

Abstract
Immunostaining is widely used in biomedical research to show the cellular expression pattern of a given protein. Multiplex immunostaining allows labeling using multiple primary antibodies. To minimize antibody cross-reactivity, multiplex immunostaining using indirect staining requires unlabeled primary antibodies from different host species. However, the appropriate combination of different species antibodies is not always available. Here, we describe a method of using unlabeled primary antibodies from the same host species (e.g., in this case both antibodies are from rabbit) for multiplex immunofluorescence on formalin-fixed paraffin-embedded (FFPE) mouse adrenal sections. This method uses the same procedure and reagents used in the antigen retrieval step to strip the activity of the previously stained primary antibody complex. Slides were stained with the first primary antibody using a general immunostaining protocol followed by a binding step with a biotinylated secondary antibody. Then, an avidin-biotin-peroxidase signal development method was used with fluorophore-tyramide as the substrate. The immunoactivity of the first primary antibody complex was stripped through immersion in a microwaved boiling sodium citrate solution for 8 min. The insoluble fluorophore-tyramide deposition remained on the sample, which allowed the slide to be stained with other primary antibodies. Although this method eliminates most false positive signals, some background from antibody cross-reactivity may remain. If the samples are enriched with endogenous biotin, a peroxidase-conjugated secondary antibody may be used to replace the biotinylated secondary antibody to avoid the false positive from recovered endogenous biotin.
Keywords: multiplex immunostaining, immunofluorescence, tyramide, microwaving, antigen retrieval, antibody stripping
INTRODUCTION:
In multiplex immunostaining, direct staining using conjugated primary antibodies can provide informative results. Without using secondary antibodies, the direct staining method has a low risk of false colocalization signals from antibody cross-reactivity. However, the conjugated reporters (fluorophore, enzymes) or biotin on the primary antibody limit its future use. Alternatively, indirect immunostaining usually provides stronger signals by using an unconjugated primary antibody with a labeled secondary antibody. Ideally, unconjugated primary antibodies used in multiplex immunostaining should come from different host species to avoid antibody cross-reactivity. However, the appropriate combination of primary antibodies from different host species is not always available.
Several methods have been established to eliminate the risk of the secondary antibody reacting with an undesired primary antibody. One common method is the use of a F(ab) monomeric antibody to block any remaining binding epitopes on the first primary antibody complex before staining with the second primary antibody1. Antibody stripping, which is similar to the strip and reprobe of a Western blot sheet, removes the previously stained antibody complex without stripping the deposition of detectable reporter molecules such as 3,3’-diaminobenzidine tetrahydrochloride (DAB)2 and the fluorescent tyramide deposition3. With this method, reporter molecules in different colors can show a multiplex result on the same slide. The multiplex staining is also achievable by the complete removal of the previously deposited layers of antibodies and the alignment of subsequently acquired images from other antibodies4,5. These methods all give reliable results, though each method has its limitations and requires either complicated procedures or a special imaging system.
The present protocol shows the application of an antibody stripping method with the use of commonly available buffers. This protocol can be used to perform multiplex immunofluorescent staining on formalin-fixed paraffin-embedded (FFPE) mouse adrenal sections with two unlabeled primary antibodies from the same host species.
PROTOCOL:
1. Staining with the First Antibody
-
Dewax and rehydrate FFPE slides with 5 min allotted to each of the following steps: xylene or equivalent reagents 3x, 100% ethanol 2x, 95% ethanol 1x, 70% ethanol 1x, 50% ethanol 1x, and distilled water 2x.
NOTE: Slides should remain moist starting from this rehydration step until mounting in the final step.
For optional antigen retrieval, place the slides in 275 mL of boiling sodium citrate solution (10 mM, pH = 6.0) for 8 min. To keep the solution boiling, place the slides flat on the bottom of a 14 × 9.5 × 9 cm3 (W × L × H) pipette tip box with a lid and microwave the solution on 70% power in a 700 W microwave oven for 8 min. Then remove the pipette tip box from the microwave oven, open the lid, and let the solution cool down at room temperature (RT) for at least 20 min.
Transfer the slides into a Coplin jar containing PBST (phosphate-buffered saline with 0.1% polysorbate 20/80). Wash with PBST for 5 min 3x. If not immediately moving to the next step, store the slides at this step with PBST in a Coplin jar at RT for a few hours or at 4 °C for 1–2 days if not immediately moving to the next step.
