

Abstract
Background
The incidence of colorectal cancer (CRC), particularly left‐sided tumors (LT), in adolescents and young adults (AYA) is rising. Epigenetic events appear to play an important role in tumorigenesis and cancer progression, especially in younger patients. We compared molecular features of LT to right‐sided tumors (RT) in AYA.
Materials and Methods
A total of 246 LT and 56 RT were identified in a cohort of 612 AYA with primary CRC. Tumors were examined by next‐generation sequencing (NGS), protein expression, and gene amplification. Tumor mutational burden (TMB) and microsatellite instability (MSI) were determined based on NGS data.
Results
RT showed higher mutation rates compared with LT in several genes including BRAF (10.3% vs. 2.8%), KRAS (64.1% vs. 45.5%), PIK3CA (27% vs. 11.2%), and RNF43 (24.2% vs. 2.9%). Notably, additional mutations in distinct genes involved in histone modification and chromatin remodeling, as well as genes associated with DNA repair and cancer‐predisposing syndromes, were characteristic of RT; most frequently KMT2D (27.8% vs. 3.4%), ARID1A (53.3% vs. 21.4%), MSH6 (11.1% vs. 2.3%), MLH1 (10.5% vs. 2.3%), MSH2 (10.5% vs. 1.2%), POLE (5.9% vs. 0.6%), PTEN (10.8% vs. 2.3%), and BRCA1 (5.4% vs. 0.6%). MSI was seen in 20.8% of RT versus 4.8% of LT. RT had a higher frequency of TMB‐high regardless of MSI status.
Conclusion
Molecular profiling of AYA CRC revealed different molecular characteristics in RT versus LT. Epigenetic mechanisms and alteration in DNA repair genes warrant further investigation and may be a promising treatment target for CRC in AYA.
Implications for Practice
Colorectal cancer (CRC) in adolescents and young adults (AYA) comprises a distinct entity with different clinicopathologic features and prognosis compared with older patients. Molecular profiling of right‐ and left‐sided tumors in AYA is needed to gain novel insight into CRC biology and to tailor targeted treatment in this age group. This study found that right‐ and left‐sided CRC show distinct molecular features in AYA, overall and in subgroups based on microsatellite instability status. Alterations in DNA double‐strand break repair and homologous recombination repair, as well as epigenetic mechanisms, appear to play a critical role. The present molecular profiling data may support the development of personalized treatment strategies in the AYA population.
Keywords: Colorectal cancer, Adolescents and young adults, Tumor sidedness, Molecular profiling
Short abstract
This article explores the molecular makeup of left‐sided and right‐sided colorectal cancers in populations aged 40 years.
Introduction
Colorectal cancer (CRC) ranks third among the most frequent malignancies in both men and women, representing a leading cause of cancer‐related death worldwide 1. Incidence and mortality of CRC vary by age. Recent statistics show a decreasing incidence and mortality in adults aged ≥50 years, offset by an increase in the incidence of CRC in adults aged <50 years, the latter frequently occurring in the descending colon and rectum 2. The reasons behind the increasing CRC rates in the younger population, which are expected to increase further over the next decade, remain unclear 3.
When considering early onset CRC, the age definition of the group referred to as “adolescent and young adults” (AYA) is still debated 4. Age cutoffs vary widely among published studies; some authors recommend an upper limit of 50 years of age, based on historically recommended CRC screening guidelines in the average‐risk population, whereas others select patients below the age of 40 years based on physiological and pathological variables 5, 6, 7. In addition, data on outcomes of younger patients with CRC are inconsistent, with reports showing either a worse or a better outcome when compared with their older counterparts 5, 6, 7.
Hereditary syndromes such as Lynch syndrome (LS), characterized by germline mutations in one of the mismatch repair (MMR) genes, or familial adenomatous polyposis, caused by a germline mutation in the APC gene, are more common among AYA and account for up to 35% of early‐onset CRC 8, 9. Sporadic tumors, however, represent the majority of CRC in AYA 10. Notably, clinicopathologic and molecular features of CRC are different between AYA and older patients. AYA patients more often present with advanced‐stage disease (stage III or IV), and their tumors are likely to appear more histologically aggressive by way of mucinous or signet ring features and/or poor differentiation 11. Nodal involvement in early stage rectal cancer is also more frequent in patients under 50 years of age compared with older individuals 12. These clinicopathologic features are likely multifactorial and may highlight underlying molecular differences in CRC biology in different age groups. There is also a probability that both patients and practitioners consider alternate causes of symptoms in younger individuals, based on the understanding that CRC incidence is a function of older age, a pattern that could lead to a late cancer diagnosis and higher staged disease at diagnosis 13, 14. Available data from AYA compared with older adults show similar frequencies of TP53, APC, BRAF, and KRAS somatic mutations 15; higher rates of MYCBP2, BRCA2, PHLPP1, TOPORS, ATR, FBXW7, and POLE mutations 16, 17; increased frequency of CpG island methylator phenotype (CIMP)‐low 18; and LINE‐1 hypomethylation 19. Microsatellite instability (MSI) characterizes 20%–40% of early‐onset CRC and is mostly associated in younger patients (<30 years) with LS, identifying a subset of CRC with distinctive features and different prognosis and therapeutic implications 20.
Tumor sidedness has emerged as a prognostic and predictive biomarker in metastatic CRC (mCRC), with evidence of poorer outcomes in right‐sided mCRC and variable responses to biological therapy based on the site of origin of the tumor 21, 22, 23, 24. Comparative molecular analyses of right‐ and left‐sided CRC reveal molecular distinctions such as different mutation rates in TP53, KRAS, PIK3CA, and BRAF; distinct methylation patterns; and different MSI rates. These molecular variations likely contribute to distinct tumor phenotypes 25, 26, 27. Interestingly, the rates of left‐ and right‐sided CRC correlate with age, and there is a higher incidence of left‐sided CRC in patients younger than 50 years. In our recent study on the molecular characterization of left‐sided CRC in AYA versus older adults, higher mutation rates in genes associated with cancer syndromes were observed in AYA. There were also greater frequencies of MSI‐high (MSI‐H) status and high tumor mutational burden (TMB). Additionally, mutations in genes involved in histone modification were found to be significantly increased compared with older patients with CRC 28.
To the best of our knowledge, no dedicated, extensive gene mutational comparisons have been reported based on tumor location in AYA. Herein, we explore and characterize the molecular makeup of left‐ and right‐sided CRCs in populations under the age of 40.
