

Abstract
Purpose:
Current standard of care for neovascular eye diseases require repeated intravitreal bolus injections of anti-vascular endothelial growth factors (anti-VEGFs). The purpose of this study was to validate a degradable microsphere-thermoresponsive hydrogel drug delivery system (DDS) capable of releasing bioactive aflibercept in a controlled and extended manner for 6 months.
Materials and Methods:
The DDS was fabricated by suspending aflibercept-loaded poly(lactic-coglycolic acid) microspheres within a biodegradable poly(ethylene glycol)-co-(L-lactic acid) diacrylate/N-isopropylacrylamide (PEG-PLLA-DA/NIPAAm) thermoresponsive hydrogel. Encapsulation efficiency of DDSs and in vitro release profiles were characterized by iodine-125 radiolabeled aflibercept. The degradation of hydrogel was determined by dry weight changes. The cytotoxicity from degraded DDS byproducts was investigated by quantifying cell viability using LIVE/DEAD® assay. In addition, dot blot and enzyme-linked immunosorbent assay were used to determine the bioactivity of released drug. Finally, morphology of microspheres and hydrogel were investigated by cryo-scanning electron microscopy before and after thermal transformation.
Results:
The microsphere-hydrogel DDS was capable of releasing bioactive aflibercept in a controlled and extended manner for 6 months. The amount and rate of aflibercept release can be controlled by both the cross-linker concentration and microspheres load amount. The initial burst (release within 24 h) was from 37.35 ± 4.92 to 74.56 ± 6.16 μg (2 and 3 mM hydrogel, each loaded with 10 and 20 mg/ml of microspheres, respectively), followed by controlled drug release of 0.07–0.15 μg/day. Higher PEG-PLLA-DA concentration (3 mM) degraded faster than the lower concentration (2 mM). No significant cytotoxicity from degraded DDS byproducts was found for all investigated time points. Bioactivity of released drug was maintained at therapeutic level over entire release period.
Conclusions:
The microsphere-hydrogel DDS is safe and can deliver bioactive aflibercept in a controlled manner. This may provide a significant advantage over current bolus injection therapies in the treatment of ocular neovascularization.
Keywords: Anti-VEGF delivery, neovascularization, intravitreal injection, thermoresponsive hydrogel, bioactivity
Introduction
Neovascular eye diseases such as wet age-related macular degeneration (AMD) and proliferative diabetic retinopathy has been identified as the leading cause of irreversible blindness in the developed nations.1 It was estimated that approximately 2.07 million Americans were diagnosed with AMD in 2010 alone, and by 2050, this number is expected to 5.44 million.2 Although wet AMD with choroidal neovascularization (CNV) consists only 10% of all AMD cases, they are recognized to be more aggressive and vision-threatening than dry AMD.3
Although the mechanisms of ocular neovascularization have not been well understood, previous studies have demonstrated that vascular endothelial growth factors (VEGFs) production is increased and serve as a major stimulator for ocular neovascularization and vascular permeability.4 These findings have led to the development of anti-VEGF therapy which is a current standard of care for wet AMD. Three anti-VEGF therapeutics have been approved by U.S. Food and Drug Administration for treatment of wet AMD, including pegaptanib (Macugen®, OSI Pharmaceuticals, Melville, NY), ranibizumab (Lucentis®, Genentech, South San Francisco, CA), and aflibercept (Eylea®, Regeneron, Tarrytown, NY). Another anti-VEGF therapeutics, bevacizumab (Avastin®, Genentech), is used as off-label drug.
Despite its great success in treatment of wet AMD, a major challenge for current anti-VEGF therapy is the repeated intravitreal (IVT) injections in a monthly/bimonthly manner. This frequent repeated injection regimen is required to maintain therapeutic effect of drugs due to their fast clearance and short half-lives.5 However, these repeated IVT injections present increased risks of potential complications including endophthalmitis, retinal detachment, IVT hemorrhage, and cataract.6 In addition, pharmacokinetic profiles of drugs are nonoptimal, since the peak level of drug after bolus injections may cause potential toxic effect while the quick clearance later may render subtherapeutic concentration.7 Finally, the significant socioeconomic burden upon patients, family, and healthcare systems cannot be ignored. Therefore, developing a drug delivery system (DDS) for anti-VEGF that will result in a controlled and extended delivery and reduce frequency of IVT injections is in great need.
Recent years have seen a variety of DDSs developed for controlled and extended delivery of anti-VEGF drugs in the form of ocular implants, cell-based systems, injectable nano-/microparticles, injectable hydrogels, and composite systems.8 Among the aforementioned systems, the composite systems that combine injectable nano-/microparticles within injectable hydrogels offer a great potential due to their applicability as a minimally invasive localized delivery platform. Issues of injectable particulate systems including unconstrained migration associated with glaucoma and ocular inflammation, fast clearance by phagocytes and large initial burst (IB) release9 can be overcome by encapsulation into hydrogel’s network. Additional barrier provided by hydrogels could also further extend the release of drugs from microspheres. On the other hand, encapsulating anti-VEGF drug directly into injectable hydrogels resulted in a shorter release time of approximately 1 month due to hydrogels’ inherent high water content.10–12 Furthermore, ability of controlling the amount and type of particles loaded within hydrogels can enhance the drug delivery potential.8
A composite ocular DDS consisting of poly(lactic-co-glycolic acid) (PLGA) microspheres suspended within a thermo-responsive injectable poly(ethylene glycol) diacrylate/N-isopropylacrylamide (PEG-DA/NIPAAm) hydrogel has been developed by our laboratory.13 The hydrogels are preformed by free-radical polymerization with drug-loaded PLGA microspheres inside. Since the hydrogel is thermoresponsive, they have a fluid-like consistency which allows injection through 28-G needle at room temperature. After injection, the body temperature triggered the hydrogel to solidify into a solid-like structure to localize microspheres. Our DDS is capable of releasing anti-VEGF drugs such as ranibizumab and aflibercept in a controlled manner for 6 months without long-term effects on the retinal function.14,15 Both in vitro bioactivity of released anti-VEGF and in vivo treatment efficacy in a rat laser CNV model by the DDS were also demonstrated.14,16 However, the PEG-DA/NIPAAm hydrogel used in the previous study was not degradable and there was a large incomplete release of drugs. Using a hydrolytically biodegradable poly(ethylene glycol)-co-(L-lactic-acid) diacrylate/N-isopropylacrylamide (PEG-PLLA-DA/NIPAAm) hydrogel12 to make the microsphere-hydrogel DDS was hypothesized to be better and enhance the anti-VEGF release.
The objective of this study was to validate a degradable micro-sphere-thermoresponsive hydrogel DDS capable of releasing bioactive aflibercept in a controlled and extended manner for 6 months. The effects of degradable cross-linker PEG-PLLA-DA concentrations and microsphere load amount on controlling aflibercept release were investigated. The degradability of hydrogels and biocompatibility of degraded byproducts were also investigated for safety. Bioactivity and stability of released aflibercept from the DDS was investigated in vitro. Additionally, morphology changes of thermoresponsive transition of DDS were studied for better understanding of initial release of aflibercept.
Materials and methods
Fabrication of microsphere-hydrogel DDS
Aflibercept (40 mg/ml) was generously provided by Dr William F. Mieler at University of Illinois at Chicago. And all subsequent chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Blank/aflibercept-loaded PLGA 75:25 microspheres were fabricated using a modified double-emulsion, solvent evaporation technique described in detail elsewhere.13 Briefly, the primary water-in-oil emulsion (w1/o) was created by vortex; this first emulsion was immediately added to the outer aqueous phase (w2) containing polyvinyl alcohol to create a (water-in-oil)-in-water (w1/o/w2) double emulsion by vortex. pH 7.4 1× phosphate buffered saline (PBS) was used as the inner water phase (w1) to make blank microspheres (control); and aflibercept stock solution (40 mg/ml) was used as the inner water phase (w1) to make aflibercept-loaded microspheres. Several excipients were used to stabilize and protect the aflibercept during fabrication, storage, and release: bovine serum albumin (BSA), PEG (MW 8 kDa), and sucrose were added in the w1 phase; Mg(OH)2 was added as a buffering salt in the oil phase (o). After solvent evaporation, microspheres were harvested by centrifugation, washed three times in deionized (DI) water, lyophilized to a dry powder, and stored at 4°C in fridge.