To prepare the blocking solution use the normal serum of the secondary antibody’s host species as the blocking reagent. Other commercial blocking reagents may also be used. If using normal serum employed for the study presented in this study, add 100 μL of normal donkey serum to 4.9 mL of PBST. The blocking solution can be stored at 4 °C for up to 3 days.
For blocking, shake off the PBST from the slides and quickly cover them with sufficient blocking solution. Incubate the slides at RT in a humidified chamber for 30 min.
- Prepare the primary antibody solution (500 μL for two slides) using the blocking solution to dilute the primary antibody to the desired concentration. The solution should be stored on ice until use.
- For 3βHSD, add 2 μL of 3βHSD into 498 μL of blocking solution.
- For TH, add 0.5 μL of TH antibody into 499 μL of blocking solution.
- For β-catenin, add 1 μL of β-catenin antibody into 499 μL of blocking solution.
- For 20αHSD, add 1 μL of 20αHSD antibody into 499 μL of blocking solution.
- For CYP2F2, add 2 μL of CYP2F2 antibody into 498 μL of blocking solution.
Incubate with the primary antibody by shaking off the blocking solution from the slides, and quickly covering them with sufficient primary antibody solution. Incubate the slides in a humidified chamber overnight at 4 °C. During incubation the slides may be covered by a small piece of paraffin film to prevent drying out.
The next morning wash the slides with PBST for 5 min 3x.
Prepare the secondary antibody solution (500 μL for two slides) using the blocking solution to dilute the biotinylated secondary antibody to the desired concentration (e.g., 1 μL of donkey anti-mouse antibody or donkey anti-rabbit antibody into 499 μL of blocking solution). The solution should be stored on ice until use.
-
Incubate with the second antibody by shaking off PBST from the slides and quickly covering them with sufficient secondary antibody solution. Incubate the slides in a humidified chamber for 1 h at RT.
NOTE: If endogenous biotin is a concern, use a peroxidase-conjugated secondary antibody instead of the biotinylated secondary antibody. Jump to step 1.14 if using a peroxidase-conjugated secondary antibody in step 1.10.
Wash the slides with PBST for 5 min 3x.
Prepare the horseradish peroxidase-conjugated streptavidin (SA-HRP) solution (500 μL for two slides) by adding 0.5 μL of SA-HRP into 499 μL of PBS. The final concentration is 1 μg/mL.
Incubate with the SA-HRP solution. Shake off PBST from the slides and quickly cover the slides with sufficient SA-HRP solution. Incubate the slides in a humidified chamber for 0.5 h at RT.
Wash the slides with PBST for 5 min 3x.
Prepare the fluorophore-tyramide solution by diluting fluorophore-tyramide with its dilution buffer according to the manufacturer’s instructions (e.g., 5 μL of fluorophore-tyramide into 496 μL of dilution buffer).
Perform the signal development with fluorophore-tyramide by shaking off PBST from the slides and quickly covering the slides with sufficient fluorophore-tyramide solution. Incubate in a humidified chamber for 1 min at RT. The incubation time may be adjusted to obtain the desired fluorescence intensity. Stop the reaction by transferring the slides to a Coplin jar containing PBST. Wash slides with PBST for 3 min 2x.
Signals may be quickly checked under a fluorescence microscope to confirm the result at this stage. If coverslips are not being used, apply a drop of glycerol:PBS (1:1) onto each section to keep the samples moist. Wash slides with PBST for 3 min 2x. Slides may be stored in PBST at 4 °C for a few days before moving to the next step.
2. Strip the first antibody
Place the slides in 275 mL of boiling sodium citrate solution (10 mM, pH = 6.0) for at least 8 min. Keep the solution boiling by placing the slides flat on the bottom of a 14 × 9.5 × 9 cm3 (W × L × H) pipette tip box with a lid and microwaving the solution on 70% power in a 700 W microwave oven for 8 min. If a longer stripping time is preferred, it may be required to top off the buffer using a boiling sodium citrate solution to keep the slides immersed in buffer solution at all times. Remove the pipette tip box from the microwave oven, open the lid, and let the solution cool down at RT for at least 20 min.
Transfer the slides into a Coplin jar containing PBST. Wash with PBST for 5 min 3x. If the next step if not performed immediately, store the slides in a Coplin jar with PBST at RT for a few hours or at 4 °C for 1–2 days.
3. Stain with the second antibody
-
Start from the blocking step and follow same procedures in step 1.5 through step 1.14. At the signal development steps (step 1.15 and step 1.16), use the fluorophore-tyramide in a different fluorescence spectrum.
