, and ALS [148]. Oxidative stress has indeed been suggested to play key roles in PD, although some parameters from clinical studies have been inconsistent [149]. Therefore, the identification in the current study of miR210 being significantly elevated in the pre-motor PD model indicates a hitherto unrecognized contribution by EV-mediated transport of this microRNA in early stages of PD.
Modifications of EV-mediated export of misfolded proteins, DNA, RNA, miRNAs, enzymes, and other EV cargo may be of considerable importance already in early stages of PD, and in neurodegenerative disease progression, and contribute to pathology. The transport of EV transcytosis across the BBB and associated release of EV cargo from brain endothelial cells into the systemic circulation has for example been suggested [150]. This correlates with strongly F95-positive vasculature observed in the pre-motor PD brains in the current study, which could also contribute to increased EV release from these sites. Therefore, the identification of early changes, such as EV numbers released and associated changes in specific EV microRNA and deiminated protein cargo, as identified here in circulating EVs in the rat pre-motor PD model, may offer novel markers indicative of early stages of pre-motor PD and be developed into useable screening tools.
4. Materials and Methods
4.1. Induction of the Rat Model of Pre-Motor PD
All procedures were approved by the Bloomsbury ethical committee and the Home Office and followed the British Home Office regulations with regard to the Animal Scientific Procedures Act 1986 (PPL PP3144142). Male Sprague-Dawley rats (200–250 g—Charles River Laboratories, U.K.) were kept under constant conditions of humidity (40–60%), temperature (18–22 °C), and a 12 h light–dark cycle. The induction of the pre-motor model was carried out as previously described [32]. In brief, intraperitoneal administration of either N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4, Sigma-Aldrich; a noradrenergic neurotoxin- pre-motor model) at a dose of 25 mg/kg, or sterile saline (sham animals) was performed 4 days prior to dopaminergic neurotoxin insult with 6-hydroxydopamine (6-OHDA). Bilateral striatal injections of either 6-OHDA (Sigma-Aldrich—dissolved in saline solution containing 0.9% ascorbic acid- pre-motor model) or saline containing 0.9% ascorbic acid for sham animals were then performed using the following coordinates from the atlas of Paxinos and Watson (1982), from Bregma: AP +1.0 mm, ML +3.0 mm, DV −6.5 mm. Animals were anaesthetised using isoflurane (5% v/v in O2 for induction and 2% v/v in O2 for maintenance) delivered through a fitted nose mask and rats were secured to a stereotaxic frame using blunt ear bars (David Kopf Instruments, Bethesda, MD, USA). Each animal received 15 μg of 6-OHDA per striatum (or vehicle) at a flow rate of 1 μL/min-1. Daily monitoring of the animals was performed following the surgical procedures. Rats were divided into two experimental groups: sham-treated animals (n = 3) and toxin-treated model (pre-motor model—n = 3). This pre-motor PD model was shown to present non-motor symptoms in the absence of motor dysfunction in a previous study [32]. The pre-motor PD models did not present any motor dysfunction prior to culling.
4.2. Immunohistochemistry
All histological procedures have been previously described [32,151]. Sham animals and pre-motor PD models were anaesthetised by inhalation of isoflurane and intraperitoneal injection of Euthatal (Merial, Harlow, UK) (60 mg/kg) and perfused transcardiacally with ice-cold oxygenated artificial cerebrospinal fluid (ACSF) containing in mM: 124 NaCl, 25.5 NaHCO3, 3.3 KCl, 1.2 KH2PO4, 1 MgSO4, 2.5 CaCl2, 15 mM D-Glucose equilibrated with 95% O2/5% CO2. Brains were removed and fixed overnight (4% paraformaldehyde, 0.2% saturated picric acid solution, 0.025% glutaraldehyde solution in 0.1 M Phosphate buffer). Fifty micrometers coronal sections containing cortex and hippocampus were cut with a vibratome (Agar Scientific Ltd, Stansted, UK). One in eight sections were collected and 4–5 slices per animal were used per staining. Sections were incubated first in 1% H2O2 for 30 min and then in 1% sodium borohydride (NaBH4) for 30 min to decrease background staining and then blocked in 10% normal goat serum (NGS) for another 30 min to block non-specific antibody binding. Sections were incubated overnight at 4 °C in the respective primary antibodies diluted in Phosphate Buffer Saline (PBS) containing 0.5% Triton X-100 (Sigma-Aldrich, Haverhill, UK): Mouse anti-peptidyl-citrulline F95 (for total deiminated proteins, MABN328, Merck, diluted 1:200), rabbit anti-Histone H3 (citrulline R2 + R8 + R17, ab5103, Abcam, diluted 1:200), rabbit anti-PAD2 (ab50257, Abcam; diluted 1:200), rabbit anti-PAD3 (ab50246; diluted 1:200) and rabbit anti-PAD4 (ab50332; diluted 1:200). Biotinylated labelled secondary anti-mouse IgM (Vector Laboratories—diluted 1:100) or anti-rabbit IgG (Diluted 1:500—Vector laboratories) were applied for 6 h. Sections were then washed and incubated in ABC (Vector Laboratories) overnight, washed, and then incubated in 3, 3′ diaminobenzidine (DAB- Sigma Aldrich) for 20 min. H2O2 was added to the DAB solution to allow the reaction until the filled cells were sufficiently labelled. Sections from the two groups were processed together and the DAB reaction was stopped at the same time to allow comparison between the shams and pre-motor models. Sections were placed onto Superfrost slides, dehydrated, cleared with Histoclear, and mounted using DPX (Sigma Aldrich). Levels of F95 immunohistochemical staining were measured by quantitative thresholding image analysis as previously described (Rahim et al., 2012; Sancandi et al., 2018). Images of both cortex and hippocampal CA1 in each section were captured using a DMR microscope and Leica Application Suite V4 (Leica Microsystems, Wetzlar, Germany) at 5× magnification with constant light intensity, microscope calibration, and video camera settings. Images were analysed using Image-Pro Premier (Media Cybernetics, Cambridge, UK) by measuring immunoreactivity using a constant threshold that was applied to all images for each respective antigen. Data are presented as the mean percentage area of immunoreactivity ± SEM.
4.3. EV isolation and Quantification by Nanoparticle Tracking Analysis
Plasma was isolated from blood collected from the heart of the anaesthetized rats via cardiac puncture prior to perfusion (described above). Two aliquots of 1.5 mL of blood were collected from each animal and plasma was isolated by centrifugation at 2000 g for 10 min, immediately aliquoted and frozen at −80 °C. EV isolation was carried out according to previously established protocols [5,7,54] as well as the recommendations of the International Society of Extracellular Vesicle Research (ISEV) [152]. Plasma aliquots were thawed before EV isolation and differential centrifugation was carried out by adding 100 µL plasma to 400 µL Dulbecco’s PBS (DPBS) and centrifuged at 4000 g for 30 min at 4 °C for removal of aggregates and apoptotic bodies. Next, centrifugation of the collected supernatant was carried out for 1 h at 4 °C at 100,000 g. The supernatant was then discarded and the isolated EV pellets were resuspended in ice-cold DPBS and centrifuged again at 100,000 g for 1 h at 4 °C. Thereafter, the final EV enriched pellet was resuspended in 100 μL sterile EV-free PBS. Nanoparticle tracking analysis (NTA) was carried out to quantify the EVs, using the NS300 Nanosight (Malvern Panalytical Ltd, Malvern, UK), equipped with a sCMOS camera and a 405 nm diode laser. EV samples were diluted 1:100 in sterile-filtered EV-free DPBS before application to the Nanosight. Particle numbers in the field of view were maintained within the rage of 40–60. The camera settings were set according to the manufacturer’s instructions (Malvern) at level 10 for recording of five 60 s videos per sample. For post-analysis of the recorded videos, the detection threshold was set at 5 and the obtained replicate histograms were averaged for each sample.
4.4. Western Blotting Analysis for EV Characterisation
For western blotting analysis, the plasma-EVs were re-constituted in 2 × Laemmli sample buffer containing 5% β-mercaptoethanol (BioRad, Kidlington, UK) and heated for 5 min at 100 °C. The proteins were separated on 4–20% Mini-Protean TGX protein gels (BioRad) by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by semi-dry western blotting. Ponceau S staining (Sigma, U.K.) was used to assess even transfer to nitrocellulose membranes (0.45 μm, BioRad), which were thereafter blocked for 1 h at room temperature (RT) in 5% bovine serum albumin (BSA) (Sigma) in Tris buffered saline (TBS) containing 0.1% Tween20 (TBS-T). Primary antibody incubation was carried out overnight at 4 °C using antibodies for two EV-specific markers: CD63 (ab68418; Abcam, Cambridge, UK; diluted 1:1000 in TBS-T) and Flotillin-1 (Flot-1; ab41927; Abcam; diluted 1:2000 in TBS-T). Thereafter, membranes were washed in TBS-T and incubated at RT for 1 h with HRP-conjugated anti-rabbit IgG secondary antibodies (BioRad, diluted 1:3000 in TBS-T). The membranes were washed for five times 10 min in TBS-T, followed by a final wash in TBS without Tween20. Protein bands were visualised using enhanced chemiluminescence (ECL; Amersham, U.K.) and the UVP BioDoc-ITTM System (Thermo Fischer Scientific, Hemel Hempstead, UK).