Degradable thermoresponsive hydrogels composed of poly (ethylene glycol)-co-(L-lactic acid) diacrylate (PEG-PLLA-DA) and N-isopropylacrylamide (NIPAAm) were synthesized by free-radical polymerization method described in detail elsewhere.12 Briefly, hydrogels precursors were prepared by dissolving 350 mM NIPAAm, 50 mM N-tert-butylacrylamide, and 13 mM ammonium persulfate in pH 7.4 1× PBS. Hydrolytically degradable copolymer PEG-PLLA-DA at two concentrations (2 and 3 mM) were then added to the precursor to synthesize hydrogels with different cross-linker densities. To make microsphere-hydrogel DDS with different microsphere load amount, 0, 10, and 20 mg/ml of microspheres were suspended in the above hydrogel precursors. Polymerization of the hydrogel was initiated by mixing 168 mM N,N,N′,N′-tetramethylethylenedia-mine—pH adjusted to 7.4 with hydrochloric acid—into the hydrogel precursors; the reaction was allowed to proceed on ice for 30 min to form microsphere-hydrogel DDS. After polymerization, DDSs were collected and washed at least three times in DI water. In this study, six different microsphere-hydrogel DDS formulations were prepared for the corresponding experiments as follows: microsphere-hydrogel DDS at PEG-PLLA-DA concentration of 2 mM with 0 mg/ml (labelled as 2 mM DDS-0), 10 mg/ml (2 mM DDS-10), and 20 mg/ml (2 mM DDS-20) microsphere load amount; microsphere-hydrogel DDS at PEG-PLLA-DA concentration of 3 mM with 0 mg/ml (3 mM DDS-0), 10 mg/ml (3 mM DDS-10), and 20 mg/ml (3 mM DDS-20) microsphere load amount.
Aflibercept radiolabeling and encapsulation efficiency
Aflibercept (Eylea®, Regeneron, Tarrytown, NY) was radiolabeled with iodine-125 (PerkinElmer, Waltham, MA) using iodination beads (Pierce, Rockford, IL) and then dialyzed against DI water using a dialysis cassette (MWCO 2 kDa, Pierce) to remove unincorporated iodine. Radiolabeled aflibercept were lyophilized, weighed, and dissolved in pH 7.4 1× PBS to create a stock solution of 40 mg/ml. The stock solution was then stored at −80°C for future use.
The encapsulation efficiency (EE) for each microsphere protocol was determined from the radioactivity measured using a gamma counter (Cobra-II, Auto-Gamma, Packard Instrument Co., Meriden, CT) before and after microsphere preparation. EE was defined as the percent-drug within the microspheres relative to the theoretical loading amount. The EE of microspheres in hydrogels was also determined by comparing radioactivity before and after hydrogel preparation to quantify final EE of aflibercept into microsphere-hydrogel DDS.
In vitro release of aflibercept
In vitro release profiles of radiolabeled aflibercept from the microsphere-hydrogel DDS formulations (2 mM DDS-10, 2 mM DDS-20, 3 mM DDS-10, and 3 mM DDS-20) were investigated to study both effects of PEG-PLLA-DA concentration and microsphere load amount on drug release. A separation method described in detail elsewhere17 was used to measure the release profiles. Briefly, 1 ml of microsphere-hydrogel DDS sample of corresponding formulation was prepared, and incubated in 1.5 ml of 1× PBS at 37°C under mild agitation throughout the release. At predetermined time intervals, 1 ml of supernatant was removed after a brief centrifugation and replaced with an equal volume of fresh buffer. Radioactivity of supernatants was measured using a gamma counter (Packard) to determine amount of drug release. Cumulative release was calculated relative to EE of aflibercept into each microsphere-hydrogel DDS. The IB release was also determined as percent drug released within the first 24 h for different DDS formulations.
In vitro degradation of hydrogels
The PEG-PLLA-DA is a hydrolytically degradable copolymer that makes NIPAAm-based hydrogels degradable. The effects of PEG-PLLA-DA concentrations (2 and 3 mM) on hydrogels’ degradation were investigated by dry weight changes during degradation. Each hydrogel sample (1 ml in volume) was incubated in 5 ml pH 7.4 1× PBS at 37°C; and the buffer was refreshed weekly. At predetermined time points, hydrogels (3 for each PEG-PLLA-DA concentration) were collected, lyophilized, and measured for dry weight. The dry weight changes of hydrogels were also normalized relative to the initial dry weight. Changes in physical appearance of hydrogels were also photographed over the entire study.
Cytotoxicity of microsphere-hydrogel DDS
After polymerization, blank (drug-free) microsphere-hydrogel DDSs of formulation 2 mM DDS-20 were subject to five consecutive washing steps using larger volume of PBS buffer (1:25 volume ratio) at room temperature with gentle agitation. Each washing step lasted for 20 min. The buffer of all five washing steps were collected for investigation of cytotoxicity. After washing, each DDS sample (1 ml in volume) was incubated in 5 ml pH 7.4 1× PBS at 37°C for in vitro degradation. Sodium azide (NaN3; 0.05% w/v) was added to the PBS to prevent bacterial contamination during degradation. At predetermined time intervals, 1 ml of degraded buffer was collected for investigation of cytotoxicity, and replaced with fresh buffer.
Human umbilical vein endothelial cells (HUVECs) were seeded in T-75 flasks and cultured using endothelial cell growth medium-2 (EGM-2, Lonza), in a 5% CO2 atmosphere at 37°C. The growth media was changed every 2–3 days. Cells were grown to confluence and harvested with trypsin/ethylenediaminetetraacetic acid solution. The cells were then suspended in growth medium and seeded in 96-well plates at 5000 cells/well (200 μl) and incubated for 48 h at 37°C to allow cell adhesion and growth. Media were then changed and 50 μl of the above buffer samples (without dilution) was added to the corresponding wells (three wells per sample). Fifty microliters of EMG-2 was used as control group. The cells were allowed to be exposed to the added samples for another 48 h before cytotoxicity test. A LIVE/DEAD cell viability assay kit (Thermo Fisher Scientific, Waltham, MA) was performed using the standard protocol recommended by manufacturer. The cells were imaged with a Carl Zeiss Axiovert 200 M confocal microscope immediately after incubation. Live cells fluoresced green when viewed with the FITC filter; and dead cells fluoresced red when viewed with the Rhod filter. Number of live and dead cells were counted using a program written in ImageJ 1.50i (National Institutes of Health, Bethesda, MD). The cell viability was defined as percent of live cells relative to the total number of cells in each well. All cell viability values from testing samples were compared to that from control for assessment of potential toxicity.
In vitro bioactivity of aflibercept
The bioactivity of aflibercept released from microsphere-hydrogel DDS was investigated both qualitatively using dot blot immunoassay, and quantitatively using enzyme-linked immunosorbent assay (ELISA) (Sigma-Aldrich). One milliliter of aflibercept-loaded 2 mM DDS-20 were incubated in 2 ml of pH 7.4 1× PBS at 37°C. One milliliter of supernatant were collected and refreshed with equal volume of PBS. Dot blot immunoassay was used to determine the bioactivity of released aflibercept qualitatively by visualizing the binding activity to human VEGF-165. Briefly, human VEGF-165 was used to coat the dots (3–4 mm in diameter) on nitrocellulose membrane, and 5% BSA in TBS-T was used to block non-specific sites. A group of reference dots were first created using different amount of aflibercept (5, 2, 0.5, and 0 ng). The release samples were diluted appropriately to a range of 0.1–10 μg/ml for dot blot assay according to the estimated release amount at corresponding time points from release profile. Two microliters of the release samples were added to the dots, followed by incubation with secondary antibody conjugated with horseradish peroxidase, which gives off chemiluminescence signal as an indication of binding activity to coating VEGF.
ELISA was used to further quantify concentration of bioactive aflibercept in release samples at corresponding time intervals. Assays were performed according to standard manufactuer’s protocol, and each sample was measured in triplicate to obtain an average released bioactive aflibercept concentration.