NOTE: Slides can be stripped using this method several times to allow multiplex staining. The last antibody does not require fluorophore- tyramide as the reporter. A fluorophore-conjugated secondary antibody might be used to show signals from the last primary antibody.
Incubate slides with 2 μg/mL 4’,6-diamidino-2-phenylindole (DAPI) or Hoechst solution for 1 min at RT if nuclear counter staining is needed. Then wash the slides with PBST for 3 min 2x.
4. Imaging: use a fluorescence microscope to detect signals
Mount a coverslip with a drop of mounting medium suitable for immunofluorescence.
For imaging, use a fluorescence microscope to detect signals of each fluorescence channel. Start with the nuclear staining (DAPI) channel and the 4x lens to locate the tissue on the slide.
Switch to the 10x lens for imaging. Adjust the exposure time (300 ms) and light source intensity (80% intensity). Adjust these settings if the signal is too bright or too dark. Take pictures of each channel without moving the stage. Refocusing may be needed for each channel. Merge the images from each channel using the microscope’s software or ImageJ (imagej.nih.gov/ij/)
REPRESENTATIVE RESULTS:
The results were obtained from samples treated with all steps described including the antigen retrieval step 1.2. All secondary antibodies used here were biotinylated. Fluorophore-tyramide was used to develop signals from the first and second primary antibodies. Images were captured using a fluorescence microscope equipped with a FITC cube (for green fluorescence), a TxRED cube (for Cy3), and a DAPI cube (for DAPI).