Subjects, Materials, and Methods
Patients
Comprehensive genomic profiles of 612 consecutive AYA CRC analyzed between 2015 and 2017 were extracted from a database in which individuals had been deidentified (Caris Life Sciences, Phoenix, AZ). Samples were categorized based on primary tumor location as follows: tumors arising from the cecum to the hepatic flexure of the transverse colon were classified as right‐sided, and tumors arising from the splenic flexure to the rectum were classified as left‐sided. Tumors without any annotation on specific location (not otherwise specified [NOS]) were excluded from the analysis. Available clinical features recorded in the database included patients’ gender and age. No information was available regarding tumor stage.
Tumor tissue used in the analysis was formalin‐fixed and paraffin‐embedded (FFPE). Both specimen and tumor quality were confirmed by a board‐certified pathologist prior to multiplex testing. All molecular techniques met Clinical Laboratory Improvement Amendments and College of American Pathology standards.
Next‐Generation Sequencing
Direct sequencing was performed on genomic DNA isolated from FFPE tumor specimens using the whole exome NextSeq platform (La Jolla, CA). An Agilent SureSelect XT assay was used to enrich 592 whole‐gene targets. All reported variants were detected with >99% confidence at an average depth of at least 700X.
MSI by Next‐Generation Sequencing and Fragment Analysis
MSI was examined using over 7,000 target microsatellite loci and compared with the reference genome hg19 from the University of California, Santa Cruz Genome Browser database. The number of microsatellite loci that were altered by somatic insertion or deletion was counted for each sample. Only insertions or deletions that increased or decreased the number of repeats were considered. MSI–next‐generation sequencing (NGS) results were compared with results from over 2,000 matching clinical cases analyzed with traditional polymerase chain reaction–based methods. The threshold to determine MSI by NGS was determined to be 46 or more loci with insertions or deletions to generate a sensitivity of >95% and specificity of >99%.
Fragment analysis (FA) included fluorescence‐labeled primers (Promega, Madison, WI) for coamplification of seven biomarkers, including five mononucleotide repeat markers (BAT‐25, BAT‐26, NR‐21, NR‐24, and MONO‐27) and two pentanucleotide repeat markers (Penta C and D). A tumor specimen was considered MMR deficient (MMR‐d) if two or more mononucleotide repeats were abnormal. If one mononucleotide repeat was abnormal or repeats were identical between the tumor and adjacent normal tissue, then the tumor sample was considered mismatch repair proficient.
TMB by NGS
TMB was measured by counting all somatic nonsynonymous missense mutations found per tumor (592 genes and 1.4 megabases [Mb] sequenced per tumor). The threshold to define TMB‐high was ≥17 mutations per Mb and was established by comparing TMB with MSI by fragment analysis in CRC cases, based on reports of TMB having high concordance with MSI‐H in CRC 27. Values below 17 mutations per Mb were considered intermediate (7–16) or low (<7) and were grouped together for chi‐square analysis.
In Situ Hybridization Methods
Using automated staining (Benchmark XT, Ventana) and imaging (BioView, Billerca, MA) techniques, chromogenic in situ hybridization assessed ERBB2 (HER2/CEP17 [chromosome 17 centromere] probe) and MET (c‐MET/CEP7 probe; Abbott Molecular/Vysis, Abbott Park, IL) gene copy alterations. The ratio of gene to pericentromeric regions of chromosome 7 (MET) and 17 (HER2) was used to determine gene amplification. Cutoffs for amplification were determined as previously described 27.
Immunohistochemistry Analysis
Automated staining techniques (Benchmark XT, Ventana, Tucson, AZ; and AutostainerLink 48, Dako, Carpinteria, CA) and commercially available detection kits were performed on FFPE tumor specimens. Positive and negative controls were included in each analysis to ensure staining efficacy and consistency across batches. Threshold values for positive expression were optimized for each antibody according to the manufacturer's recommendations. Details on immunohistochemistry (IHC) interpretations have been described previously 27.
Statistical Analysis
Standard descriptive statistics were used for this retrospective analysis. Pearson's chi‐square test was used to obtain p values (IBM SPSS Statistics, Version 25.0., IBM, Armonk, NY). Only p values <.05 were considered statistically significant.
Results
Patient Demographics and Tumor Characteristics
Out of 612 available primary CRCs arising in patients younger than 40 years of age (AYA), tumor location was annotated in 302 cases. Overall, 246 left‐sided primary tumors (LT) and 56 right‐sided tumors (RT) were included in the analysis (Fig. 1). Looking at the distribution of LT, 42.6% had a rectal primary, 32.9% had a sigmoid primary, 13.4% were from the rectosigmoid colon, 5.7% were from the descending colon, 2.4% were from the splenic flexure, and the rest were LT with no specific location reported. RT distribution was as follows: 50.0% from the cecum, 30.4% from the ascending colon, 12.5% from the hepatic flexure, and the rest unspecified.
Figure 1.
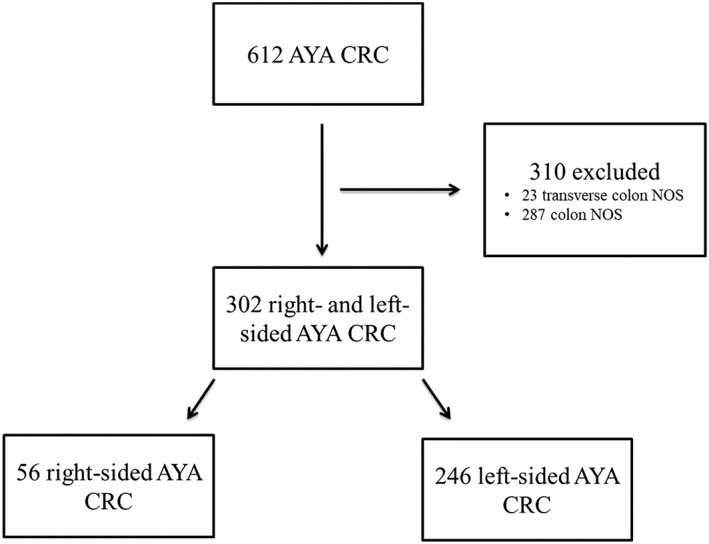
Study diagram. Flow chart showing the inclusion and exclusion of samples in the study.Abbreviations: AYA, adolescent and young adult; CRC, colorectal cancer; NOS, not otherwise specified.