Morphology of microsphere-hydrogel DDS
The morphology of microspheres distributed in the hydrogels both at room temperature and after 1-day incubation at body temperature were investigated using low temperature scanning electron microscopy (cryo-SEM) at Atomic and Nanoscale Characterization Experimental Center at Northwestern University (NUANCE) (Chicago, IL). Blank 2 mM DDS-20 (drug-free) samples were preconditioned, and thin slices were mounted onto standard specimen carriers (Type A, Technotrade #241). The samples were cryo-fixed in a high-pressure freezer (Leica HPM100, Buffalo Grove, IL, US) and stored in liquid nitrogen. For freeze fracturing, the sample carriers were transferred into a precooled loading station (Leica) and mounted under liquid nitrogen onto a cryo-SEM sample holder and freeze fractured with a cold scalpel blade. The sample holder with the mounted samples was loaded into a liquid nitrogen-cooled shuttle (Leica VCT100) and transferred to a cryo coating system (Leica ACE600). The samples were freeze etched for 15 min at −95°C, and sputter coated with 5 nm of platinum and 4 nm of carbon. After coating, the samples were transferred with the cooled VCT100 under vacuum to the Hitachi S-4800 FE-SEM mounted on the cryostage and observed at −120°C.
Statistical analysis
All values were reported as mean ± standard error in tables, and error bars in all graphs represented standard error. All statistical comparisons between different experimental groups were performed using Student’s t-test, and a p-value less than 0.05 was determined to be significantly different.
Results
Drug-free and aflibercept-loaded PLGA microspheres were successfully formed and embedded within biodegradable thermoresponsive PEG-PLLA-DA/NIPAAm hydrogels at different cross-linker densities (PEG-PLLA-DA concentration of 2 and 3 mM). All composite microsphere-hydrogel DDS formulations investigated in this study were able to be injected through 28-G needles at room temperature (23°C) with ease. This is of great importance for minimally invasive delivery of the DDS into the vitreous humor.
Aflibercept EE and in vitro release
Aflibercept was successfully radiolabeled and encapsulated into PLGA microspheres, and the mean EE of aflibercept in microsphere was 52.78 ± 5.84% (Table 1), which was consistent with previously reported.14 The average diameter of microspheres was 7.0 ± 1.3 μm by examining under microscope, which was also consistent with our previous studies (7.5 ± 0.4 μm).14 Approximately 30% of the microspheres were lost during encapsulation into hydrogels after washing.
Table 1.
Encapsulation efficiency of microsphere-hydrogel DDSs.
| DDS formulation | EE of aflibercept in microsphere | EE of microsphere in hydrogel |
|---|---|---|
| 2 mM DDS-10 | 52.78 ± 5.84% | 72.33 ± 1.35% |
| 2 mM DDS-20 | 52.78 ± 5.84% | 69.68 ± 2.32% |
| 3 mM DDS-10 | 52.78 ± 5.84% | 70.98 ± 1.64% |
| 3 mM DDS-20 | 52.78 ± 5.84% | 68.69 ± 3.42% |
“2” and “3 mM” represents PEG-PLLA-DA concentrations; and numbers following “DDS-” refer to microsphere load amount in units of mg/ml. Data presented as mean ± SEM (n = 3).
Figure 1 shows in vitro release profiles of aflibercept from the four microsphere-hydrogel DDS formulations, where release profiles were grouped according to PEG-PLLA-DA concentration. As shown in the figure, aflibercept released rapidly from all investigated microsphere-hydrogel DDSs during the first week, then it released steadily in a linear manner. The linear regression results for all release profiles after first week were also shown in the figure. Regardless of PEG-PLLA-DA concentrations, DDSs with 20 mg/ml microsphere load amount released more drug during the first week than 10 mg/ml DDSs; and they also released faster thereafter. And regardless of the microsphere load amount, DDSs with 2 mM PEG-PLLA-DA concentration exhibited slightly faster daily release after the first week than those with 3 mM PEG-PLLA-DA concentration. However, the overall release profiles for DDSs with 2 and 3 mM PEG-PLLA-DA concentrations were similar. The release characteristics for all investigated DDSs are also summarized in Table 2. A total release time of ~204 days was detected for all the DDSs before termination of experiment due to the limitation of half-life of I-125 and decaying of radioactivity signal close to background level. All investigated DDSs were capable of releasing aflibercept in a controlled manner for more than 6 months. As shown in Table 2, a total drug load amount ranging from 98.70 ± 4.81 to 193.93 ± 5.56 μg were achieved for all investigated DDSs. The IB release (release within first 24 h) were ranged from 37.35 ± 4.92 to 74.56 ± 6.16 μg, followed by controlled drug release of 0.07–0.15 μg/day. A range of 62–71% of release was achieved by the degradable DDSs. Regardless of microsphere load amount, DDSs with higher PEG-PLLA-DA concentration (3 mM) exhibited more complete release than those with lower PEG-PLLA-DA concentration (2 mM). DDSs with higher microsphere load amount (20 mg/ml) exhibited less complete release than those with lower load amount (10 mg/ml) at the end of release regardless of PEG-PLLA-DA concentrations.
Figure 1.
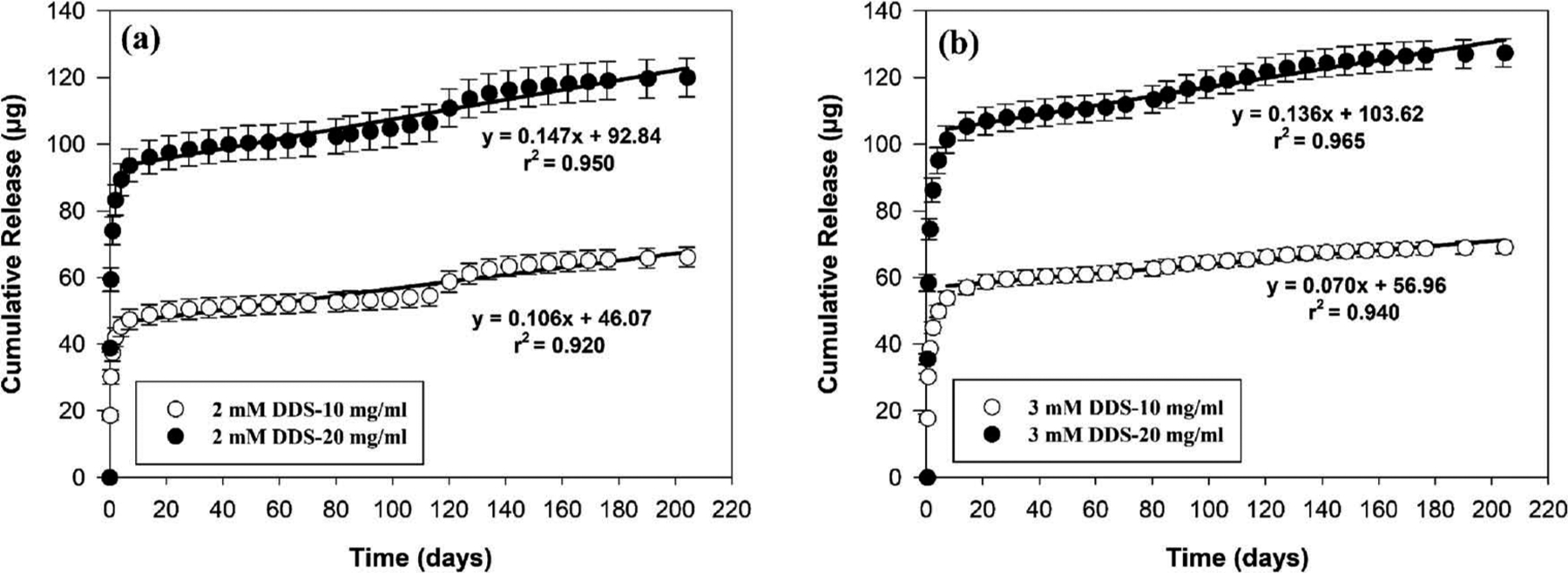
In vitro release of aflibercept from degradable microsphere-hydrogel DDS with varied PEG-PLLA-DA concentrations and different microsphere load amount. (a) Cumulative release (μg) of aflibercept from 2 mM PEG-PLLA-DA concentration DDS with 10 and 20 mg/ml microsphere load amount. (b) Cumulative release (μg) of aflibercept from 3 mM PEG-PLLA-DA concentration DDS with 10 mg/ml and 20 mg/ml microsphere load amount. Error bars represent standard error (n = 3). Equations were obtained by applying linear regression analysis to release profiles after 7 days.
Table 2.