Microwaving-mediated stripping eliminates the cross-reactivity.
Mouse FFPE adrenal sections were stained using two primary rabbit antibodies. Without stripping (Figure 1A–C), the secondary antibody for TH picked up 3βHSD(+) sites and gave red signals in the adrenal cortex (Figure 1B). The lack of stripping led to the false colocalization signals seen in yellow (Figure 1C). This cross-reactivity comes from (1) the HRP enzyme that catalyzed the first tyramide reaction and (2) the antibody species cross-reactivity. To remove the antibody cross-reactivity and the HRP from the first immunostaining, we tested a 1 min (Figure 1D–F) and an 8 min microwave-mediated stripping (Figure 1G–I). Both treatments were sufficient to eliminate the nonspecific signal, giving clean double staining results (Figure 1F,I). The negative control followed all of the same steps with the exception of incubation with primary antibodies (Figure 1J). No significant signal was picked up in the negative control. We found that an 8 min stripping in a boiling citrate buffer was sufficient to eliminate antibody cross-reactivity for many antibodies commonly used in the adrenal gland (Figure 2).
Figure 1. Representative double immunostaining on P1 FFPE mouse adrenals.
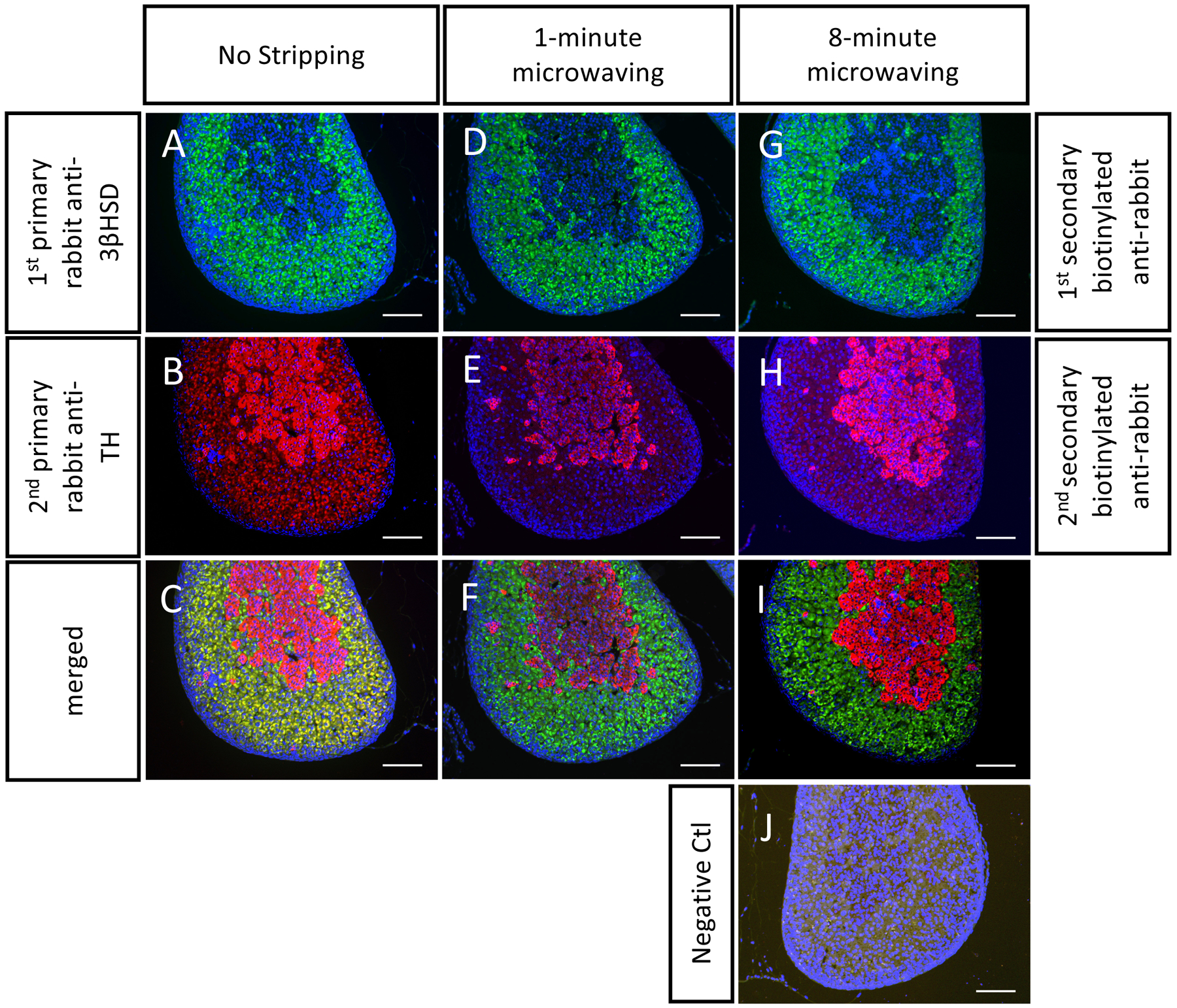
FFPE mouse adrenal sections were stained with two primary antibodies from rabbits: anti-3βHSD (in green, stains the cortex), anti-TH (in red, stains the medulla). The three groups of stains include the three different stripping procedures used: (A-C) no-stripping, (D-F) 1-min stripping, and (G-I) 8-min stripping. Note