Available patient characteristics were comparable in the LT and RT groups. Median age was 35 years (range, 18–40) for LT and 36 years (range, 18–40) for RT. In addition, gender distribution was balanced between the two cohorts, with female patients comprising 50% in the LT group and 47.1% in the RT cohort.
Mutational Profile via NGS in LT Versus RT
Pathogenic and presumed pathogenic mutation rates were compared between LT and RT AYA CRC (Figs. 2 and 3). LT had significantly higher mutation rates in APC (74.6% vs. 51.4%, p = .005) and TP53 (79% vs. 51.3%, p < .001). RT, in contrast, showed significantly higher mutation rates in BRAF (10.3% vs. 2.8%, p = .036), KRAS (64.1% vs. 45.5%, p = .035), PIK3CA (27% vs. 11.2%, p = .012), RNF43 (24.2% vs. 2.9%, p < .001), and several other genes. Additionally, some mutations were restricted to RT including CDH1 (7.9%), MRE11 (6.5%), SMAD2 (5.7%), and NOTCH1 (5.6%). Also found on RT were lone alterations in PRKDC, JAK1, POT1, AKT1, and EGFR.
Figure 2.
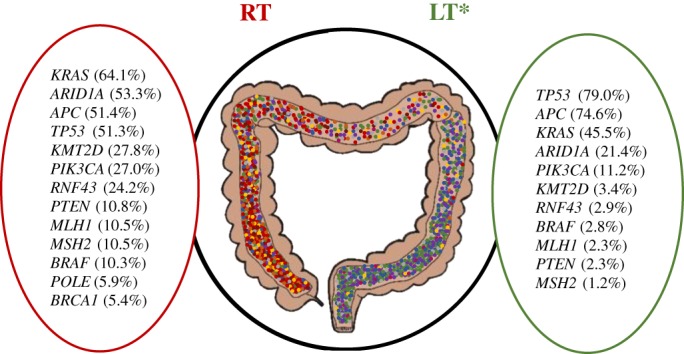
Main significant gene mutation rates observed between RT and LT in adolescent and young adult (AYA) colorectal cancer (CRC) using next‐generation sequencing (NGS). Summary of main significant differences (p < .05) in mutation rates by NGS in RT versus LT AYA CRC.*LT genes not mentioned here but highlighted for RT had a frequency < 1%. Abbreviations: LT, left‐sided tumor; RT, right‐sided tumor.
Figure 3.
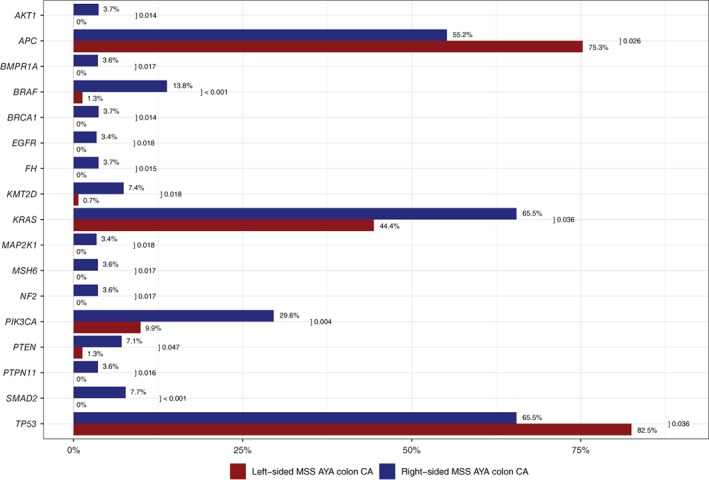
Mutation rates by next‐generation sequencing (NGS) in right‐sided tumors (RT) versus left‐sided tumors (LT) in adolescent and young adult (AYA) colorectal cancer (CRC). Complete listing of significant differences (p < .05) in mutations rates by NGS in RT versus LT AYA CRC.Abbreviation: CA, cancer.
Numerous genes associated with cancer‐predisposing syndromes, such as LS, juvenile polyposis syndrome (JPS), PTEN hamartoma tumor syndrome (PHTS), neurofibromatosis type 2 (NF2), and hereditary breast ovarian cancer (HBOC) syndrome, were found to have a significantly higher incidence of mutations in RT than LT: MSH6 (11.1% vs. 2.3%, p = .013), MLH1 (10.5% vs. 2.3%, p = .015), MSH2 (10.5% vs. 1.2%, p = .002), POLE (5.9% vs. 0.6%, p = .02, comprising two P286R mutations and one A465V mutation), PTEN (10.8% vs. 2.3%, p = .014), BMPR1A (5.3% vs. 0%, p = .002), BRCA1 (5.4% vs. 0.6%, p = .023), BAP1 (2.6% vs. 0%, p = .033), BRIP1 (3.2% vs. 0%, p = .022), NF2 (2.6% vs. 0%, p = .031), and MEN1 (2.6% vs. 0%, p = .032).
Furthermore, two genes responsible for histone modification and chromatin remodeling, respectively, were also significantly more commonly mutated in RT: KMT2D (27.8% vs. 3.4%, p < .001) and ARID1A (53.3% vs. 21.4%, p = .02).
As distal tumors are more prevalent in young patients with onset CRC, we investigated whether there were distinct properties associated with rectal cancers. In total, 105 rectal cancers were identified based on a conservative assessment (42.6% of LT, 105/246). The highest gene mutation rates were as follows: TP53, 80.8%; APC, 71.2%; KRAS, 41.9%; ARID1A, 20.0%; and FBXW7, 20.0%. With the exception of FBXW7, these gene mutation rates were comparable to those seen in the LT cohort as a whole. The incidence of MSI‐H and ERBB2 (HER2) amplification in rectal tumors, 4.8% and 4.1%, respectively, were also comparable to overall LT findings.
MSI via NGS or FA, TMB by NGS
Among tumors with available MSI status, 20.8% (10/48) of RT were MSI‐H compared with 4.8% (10/207) of LT (p = .05).
We further evaluated TMB according to primary site of origin in 206 tumors (36 RT and 170 LT; Fig. 4). TMB‐high was of greater frequency in RT compared with LT (30.6% vs. 6.5%, p < .001). A higher TMB rate was also observed in RT when restricting the analysis to microsatellite‐stable (MSS) tumors, albeit at lower rates (7.4% [2/27] vs. 1.2% [2/161], p = .04), supporting a higher TMB in RT compared with LT, independent of MSI status.