Characteristics of aflibercept release.
| DDS formulation | Drug total load amount (μg) | Initial burst release (μg) | Release rate after 7 days (μg/day) | Release time (days) | Final percent release (%) |
|---|---|---|---|---|---|
| 2 mM DDS-10 | 99.45 ± 10.06 | 37.35 ± 4.92 | 0.11(r2 = 0.92) | 204 | 66.56 ± 1.35 |
| 2 mM DDS-20 | 193.93 ± 5.56 | 74.03 ± 8.23 | 0.15(r2 = 0.95) | 204 | 61.84 ± 2.31 |
| 3 mM DDS-10 | 98.70 ± 4.81 | 38.62 ± 2.78 | 0.07(r2 = 0.94) | 204 | 70.02 ± 1.91 |
| 3 mM DDS-20 | 190.72 ± 7.77 | 74.56 ± 6.16 | 0.14(r2 = 0.97) | 204 | 66.78 ± 1.56 |
In vitro degradation of hydrogels
PEG-PLLA-DA is a hydrolytically biodegradable polymer used to cross-link with NIPAAm to make the hydrogels degradable. The degradation of hydrogels with PEG-PLLA-DA concentration of 2 and 3 mM were measured by dry weight changes as depicted in Figure 2(a) and (b). The net dry weight change was shown in Figure 2(a) and the normalized percent dry weight change relative to the initial dry weight was presented in Figure 2(b). It was observed that both 2 and 3 mM hydrogels degraded faster during the first month; then their dry weight loss (%) slowed down. At the end of study (220 days), the percent dry weight loss for hydrogels with 2 and 3 mM PEG-PLLA-DA concentrations were 54.18% and 62.51%, respectively; hydrogels with 3 mM PEG-PLLA-DA concentration exhibited 8.33% more dry weight loss than those with 2 mM PEG-PLLA-DA. The above results indicate that hydrogels with higher PEG-PLLA-DA concentration degraded more than those with lower PEG-PLLA-DA concentration. Changes in physical appearance of hydrogels over the entire degradation study were also presented in Figure 2(c). It was observed that these hydrogels collapsed and turned opaque after incubation under room temperature; and after 220 days of degradation, hydrogels lost their shape integrity and decreased in size, with a small amount of undissolved white polymer residues. It was worth noting that when the temperature was brought down to room temperature at 220 days of degradation, the remaining unsolved polymers became soluble and transparent.
Figure 2.

Degradation of thermoresponsive PEG-PLLA-DA/NIPAAm hydrogels. (a) Dry weight changes of hydrogels with 2 and 3 mM PEG-PLLA-DA over 220 days. Error bars represent standard error (n = 3). (b) Normalized percent dry weight changes of hydrogels. (c) Physical appearance of 2 and 3 mM hydrogels over the entire degradation study.
Cytotoxicity of microsphere-hydrogel DDS
The cell viability of cultured HUVECs after incubation with corresponding testing samples were shown in Figure 3: endothelial cell growth media (EGM-2) as control (white bar); 5-step washing buffer (gray bars); and degraded samples at corresponding time points (black bars). Comparing to the control (EGM-2), only the first- and second-step washing buffer generated significantly lower (p < 0.025) cell viability indicating potential toxic effect on cultured HUVECs. No significant difference (p > 0.05) in cell viability between control and the following three-step washing buffer was found. The data indicate that at least three times of washing are necessary to eliminate any potential toxic catalyst and unreacted polymers after hydrogel polymerization.18,19 All degraded samples over the entire investigated degradation timeframe were shown to have comparable cell viability to the control which indicate they are not toxic to the cultured cells. Our data suggest that degraded byproducts of microsphere-hydrogel DDS are safe and biocompatible to human cells.
Figure 3.
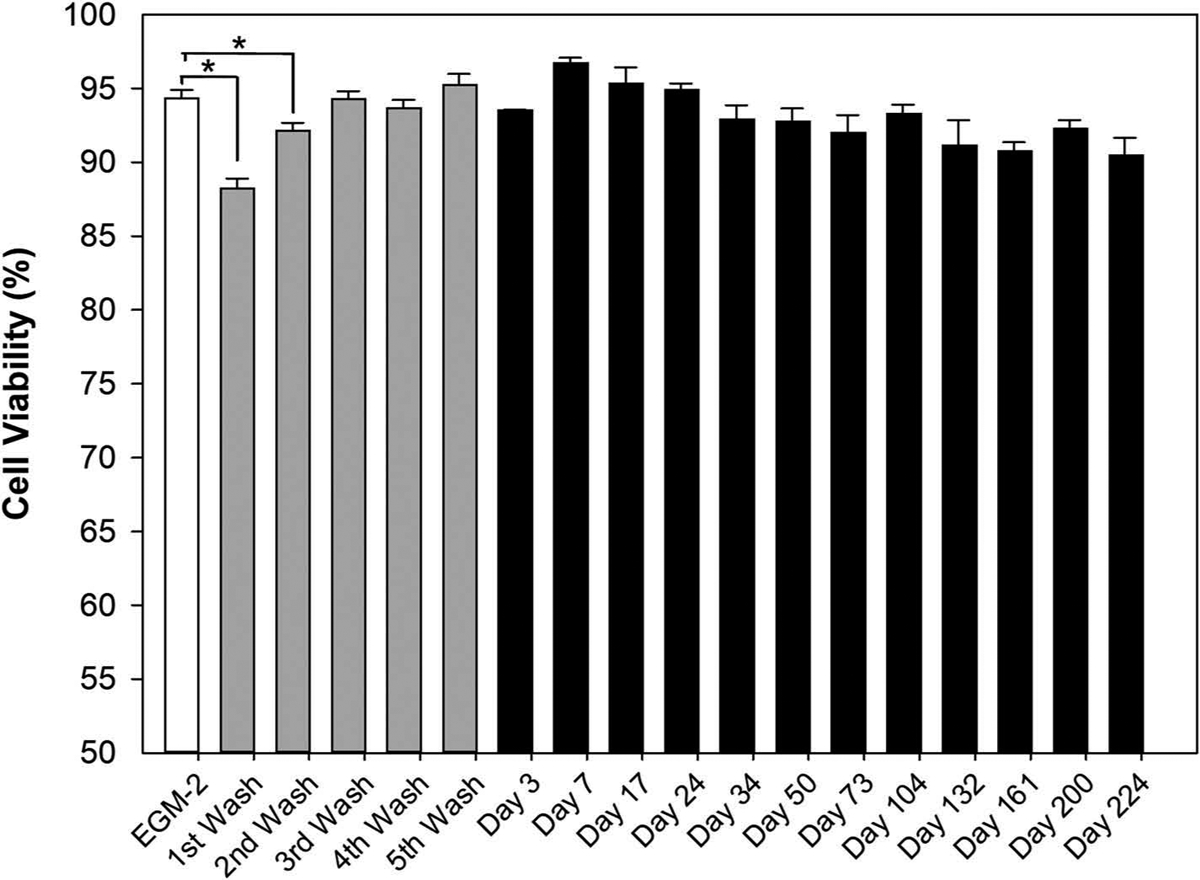
LIVE/DEAD cell viability results. The white bar represents cell viability from endothelial cell growth media-2 (EGM-2) as control; the gray bars stand for cell viability from five times washing buffer; and the black bars signifies cell viability from degraded buffer of 2 mM DDS-20 incubated under body temperature at corresponding time points. Error bars represents standard error (n = 3). And * indicates statistically significant difference between groups (p < 0.05).
In vitro bioactivity of released aflibercept
Aflibercept is a recombinant fusion protein, and its stability and bioactivity is a crucial factor to be considered for long-term controlled-release DDS. Using dot blot immunoassay, direct binding activity of released aflibercept to coated human VEGF-165 over the entire release timeframe can be observed in Figure 4. The intensity of signal (darkness and area) from the dots are proportional to the binding activity (bioactivity) of released drugs to VEGF-165. As displayed in Figure 4, released samples exhibited stronger binding activity to coated VEGF-165 at earlier stages of release timeframe, and this binding activity became weaker for later time points. At the end of release (226 days), the signal from the dot was similar to control indicating the bioactivity of released drug is negligible.
Figure 4.