that without microwaving, the secondary antibody with TH still picked up 3βHSD signals, whereas specific staining results were obtained in the 1-min and 8-min microwave treated groups. There is no positive signal in the negative control (without incubation with primary antibodies). Scale bar 100 μm. DAPI stains cell nuclei, in blue.
Figure 2. Representative double immunostaining on P21 FFPE mouse adrenals.
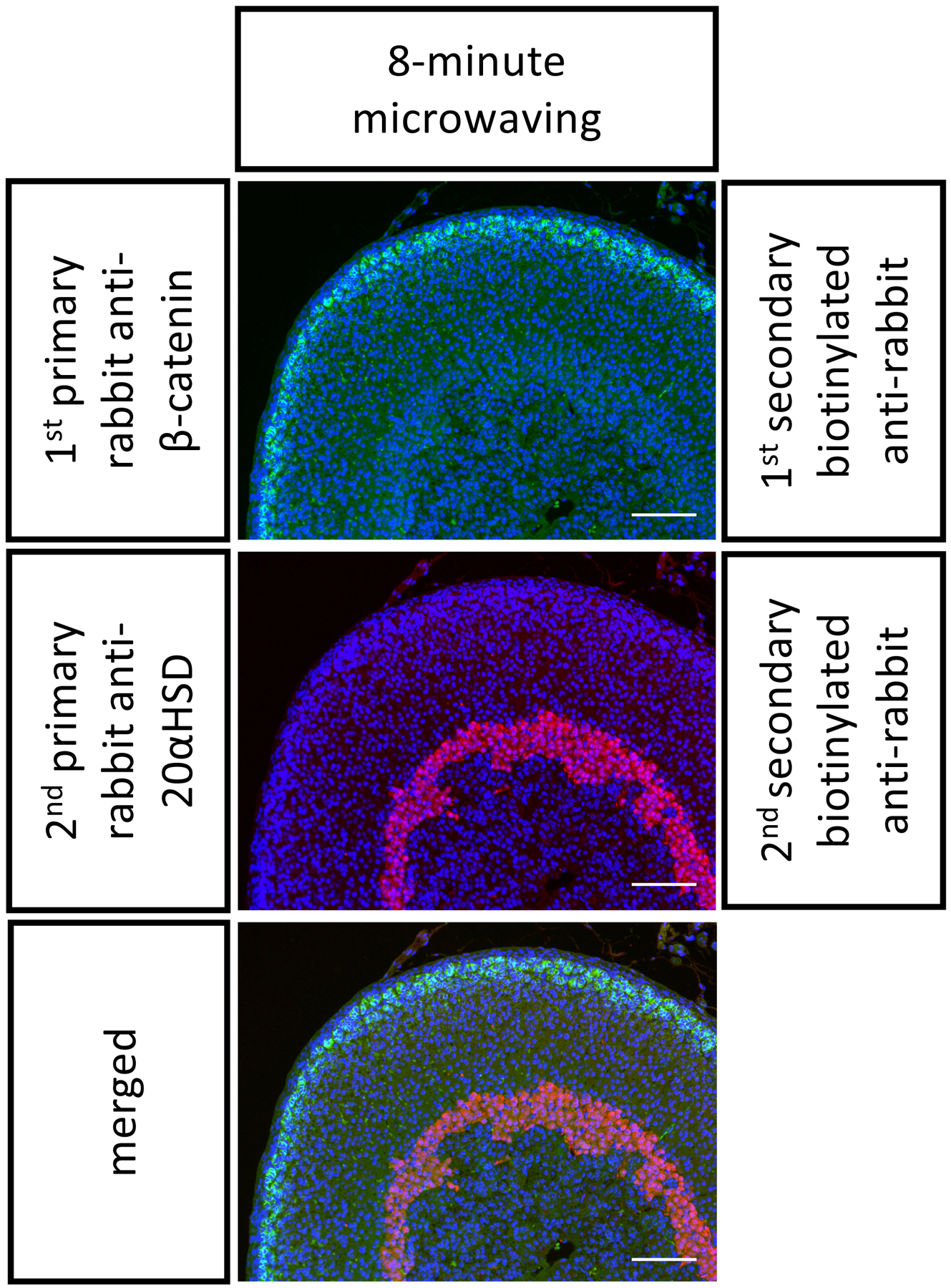
The FFPE mouse adrenal sections were stained with two primary antibodies from rabbits in the following order: anti-β-catenin (in green, stains the outer cortex) and then anti-20αHSD (in red, stains the inner cortex). An 8-min stripping step was performed in between. Scale bar 100 μm. DAPI stains cell nuclei, in blue.
The limitation — the efficacy of the microwave-mediated stripping is antibody-dependent.
Although the microwave-mediated stripping used in this protocol works for most cases, there are limitations to this method. For some antibodies, a microwave-mediated stripping method with a citrate buffer may not completely remove the antibody cross-reactivity. For example, an 8 min stripping removed most nonspecific signals from the mouse anti-TH antibody and the mouse anti-CYP2F2 antibody, but a weak false positive signal was still detectable (the medulla in Figure 3E and the inner cortex in Figure 3K). Note that an increase of the microwaving time to 20 min still did not completely remove antibody cross-reactivity (Figure 3H,K).
Figure 3. Some antibodies may not be fully stripped which could lead to weak non-specific signals.