Figure 4.
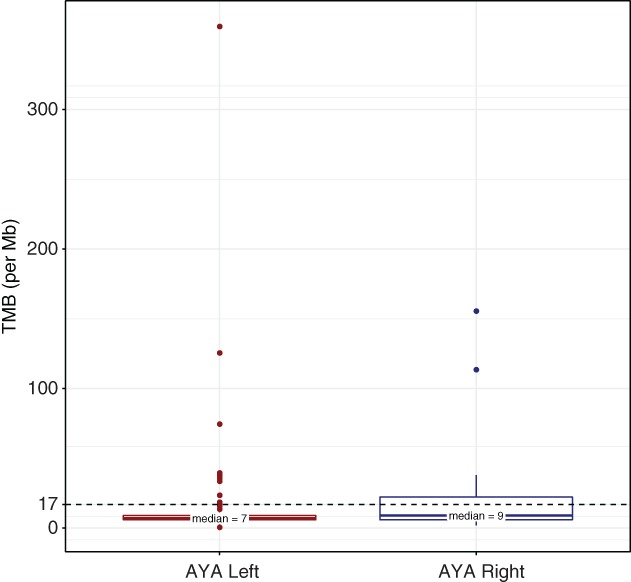
Tumor mutational burden in RT and LT AYA colorectal cancer (CRC). Comparison of TMB in RT and LT AYA CRC.Abbreviations: AYA, adolescent and young adult; Mb, megabase; TMB, tumor mutational burden.
Mutational Profile in MSS and MSI‐H Tumors
Considering the higher incidence of MMR‐d in AYA and the distinctive clinicopathological and molecular features of MSI‐H CRC, we conducted another comparison between RT and LT based on MSI status.
The highest mutational rates in the aggregated MSS group (n = 235), irrespective of tumor site of origin, were observed in TP53 (79.9%), APC (72.3%), KRAS (47.6%), ARID1A (17.1%), PIK3CA (12.7%), SMAD4 (12.2%), and FBXW7 (11%; Fig. 5). RT (n = 38) and LT (n = 197) showed significant differences in mutational rates comparable to findings in the global population such as APC, TP53, BRAF, KRAS, PIK3CA, SMAD2, AKT1, EGFR, PTEN, BRCA1, and NF2 (Fig. 6). Notably, no significant differences were observed in ARID1A mutations according to tumor side in the MSS group, whereas KMT2D mutations were still significantly higher in RT compared with LT but at a lower frequency (7.4% vs. 0.7%, p = .018).
Figure 5.
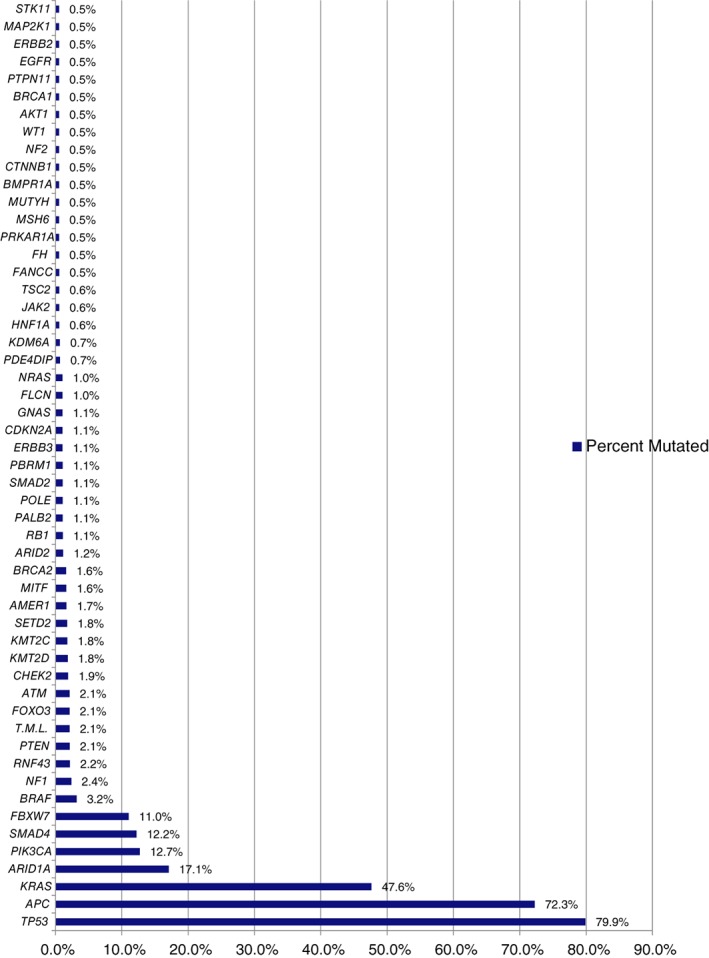
Global mutation rates in microsatellite‐stable adolescent and young adult colorectal cancer.
Figure 6.
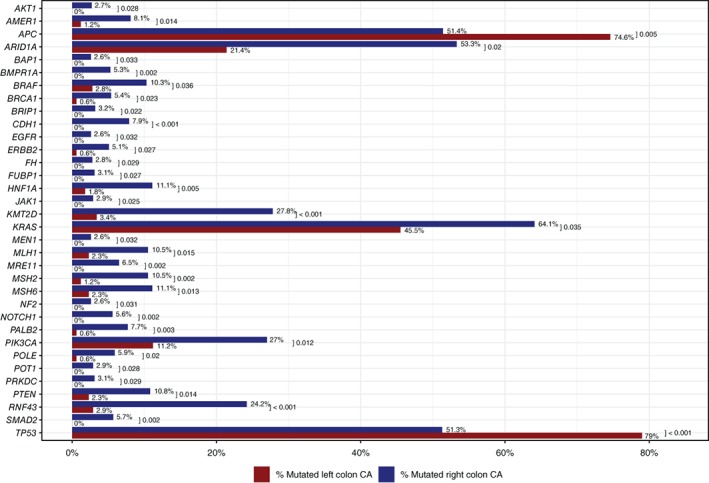
Mutation rates by next‐generation sequencing (NGS) in right‐sided tumor (RT) versus left‐sided tumor (LT) MSS AYA colorectal cancer (CRC). Significant differences in mutation rates by NGS in RT versus LT MSS AYA CRC.Abbreviations: AYA, adolescent and young adult; CA, cancer; MSS, microsatellite‐stable.