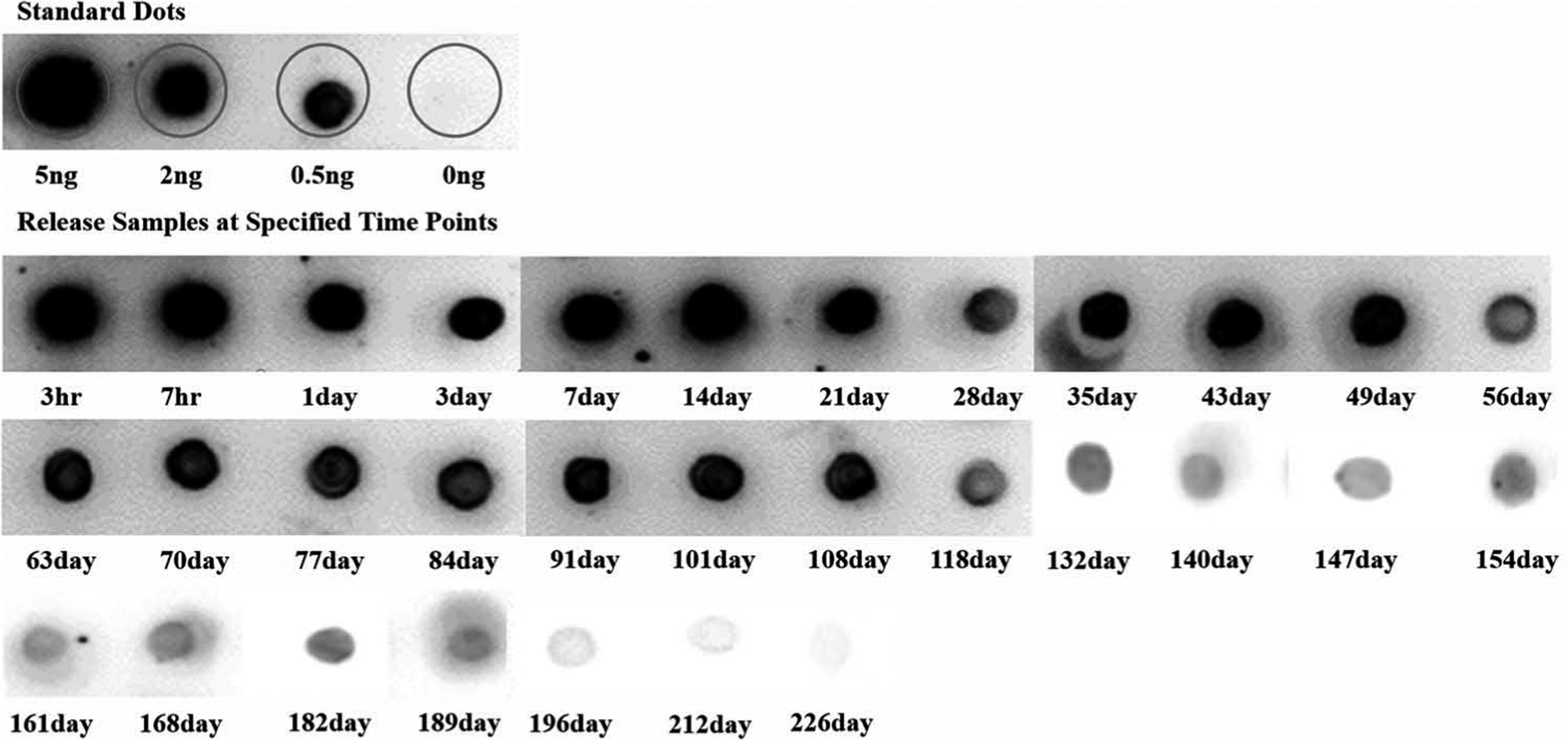
In vitro bioactivity from dot blot assays. Images of chemiluminescence signal from spotted dots showing binding activity of standard/released aflibercept to coated human VEGF-165. Release samples from first week (3 h, 7 h, 1 day, 3 day, and 7 day) were diluted 5–10 times accordingly.
Since dot blot immunoassay provides qualitative detection of bioactive aflibercept released from DDS at specified time points, the concentrations of bioactive aflibercept at these time points were further quantified using ELISA. Figure 5 shows the bioactive released aflibercept concentration from 2 mM DDS-20 measured at different time points over the entire release timeframe. As shown in the figure, a high concentration of bioactive aflibercept was measured during first 3 days (> 1000 ng/ml) which corresponded to the IB release. After IB release, the concentration of bioactive aflibercept continued to decrease with elapse of release time, and finally decreased to a minimum level of 5–10 ng/ml after 2 months. No bioactive aflibercept can be detected after 200 days. This decrease in bioactive aflibercept concentration with longer release time agreed with the weakening of binding activity also seen in dot blot assay at later time points. However, it was worth noting that all the detected bioactive aflibercept concentration during the release timeframe, even the lowest level (5 ng/ml), were well above both reported aflibercept’s binding affinity concentration (0.06 ng/ml)5 and its IC50 for HUVEC in vitro (3 ng/ml).20 This indicates that the released bioactive aflibercept should be maintained effective to block the VEGF-induced angiogenesis over the entire release.
Figure 5.
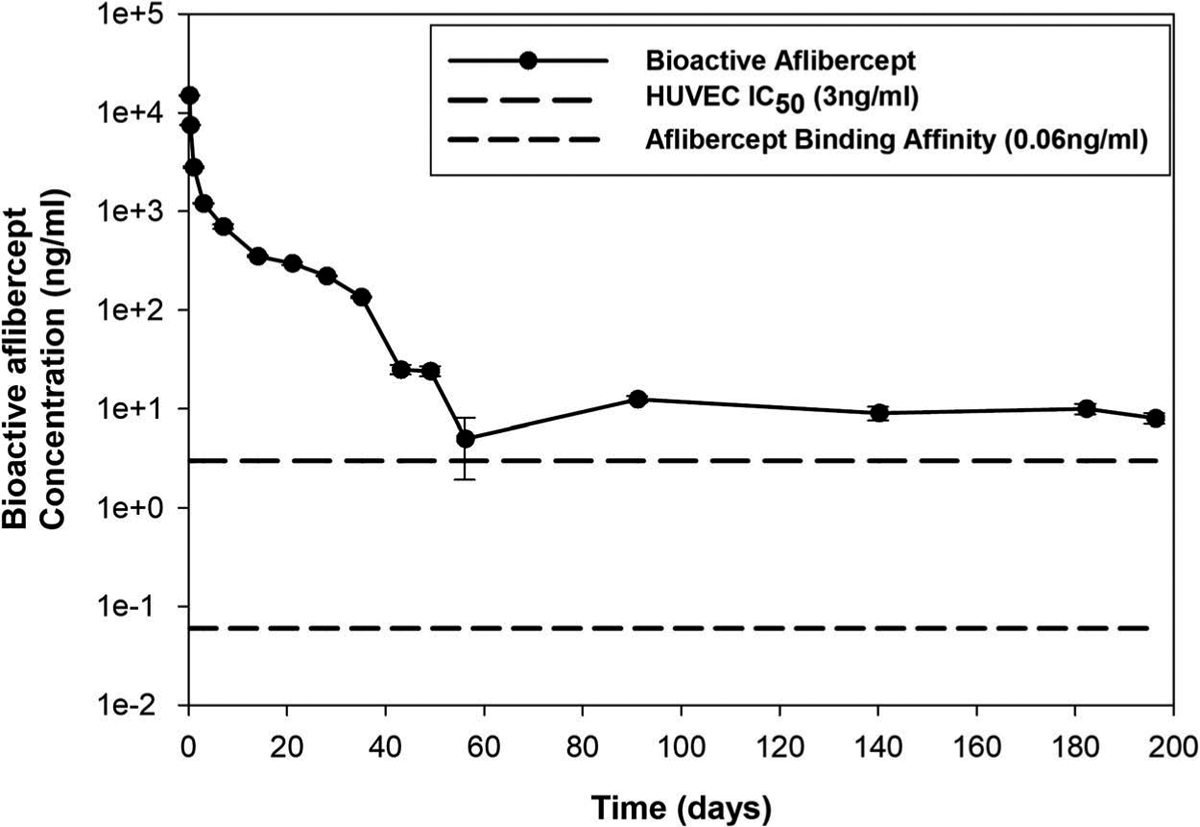
In vitro bioactivity from ELISA. Released bioactive aflibercept concentration from 2 mM DDS-20 at predetermined time points over the release time frame, with reference lines indicating aflibercept in vitro binding affinity (0.06 ng/ml) and half maximal inhibitory concentration (IC50 = 3 ng/ml).5,18 Y-axis was plotted in common log scale for better demonstration, and error bars represent standard error (n = 3).