4.5. EV Characterisation by Transmission Electron Microscopy
TEM imaging of EVs was carried out according to previously described protocols [7,53]. The plasma-EVs were resuspended in 100 mM sodium cacodylate buffer (pH 7.4). One drop (~3–5 μL) of the EV suspension was placed onto a grid carrying a carbon support film, which previously had been glow discharged. After the suspension had partly dried, the grid was placed on to a drop of a 2.5% glutaraldehyde solution in 100 mM sodium cacodylate buffer (pH 7.4) for 1 min and thereafter washed by touching it to the surface of three drops of distilled water. Excess water was carefully removed using a filter paper. A drop of 2% aqueous Uranyl Acetate stain (Sigma-Aldrich) was next applied to the grid. Following 1 min, any excess stain was removed using a filter paper. The grid was dried at room temperature, whereafter the samples were imaged in TEM using a JEOL JEM 1400 transmission electron microscope (JEOL, Tokyo, Japan), which was operated at 80 kV at a magnification of 30,000 to 60,000. Digital images of EVs were recorded with an AMT XR60 CCD camera (Deben UK Limited, Bury Saint Edmunds, UK).
4.6. Analysis of microRNAs miR21, miR155, and miR210 in Plasma-EVs
For assessment of microRNA cargo in plasma-derived EVs of the pre-motor PD models and sham treated rats, EVs were isolated as described above and further processed for RNA isolation, cDNA translation, and assessment for expression of miR21, miR155, and miR210, according to previously described protocols [7]. First, RNA was extracted using Trizol (Sigma Aldrich, Gillingham, UK) and thereafter the RNA concentration and purity were measured by NanoDrop Spectrophotometer at 260 nm and 280 nm absorbance. The RNA was then reverse transcribed to cDNA, using the qScript microRNA cDNA Synthesis Kit (Quantabio, Beverly, MA, USA), following the manufacturer’s instructions. The resulting cDNA was utilised for assessment of relative expression levels of miR21, miR155, and miR210. Reference RNAs used for normalisation of miR expressions were U6 and hsa-let-7a-5p. MystiCq microRNA qPCR primers for miR21 (hsa-miR-21-5p), mir155 (hsa-miR-155-5p), and miR210 (hsa-miR-210-5p), all obtained from Sigma (U.K.), were used together with the PerfeCTa SYBR® Green SuperMix (Quantabio). The sequences for U6-snRNA primers were as follows: U6 forward, 5′-GCTTCGGCAGCACATATACTAAAAT-3′ and hsa-let-7a-5p forward 5′-CCGAGCTGAGGTAGTAGGTTGTATA-3′ reverse 5′-CGCTTCACGAATTTGCGTGTCAT-3′ for both. The following thermocycling conditions were applied: Denaturation: 95 °C for 2 min; followed by 40 cycles at 95 °C for 2 s and 60 °C for 15 s; extension was carried out at 72° C for 15 s. Expression levels of miR21, miR155, and miR210 were thereafter normalized to that of U6, according to the 2ΔΔCT method [153]. Each experiment was repeated in biological and technical triplicates.
4.7. Assessment of KEGG Pathways for Deiminated Proteins in Plasma and Plasma-EVs
F95 enrichment of deiminated proteins was carried out according to previously described protocols [7,53,54]. Rat plasma (a pool of 3 × 30 μL, using 30 μL aliquots from 3 individual animals per experimental group) and plasma-EVs (a pool of 3 × 30 μL extracted EVs from 3 individual animals per experimental group) respectively, were added to mini-prep sepharose columns in the presence of the pan-deimination F95 antibody (MABN328, Merck, Feltham, UK; [154]) for immunoprecipitation of total deiminated proteins, using the Catch and Release® v2.0 Immunoprecipitation Kit according to the manufacturer’s instructions (Merck). F95-enrichment was carried out overnight at 4 °C on a rotating platform and the F95 bound proteins were thereafter eluted using denaturing elution buffer, according to the manufacturer’s instructions (Merck). F95-enriched eluates from the plasma and plasma-EVs were thereafter analysed both by SDS-PAGE electrophoresis followed by silver staining as well as by liquid chromatography with tandem mass spectrometry (LC-MS/MS; Cambridge Proteomics, Cambridge, UK), according to previously described methods [7,53]. For LC-MS/MS, the F95-enriched eluates were first run 0.5 cm into a 12% TGX gel (BioRad) and thereafter cut out as one band each, respectively. The 1D gel bands were then transferred into a 96-well PCR plate. The gel-bands were cut into 1 mm2 pieces, destained, reduced using DTT, and alkylated using iodoacetamide, and thereafter digested with trypsin overnight at 37 °C. Thereafter, the supernatant was pipetted into a sample vial, following by loading onto an autosampler for automated LC-MS/MS analysis using a Dionex Ultimate 3000 RSLC nanoUPLC (Thermo Fisher Scientific Inc., Waltham, MA, USA) system in conjunction with a QExactive Orbitrap mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Peptide separation was carried out using reverse-phase chromatography (flow rate 300 nL/min) and a Thermo Scientific reverse-phase nano Easy-spray column (Thermo Scientific PepMap C18, 100A pore size, 2 µm particle size, 75 µm i.d. ×50 cm length). The peptides were loaded onto a pre-column (Thermo Scientific PepMap 100 C18, 5 µm particle size, 100A pore size, 300 µm i.d. ×5 mm length) from the Ultimate 3000 autosampler together with 0.1% formic acid for 3 min (flow rate 10 µL/min). Thereafter, the column valve was switched for elution of peptides from the pre-column onto the analytical column. Solvent A was water containing 0.1% formic acid, while solvent B contained 20% water and 80% acetonitrile, as well as 0.1% formic acid. A linear gradient of 2–40% B was employed for 30 min. The LC eluent was sprayed into the mass spectrometer using an Easy-Spray source (Thermo Fisher Scientific Inc.). The m/z values of all eluting ions were measured in an Orbitrap mass analyzer, which was set at a resolution of 70,000, while scanning was performed between m/z 380–1500. Data dependent scans (selecting top 20) were employed for automatic isolation and generation of fragment ions by higher energy collisional dissociation (HCD, NCE:25%) using the HCD collision cell. The resulting fragment ions were measured using the Orbitrap analyser, which was set at a resolution of 17,500. Both singly charged ions as well as ions with unassigned charge states were excluded from selection for MS/MS. A dynamic exclusion window of 20 sec was also applied. The data was processed post-run using Protein Discoverer (version 2.1., Thermo Scientific). In brief, MS/MS data was converted to mgf files which were submitted Mascot (Mascot search algorithm; Matrix Science, London, UK). Search for hits was carried out against the UniProt Rattus_norvegicus_20181203 (31,558 sequences; 17,280,660 residues) database as well as common contaminant sequences database (123 sequences; 40,594 residues). Peptide and fragment mass tolerances were respectively set at 20 ppm and 0.1 Da. The threshold value for significance was set at p < 0.05, while the peptide cut-off score was set at 20. To identify protein–protein interaction networks for deiminated proteins identified in whole plasma and plasma-EVs, respectively, STRING analysis (Search Tool for the Retrieval of Interacting Genes/Proteins; https://string-db.org/) was used. The protein networks were built as follows: function applied in STRING was “search multiple proteins”, the species database chosen was “Rattus norvegicus”, and “basic settings and medium confidence were applied. Colour lines between the nodes indicate the following evidence-based interactions for network edges: “known interactions” (these are based on curated databases, experimentally determined), as well as “predicted interactions” (these are based on gene neighbourhood, gene fusion, gene co-occurrence, or via text mining, co-expression, or protein homology).
4.8. Silver Staining
Following electrophoresis under reducing conditions as described above, using SDS-PAGE 4–20% TGX gels (BioRad), the F95-enriched eluted proteins from rat plasma and plasma-EVs were stained by silver staining. Staining was carried out using the BioRad Silver Stain Plus Kit (1610449, BioRad, UK), according to the manufacturer’s instructions (BioRad).
4.9. Statistical Analysis
All graphs were prepared and statistical analysis was performed using GraphPad Prism version 7 (GraphPad Software, San Diego, CA, USA). The experiments were repeated in triplicates, all histograms represent mean of data, while the error bars indicate standard deviation (SD). Significant differences were considered as p ≤ 0.05, following Student’s t-test. The NTA curves were generated by the NanoSight 3.0 software (Malvern, UK) with the black line representing the mean of the 5 repetitive readings per sample and the red line representing standard error (+/−).