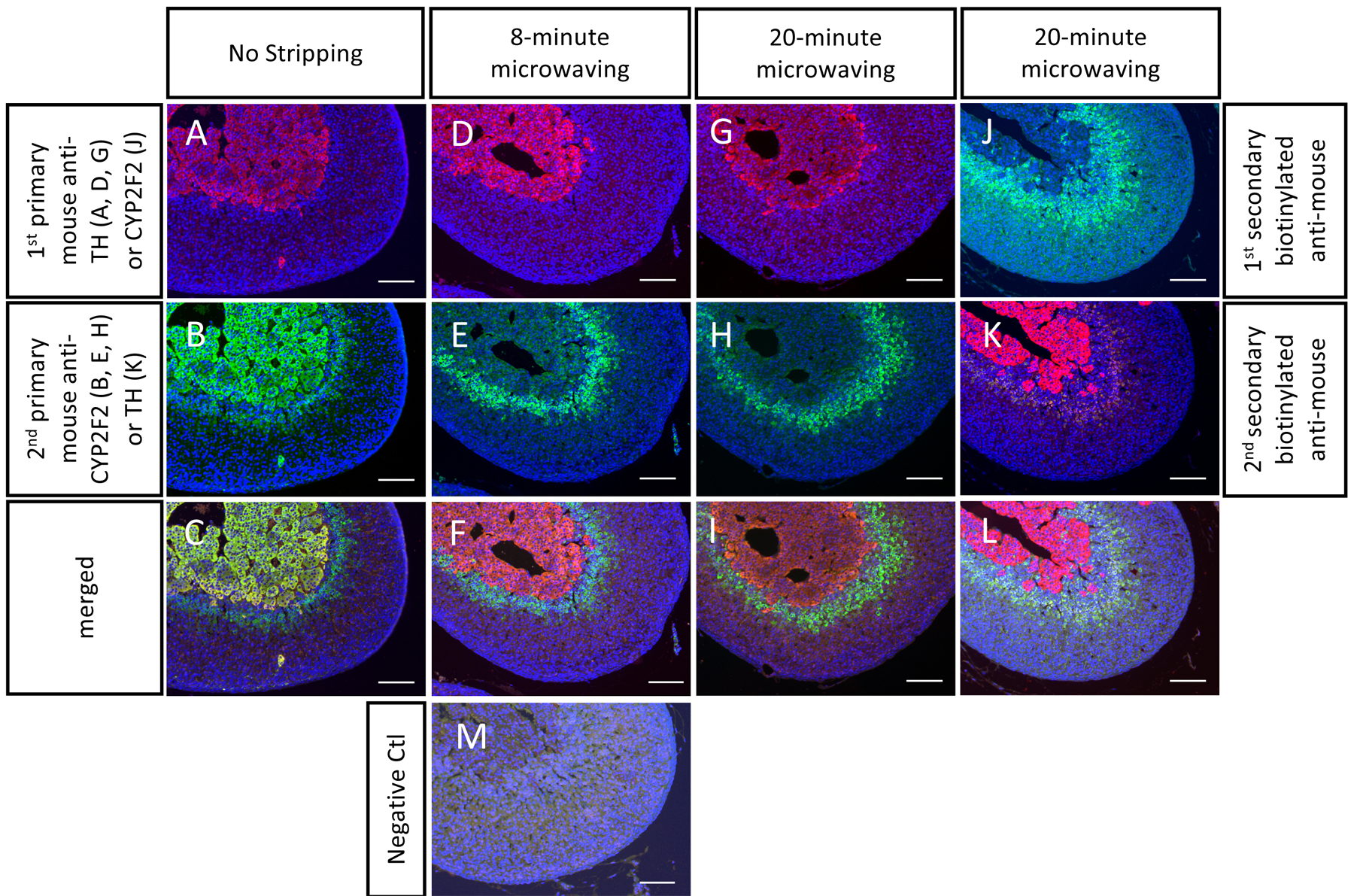
The FFPE mouse adrenal sections were stained with two primary antibodies from mice: anti-CYP2F2 (in green, stains the inner cortex) and anti-TH (in red, stains the medulla). The results from the no stripping group (A-C) showed the reactions for detecting the CYP2F2 protein still stained the TH(+) area. A false colocalization was seen in the adrenal medulla (C). Although an 8-min and a 20-min microwave treatment reduced the green fluorescence intensity in the medulla, a noticeable background is still present (D-I). The anti-CYP2F2 antibody is another example showing that the cross-reactivity cannot be fully removed even after 20 min of microwaving (J-L). Note that there was no positive signal in the negative control (M, without incubation with primary antibodies), indicating the false-positive signal was from the antibody cross-reactivity. Scale bar 100 μm. DAPI stains cell nuclei, in blue.
DISCUSSION:
Multiplex immunostaining is useful to examine the cellular colocalization of two or more antigens. This widely used technique gives convincing colocalization results when primary antibodies are conjugated with different reporters (direct staining). However, direct staining usually provides weaker signals compared to indirect staining, which involves conjugated secondary antibodies to detect the primary antibodies. In indirect staining, a high-quality multiplex immunostaining result relies on whether the secondary antibodies can distinguish between the different primary antibodies. Due to the possible antibody cross-reactivity, multiplex staining utilizing the indirect staining method is seen more clearly with primary antibodies from different host species. Carl and others developed a protocol using an excess of unconjugated IgG F(ab) fragment to block antibody cross-reactivity1. A similar strategy is used in the “mouse-on-mouse” staining which uses a F(ab) monomeric anti-mouse antibody to prevent the anti-mouse secondary antibody from detecting any endogenous mouse immunoglobulin in the tissue. However, this blocking method is time consuming and may not completely block antibodies from previous steps especially if the detecting antigens are of high abundance2.