Morphology of microsphere-hydrogel DDS
Morphology of microsphere and surrounding hydrogels were imaged by cryo-SEM both at room temperature (Figure 6 A1, A2) and after incubation at body temperature for 24 h (B1, B2). The morphological changes of microsphere-hydrogel DDS due to thermoresponsive transformation can be seen from these images. At room temperature, we found that the mesh of the thermoresponsive hydrogels (2 mM) was homogeneous, and their sizes were approximately 100 nm in diameter. The microspheres were sparsely distributed inside the hydrogels and their surfaces were smooth and attached to surrounding hydrogel matrix. The internal structure of microsphere was hollow giving the microsphere more like a capsule appearance. After incubation at body temperature, the meshes of hydrogels were collapsed and difficult to visualize under electron microscope. The distribution of microspheres became denser due to the collapsing of hydrogel. The collapse of hydrogels resulted in gap between microspheres and nearby hydrogel matrix indicated by the white triangles in Figure 6 B2. The surface of microsphere became rougher than that at room temperature.
Figure 6.
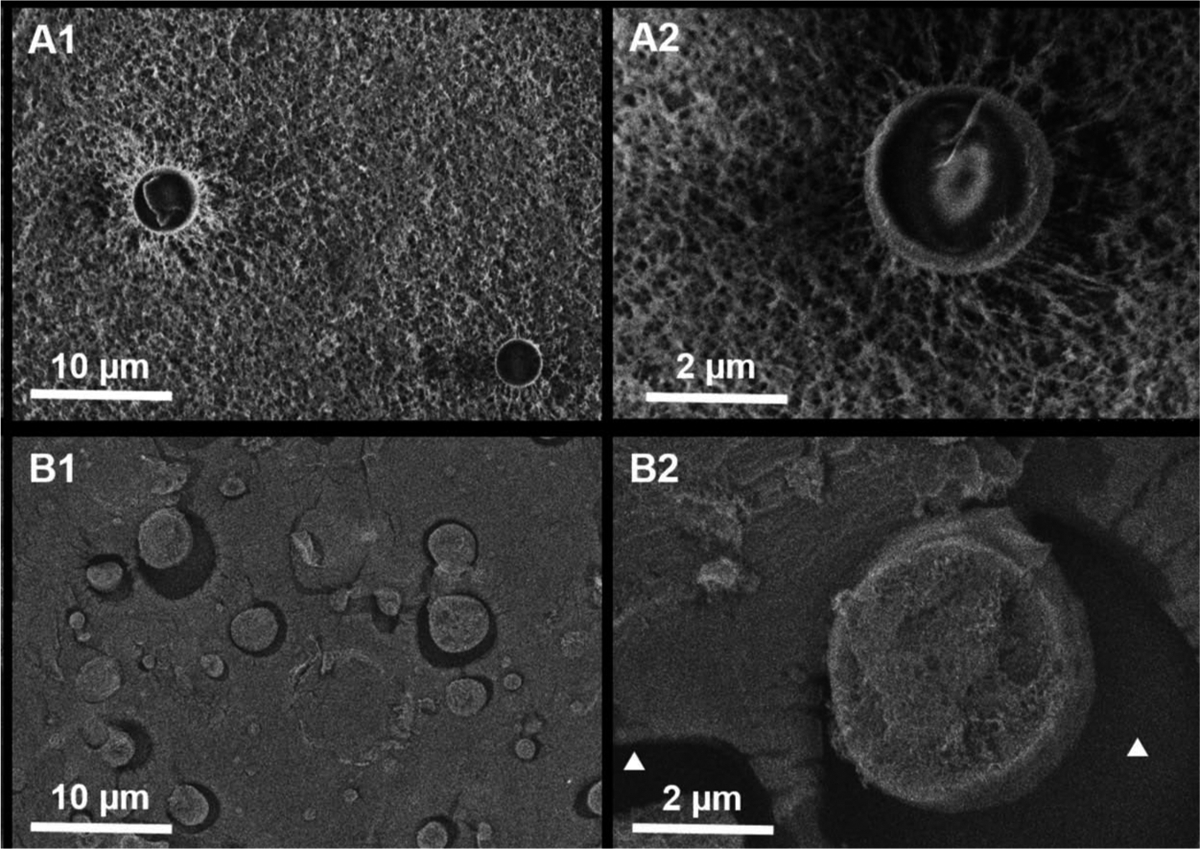
Morphology by cryo-SEM. Cryo-SEM images showing the morphology of microspheres embedded in hydrogels from 2 mM DDS-20 at room temperature (A1, A2) and incubated at body temperature for 24 hours (B1, B2). White triangles in B2 indicate the hollow space between the microsphere and hydrogel created after thermal transformation.
Discussion
In this study, we have shown that all investigated degradable PLGA microspheres suspended in degradable PEG-PLLA-DA/NIPAAm hydrogels were capable of releasing aflibercept in a controlled and extended manner for ~200 days. The introduction of hydrolytically degradable cross-linker PEG-PLLA-DA to hydrogel allows for slow degradation of hydrogel. The slower degradation of hydrogel (>200 days) than PLGA microspheres (~154 days)13 allows more complete release of drug and localization of microspheres to the injection site. The degraded byproducts from the DDS were found to be biocompatible. Additionally, bioactivity of aflibercept was detected throughout the release period for 6 months which we believe to be an important discovery for anti-VEGF sustained delivery. This long-term controlled release of bioactive aflibercept provided by the degradable microsphere-hydrogel DDS in this study holds a great potential toward replacing the current monthly/bimonthly IVT injection treatment regimens.
To make aflibercept-loaded PLGA microspheres, we used aflibercept stock solution at clinical concentration of 40 mg/ml. An EE of 52.78 ± 5.84% was achieved for microspheres after washing. It has been reported that factors such as polymer concentration, polymer molecular weight, lactide/glycolide ratio, and solvent evaporation procedure have major effects on drug EE.21,22 Although the above factors have been investigated and optimized in our previous study using BSA as a model protein for encapsulation into PLGA microspheres,13,23 aflibercept is different from BSA in various aspects, such as molecular weight, isoelectric point, which may influence the results. Therefore, further improvement on aflibercept’s encapsulation into microspheres is still possible. We also experienced approximately 30% of the microspheres lost during loading into hydrogels, this could be attributed to a portion of the microspheres (surface bound) were not fully embedded inside of hydrogels and got washed away after washing the hydrogel to reduce cytotoxicity.
Both cross-linker concentration of PEG-PLLA-DA (2 and 3 mM) and microsphere load amount (10 and 20 mg/ml) were investigated for effects on controlling aflibercept release. We observed a rapid release of radiolabeled aflibercept from all investigated microsphere-hydrogel DDSs during the first week, followed by a consistent near zero-order release thereafter until 6 months. The length of release timeframe (204 days) for aflibercept was comparable to that of our previous nondegradable systems (196 days).14 It is worth noting that we terminated the release profiles on day 204 due to the limitation of I-125 signal decaying, thus, it is possible that a longer and more complete release can be achieved. However, the released aflibercept at day 204 has minimal bioactivity based on the dot blot and ELISA results.
The percentage IB (release within first 24 h) for all investigated microsphere-hydrogel DDSs were on average ~38%. It is well known that fast diffusion of surface-bound drugs away from microspheres contributes mostly to IB.24,25 Comparing to aflibercept-loaded microspheres alone, which was reported in our previous studies, a significant reduction of IB from 83.3%14 to 38% for aflibercept was achieved by our degradable microsphere-hydrogel DDS. This reduction of IB may be due to hydrogels acting as a barrier to provide resistance to diffusion of surface-bound drugs. However, the IB release can only be reduced but cannot be eliminated by our hydrogels. This can be explained by the hydrogels’ thermoresponsive transformation behavior. As temperature increased from room temperature to body temperature, the hydrogels collapsed and the volume shrunk (Figure 2(c)), which corresponds to a much denser polymer network seen at the microscopic scale (Figure 6). This thermoresponsive transformation behavior results from a hydrophobic-hydrophilic balance established by polar and nonpolar moieties in the polymer chain, that is, the hydrophobic interactions between polymer will become more dominant at body temperature comparing to room temperature causing hydrogels to lose water and become solidified.26,27 We speculate that this loss of water from hydrogels network during thermal transformation would result in convective mass transport of mainly surface-bound aflibercept on the microspheres, and few free aflibercept in the hydrogels network if any. We believe that this initial convective flow is a major contributor to IB release of aflibercept from the system. We believe that the IB release of anti-VEGF can be beneficial since it can act as a loading dose to neutralize VEGF in the beginning of treatment for ocular neovascularization.28
After IB release, we also observed a relative fast release of aflibercept before becoming into slow and zero-order release for all investigated DDSs. This release phase corresponded to the degradation of hydrogels as shown in Figure 2(a) and (b), where hydrogels with 2 and 3 mM PEG-PLLA-DA both degraded faster during first 2 weeks followed by slower degradation. Both PEG-PLLA-DA concentration and microsphere load amount contribute to the release of aflibercept. However, effects of PEG-PLLA-DA concentration on release profile seem to be smaller, since two different PEG-PLLA-DA concentrations regardless of microsphere load amount yielded comparable overall release profiles of aflibercept. Regardless of PEG-PLLA-DA concentrations, the IB release, first-week release, and daily release rate were found to be proportional to microsphere load amount. Our results suggest that the drug release rate and amount can be easily adjusted by microsphere load amount than PEG-PLLA-DA polymer concentration. It is important to point out that increase of microsphere load amount may influence DDS’s injectability through small-gauge needles,29 further test on effects of microsphere load amount on mechanical properties of DDS should be done in future.