5. Conclusions
In summary, the findings of the current study highlight PAD-mediated communication in early pre-motor stages of PD, both with respect to cerebrovascular changes as well as post-translational changes in protein networks relating to neurodegenerative diseases in plasma and EV-cargo. Changes of EV-mediated microRNA export were also verified, with increased expression of inflammatory and hypoxia related microRNAs in circulating plasma-EVs in the pre-motor PD model. Overall, an increase in F95 positive proteins, both in brain tissue and plasma, as well as in circulating plasma-EVs observed here in the pre-motor PD model, highlights hitherto under-recognized roles for these parameters in early stages of PD. Our findings emphasise the need to further assess and refine protein deimination and EV related microRNA and deimination biomarkers for early pre-motor PD diagnosis.
Acknowledgments
The authors would like to thank Yagnesh Umrania and Michael Deery at the Cambridge Centre for Proteomics for the LC–MS/MS analysis. Thanks are due to The Guy Foundation for funding the purchase of equipment utilised in this work.
Abbreviations
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| BSA | Bovine serum albumin |
| CD63 | CD63 antigen; granulophysin; lysosomal-associated membrane protein 3 |
| ECL | Enhanced chemiluminescence |
| EVs | Extracellular vesicles |
| F95 | Pan-deimination/citrullination antibody |
| FBS | Foetal bovine serum |
| Flot-1 | Flotillin-1 |
| HD | Huntington’s disease |
| KEGG | Kyoto encyclopedia of genes and genomes |
| LC-MS/MS | Liquid chromatography mass spectrometry |
| miR | microRNA |
| MS | Multiple sclerosis |
| NAFLD | Non-alcoholic fatty liver disease |
| NTA | Nanoparticle tracking analysis |
| PAD | Peptidylarginine deiminase |
| PD | Parkinson’s disease |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SLE | Systemic lupus erythematosus |
| TBS | Tris buffered saline |
| TEM | Transmission electron microscopy |
| WB | Western blotting |
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/21/8/2743/s1, Table S1. LC-MS/MS analysis of F95-enriched proteins in plasma of sham-treated (control) rats. The detailed list of protein hits identified following protein immunoprecipitation using the pan-deimination F95 antibody in control rat plasma is presented. Table S2. LC-MS/MS analysis of F95-enriched proteins in plasma of pre-motor PD-model rats. The detailed list of protein hits identified following protein immunoprecipitation using the pan-deimination F95 antibody in control rat plasma is presented. Table S3. LC-MS/MS analysis of F95-enriched proteins in plasma-EVs of sham-treated (control) rats. The detailed list of protein hits identified following protein immunoprecipitation using the pan-deimination F95 antibody in control rat plasma is presented. Table S4. LC-MS/MS analysis of F95-enriched proteins in plasma-EVs of pre-motor PD-model rats. The detailed list of protein hits identified following protein immunoprecipitation using the pan-deimination F95 antibody in control rat plasma is presented. Figure S1. Deiminated histone H3 (citH3) is not detected in the substantia nigra of pre-motor PD rat brains. Immunostaining of substantia nigra is shown in sham and pre-motor rat brains indicating no positive immunodetection for citH3. A similar result was seen for F95 which also showed negative in SN both in sham and pre-motor PD brains (not shown); the scale-bar represents 100 µm.
Author Contributions
Conceptualization, S.L.; methodology, M.S., P.U.-O., A.M., I.K. and S.L.; validation, M.S., P.U.-O., A.M. and S.L.; formal analysis, M.S., P.U-O., S.L.; investigation, M.S., P.U.-O., and S.L.; resources, A.M., P.U.-O., I.K. and S.L.; data curation, S.L.; writing—original draft preparation, S.L.; writing—review and editing, S.M., P.U.-O., A.M. and S.L.; visualization, M.S., P.U.-O., I.K., and S.L.; supervision, A.M. and S.L.; project administration, S.L.; funding acquisition, A.M., P.U.-O and S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partly supported by a University of Westminster start-up grants to S.L. and P.U.-O., and by internal funds from the UCL School of Pharmacy (A.M).
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Vossenaar E.R., Zendman A.J., van Venrooij W.J., Pruijn G.J. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. Bioessays. 2003;25:1106–1118. doi: 10.1002/bies.10357. [DOI] [PubMed] [Google Scholar]
- 2.György B., Toth E., Tarcsa E., Falus A., Buzas E.I. Citrullination: A posttranslational modification in health and disease. Int. J. Biochem. Cell Biol. 2006;38:1662–1677. doi: 10.1016/j.biocel.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Kholia S., Jorfi S., Thompson P.R., Causey C.P., Nicholas A.P., Inal J., Lange S. A Novel Role for Peptidylarginine Deiminases (PADs) in Microvesicle Release: A Therapeutic Potential for PAD Inhibitors to Sensitize Prostate Cancer Cells to Chemotherapy. J. Extracell. Vesicles. 2015;4:26192. doi: 10.3402/jev.v4.26192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kosgodage U.S., Trindade R.P., Thompson P.R., Inal J.M., Lange S. Chloramidine/Bisindolylmaleimide-I-Mediated Inhibition of Exosome and Microvesicle Release and Enhanced Efficacy of Cancer Chemotherapy. Int. J. Mol. Sci. 2017;18:1007. doi: 10.3390/ijms18051007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kosgodage U.S., Uysal-Onganer P., MacLatchy A., Kraev I., Chatterton N.P., Nicholas A.P., Inal J.M., Lange S. Peptidylarginine Deiminases Post-Translationally Deiminate Prohibitin and Modulate Extracellular Vesicle Release and MicroRNAs in Glioblastoma Multiforme. Int. J. Mol. Sci. 2018;20:103. doi: 10.3390/ijms20010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosgodage U.S., Matewele P., Mastroianni G., Kraev I., Brotherton D., Awamaria B., Nicholas A.P., Lange S., Inal J.M. Peptidylarginine Deiminase Inhibitors Reduce Bacterial Membrane Vesicle Release and Sensitize Bacteria to Antibiotic Treatment. Front. Cell Infect. Microbiol. 2019;9:227. doi: 10.3389/fcimb.2019.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uysal-Onganer P., MacLatchy A., Mahmoud R., Kraev I., Thompson P.R., Inal J.M., Lange S. Peptidylarginine Deiminase Isozyme-Specific PAD2, PAD3 and PAD4 Inhibitors Differentially Modulate Extracellular Vesicle Signatures and Cell Invasion in Two Glioblastoma Multiforme Cell Lines. Int. J. Mol. Sci. 2020;21:1495. doi: 10.3390/ijms21041495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lange S., Gallagher M., Kholia S., Kosgodage U.S., Hristova M., Hardy J., Inal J.M. Peptidylarginine Deiminases—Roles in Cancer and Neurodegeneration and Possible Avenues for Therapeutic Intervention via Modulation of Exosome and Microvesicle (EMV) Release? Int. J. Mol. Sci. 2017;18:1196. doi: 10.3390/ijms18061196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lange S., Wray S., Devine M., Matarin M., Hardy J. Protein Deimination in Protein Misfolding Disorders–Modelled in Human Induced Pluripotent Stem Cells (iPSCs) In: Nicholas A.P., Bhattacharya S.K., editors. Protein Deimination in Human Health and Disease. Volume 2. Springer Science and Business Media; New York, NY, USA: 2017. Chapter 24. [Google Scholar]
- 10.Nicholas A.P. Dual immunofluorescence study of citrullinated proteins in Parkinson diseased substantia nigra. Neurosci. Lett. 2011;495:26–29. doi: 10.1016/j.neulet.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 11.Nicholas A.P. Dual immunofluorescence study of citrullinated proteins in Alzheimer diseased frontal cortex. Neurosci. Lett. 2013;545:107–111. doi: 10.1016/j.neulet.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicholas A.P., Lu L., Heaven M., Kadish I., van Groen T., Accavitti-Loper M.A., Wewering S., Kofskey D., Gambetti P., Brenner M. Ongoing studies of deimination in neurodegenerative diseases using the F95 antibody. In: Nicholas A.P., Bhattacharya S.K., editors. Protein Deimination in Human Health and Disease. Springer-Verlag; New York, NY, USA: 2014. pp. 257–280. [Google Scholar]