Comparison by tumor site of origin in the MSI‐H subgroup (n = 20, 10 LT and 10 RT) showed that RNF43 mutations were associated with RT, whereas SMARCA4 mutations with LT (p < .05). Mutations in KMT2D showed a rate of 100% (8/8) in RT compared with 50% (3/6) in LT (p = .024; Table 1).
Table 1.
Significant differences in RT versus LT MSI‐H AYA CRC
| Biomarker | Platform | Right‐sided MSI‐H CRC, n (%) | Left‐sided MSI‐H CRC, n (%) | p value |
|---|---|---|---|---|
| MGMT | IHC | 0/2 (0.0) | 2/2 (100) | .046 |
| KMT2D | NGS | 8/8 (100.0) | 3/6 (50.0) | .024 |
| RNF43 | NGS | 7/8 (87.5) | 2/8 (25.0) | .012 |
| SMARCA4 | NGS | 0/9 (0.0) | 3/6 (50.0) | .018 |
Abbreviations: CRC, colorectal cancer; IHC, immunohistochemistry; MSI‐H, microsatellite instability‐high; NGS, next‐generation sequencing.
Protein Expression and Gene Amplification
When examining protein expression by means of IHC, higher rates of protein loss of MLH1 (12.2% vs. 1.8% p = .001) and PMS2 (14.3% vs. 2.8%, p = .001) in RT compared with LT were noted, which is consistent with the higher mutation rate of MMR genes observed in RT. Additionally, RT showed a significantly higher rate of PTEN loss compared with LT, both in the global study population (48.0% vs. 30.1%, p = .016) and in the MSS group (45.7% vs. 28.8%).
RT showed a higher rate of copy number alterations compared with LT, although significant differences were only found in KRAS (5.6% [2/36] vs. 0.6% [1/170], p = .024), ERCC4 (2.8% [1/36] vs. 0.0% [0/169], p = .03), and MNX1 (2.8% [1/36] vs. 0.0% [0/170], p = .029). ERBB2 was amplified at a higher frequency in RT compared with LT (5.4% vs. 2.9%), although the difference was not statistically significant.
Finally, in the MSI‐H subgroup, MMR protein staining showed a lower expression of MLH1 in RT (40.0% vs. 70.0%, p = .178) and lower expression of MSH2 in LT (44.0% vs. 70.0%, p = .178), although not statistically significant.
Discussion
The rising prevalence of early onset CRC represents a global health issue. The increasing rates of CRC in AYA are primarily driven by distal tumors, yet the underlying etiology remains unknown. Because of their young age, AYA patients often receive more aggressive treatments, but this approach has shown no corresponding improvement in survival 13. Recent evidence has revealed distinct clinicopathological features and underlying molecular characteristics (e.g. methylation status, MSI, and somatic mutations) between CRC in AYA and older patients, highlighting the need for broader genomic profiling and a better understanding of tumor biology in younger patients. In our study, we show distinct mutational profiles characterizing RT and LT CRC in AYA, which may provide novel insights into tumor biology and hence inform the tailoring of treatment to fit this individual subgroup of patients.
Consistent with previous reports in unselected, age‐independent populations, differences in oncogenic drivers were observed between LT and RT in our younger population, with a markedly increased rate of oncogenic mutations in RT compared with LT. Specifically, LT had higher APC and TP53 mutations rates, whereas RT showed higher mutation rates in BRAF, KRAS, PIK3CA, and RNF43. Interestingly, AYA RT also showed a higher incidence of mutations in several cancer‐related genes such as SMAD2, PTEN, CDH1, AMER1 (encoding for a key regulator of the WNT pathway), NOTCH1, AKT1, EGFR, JAK1, and BMPR1A. These higher alteration rates suggest the critical role of specific pathways in the biology of AYA RT and highlight potentially actionable targets for the development of tailored treatments in this patient population. In addition, growing evidence is accumulating on the negative predictive impact of PIK3CA mutations, loss of PTEN function, and activating mutations of the MAPKs or PI3 K/AKT axis on anti‐epidermal growth factor (EGFR) drugs activity 29, 30, which may partially explain the observed absence of benefit from anti‐EGFRs in RAS wild‐type RT. Our results, in line with recent evidence, suggest caution in the use of anti‐EGFRs in AYA with RT, particularly as first‐line therapy.
Higher mutational rates were observed in DNA damage repair genes in RT compared with LT, including MRE11, PALB2, and PRKDC, along with BRCA1 and BAP1. Hence, alterations in the mechanisms of DNA double‐strand break and homologous recombination (HR) repair appear to contribute to RT carcinogenesis in AYA. Notably, inherited mutations in HR genes (i.e., ATM and PALB2), have recently been associated with a relative 60%–80% increase in the baseline risk of CRC 31. Hence, mutations in HR genes might represent actionable targets using novel therapies in CRC 32. Indeed, individuals with breast, ovarian, and prostate cancers with underlying germline mutations in canonical HR genes have been shown to derive significant benefit from PARP inhibitors (PARPi) and platinum‐based chemotherapy 33, 34, 35. In vitro studies have shown sensitivity of HR‐deficient CRC cell lines to these agents 36 and the clinical efficacy of PARPi in patients with CRC is under study in several trials (NCT00912743, NCT02305758, NCT01589419, and NCT02921256).
Furthermore, genes responsible for histone modification and chromatin remodeling, such as KMT2D and ARID1A, were more frequently mutated in RT compared with LT. Interestingly, in our previous analysis focused on the molecular characterization of left‐sided CRC in AYA versus older adults, we highlighted an increased incidence of mutations in genes involved in histone modification in AYA compared with older patients with CRC, including KDM5C, KMT2A, KMT2C, KMT2D, and SETD2 28. Hence, disruption of the histone modification pathway appears to be a distinctive feature of CRC in AYA and to be more prominent in RT in our population. Mechanisms of epigenetic regulation in CRC have recently been the focus of extensive research, and these findings may identify promising candidate targets for anticancer therapy. Indeed, multiple DNA methyltransferase and histone deacetylase inhibitors have been approved by the Food and Drug Administration for the treatment of hematologic malignancies, with several phase I–II trials underway to test the safety and efficacy of drugs targeting histone modifiers in solid tumors, including CRC 37.