More complete release of aflibercept (average of 66%) were achieved for microsphere-hydrogel DDS using degradable hydrogels than previous nondegradable DDS (~47%).14 In addition, higher degradable PEG-PLLA-DA cross-linker concentration (comparing 3–2 mM) resulted in ~5% more (p < 0.05) complete drug release. The above results suggest that the degradation of hydrogels enhance release of drugs. It is expected that increase in PEG-PLLA-DA concentration would generate more complete drug release; however, the disadvantage of higher cross link concentration is a decrease in injectability of hydrogels via small gauge needles. It is possible to accelerate the degradation of hydrogels by adding chain transfer agents (CTA) such as glutathione.12 However, addition of CTA resulted in fast complete degradation in less than a month. This fast degradation is not desirable for long-term controlled release of anti-VEGF from microspheres since we need hydrogels to degrade slower to secure microspheres to localized injection site.13 Although there was a small amount of undissolved polymer residuals left at the end of degradation studies, these remaining polymers lost hydrogels’ original shape integrity and decreased significantly in size (Figure 2(c)). In fact, the remaining polymers became immediately dissolved once brought back to room temperature. This indicates that the chemical cross-links which is critical for hydrogels’ shape integrity were actually degraded, the undissolved polymers were kept together through weak physical cross-links due to hydrophobic interactions between PNIPAAm chains. For our future study, the hydrogels’ degradability in vivo will be further characterized, since the environment in vivo may influence the degradation of materials by various factors such as pH, enzymes, and macrophages.30
It is well-documented that free radical initiator, N,N,N′,N ′-tetramethylenediamine (TEMED), used in free-radical polymerization to form PEG-PLLA-DA/NIPAAm hydrogels can cause oxidative damage to cells.18,19 From cell viability test results in Figure 3, we conclude that at least three times of wash using larger volume of PBS buffer (1:25 volume ratio) are necessary to remove TEMED and other potential toxic unreacted monomers. All the degraded samples collected throughout the entire degradation timeframe had no cytotoxic effects on cultured HUVECs, and these findings demonstrate that our degradable microsphere-hydrogel DDSs are safe and biocompatible.
As the bioactivity of protein drug is heavily dependent on their complex structures which are susceptible to environmental conditions, such as pH; temperature; and presence of oxidants, salts, or surfactants,1,31 maintaining protein bioactivity during release remains as a big challenge for controlled protein DDS. Based on previous studies, aflibercept exhibited detectable strong binding affinity to VEGF at concentration as low as 0.06 ng/ml (0.5 pM);5 and its IC50 for HUVEC was reported to be 3 ng/ml in vitro.20 Despite a considerably decreased aflibercept bioactivity after 2 months of release (Figures 4 and 5), the lowest concentration of bioactive aflibercept detectable across the release timeframe (5 ng/ml) was well above these thresholds. Therefore, we believe that this sustained released bioactive drug from our DDS should be sufficient to block VEGF-induced angiogenesis over the entire length of release. Nevertheless, in vivo efficacy in animal models should be further confirmed in future. It is not surprising to see the low bioactivity of aflibercept at later time points in this study, as decreased stability of protein drug released from controlled DDSs like PLGA micro-/nanoparticles and hydrogels has been well documented.17,33–35 There are various factors contributing to loss of bioactivity of protein drug during release from degradable polymer DDS, such as hydrophobic polymer–protein interaction, moisture-induced aggregation, and non-covalent aggregation due to acidic microenvironment.1 We speculate that the low bioactivity of released drug from our system at later time points is due to environmental pH change as a result of acidic degraded byproducts accumulation. Both hydrolysis of PLLA cross-linkers of hydrogels and bulk erosion of PLGA microspheres will generate hydroxyl-carboxylic acid through ester bond cleavage and these acidic products can be finally metabolized into water and carbon dioxide through a citric acid cycle in vivo.36 In order to address this challenge, basic additive like Mg(OH)2 was added into the microsphere formulation to counter pH drop during polymer degradation.37 We believe that several excipients introduced in our manufacture process preserved bioactivity of the drug for 6 months. To our knowledge, our system is the first system to maintain bioactivity of aflibercept released from DDS for 6 months. In addition, the sampling and separation method was used in this study for collecting release samples in vitro due to its low cost and simplicity. However, this in vitro drug release method lacks continuous flow and clearance system of in vivo physiological condition. Without continuous flow and clearance, it is possible that the acidic products accumulated more which contribute to protein degradation and loss of bioactivity.38
In conclusion, our degradable microsphere-hydrogel DDS is safe and capable of releasing bioactive aflibercept in a controlled and extended manner for 6 months. The amount and rate of drug release can be easily controlled by microsphere load amount within the hydrogels. Our degradable microsphere-hydrogel DDS holds a great potential to reduce frequency of IVT injections in current anti-VEGF therapy and lower the socioeconomic burdens on patients, family, and healthcare systems.
Acknowledgments
The authors would like to thank Dr Brianna Roux and Mr Feipeng Yang for providing and assistance in culturing the HUVECs. In addition, the cryo-SEM images in this work made use of the BioCryo facility of Northwestern University’s NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1720139) at the Materials Research Center; the International Institute for Nanotechnology (IIN); and the State of Illinois, through the IIN. It also made use of the CryoCluster equipment, which has received support from the MRI program (NSF DMR-1229693). This research was supported by the NIH/NEI (EY025434) research grant (JJKM).
No commercial relationship exists in the form of financial support. Dr Kang-Mieler has a US Patent pending on “Microsphere-thermo-responsive drug delivery system”.