- 13.Bicker K.L., Thompson P.R. The protein arginine deiminases: Structure, function, inhibition, and disease. Biopolymers. 2013;99:155–163. doi: 10.1002/bip.22127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang S., Wang Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim. Biophys. Acta. 2013;1829:1126–1135. doi: 10.1016/j.bbagrm.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Devine M.J., Ryten M., Vodicka P., Thomson A.J., Burdon T., Houlden H., Cavaleri F., Nagano M., Drummond N.J., Taanman J.W., et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2011;2:440. doi: 10.1038/ncomms1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pegtel D.M., Peferoen L., Amor S. Extracellular vesicles as modulators of cell-to-cell communication in the healthy and diseased brain. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014;369:20130516. doi: 10.1098/rstb.2013.0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill A.F. Extracellular Vesicles and Neurodegenerative Diseases. J. Neurosci. 2019;39:9269–9273. doi: 10.1523/JNEUROSCI.0147-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W., Bai X., Zhang A., Huang J., Xu S., Zhang J. Role of Exosomes in Central Nervous System Diseases. Front. Mol. Neurosci. 2019;12:240. doi: 10.3389/fnmol.2019.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longoni B., Fasciani I., Kolachalam S., Pietrantoni I., Marampon F., Petragnano F., Aloisi G., Coppolino M.F., Rossi M., Scarselli M., et al. Neurotoxic and Neuroprotective Role of Exosomes in Parkinson’s Disease. Curr. Pharm. Des. 2019;25:4510–4522. doi: 10.2174/1381612825666191113103537. [DOI] [PubMed] [Google Scholar]
- 20.Porro C., Panaro M.A., Lofrumento D.D., Hasalla E., Trotta T. The multiple roles of exosomes in Parkinson’s disease: An overview. Immunopharmacol. Immunotoxicol. 2019;41:469–476. doi: 10.1080/08923973.2019.1650371. [DOI] [PubMed] [Google Scholar]
- 21.Gualerzi A., Picciolini S., Carlomagno C., Terenzi F., Ramat S., Sorbi S., Bedoni M. Raman profiling of circulating extracellular vesicles for the stratification of Parkinson’s patients. Nanomedicine. 2019;22:102097. doi: 10.1016/j.nano.2019.102097. [DOI] [PubMed] [Google Scholar]
- 22.Picca A., Guerra F., Calvani R., Marini F., Biancolillo A., Landi G., Beli R., Landi F., Bernabei R., Bentivoglio A.R., et al. Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. J. Clin. Med. 2020;9:504. doi: 10.3390/jcm9020504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thome A.D., Harms A.S., Volpicelli-Daley L.A., Standaert D.G. MicroRNA-155 Regulates Alpha-Synuclein-Induced Inflammatory Responses in Models of Parkinson Disease. J. Neurosci. 2016;36:2383–2390. doi: 10.1523/JNEUROSCI.3900-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez B., Peplow P. MicroRNAs as Diagnostic Markers and Therapeutic Targets for Traumatic Brain Injury. Neural Regen. Res. 2017;12:1749. doi: 10.4103/1673-5374.219025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slota J.A., Booth S.A. MicroRNAs in Neuroinflammation: Implications in Disease Pathogenesis, Biomarker Discovery and Therapeutic Applications. Noncoding RNA. 2019;5:35. doi: 10.3390/ncrna5020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X., Chen J., Guan T., Yao H., Zhang W., Guan Z., Wang Y. miRNAs and target genes in the blood as biomarkers for the early diagnosis of Parkinson’s disease. BMC Syst. Biol. 2019;13:10. doi: 10.1186/s12918-019-0680-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Renani P.G., Taheri F., Rostami D., Farahani N., Abdolkarimi H., Abdollahi E., Taghizadeh E., Gheibi Hayat S.M. Involvement of aberrant regulation of epigenetic mechanisms in the pathogenesis of Parkinson’s disease and epigenetic-based therapies. J. Cell Physiol. 2019;234:19307–19319. doi: 10.1002/jcp.28622. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y., Chen K., Sloan S.A., Bennett M.L., Scholze A.R., O’Keeffe S., Phatnani H.P., Guarnieri P., Caneda C., Ruderisch N., et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishigami A., Ohsawa T., Hiratsuka M., Taguchi H., Kobayashi S., Saito Y., Murayama S., Asaga H., Toda T., Kimura N., et al. Abnormal accumulation of citrullinated proteins catalyzed by peptidylarginine deiminase in hippocampal extracts from patients with Alzheimer’s disease. J. Neurosci. Res. 2005;80:120–128. doi: 10.1002/jnr.20431. [DOI] [PubMed] [Google Scholar]
- 30.Ishigami A., Choi E.-K., Kim S., Maruyama N. Deimination in Alzheimer’s Disease. In: Nicholas A.P., Bhattacharya S.K., editors. Protein Deimination in Human Health and Disease. Springer Science and Business Media; New York, NY, USA: 2014. Chapter 13. [Google Scholar]
- 31.Ishigami A., Masutomi H., Handa S., Nakamura M., Nakaya S., Uchida Y., Saito Y., Murayama S., Jang B., Jeon Y.C., et al. Mass spectrometric identification of citrullination sites and immunohistochemical detection of citrullinated glial fibrillary acidic protein in Alzheimer’s disease brains. J. Neurosci. Res. 2015;93:1664–1674. doi: 10.1002/jnr.23620. [DOI] [PubMed] [Google Scholar]
- 32.Sancandi M., Schul E.V., Economides G., Constanti A., Mercer A. Structural Changes Observed in the Piriform Cortex in a Rat Model of Pre-motor Parkinson’s Disease. Front. Cell Neurosci. 2018;12:479. doi: 10.3389/fncel.2018.00479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang B., Jin J.K., Jeon Y.C., Cho H.J., Ishigami A., Choi K.C., Carp R.I., Maruyama N., Kim Y.S., Choi E.K. Involvement of peptidylarginine deiminase-mediated post-translational citrullination in pathogenesis of sporadic Creutzfeldt-Jakob disease. Acta Neuropathol. 2009;119:199–210. doi: 10.1007/s00401-009-0625-x. [DOI] [PubMed] [Google Scholar]
- 34.Jang B., Ishigami A., Maruyama N., Carp R.I., Kim Y.S., Choi E.K. Peptidylarginine deiminase and protein citrullination in prion diseases: Strong evidence of neurodegeneration. Prion. 2013;7:42–46. doi: 10.4161/pri.22380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jang B., Jeon Y.C., Shin H.Y., Lee Y.J., Kim H., Kondo Y., Ishigami A., Kim Y.S., Choi E.K. Myelin Basic Protein Citrullination, a Hallmark of Central Nervous System Demyelination, Assessed by Novel Monoclonal Antibodies in Prion Diseases. Mol. Neurobiol. 2018;55:3172–3184. doi: 10.1007/s12035-017-0560-0. [DOI] [PubMed] [Google Scholar]
- 36.Li P., Nie Y., Yu J. An Effective Method to Identify Shared Pathways and Common Factors among Neurodegenerative Diseases. PLoS ONE. 2015;10:e0143045. doi: 10.1371/journal.pone.0143045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo S., Kang S.S., Wang Z.H., Liu X., Day J.X., Wu Z., Peng J., Xiang D., Springer W., Ye K. Akt Phosphorylates NQO1 and Triggers its Degradation, Abolishing Its Antioxidative Activities in Parkinson’s Disease. J. Neurosci. 2019;39:7291–7305. doi: 10.1523/JNEUROSCI.0625-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deus C.M., Pereira S.P., Cunha-Oliveira T., Pereira F.B., Raimundo N., Oliveira P.J. Mitochondrial remodeling in human skin fibroblasts from sporadic male Parkinson’s disease patients uncovers metabolic and mitochondrial bioenergetic defects. Biochim. Biophys. Acta Mol. Basis Dis. 2020;1866:165615. doi: 10.1016/j.bbadis.2019.165615. [DOI] [PubMed] [Google Scholar]
- 39.Hikosaka K., Yaku K., Okabe K., Nakagawa T. Implications of NAD metabolism in pathophysiology and therapeutics for neurodegenerative diseases. Nutr. Neurosci. 2019:1–13. doi: 10.1080/1028415X.2019.1637504. [DOI] [PubMed] [Google Scholar]