Heat-mediated stripping is another method to prevent antibody cross-reactivity in multiplex immunostaining. The concept is similar to the strip and reprobe method of a Western blot. The use of a microwave to boil the slides in a citrate buffer had a marked positive effect on blocking the antibody cross-reactivity. Two 5-min microwave treatments between sequential rounds of colorimetric immunostaining allow the detection of multiple antigens on the same slide using mouse monoclonal antibodies2. Microwave treatments combined with the thyramide signal amplification (TSA) method, which uses fluorophore-tyramide as the substrate of HRP, are also useful for immunofluorescence double staining3,6,7. The microwave treatment between the first and second staining cycles blocks the activity of the first immunocomplex without washing out its fluorescent tyramide precipitation. However, a microwave treatment may not be able to fully prevent contaminations in staining8. Although some methods using different types of stripping buffers may provide a better stripping efficacy, specialized buffers with longer incubation times are needed, or samples need to undergo image acquisition before another stain is applied4,5,7,9–11. Here we demonstrated a simple method with a less complicated procedure and a common buffer for microwave-mediated antigen retrieval in multiplex immunofluorescent staining. Although this method eliminates most cross-reactivity, users should be aware of a possible weak false colocalization seen with some antibodies even after a 20-min microwave treatment.
Endogenous biotin can be used as a marker of steroidogenic cells in the adrenal cortex12. Streptavidin-conjugated fluorophore alone is sufficient to detect endogenous biotin and lights up the entire adrenal cortex, especially in frozen sections. Since endogenous biotin is usually blocked in FFPE samples, the avidin-biotin-peroxidase procedure (e.g. ABC kit) is still widely used to amplify targets in FFPE adrenal samples. It is important to know that the antigen retrieval step also unmasks endogenous biotin and induces its immunoactivity13. Since our method combines microwave-mediated antigen retrieval and the use of streptavidin-conjugated HRP, it is possible that endogenous biotin will lead to false positive signals especially in tissues rich with endogenous biotin such as the liver, kidneys and some tumors13. If the avidin-biotin-peroxidase procedure is needed to amplify the signal, an additional blocking step such as pre-incubation with free avidin and free biotin may help to reduce the endogenous biotin signal14. While our method gives a low endogenous biotin background on FFPE mouse adrenals, it is important to always include a negative control with each sample at all times when using the avidin-biotin-peroxidase procedure. The alternative approach is to use a peroxidase-conjugated secondary antibody instead of the biotinylated secondary antibody.
Our results demonstrate that microwave treatment with a citrate buffer is a useful method for multiplex immunofluorescence staining. However, it should be noted that not all cross-reactivities can be fully blocked. A weak false colocalization is possible. This protocol can be used as an alternative approach when an appropriate combination of primary antibodies from different host species is not available. Users should be aware of possible false-positive from the remaining cross-reactivity as well as the recovered endogenous biotin. Proper negative controls should be included at all times.
Table 1.
Table of Materials
| Name of Material/Equipment | Company | Catalog Number | Comments/Description |
|---|---|---|---|
| Antifade Mounting Medium | Vector Laboratories | H-1000 | |
| Biotinylated donkey anti-mouse | JacksonImmuno | 715-066-151 | 1:500 dilution |
| Biotinylated donkey anti-rabbit | JacksonImmuno | 711-066-152 | 1:500 dilution |
| DAPI | BioLegend | 422801 | 2 μg/mL in distilled water |
| Fluorescence microscope | ECHO | Revolve 4 | |
| Horseradish peroxidase-conjugated streptavidin | JacksonImmuno | 016-030-084 | 1:1000 dilution |
| Microwave oven, 700W | General Electric | JEM3072DH1BB | |
| Mouse anti-CYP2F2 | Santa Cruz, | SC-374540 | 1:250 dilution |
| Mouse anti-TH | Santa Cruz, | SC-25269 | 1:1000 dilution |
| Normal donkey serum | JacksonImmuno | 017-000-121 | 2% serum in PBST |
| Rabbit anti-20αHSD | Kerafast, | EB4002 | 1:500 dilution |
| Rabbit anti-3βHSD | TransGenic, | KO607 | 1:250 dilution |
| Rabbit anti-TH | NOVUS, | NB300–109 | 1:1000 dilution |
| Rabbit anti-β-catenin | Abcam, | ab32572 | 1:500 dilution |
| Streptavidin Horseradish Peroxidase (SA-HRP) | JacksonImmuno | 016-303-084 | 1:1000 dilution |
| TSA Cy3 Tyramide | PerkinElmer | SAT704B001EA | 1:100 dilution |
| TSA Fluorescein Tyramide | PerkinElmer | SAT701001EA | 1:100 dilution |
ACKNOWLEDGMENTS:
This work is supported by NIH R00 HD032636.