We previously reported higher mutation rates in genes related to cancer‐predisposing syndromes such as BRCA2, MSH2, and TSC2 in the younger CRC population compared with older patients 28. In the current study, the highest incidence of mutations in these genes among AYA CRCs was observed in RT. Specifically, we observed higher mutation rates in MMR genes (MSH6, MLH1, and MSH2), POLE, PTEN, BMPR1A, BRCA1, BAP1, BRIP1, NF2, and MEN1. Germline mutations in these genes are known to be associated with hereditary cancer syndromes; for example, MMR genes with LS, PTEN with PHTS, BMPR1A with JPS, POLE with colorectal cancer 12, NF2 with neurofibromatosis type 2, MEN1 with multiple endocrine neoplasia type 1, BRCA1 and BRCA2 with HBOC, and BAP1 with Bap1 tumor predisposition syndrome. This may have relevant implications, not only in terms of tumor biology and potential targeted treatments but also in terms of genetic counseling and patient surveillance. Interestingly, POLE mutations and mutations in MMR genes, which are considered to be mutually exclusive 38, were more frequent in RT, although the rate of POLE mutations in our series was lower than that previously reported for AYA CRC (5.9% vs. 9.8%) 16. It is well known that MSI‐H status and POLE mutations, whether somatic or germline, are associated with a hypermutated cancer phenotype 39. As expected, in our series, the TMB‐high rate in RT was over fourfold greater than that seen in LT (30.6% vs. 6.5%, p < .001). This observation has relevant implications as TMB‐high status recently emerged as an independent prognostic biomarker 40 as well as a predictive biomarker for response to immunotherapy 41.
In our subanalysis of MSS tumors, results were comparable to findings in the whole study population, except for a lower absolute incidence of mutations in KMT2D along with a loss of significance of ARID1A mutation rates according to tumor side. These findings may imply a major role for epigenetic mechanisms in the biology of MSI‐H tumors compared with MSS tumors and are consistent with recent evidence highlighting the association of histone modifier gene mutations and ARID1A mutations with MSI status 42, 43.
Finally, we performed an exploratory analysis comparing the molecular profiles of a small cohort of AYA MSI‐H CRCs according to tumor sidedness. As expected, in our global population, the MSI‐H rate was significantly higher in RT compared with LT (20.8% vs. 4.8%), and IHC showed lower protein expression of MLH1 and PMS2 in RT compared with LT (p = .05). No significant differences were observed in MSH2 and MSH6 levels. However, when comparing right‐ and left‐sided MSI‐H tumors, we found a lower expression of MLH1 in RT and MSH2 in LT, although not statistically significant. Of note, the small absolute number of MSI‐H tumors in our series warrants some caution, making it hard to draw any significant conclusions. Therefore, further validation is needed.
Consistent with published evidence 44, 45, RNF43 mutations were more prevalent in MSI‐H tumors, and higher rates of RNF43 mutations were associated with RT. Notably, loss‐of‐function mutations in RNF43 have been shown to confer Wnt dependence and to be associated with enhanced sensitivity to porcupine inhibitors in preclinical cancer models 46, 47. Porcupine inhibitors are currently being developed to suppress paracrine Wnt‐driven growth in RNF43 mutant tumors, including BRAF mutant mCRC (NCT02278133, NCT01351103). Furthermore, dysregulated Wnt signaling has been linked to T‐cell exclusion in MSI‐H CRC tissue and resistance to immunotherapy 48, suggesting that Wnt signaling inhibitors may be used to reverse immune escape in immunotherapy‐resistant tumors. Among MSI‐H tumors, alterations in epigenetic genes showed a differential distribution; mutations in SMARCA4 (a gene involved in chromatin remodeling) were limited to LT, whereas the frequency of KMT2D mutations was greater in RT than LT (100% vs. 50%, p = .024), which has implications for histone modification. MSI‐H tumors represent a distinct entity in terms of clinicopathological features and treatment options. As noted, MSI is caused by a defect in the MMR pathway, which can derive from one or more mutations in the MMR genes (MLH1, PMS2, MSH2, MSH6, and EPCAM) as part of hereditary LS or from sporadic silencing of MLH1 by promoter methylation. The MSI‐H phenotype is associated with consensus molecular subtype 1, CIMP positive (CIMP+), and BRAF V600E mutation, the latter being restricted to sporadic MSI‐H. Of note, distinct patterns characterize germline and sporadic MSI‐H mCRC 20. One of the major groundbreaking advances in mCRC treatment in recent years has been the approval of immunotherapy for patients with MSI‐H tumors following evidence of efficacy of immune checkpoint inhibitors like pembrolizumab and nivolumab in this patient subset 49, 50. Although molecular profiling studies of MSI‐H CRC are limited, a recent study highlighted distinctive genetic alterations in MSI‐H/hypermutated tumors in comparison to MSS tumors, including BRAF V600E mutations, BRCA1 and 2 alterations, NTRK fusions, and enrichment of PIK3CA and PTEN oncogenic mutations 45. Our results address the need for a thorough characterization of MSI‐H tumors and appear to show potentially meaningful differences in underlying biological mechanisms and actionable targets according to primary tumor site in AYA. However, as noted, the small absolute number of MSI‐H tumors in our series hinders our ability to draw definitive conclusions, and further validation is needed. Nevertheless, our findings may suggest relevant implications for the treatment of MSI‐H CRC as we move toward the development of treatment combinations to overcome immune‐escape mechanisms. Indeed, epigenetic immunomodulation plays a key role in the ability of tumors to elude the host immune response. Several ongoing trials are testing immune checkpoint inhibitors combined with epigenetic targeted therapy, such as modifiers of histone acetylation and DNA methylation, to improve outcomes in patients with various cancer types 51.
Overall, in accordance with data from the adult population in The Cancer Genome Atlas (TCGA) data set 52 and our previously reported data 27, RT showed a significantly higher rate of oncogenic mutations compared with LT. Relative distribution of mutations between RT and LT in the core oncogenes and tumor suppressor genes, APC, TP53, BRAF, KRAS, PIK3CA, and RNF43, as well as MSI status, was consistent with previous findings and recent literature 45. However, the absolute frequency of mutations in our study varied compared with the TCGA adult population, an example being BRAF mutation incidence in AYA RT. In fact, in our study, similar BRAF mutation rates were observed in MSS RT compared with “unselected” RT in AYA (13.8% and 10.3%, respectively), suggesting that BRAF mutation rates are not influenced by MSI‐H status. These results contrast with previously reported mutation rates of 24%–25% for BRAF in right‐sided CRC (unselected for age or MSI status) and the known association between BRAF mutation and MSI‐H status 27, 52, 53. However, a previous study on 39 early‐onset CRCs (including patients younger than 45 years of age) found that sporadic tumors neither contained higher rates of BRAF mutations nor displayed a CIMP+ profile in comparison with tumors from older patients (aged >60 years), in both the MSI‐H and the MSS subgroups 54. According to the authors, these data suggest a distinctive carcinogenesis pathway of early‐onset CRC involving a subtype of the chromosomal instability pathway, without involvement of the methylator pathway and BRAF mutation. Further studies are warranted to verify this hypothesis. Furthermore, AYA CRCs were characterized by high mutation rates in histone modification and chromatin remodeling genes and genes related to cancer‐predisposing syndromes.