- 40.Mashima K., Takahashi S., Minami K., Izawa Y., Abe T., Tsukada N., Hishiki T., Suematsu M., Kajimura M., Suzuki N. Neuroprotective Role of Astroglia in Parkinson Disease by Reducing Oxidative Stress Through Dopamine-Induced Activation of Pentose-Phosphate Pathway. ASN Neuro. 2018;10:1759091418775562. doi: 10.1177/1759091418775562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Area-Gomez E., Guardia-Laguarta C., Schon E.A., Przedborski S. Mitochondria, OxPhos, and neurodegeneration: Cells are not just running out of gas. J. Clin. Investig. 2019;129:34–45. doi: 10.1172/JCI120848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karki R., Kodamullil A.T., Hoyt C.T., Hofmann-Apitius M. Quantifying mechanisms in neurodegenerative diseases (NDDs) using candidate mechanism perturbation amplitude (CMPA) algorithm. BMC Bioinform. 2019;20:494. doi: 10.1186/s12859-019-3101-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Depboylu C., Schäfer M.K., Arias-Carrión O., Oertel W.H., Weihe E., Höglinger G.U. Possible involvement of complement factor C1q in the clearance of extracellular neuromelanin from the substantia nigra in Parkinson disease. J. Neuropathol. Exp. Neurol. 2011;70:125–132. doi: 10.1097/NEN.0b013e31820805b9. [DOI] [PubMed] [Google Scholar]
- 44.Chao Y., Wong S.C., Tan E.K. Evidence of inflammatory system involvement in Parkinson’s disease. Biomed. Res. Int. 2014;2014:308654. doi: 10.1155/2014/308654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carpanini S.M., Torvell M., Morgan B.P. Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol. 2019;10:362. doi: 10.3389/fimmu.2019.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pal R., Tiwari P.C., Nath R., Pant K.K. Role of neuroinflammation and latent transcription factors in pathogenesis of Parkinson’s disease. Neurol. Res. 2016;38:1111–1122. doi: 10.1080/01616412.2016.1249997. [DOI] [PubMed] [Google Scholar]
- 47.Sun C., Yu W., Zhao Z., Song C., Liu Y., Jia G., Wang X., Liu Y. Peripheral Humoral Immune Response Is Associated With the Non-motor Symptoms of Parkinson’s Disease. Front. Neurosci. 2019;13:1057. doi: 10.3389/fnins.2019.01057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y., Hancock A.M., Bradner J., Chung K.A., Quinn J.F., Peskind E.R., Galasko D., Jankovic J., Zabetian C.P., Kim H.M., et al. Complement 3 and factor h in human cerebrospinal fluid in Parkinson’s disease, Alzheimer’s disease, and multiple-system atrophy. Am. J. Pathol. 2011;178:1509–1516. doi: 10.1016/j.ajpath.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Magnadóttir B., Bragason B.T., Bricknell I.R., Bowden T., Nicholas A.P., Hristova M., Guðmundsdóttir S., Dodds A.W., Lange S. Peptidylarginine deiminase and deiminated proteins are detected throughout early halibut ontogeny—Complement components C3 and C4 are post-translationally deiminated in halibut (Hippoglossus hippoglossus L.) Dev. Comp. Immunol. 2019;92:1–19. doi: 10.1016/j.dci.2018.10.016. [DOI] [PubMed] [Google Scholar]
- 50.Magnadóttir B., Kraev I., Guðmundsdóttir S., Dodds A.W., Lange S. Extracellular vesicles from cod (Gadus morhua L.) mucus contain innate immune factors and deiminated protein cargo. Dev. Comp. Immunol. 2019;99:103397. doi: 10.1016/j.dci.2019.103397. [DOI] [PubMed] [Google Scholar]
- 51.Magnadóttir B., Uysal-Onganer P., Kraev I., Svansson V., Hayes P., Lange S. Deiminated proteins and extracellular vesicles—Novel serum biomarkers in whales and orca. Comp. Biochem. Physiol. Part D Genom. Proteom. 2020;34:100676. doi: 10.1016/j.cbd.2020.100676. [DOI] [PubMed] [Google Scholar]
- 52.Lange S., Kraev I., Magnadóttir B., Dodds A.W. Complement component C4-like protein in Atlantic cod (Gadus morhua L.)—Detection in ontogeny and identification of post-translational deimination in serum and extracellular vesicles. Dev. Comp. Immunol. 2019;101:103437. doi: 10.1016/j.dci.2019.103437. [DOI] [PubMed] [Google Scholar]
- 53.Phillips R.A., Kraev I., Lange S. Protein Deimination and Extracellular Vesicle Profiles in Antarctic Seabirds. Biology (Basel) 2020;9:15. doi: 10.3390/biology9010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pamenter M.E., Uysal-Onganer P., Huynh K.W., Kraev I., Lange S. Post-Translational Deimination of Immunological and Metabolic Protein Markers in Plasma and Extracellular Vesicles of Naked Mole-Rat (Heterocephalus glaber) Int. J. Mol. Sci. 2019;20:5378. doi: 10.3390/ijms20215378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Y., Su Y., Xu W., Wang W., Yao S. Constipation Increases Disability and Decreases Dopamine Levels in the Nigrostriatal System through Gastric Inflammatory Factors in Parkinson’s Disease. Curr. Neurovasc. Res. 2019;16:241–249. doi: 10.2174/1567202616666190618170103. [DOI] [PubMed] [Google Scholar]
- 56.Wang W., Meng X., Yang C., Fang D., Wang X., An J., Zhang J., Wang L., Lu T., Ruan H.B., et al. Brown adipose tissue activation in a rat model of Parkinson’s disease. Am. J. Physiol. Endocrinol. Metab. 2017;313:E731–E736. doi: 10.1152/ajpendo.00049.2017. [DOI] [PubMed] [Google Scholar]
- 57.Carreras M.C., Franco M.C., Peralta J.G., Poderoso J.J. Nitric oxide, complex I, and the modulation of mitochondrial reactive species in biology and disease. Mol. Aspects Med. 2004;25:125–139. doi: 10.1016/j.mam.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 58.Klotzsch E., Smorodchenko A., Löfler L., Moldzio R., Parkinson E., Schütz G.J., Pohl E.E. Superresolution microscopy reveals spatial separation of UCP4 and F0F1-ATP synthase in neuronal mitochondria. Proc. Natl. Acad. Sci. USA. 2015;112:130–135. doi: 10.1073/pnas.1415261112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andrews Z.B., Diano S., Horvath T.L. Mitochondrial uncoupling proteins in the CNS: In support of function and survival. Nat. Rev. Neurosci. 2005;6:829–840. doi: 10.1038/nrn1767. [DOI] [PubMed] [Google Scholar]
- 60.Akhter F., Chen D., Yan S.F., Yan S.S. Mitochondrial Perturbation in Alzheimer’s Disease and Diabetes. Prog. Mol. Biol. Transl. Sci. 2017;146:341–361. doi: 10.1016/bs.pmbts.2016.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arendt T., Bullmann T. Neuronal plasticity in hibernation and the proposed role of the microtubule-associated protein tau as a “master switch” regulating synaptic gain in neuronal networks. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013;305:R478–R489. doi: 10.1152/ajpregu.00117.2013. [DOI] [PubMed] [Google Scholar]
- 62.Laursen W.J., Mastrotto M., Pesta D., Funk O.H., Goodman J.B., Merriman D.K., Ingolia N., Shulman G.I., Bagriantsev S.N., Gracheva E.O. Neuronal UCP1 expression suggests a mechanism for local thermogenesis during hibernation. Proc. Natl. Acad. Sci. USA. 2015;112:1607–1612. doi: 10.1073/pnas.1421419112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang X., Jin T., Zhang H., Miao J., Zhao X., Su Y., Zhang Y. Current Progress of Mitochondrial Quality Control Pathways Underlying the Pathogenesis of Parkinson’s Disease. Oxid. Med. Cell Longev. 2019;2019:4578462. doi: 10.1155/2019/4578462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Limphaibool N., Iwanowski P., Holstad M.J.V., Perkowska K. Parkinsonism in Inherited Metabolic Disorders: Key Considerations and Major Features. Front. Neurol. 2018;9:857. doi: 10.3389/fneur.2018.00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Özcan E., Çakır T. Genome-Scale Brain Metabolic Networks as Scaffolds for the Systems Biology of Neurodegenerative Diseases: Mapping Metabolic Alterations. Adv. Neurobiol. 2018;21:195–217. doi: 10.1007/978-3-319-94593-4_7. [DOI] [PubMed] [Google Scholar]
- 66.Ferrer I., Martinez A., Blanco R., Dalfó E., Carmona M. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: Preclinical Parkinson disease. J. Neural Transm. (Vienna) 2011;118:821–839. doi: 10.1007/s00702-010-0482-8. [DOI] [PubMed] [Google Scholar]
- 67.Tan C., Liu X., Chen J. Microarray Analysis of the Molecular Mechanism Involved in Parkinson’s Disease. Parkinsons Dis. 2018;2018:1590465. doi: 10.1155/2018/1590465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kusbeci T., Kusbeci O.Y., Aktepe O.C., Yavas G., Ermis S.S. Conjunctival flora in patients with Parkinson’s disease. Curr. Eye Res. 2009;34:251–256. doi: 10.1080/02713680902725970. [DOI] [PubMed] [Google Scholar]