Footnotes
DISCLOSURES:
The authors have nothing to disclose.
Video Link
The video component of this article can be found at https://www.jove.com/video/60868
REFERENCES:
- 1.Lewis Carl SA, Gillete-Ferguson I & Ferguson DG An indirect immunofluorescence procedure for staining the same cryosection with two mouse monoclonal primary antibodies. Journal of Histochemistry and Cytochemistry. 41 (8), 1273–1278, doi: 10.1177/41.8.7687266, (1993). [DOI] [PubMed] [Google Scholar]
- 2.Lan HY, Mu W, Nikolic-Paterson DJ & Atkins RC A novel, simple, reliable, and sensitive method for multiple immunoenzyme staining: use of microwave oven heating to block antibody crossreactivity and retrieve antigens. Journal of Histochemistry and Cytochemistry. 43 (1), 97–102, doi: 10.1177/43.1.7822770, (1995). [DOI] [PubMed] [Google Scholar]
- 3.Tornehave D, Hougaard DM & Larsson L Microwaving for double indirect immunofluorescence with primary antibodies from the same species and for staining of mouse tissues with mouse monoclonal antibodies. Histochemistry and Cell Biology. 113 (1), 19–23 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Kim M, Soontornniyomkij V, Ji B & Zhou X System-wide immunohistochemical analysis of protein co-localization. PLoS One. 7 (2), e32043, doi: 10.1371/journal.pone.0032043, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolognesi MM et al. Multiplex Staining by Sequential Immunostaining and Antibody Removal on Routine Tissue Sections. Journal of Histochemistry and Cytochemistry. 65 (8), 431–444, doi: 10.1369/0022155417719419, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toth ZE & Mezey E Simultaneous visualization of multiple antigens with tyramide signal amplification using antibodies from the same species. Journal of Histochemistry and Cytochemistry. 55 (6), 545–554, doi: 10.1369/jhc.6A7134.2007, (2007). [DOI] [PubMed] [Google Scholar]
- 7.Buchwalow I, Samoilova V, Boecker W & Tiemann M Multiple immunolabeling with antibodies from the same host species in combination with tyramide signal amplification. Acta Histochemica. 120 (5), 405–411, doi: 10.1016/j.acthis.2018.05.002, (2018). [DOI] [PubMed] [Google Scholar]
- 8.Bauer M, Schilling N & Spanel-Borowski K Limitation of microwave treatment for double immunolabelling with antibodies of the same species and isotype. Histochemistry and Cell Biology. 116 (3), 227–232, doi: 10.1007/s004180100309, (2001). [DOI] [PubMed] [Google Scholar]
- 9.Pirici D et al. Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. Journal of Histochemistry and Cytochemistry. 57 (6), 567–575, doi: 10.1369/jhc.2009.953240, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glass G, Papin JA & Mandell JW SIMPLE: a sequential immunoperoxidase labeling and erasing method. Journal of Histochemistry and Cytochemistry. 57 (10), 899–905, doi: 10.1369/jhc.2009.953612, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W et al. Fully automated 5-plex fluorescent immunohistochemistry with tyramide signal amplification and same species antibodies. Laboratory Investigation. 97 (7), 873–885, doi: 10.1038/labinvest.2017.37, (2017). [DOI] [PubMed] [Google Scholar]
- 12.Paul A & Laufer E Endogenous biotin as a marker of adrenocortical cells with steroidogenic potential. Molecular and Cellular Endocrinology. 336 (1–2), 133–140, doi: 10.1016/j.mce.2011.01.015, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bussolati G, Gugliotta P, Volante M, Pace M & Papotti M Retrieved endogenous biotin: a novel marker and a potential pitfall in diagnostic immunohistochemistry. Histopathology. 31 (5), 400–407, doi: 10.1046/j.1365-2559.1997.3020895.x, (1997). [DOI] [PubMed] [Google Scholar]
- 14.Wang H & Pevsner J Detection of endogenous biotin in various tissues: novel functions in the hippocampus and implications for its use in avidin-biotin technology. Cell and Tissue Research. 296 (3), 511–516, doi:DOI 10.1007/s004410051311, (1999). [DOI] [PubMed] [Google Scholar]