We acknowledge that there are several limitations to our study, including the retrospective nature of the analysis; the exclusion of a large number of samples due to the lack of annotation of primary tumor location; the heterogeneity of the study population unselected for tumor stage; the absence of clinical data associating our findings to outcomes; the lack of a DNA methylation evaluation, CMS classification, and gene expression data; and the lack of data on hereditary cancer syndromes (i.e. Lynch syndrome). Furthermore, the small absolute number of patients in the RT group and the MSI‐H subgroup warrants caution when interpreting results. Future validation of current findings and use of broader molecular profiling of AYA in prospective and larger cohorts are needed.
Conclusion
Our study highlights significant molecular differences in AYA CRC based on tumor sidedness. Distinct features emerged as characteristic of AYA CRC, such as high mutation rates in histone modification and chromatin remodeling genes, as well as in genes related to DNA repair and cancer‐predisposing syndromes, all of which showed a different distribution pattern across RT and LT and across MSS and MSI tumors. Although warranting further validation, our results provide supportive evidence for the development of novel, tailored, therapeutic strategies to improve outcomes in younger patients with CRC.
Author Contributions
Conception and design: Mohamed E. Salem, Francesca Battaglin
Development of methodology, collection and assembly of data: Mohamed E. Salem, Francesca Battaglin, David Arguello
Analysis and interpretation of data: Mohamed E. Salem, Francesca Battaglin, Richard M. Goldberg, Alberto Puccini, Anthony F. Shields, David Arguello, W. Michael Korn, John L. Marshall, Axel Grothey, Heinz‐Josef Lenz
Writing, review, and/or revision of the manuscript: Mohamed E. Salem, Francesca Battaglin, Richard M. Goldberg, Alberto Puccini, Anthony F. Shields, David Arguello, W. Michael Korn, John L. Marshall, Axel Grothey, Heinz‐Josef Lenz
Final approval of manuscript: Mohamed E. Salem, Francesca Battaglin, Richard M. Goldberg, Alberto Puccini, Anthony F. Shields, David Arguello, W. Michael Korn, John L. Marshall, Axel Grothey, Heinz‐Josef Lenz
Disclosures
Mohamed E. Salem: Caris Life Sciences (RF‐travel support); Richard M. Goldberg: Taiho, Novartis, Merck, Merck KGaA (C/A through SAB), Amgen (H); David Arguello: Caris Life Sciences (E); W. Michael Korn: Merck, Sharp & Dohme (C/A), Caris Life Sciences (OI); John L. Marshall: Caris Life Sciences (E‐served as CMO). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Acknowledgments
We thank all of the patients and physicians contributing to the establishment of the analyzed data set.
Human subjects were deidentified prior to this retrospective analysis, and accordingly, this study was exempt per the Western Institutional Review Board.
Use of standardized official symbols: in this study, HUGO (Human Genome Organization)‐approved official symbols for genes and gene products were used, all of which are described at http://www.genenames.org.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Fedewa SA, Anderson WF et al. Colorectal cancer incidence patterns in the United States, 1974‐2013. J Natl Cancer Inst 2017;109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bailey CE, Hu CY, You YN et al. Increasing disparities in the age‐related incidences of colon and rectal cancers in the United States, 1975‐2010. JAMA Surg 2015;150:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fidler MM, Gupta S, Soerjomataram I et al. Cancer incidence and mortality among young adults aged 20‐39 years worldwide in 2012: A population‐based study. Lancet Oncol 2017;18:1579–1589. [DOI] [PubMed] [Google Scholar]
- 5. Li Q, Zhuo C, Cai G et al. Pathological features and survival outcomes of young patients with operable colon cancer: Are they homogeneous? PLoS One 2014;9:e102004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heeke AC, Xiu J, Reddy SK et al. Molecular characterization of colorectal tumors in young patients compared with older patients and impact on outcome. J Clin Oncol 2016;34(suppl 4S):505a. [Google Scholar]
- 7. Abdelsattar ZM, Wong SL, Regenbogen SE et al. Colorectal cancer outcomes and treatment patterns in patients too young for average‐risk screening. Cancer 2016;122:929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pearlman R, Frankel WL, Swanson B et al.; Ohio Colorectal Cancer Prevention Initiative Study Group. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early‐onset colorectal cancer. JAMA Oncol 2017;3:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mork ME, You YN, Ying J et al. High prevalence of hereditary cancer syndromes in adolescents and young adults with colorectal cancer. J Clin Oncol 2015;33:3544–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stigliano V, Sanchez‐Mete L, Martayan A et al. Early‐onset colorectal cancer: A sporadic or inherited disease? World J Gastroenterol 2014;20:12420–12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang R, Wang MJ, Ping J. Clinicopathological features and survival outcomes of colorectal cancer in young versus elderly: A population‐based cohort study of SEER 9 registries data (1988‐2011). Medicine (Baltimore) 2015;94:e1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meyer JE, Cohen SJ, Ruth KJ et al. Young age increases risk of lymph node positivity in early‐stage rectal cancer. J Natl Cancer Inst 2016;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weinberg BA, Marshall JL, Salem ME. The growing challenge of young adults with colorectal cancer. Oncology (Williston Park) 2017;31:381–389. [PubMed] [Google Scholar]
- 14. Ballester V, Rashtak S, Boardman L. Clinical and molecular features of young‐onset colorectal cancer. World J Gastroenterol 2016;22:1736–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vatandoust S, Price TJ, Ullah S et al. Metastatic colorectal cancer in young adults: A study from the South Australian population‐based registry. Clin Colorectal Cancer 2016;15:32–36. [DOI] [PubMed] [Google Scholar]
- 16. Kothari N, Teer JK, Abbott AM et al. Increased incidence of FBXW7 and POLE proofreading domain mutations in young adult colorectal cancers. Cancer 2016;122:2828–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tricoli JV, Boardman LA, Patidar R et al. A mutational comparison of adult and adolescent and young adult (AYA) colon cancer. Cancer 2018;124:1070–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Magnani G, Furlan D, Sahnane N et al. Molecular features and methylation status in early onset (≤40 years) colorectal cancer: A population based, case‐control study. Gastroenterol Res Pract 2015;2015:132190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Antelo M, Balaguer F, Shia J et al. A high degree of LINE‐1 hypomethylation is a unique feature of early‐onset colorectal cancer. PLoS One 2012;7:e45357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cohen R, Buhard O, Cervera P et al. Clinical and molecular characterisation of hereditary and sporadic metastatic colorectal cancers harbouring microsatellite instability/DNA mismatch repair deficiency. Eur J Cancer 2017;86:266–274. [DOI] [PubMed] [Google Scholar]