Footnotes
Color versions of one or more of the figures in the article can be found online at www.tandfonline.com/icey.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Radhakrishnan K, Sonali N, Moreno M, Nirmal J, Fernandez AA, Venkatraman S, Agrawal R. Protein delivery to the back of the eye: barriers, carriers and stability of anti-VEGF proteins. Drug Discov Today. 2017;22:416–23. doi: 10.1016/j.drudis.2016.10.015. [DOI] [PubMed] [Google Scholar]
- 2.National Eye Institute, Age-related macular degeneration (AMD). 2017. [accessed 2018 May 26]. http://www.nei.nih.gov/eyedata/amd
- 3.Foundation AMD. Dry vs wet age-related macular degeneration. 2017. [accessed 2018 May 26].http://www.macular.org/dry-vs-wetmacular-degeneration
- 4.Kwak N, Okamoto N, Wood JM, Campochiaro PA. VEGF is major stimulator in model of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2000;41:3158–64. [PubMed] [Google Scholar]
- 5.Kim LA, D’Amore PA. A brief history of anti-VEGF for the treatment of ocular angiogenesis. Am J Pathol. 2012;181:376–79. doi: 10.1016/j.ajpath.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falavarjani KG, Nguyen QD. Adverse events and complications associated with intravitreal injection of anti-VEGF agents: a review of literature. Eye. 2013;27:787–94. doi: 10.1038/eye.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El Sanharawi M, Kowalczuk L, Touchard E, Omri S, de Kozak Y, Behar-Cohen F. Protein delivery for retinal diseases: from basic considerations to clinical applications. Prog Retin Eye Res. 2010;29:443–65. doi: 10.1016/j.preteyeres.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Kang-Mieler JJ, Dosmar E, Liu W, Mieler WF. Extended ocular drug delivery systems for the anterior and posterior segments: biomaterial options and applications. Expert Opin Drug Deliv. 2017;14:611–20. doi: 10.1080/17425247.2016.1227785. [DOI] [PubMed] [Google Scholar]
- 9.Adamson P, Wilde T, Dobrzynski E, Sychterz C, Polskyc R, Kurali E, Haworth R, Tang CM, Korczynska J, Cook F, et al. Single ocular injection of a sustained-release anti-VEGF delivers 6 months pharmacokinetics and efficacy in a primate laser CNV model. J Control Release. 2016;244:1–13. doi: 10.1016/j.jconrel.2016.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie B, Jin L, Luo Z, Yu J, Shi S, Zhang Z, Shen M, Chen H, Li X, Song Z. An injectable thermosensitive polymeric hydrogel for sustained release of Avastin® to treat posterior segment disease. Int J Pharm. 2015;490:375–83. doi: 10.1016/j.ijpharm.2015.05.071. [DOI] [PubMed] [Google Scholar]
- 11.Huang J, Wang W, Yu J, Yu X, Zheng Q, Peng F, He Z, Zhao W, Zhang Z, Li X, et al. Combination of dexamethasone and Avastin® by supramolecular hydrogel attenuates the inflammatory corneal neovascularization in rat alkali burn model. Colloids Surf B Biointerfaces. 2017;159:241–50. doi: 10.1016/j.colsurfb.2017.07.057. [DOI] [PubMed] [Google Scholar]
- 12.Drapala PW, Jiang B, Chiu YC, Mieler WF, Brey EM, Kang-Mieler JJ, Perez-Luna VH. The effect of glutathione as chain transfer agent in PNIPAAm-based thermo-responsive hydrogels for controlled release of proteins. Pharm Res. 2014;31:742–53. doi: 10.1007/s11095-013-1195-0. [DOI] [PubMed] [Google Scholar]
- 13.Osswald CR, Kang-Mieler JJ. Controlled and extended release of a model protein from a microsphere-hydrogel drug delivery system. Ann Biomed Eng. 2015;43:2609–17. doi: 10.1007/s10439-015-1314-7. [DOI] [PubMed] [Google Scholar]
- 14.Osswald CR, Kang-Mieler JJ. Controlled and extended in vitro release of bioactive anti-vascular endothelial growth factors from a microsphere-hydrogel drug delivery system. Curr Eye Res. 2016;41:1216–22. doi: 10.3109/02713683.2015.1101140. [DOI] [PubMed] [Google Scholar]
- 15.Turturro SB, Guthrie MJ, Appel AA, Drapala PW, Brey EM, Perez-Luna VH, Mieler WF, Kang-Mieler JJ. The effects of cross-linked thermo-responsive PNIPAAm-based hydrogel injection on retinal function. Biomaterials. 2011;32:3620–26. doi: 10.1016/j.biomaterials.2011.01.058. [DOI] [PubMed] [Google Scholar]
- 16.Osswald CR, Guthrie MJ, Avila A, Valio JA, Mieler WF, Kang-Mieler JJ. In vivo efficacy of an injectable microsphere-hydrogel ocular drug delivery system. Curr Eye Res. 2017;42:1293–301. doi: 10.1080/02713683.2017.1302590. [DOI] [PubMed] [Google Scholar]
- 17.Giteau A, Venier-Julienne MC, Aubert-Pouessel A, Benoit JP. How to achieve sustained and complete protein release from PLGA-based microparticles. Int J Pharm. 2008;350:14–26. doi: 10.1016/j.ijpharm.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 18.Friedman M Chemistry, biochemistry, and safety of acrylamide. Review J Agric Food Chem. 2003;51:4504–26. [DOI] [PubMed] [Google Scholar]
- 19.Moreau MF, Chappard D, Lesourd M, Montheard JP, Basle MF. Free radicals and side products released during methylmethacrylate polymerization are cytotoxic for osteoblastic cells. J Biomed Mater Res. 1998;40:124–31. [DOI] [PubMed] [Google Scholar]
- 20.Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, Pyles EA, Yancopoulos GD, Stahl N, Wiegand SJ. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis. 2012;15:171–85. doi: 10.1007/s10456-011-9249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vila A, Sanchez A, Tobio M, Calvo P, Alonso MJ. Design of biodegradable particles for protein delivery. J Control Release. 2002;78:15–24. [DOI] [PubMed] [Google Scholar]
- 22.Fu X, Ping Q, Gao Y. Effects of formulation factors on encapsulation efficiency and release behavior in vitro of huperzine A-PLGA microspheres. J Microencapsul. 2005;22:705–14. doi: 10.1080/02652040500162196. [DOI] [PubMed] [Google Scholar]
- 23.Jiang B, Zhang G, Brey EM. Dual delivery of chlorhexidine and platelet-derived growth factor-BB for enhanced wound healing and infection control. Acta Biomaterialia. 2013;9:4976–84. doi: 10.1016/j.actbio.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 24.Allison SD. Analysis of initial burst in PLGA microparticles. Expert Opin Drug Deliv. 2008;5:615–28. doi: 10.1517/17425247.5.6.615. [DOI] [PubMed] [Google Scholar]
- 25.Yeo Y, Park K. Control of encapsulation efficiency and initial burst in polymeric microparticle systems. Arch Pharm Res. 2004;27:1–12. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida R, Sakai K, Okano T, Sakurai Y. Modulating the phase transition temperature and thermosensitivity in N-isopropylacrylamide copolymer gels. J Biomater Sci Polym Ed. 1994;6:585–98. [DOI] [PubMed] [Google Scholar]
- 27.Geever LM, Devine DM, Nugent MJD, Kennedy JE, Lyons JG, Higginbotham CL. The synthesis, characterization, phase behavior and swelling of temperature sensitive physically crosslinked poly (1-vinyl-2-pyrrolidinone)/poly(N-isopropylacrylamide) hydrogels. Eur Polym J. 2006;42:69–80. doi: 10.1016/j.eurpolymj.2005.09.027. [DOI] [Google Scholar]
- 28.Semeraro F, Morescalchi F, Duse S, Parmeggiani F, Gambicorti E, Costagliola C. Aflibercept in wet AMD: specific role and optimal use. Drug Des Dev Ther. 2013;7:711–22. doi: 10.2147/DDDT.S40215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Tomme SR, van Nostrum CF, Dijkstra M, de Smedt SC, Hennink WE. Effect of particle size and charge on the network properties of microsphere-based hydrogels. Eur J Pharm Biopharm. 2008;70:522–30. doi: 10.1016/j.ejpb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 30.Spenlehauer G, Vert M, Benoit JP, Boddaert A. In vitro and in vivo degradation of poly(D,L lactide/glycolide) type microspheres made by solvent evaporation method. Biomaterials. 1989;10:557–63. [DOI] [PubMed] [Google Scholar]
- 31.Balcao VM, Vila MM. Structural and functional stabilization of protein entities: state-of-the-art. Adv Drug Deliv Rev. 2015;93:45–41. doi: 10.1016/j.addr.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Instability Wang W., stabilization, and formulation of liquid protein pharmaceuticals. Int J Pharm. 1999;185:129–88. [DOI] [PubMed] [Google Scholar]
- 33.Mohammadi-Samani S, Taghipour B. PLGA micro and nanoparticles in delivery of peptides and proteins: problems and approaches. Pharm Dev Technol. 2015;20:385–93. doi: 10.3109/10837450.2014.882940. [DOI] [PubMed] [Google Scholar]
- 34.Varschochian R, Jeddi-Tehrani M, Mahmoudi AR, Khoshatand MR, Atyabi F, Sabzevari A, Esfahani MR, Dinarvand R. The protective effect of albumin on bevacizumab activity and stability in PLGA nanoparticles intended for retinal and choroidal neovascularization treatments. Eur J Pharm Sci. 2013;50:341–52. doi: 10.1016/j.ejps.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 35.Marquette S, Peerboom C, Yates A, Denis L, Langer I, Amighi K, Goole J. Stability study of full-length antibody (anti-TNF alpha) loaded PLGA microspheres. Int J Pharm. 2014;470:41–50. doi: 10.1016/j.ijpharm.2014.04.063. [DOI] [PubMed] [Google Scholar]
- 36.Park TG. Degradation of poly(lactic-co-glycolic acid) microspheres: effect of copolymer composition. Biomaterials. 1995;16:1123–30. [DOI] [PubMed] [Google Scholar]
- 37.Kang JC, Schwendeman SP. Comparison of the effects of Mg(OH)2 and sucrose on the stability of bovine serum albumin encapsulated in injectable poly(D,L-lactide-co-glycolide) implants. Biomaterials. 2002;23:239–45. [DOI] [PubMed] [Google Scholar]
- 38.D’Souza S A review of in vitro drug release test methods for nano-sized dosage forms. Adv Pharm. 2014;2014:1–12. [Google Scholar]