- 69.Park S.C., Moon J.C., Shin S.Y., Son H., Jung Y.J., Kim N.H., Kim Y.M., Jang M.K., Lee J.R. Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem. Biophys. Res. Commun. 2016;478:924–928. doi: 10.1016/j.bbrc.2016.08.052. [DOI] [PubMed] [Google Scholar]
- 70.Davydova T.V., Fomina V.G., Voskresenskaya N.I., Doronina O.A. Phagocytic activity and state of bactericidal systems in polymorphonuclear leukocytes from patients with Alzheimer’s disease. Bull. Exp. Biol. Med. 2003;136:355–357. doi: 10.1023/B:BEBM.0000010950.53560.e2. [DOI] [PubMed] [Google Scholar]
- 71.Bielecka E., Scavenius C., Kantyka T., Jusko M., Mizgalska D., Szmigielski B., Potempa B., Enghild J.J., Prossnitz E.R., Blom A.M., et al. Peptidyl arginine deiminase from Porphyromonas gingivalis abolishes anaphylatoxin C5a activity. J. Biol. Chem. 2014;289:32481–32487. doi: 10.1074/jbc.C114.617142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ilie O.D., Ciobica A., McKenna J., Doroftei B., Mavroudis I. Minireview on the Relations between Gut Microflora and Parkinson’s Disease: Further Biochemical (Oxidative Stress), Inflammatory, and Neurological Particularities. Oxid. Med. Cell Longev. 2020;2020:4518023. doi: 10.1155/2020/4518023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Loffredo L., Ettorre E., Zicari A.M., Inghilleri M., Nocella C., Perri L., Spalice A., Fossati C., De Lucia M.C., Pigozzi F., et al. Oxidative Stress and Gut-Derived Lipopolysaccharides in Neurodegenerative Disease: Role of NOX2. Oxid. Med. Cell Longev. 2020;2020:8630275. doi: 10.1155/2020/8630275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lim K.M., Kim H.H., Bae O.N., Noh J.Y., Kim K.Y., Kim S.H., Chung S.M., Shin S., Kim H.Y., Chung J.H. Inhibition of platelet aggregation by 1-methyl-4-phenyl pyridinium ion (MPP+) through ATP depletion: Evidence for the reduced platelet activities in Parkinson’s disease. Platelets. 2009;20:163–170. doi: 10.1080/09537100902721746. [DOI] [PubMed] [Google Scholar]
- 75.Meşe G., Richard G., White T.W. Gap junctions: Basic structure and function. J. Investig. Dermatol. 2007;127:2516–2524. doi: 10.1038/sj.jid.5700770. [DOI] [PubMed] [Google Scholar]
- 76.Schwab B.C., Heida T., Zhao Y., van Gils S.A., van Wezel R.J. Pallidal gap junctions-triggers of synchrony in Parkinson’s disease? Mov. Disord. 2014;29:1486–1494. doi: 10.1002/mds.25987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Phookan S., Sutton A.C., Walling I., Smith A., O’Connor K.A., Campbell J.C., Calos M., Yu W., Pilitsis J.G., Brotchie J.M, et al. Gap junction blockers attenuate beta oscillations and improve forelimb function in hemiparkinsonian rats. Exp. Neurol. 2015;265:160–170. doi: 10.1016/j.expneurol.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 78.Ahmadian E., Eftekhari A., Samiei M., Maleki Dizaj S., Vinken M. The role and therapeutic potential of connexins, pannexins and their channels in Parkinson’s disease. Cell Signal. 2019;58:111–118. doi: 10.1016/j.cellsig.2019.03.010. [DOI] [PubMed] [Google Scholar]
- 79.Sorli S.C., van den Berghe L., Masri B., Knibiehler B., Audigier Y. Therapeutic potential of interfering with apelin signalling. Drug. Discov. Today. 2006;11:1100–1106. doi: 10.1016/j.drudis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 80.Reaux A., De Mota N., Skultetyova I., Lenkei Z., El Messari S., Gallatz K., Corvol P., Palkovits M., Llorens-Cortès C. Physiological role of a novel neuropeptide, apelin, and its receptor in the rat brain. J. Neurochem. 2001;77:1085–1096. doi: 10.1046/j.1471-4159.2001.00320.x. [DOI] [PubMed] [Google Scholar]
- 81.Reaux-Le Goazigo A., Bodineau L., De Mota N., Jeandel L., Chartrel N., Knauf C., Raad C., Valet P., Llorens-Cortes C. Apelin and the proopiomelanocortin system: A new regulatory pathway of hypothalamic α-MSH release. Am. J. Physiol. Endocrinol. Metab. 2011;301:E955–E966. doi: 10.1152/ajpendo.00090.2011. [DOI] [PubMed] [Google Scholar]
- 82.De Mota N., Lenkei Z., Llorens-Cortès C. Cloning, pharmacological characterization and brain distribution of the rat apelin receptor. Neuroendocrinology. 2000;72:400–407. doi: 10.1159/000054609. [DOI] [PubMed] [Google Scholar]
- 83.Sauvant J., Delpech J.C., Palin K., De Mota N., Dudit J., Aubert A., Orcel H., Roux P., Layé S., Moos F., et al. Mechanisms involved in dual vasopressin/apelin neuron dysfunction during aging. PLoS ONE. 2014;9:e87421. doi: 10.1371/journal.pone.0087421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Iturrioz X., Alvear-Perez R., De Mota N., Franchet C., Guillier F., Leroux V., Dabire H., Le Jouan M., Chabane H., Gerbier R., et al. Identification and pharmacological properties of E339-3D6, the first nonpeptidic apelin receptor agonist. FASEB J. 2010;24:1506–1517. doi: 10.1096/fj.09-140715. [DOI] [PubMed] [Google Scholar]
- 85.Heras-Sandoval D., Pérez-Rojas J.M., Pedraza-Chaverri J. Novel compounds for the modulation of mTOR and autophagy to treat neurodegenerative diseases. Cell Signal. 2020;65:109442. doi: 10.1016/j.cellsig.2019.109442. [DOI] [PubMed] [Google Scholar]
- 86.Masoumi J., Abbasloui M., Parvan R., Mohammadnejad D., Pavon-Djavid G., Barzegari A., Abdolalizadeh J. Apelin, a promising target for Alzheimer disease prevention and treatment. Neuropeptides. 2018;70:76–86. doi: 10.1016/j.npep.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 87.Aminyavari S., Zahmatkesh M., Farahmandfar M., Khodagholi F., Dargahi L., Zarrindast M.R. Protective role of Apelin-13 on amyloid β25-35-induced memory deficit; Involvement of autophagy and apoptosis process. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2019;89:322–334. doi: 10.1016/j.pnpbp.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 88.Aminyavari S., Zahmatkesh M., Khodagholi F., Sanati M. Anxiolytic impact of Apelin-13 in a rat model of Alzheimer’s disease: Involvement of glucocorticoid receptor and FKBP5. Peptides. 2019;118:170102. doi: 10.1016/j.peptides.2019.170102. [DOI] [PubMed] [Google Scholar]
- 89.Zou D., Li R., Huang X., Chen G., Liu Y., Meng Y., Wang Y., Wu Y., Mao Y. Identification of molecular correlations of RBM8A with autophagy in Alzheimer’s disease. Aging (Albany N.Y.) 2019;11:11673–11685. doi: 10.18632/aging.102571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhu J., Dou S., Jiang Y., Bai B., Chen J., Wang C., Cheng B. Apelin-36 exerts the cytoprotective effect against MPP+-induced cytotoxicity in SH-SY5Y cells through PI3K/Akt/mTOR autophagy pathway. Life Sci. 2019;224:95–108. doi: 10.1016/j.lfs.2019.03.047. [DOI] [PubMed] [Google Scholar]
- 91.Zhu J., Dou S., Wang C., Jiang Y., Wang C., Cheng B. Apelin-36 mitigates MPTP/MPP+-induced neurotoxicity: Involvement of α-synuclein and endoplasmic reticulum stress. Brain Res. 2019;1721:146334. doi: 10.1016/j.brainres.2019.146334. [DOI] [PubMed] [Google Scholar]
- 92.Zhu J., Dou S., Jiang Y., Chen J., Wang C., Cheng B. Apelin-13 protects dopaminergic neurons in MPTP-induced Parkinson’s disease model mice through inhibiting endoplasmic reticulum stress and promoting autophagy. Brain Res. 2019;1715:203–212. doi: 10.1016/j.brainres.2019.03.027. [DOI] [PubMed] [Google Scholar]
- 93.Zhu J., Gao W., Shan X., Wang C., Wang H., Shao Z., Dou S., Jiang Y., Wang C., Cheng B. Apelin-36 mediates neuroprotective effects by regulating oxidative stress, autophagy and apoptosis in MPTP-induced Parkinson’s disease model mice. Brain Res. 2020;1726:146493. doi: 10.1016/j.brainres.2019.146493. [DOI] [PubMed] [Google Scholar]
- 94.Berghuis P., Dobszay M.B., Ibanez R.M., Ernfors P., Harkany T. Turning the heterogeneous into homogeneous: Studies on selectively isolated GABAergic interneuron subsets. Int. J. Dev. Neurosci. 2004;22:533–543. doi: 10.1016/j.ijdevneu.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 95.Melis M., Pistis M. Endocannabinoid signaling in midbrain dopamine neurons: More than physiology? Curr. Neuropharmacol. 2007;5:268–277. doi: 10.2174/157015907782793612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Orgado J.M., Fernández-Ruiz J., Romero J. The endocannabinoid system in neuropathological states. Int. Rev. Psychiatry. 2009;21:172–180. doi: 10.1080/09540260902782828. [DOI] [PubMed] [Google Scholar]