- 21. Arnold D, Lueza B, Douillard JY et al. Prognostic and predictive value of primary tumour side in patients with RAS wild‐type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol 2017;28:1713–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stintzing S, Tejpar S, Gibbs P et al. Understanding the role of primary tumour localisation in colorectal cancer treatment and outcomes. Eur J Cancer 2017;84:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holch JW, Ricard I, Stintzing S et al. The relevance of primary tumour location in patients with metastatic colorectal cancer: A meta‐analysis of first‐line clinical trials. Eur J Cancer 2017;70:87–98. [DOI] [PubMed] [Google Scholar]
- 24. Boeckx N, Koukakis R, Op de Beeck K et al. Effect of primary tumor location on second‐ or later‐line treatment outcomes in patients with RAS wild‐type metastatic colorectal cancer and all treatment lines in patients with RAS mutations in four randomized panitumumab studies. Clin Colorectal Cancer 2018;17:170–178.e173. [DOI] [PubMed] [Google Scholar]
- 25. Hu W, Yang Y, Li X et al. Multi‐omics approach reveals distinct differences in left‐ and right‐sided colon cancer. Mol Cancer Res 2018;16:476–485. [DOI] [PubMed] [Google Scholar]
- 26. Lee MS, Menter DG, Kopetz S. Right versus left colon cancer biology: Integrating the consensus molecular subtypes. J Natl Compr Canc Netw 2017;15:411–419. [DOI] [PubMed] [Google Scholar]
- 27. Salem ME, Weinberg BA, Xiu J et al. Comparative molecular analyses of left‐sided colon, right‐sided colon, and rectal cancers. Oncotarget 2017;8:86356–86368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Puccini A, Lenz HJ, Marshall JL et al. Impact of patient age on molecular alterations of the left‐sided colorectal tumors. The Oncologist 2019;24:319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bertotti A, Papp E, Jones S et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015;526:263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cremolini C, Morano F, Moretto M et al. Dissecting primary resistance to anti‐EGFRs in RAS and BRAF wt metastatic colorectal cancer (mCRC): A case‐control study. J Clin Oncol 2017;35(suppl):11508a. [Google Scholar]
- 31. AlDubayan SH, Giannakis M, Moore ND et al. Inherited DNA‐repair defects in colorectal cancer. Am J Hum Genet 2018;102:401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jonsson P, Bandlamudi C, Cheng ML et al. Tumour lineage shapes BRCA‐mediated phenotypes. Nature 2019;571:576–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tutt A, Robson M, Garber JE et al. Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof‐of‐concept trial. Lancet 2010;376:235–244. [DOI] [PubMed] [Google Scholar]
- 34. Mateo J, Carreira S, Sandhu S et al. DNA‐repair defects and olaparib in metastatic prostate cancer. N Engl J Med 2015;373:1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fong PC, Boss DS, Yap TA et al. Inhibition of poly(ADP‐ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009;361:123–134. [DOI] [PubMed] [Google Scholar]
- 36. Wang C, Jette N, Moussienko D et al. ATM‐deficient colorectal cancer cells are sensitive to the PARP inhibitor olaparib. Transl Oncol 2017;10:190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang T, Lin C, Zhong LL et al. Targeting histone methylation for colorectal cancer. Therap Adv Gastroenterol 2017;10:114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Domingo E, Freeman‐Mills L, Rayner E et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol 2016;1:207–216. [DOI] [PubMed] [Google Scholar]
- 39. Briggs S, Tomlinson I. Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol 2013;230:148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Innocenti F, Ou FS, Zemla T et al. Somatic DNA mutations, MSI status, mutational load (ML): Association with overall survival (OS) in patients (pts) with metastatic colorectal cancer (mCRC) of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2017;35(suppl):3504a. [Google Scholar]
- 41. Goodman AM, Kato S, Bazhenova L et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 2017;16:2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sarshekeh AM, Loree JM, Manyam GC et al. The characteristics of ARID1A mutations in colorectal cancer. J Clin Oncol 2018;36(suppl):3595a. [Google Scholar]
- 43. Puccini A, Poorman K, Goldberg R et al. Molecular differences between colorectal cancers with mutations in histone modifiers genes vs wild‐type (WT) tumors. Ann Oncol 2018;29(suppl 8):1831PDa. [Google Scholar]
- 44. Giannakis M, Hodis E, Jasmine Mu X et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet 2014;46:1264–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yaeger R, Chatila WK, Lipsyc MD et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 2018;33:125–136.e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jiang X, Hao HX, Growney JD et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA 2013;110:12649–12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van de Wetering M, Francies HE, Francis JM et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015;161:933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grasso CS, Giannakis M, Wells DK et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov 2018;8:730–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Le DT, Durham JN, Smith KN et al. Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 2017;357:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Overman MJ, McDermott R, Leach JL et al. Nivolumab in patients with metastatic DNA mismatch repair‐deficient or microsatellite instability‐high colorectal cancer (CheckMate 142): An open‐label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dunn J, Rao S. Epigenetics and immunotherapy: The current state of play. Mol Immunol 2017;87:227–239. [DOI] [PubMed] [Google Scholar]
- 52. Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guinney J, Dienstmann R, Wang X et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kirzin S, Marisa L, Guimbaud R et al. Sporadic early‐onset colorectal cancer is a specific sub‐type of cancer: A morphological, molecular and genetics study. PLoS One 2014;9:e103159. [DOI] [PMC free article] [PubMed] [Google Scholar]