- 97.Wang Y., Zhang Q.J., Wang H.S., Wang T., Liu J. Genome-wide microarray analysis identifies a potential role for striatal retrograde endocannabinoid signaling in the pathogenesis of experimental L-DOPA-induced dyskinesia. Synapse. 2014;68:332–343. doi: 10.1002/syn.21740. [DOI] [PubMed] [Google Scholar]
- 98.Aymerich M.S., Aso E., Abellanas M.A., Tolon R.M., Ramos J.A., Ferrer I., Romero J., Fernández-Ruiz J. Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem. Pharmacol. 2018;157:67–84. doi: 10.1016/j.bcp.2018.08.016. [DOI] [PubMed] [Google Scholar]
- 99.Talarico G., Trebbastoni A., Bruno G., de Lena C. Modulation of the Cannabinoid System: A New Perspective for the Treatment of the Alzheimer’s Disease. Curr. Neuropharmacol. 2019;17:176–183. doi: 10.2174/1570159X16666180702144644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Estrada J.A., Contreras I. Endocannabinoid receptors in the CNS: Potential drug targets for the prevention and treatment of neurologic and psychiatric disorders. Curr. Neuropharmacol. 2020 doi: 10.2174/1570159X18666200217140255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dennis M.S., Byrne E.J., Hopkinson N., Bendall P. Neuropsychiatric systemic lupus erythematosus in elderly people: A case series. J. Neurol. Neurosurg. Psychiatry. 1992;55:1157–1161. doi: 10.1136/jnnp.55.12.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chiewthanakul P., Sawanyawisuth K., Foocharoen C., Tiamkao S. Clinical features and predictive factors in neuropsychiatric lupus. Asian Pac. J. Allergy Immunol. 2012;30:55–60. [PubMed] [Google Scholar]
- 103.Kivity S., Agmon-Levin N., Zandman-Goddard G., Chapman J., Shoenfeld Y. Neuropsychiatric lupus: A mosaic of clinical presentations. BMC Med. 2015;13:43. doi: 10.1186/s12916-015-0269-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tan E.K., Chan L.L., Auchus A.P. Reversible Parkinsonism in systemic lupus erythematosus. J. Neurol. Sci. 2001;193:53–57. doi: 10.1016/S0022-510X(01)00604-9. [DOI] [PubMed] [Google Scholar]
- 105.Miyoshi Y., Atsumi T., Kitagawa H., Ogura N., Amasaki Y., Tsutsumi A., Ohnishi K., Fujisaku A., Sagawa A., Koike T. Parkinson-like symptoms as a manifestation of systemic lupus erythematosus. Lupus. 1993;2:199–201. doi: 10.1177/096120339300200314. [DOI] [PubMed] [Google Scholar]
- 106.Shahar E., Goshen E., Tauber Z., Lahat E. Parkinsonian syndrome complicating systemic lupus erythematosus. Pediatr. Neurol. 1998;18:456–458. doi: 10.1016/S0887-8994(97)00229-4. [DOI] [PubMed] [Google Scholar]
- 107.Liu F.C., Huang W.Y., Lin T.Y., Shen C.H., Chou Y.C., Lin C.L., Lin K.T., Kao C.H. Inverse Association of Parkinson Disease With Systemic Lupus Erythematosus: A Nationwide Population-based Study. Medicine (Baltimore) 2015;94:e2097. doi: 10.1097/MD.0000000000002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jha S.K., Jha N.K., Kumar D., Ambasta R.K., Kumar P. Linking mitochondrial dysfunction, metabolic syndrome and stress signaling in Neurodegeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2017;1863:1132–1146. doi: 10.1016/j.bbadis.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 109.Chi J., Xie Q., Jia J., Liu X., Sun J., Deng Y., Yi L. Integrated Analysis and Identification of Novel Biomarkers in Parkinson’s Disease. Front. Aging Neurosci. 2018;10:178. doi: 10.3389/fnagi.2018.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Picca A., Calvani R., Landi G., Marini F., Biancolillo A., Gervasoni J., Persichilli S., Primiano A., Urbani A., Bossola M., et al. Circulating amino acid signature in older people with Parkinson’s disease: A metabolic complement to the EXosomes in PArkiNson Disease (EXPAND) study. Exp. Gerontol. 2019;128:110766. doi: 10.1016/j.exger.2019.110766. [DOI] [PubMed] [Google Scholar]
- 111.Magnadóttir B., Hayes P., Gísladóttir B., Bragason B.Þ., Hristova M., Nicholas A.P., Guðmundsdóttir S., Lange S. Pentraxins CRP-I and CRP-II are post-translationally deiminated and differ in tissue specificity in cod (Gadus morhua L.) ontogeny. Dev. Comp. Immunol. 2018;87:1–11. doi: 10.1016/j.dci.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 112.Jiang R., Rong C., Ke R., Meng S., Yan X., Ke H., Wu S. Differential proteomic analysis of serum exosomes reveals alterations in progression of Parkinson disease. Medicine (Baltimore) 2019;98:e17478. doi: 10.1097/MD.0000000000017478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li N., Wu Y., Zhu L., Huang Y., Liu Z., Shi M., Soltys D., Zhang J., Chang Q. Extracellular microvesicles-derived from microglia treated with unaggregated α-synuclein attenuate mitochondrial fission and toxicity-induced by Parkinsonian toxin MPP. Biochem. Biophys. Res. Commun. 2019;517:642–647. doi: 10.1016/j.bbrc.2019.07.084. [DOI] [PubMed] [Google Scholar]
- 114.Nemmara V.V., Tilvawala R., Salinger A.J., Miller L., Nguyen S.H., Weerapana E., Thompson P.R. Citrullination Inactivates Nicotinamide- N-methyltransferase. ACS Chem. Biol. 2018;13:2663–2672. doi: 10.1021/acschembio.8b00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Surmeier D.J., Schumacker P.T., Guzman J.D., Ilijic E., Yang B., Zampese E. Calcium and Parkinson’s disease. Biochem. Biophys. Res. Commun. 2017;483:1013–1019. doi: 10.1016/j.bbrc.2016.08.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Enders M., Heider T., Ludwig A., Kuerten S. Strategies for Neuroprotection in Multiple Sclerosis and the Role of Calcium. Int. J. Mol. Sci. 2020;21:1663. doi: 10.3390/ijms21051663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Acharya N.K., Nagele E.P., Han M., Coretti N.J., DeMarshall C., Kosciuk M.C., Boulos P.A., Nagele R.G. Neuronal PAD4 expression and protein citrullination: Possible role in production of autoantibodies associated with neurodegenerative disease. J. Autoimmun. 2012;38:369–380. doi: 10.1016/j.jaut.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 118.Kim S.W., Lee H., Lee H.K., Kim I.D., Lee J.K. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol. Commun. 2019;7:94. doi: 10.1186/s40478-019-0747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lange S., Gögel S., Leung K.Y., Vernay B., Nicholas A.P., Causey C.P., Thompson P.R., Greene N.D., Ferretti P. Protein deiminases: New players in the developmentally regulated loss of neural regenerative ability. Dev. Biol. 2011;355:205–214. doi: 10.1016/j.ydbio.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lange S., Rocha-Ferreira E., Thei L., Mawjee P., Bennett K., Thompson P.R., Subramanian V., Nicholas A.P., Peebles D., Hristova M., et al. Peptidylarginine deiminases: Novel drug targets for prevention of neuronal damage following hypoxic ischemic insult (HI) in neonates. J. Neurochem. 2014;130:555–562. doi: 10.1111/jnc.12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lange S. Peptidylarginine Deiminases as Drug Targets in Neonatal Hypoxic-Ischemic Encephalopathy. Front. Neurol. 2016;22:22. doi: 10.3389/fneur.2016.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hao B., Chen X., Dai D., Zou C., Wu X., Chen J. Bioinformatic analysis of microRNA expression in Parkinson’s disease. Mol. Med. Rep. 2015;11:1079–1084. doi: 10.3892/mmr.2014.2837. [DOI] [PubMed] [Google Scholar]
- 123.Ramaswamy P., Christopher R., Pal P.K., Yadav R. MicroRNAs to differentiate Parkinsonian disorders: Advances in biomarkers and therapeutics. J. Neurol. Sci. 2018;394:26–37. doi: 10.1016/j.jns.2018.08.032. [DOI] [PubMed] [Google Scholar]
- 124.Mushtaq G., Greig N.H., Shaik M.M., Tamargo I.A., Kamal M.A. MiRNAs as Circulating Biomarkers for Alzheimer’s Disease and Parkinson’s Disease. Med. Chem. 2016;12:217–225. doi: 10.2174/1573406411666151030112140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Johnson M.E., Stecher B., Labrie V., Brundin L., Brundin P. Triggers, facilitators and aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2018;42:4–13. doi: 10.1016/j.tins.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sheedy F.J. Turning 21: Induction of miR-21 as a Key Switch in the Inflammatory Response. Front. Immunol. 2015;6:19. doi: 10.3389/fimmu.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu B., Wei H., Lan M., Jia N., Liu J., Zhang M. MicroRNA-21 mediates the protective effects of salidroside against hypoxia/reoxygenation-induced myocardial oxidative stress and inflammatory response. Exp. Ther. Med. 2020;19:1655–1664. doi: 10.3892/etm.2020.8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang L., Dong L.Y., Li Y.J., Hong Z., Wei W.S. MiR-21 Represses FasL in Microglia and Protects against Microglia-Mediated Neuronal Cell Death Following Hypoxia/Ischemia. Glia. 2012;60:1888–1895. doi: 10.1002/glia.22404. [DOI] [PubMed] [Google Scholar]
- 129.Han Z., Chen F., Ge X., Tan J., Lei P., Zhang J. MiR-21 Alleviated Apoptosis of Cortical Neurons through Promoting PTEN-Akt Signaling Pathway in Vitro after Experimental Traumatic Brain Injury. Brain Res. 2014;1582:12–20. doi: 10.1016/j.brainres.2014.07.045. [DOI] [PubMed] [Google Scholar]
- 130.Gruner K., Sahni V., Pan L., McGuire T.L., Bhalala O.G., Kessler J.A., Tourtellotte W.G. MicroRNA-21 Regulates Astrocytic Response Following Spinal Cord Injury. J. Neurosci. 2012;32:17935–17947. doi: 10.1523/JNEUROSCI.3860-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Junker A., Krumbholz M., Eisele S., Mohan H., Augstein F., Bittner R., Lassmann H., Wekerle H., Hohlfeld R., Meinl E. MicroRNA Profiling of Multiple Sclerosis Lesions Identifies Modulators of the Regulatory Protein CD47. Brain. 2009;132:3342–3352. doi: 10.1093/brain/awp300. [DOI] [PubMed] [Google Scholar]
- 132.Alvarez-Erviti L., Seow Y., Schapira A.H., Rodriguez-Oroz M.C., Obeso J.A., Cooper J.M. Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson’s disease. Cell Death Dis. 2013;4:e545. doi: 10.1038/cddis.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Burgos K., Malenica I., Metpally R., Courtright A., Rakela B., Beach T., Shill H., Adler C., Sabbagh M., Villa S., et al. Profiles of Extracellular MiRNA in Cerebrospinal Fluid and Serum from Patients with Alzheimer’s and Parkinson’s Diseases Correlate with Disease Status and Features of Pathology. PLoS ONE. 2014;9:e94839. doi: 10.1371/journal.pone.0094839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kanata E., Thüne K., Xanthopoulos K., Ferrer I., Dafou D., Zerr I., Sklaviadis T., Llorens F. MicroRNA Alterations in the Brain and Body Fluids of Humans and Animal Prion Disease Models: Current Status and Perspectives. Front. Aging Neurosci. 2018;10:220. doi: 10.3389/fnagi.2018.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Harrison E.B., Hochfelder C.G., Lamberty B.G., Meays B.M., Morsey B.M., Kelso M.L., Fox H.S., Yelamanchili S.V. Traumatic Brain Injury Increases Levels of MiR-21 in Extracellular Vesicles: Implications for Neuroinflammation. FEBS Open Bio. 2016;6:835–846. doi: 10.1002/2211-5463.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Worm J., Stenvang J., Petri A., Frederiksen K.S., Obad S., Elmén J., Hedtjärn M., Straarup E.M., Hansen J.B., Kauppinen S. Silencing of MicroRNA-155 in Mice during Acute Inflammatory Response Leads to Derepression of c/Ebp Beta and down-Regulation of G-CSF. Nucleic Acids Res. 2009;37:5784–5792. doi: 10.1093/nar/gkp577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lopez-Ramirez M.A., Wu D., Pryce G., Simpson J.E., Reijerkerk A., King-Robson J., Kay O., de Vries H.E., Hirst M.C., Sharrack B., et al. MicroRNA-155 Negatively Affects Blood-Brain Barrier Function during Neuroinflammation. FASEB J. 2014;28:2551–2565. doi: 10.1096/fj.13-248880. [DOI] [PubMed] [Google Scholar]
- 138.Gandhi R. MiRNA in Multiple Sclerosis: Search for Novel Biomarkers. Mult. Scler. J. 2015;21:1095–1103. doi: 10.1177/1352458515578771. [DOI] [PubMed] [Google Scholar]
- 139.Guedes J.R., Custódia C.M., Silva R.J., de Almeida L.P., de Lima M.C.P., Cardoso A.L. Early MiR-155 Upregulation Contributes to Neuroinflammation in Alzheimer’s Disease Triple Transgenic Mouse Model. Hum. Mol. Genet. 2014;23:6286–6301. doi: 10.1093/hmg/ddu348. [DOI] [PubMed] [Google Scholar]
- 140.Prajapati P., Sripada L., Singh K., Bhatelia K., Singh R., Singh R. TNF-alpha regulates miRNA targeting mitochondrial complex-I and induces cell death in dopaminergic cells. Biochim. Biophys. Acta. 2015;1852:451–461. doi: 10.1016/j.bbadis.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 141.Fasanaro P., D’Alessandra Y., Di Stefano V., Melchionna R., Romani S., Pompilio G., Capogrossi M.C., Martelli F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand ephrin-A3. J. Biol. Chem. 2008;283:15878–15883. doi: 10.1074/jbc.M800731200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Huang X., Le Q.-T., Giaccia A.J. MiR-210—Micromanager of the hypoxia pathway. Trends Mol. Med. 2010;16:230–237. doi: 10.1016/j.molmed.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bavelloni A., Ramazzotti G., Poli A., Piazzi M., Focaccia E., Blalock W., Faenza I. MiRNA-210: A Current Overview. Anticancer Res. 2017;37:6511–6521. doi: 10.21873/anticanres.12107. [DOI] [PubMed] [Google Scholar]
- 144.Voloboueva L.A., Sun X., Xu L., Ouyang Y.-B., Giffard R.G. Distinct effects of miR-210 reduction on neurogenesis: Increased neuronal survival of inflammation but reduced proliferation associated with mitochondrial enhancement. J. Neurosci. 2017;37:3072–3084. doi: 10.1523/JNEUROSCI.1777-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 146.Chen Z., Li Y., Zhang H., Huang P., Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene. 2010;29:4362–4368. doi: 10.1038/onc.2010.193. [DOI] [PubMed] [Google Scholar]
- 147.Harischandra D.S., Ghaisas S., Rokad D., Zamanian M., Jin H., Anantharam V., Kimber M., Kanthasamy A., Kanthasamy AG. Environmental neurotoxicant manganese regulates exosome-mediated extracellular miRNAs in cell culture model of Parkinson’s disease: Relevance to α-synuclein misfolding in metal neurotoxicity. Neurotoxicology. 2018;64:267–277. doi: 10.1016/j.neuro.2017.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Konovalova J., Gerasymchuk D., Parkkinen I., Chmielarz P., Domanskyi A. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019;20:6055. doi: 10.3390/ijms20236055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wei Z., Li X., Li X., Liu Q., Cheng Y. Oxidative stress in Parkinson’s disease: A systematic review and meta-analysis. Front. Mol. Neurosci. 2018;11:236. doi: 10.3389/fnmol.2018.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Haqqani A.S., Delaney C.E., Tremblay T.L., Sodja C., Sandhu J.K., Stanimirovic D.B. Method for isolation and molecular characterization of extracellular microvesicles released from brain endothelial cells. Fluids Barriers CNS. 2013;10:4. doi: 10.1186/2045-8118-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Botcher N.A., Falck J.E., Thomson A.M., Mercer A. Distribution of interneurons in the CA2 region of the rat hippocampus. Front. Neuroanat. 2014;8:124. doi: 10.3389/fnana.2014.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Théry C., Witwer K.W., Aikawa E., Alcaraz M.J., Anderson J.D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin-Smith G.K., et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles. 2018;7:1535750. doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 154.Nicholas A.P., Whitaker J.N. Preparation of a monoclonal antibody to citrullinated epitopes: Its characterization and some applications to immunohistochemistry in human brain. Glia. 2002;37:328–336. doi: 10.1002/glia.10039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.