

Abstract
Background
Appendiceal cancers (ACs) are rare. The genomic landscape of ACs has not been well studied. The aim of this study was to confirm the feasibility of next‐generation sequencing (NGS) using circulating tumor DNA (ctDNA) in ACs and characterize common genomic alterations.
Materials and Methods
Molecular alterations in 372 plasma samples from 303 patients with AC using clinical‐grade NGS of ctDNA (Guardant360) across multiple institutions were evaluated. Test detects single nucleotide variants in 54–73 genes, copy number amplifications, fusions, and indels in selected genes.
Results
A total of 303 patients with AC were evaluated, of which 169 (56%) were female. Median age was 56.8 (25–83) years. ctDNA NGS testing was performed on 372 plasma samples; 48 patients had testing performed twice, 9 patients had testing performed three times, and 1 patient had testing performed four times. Genomic alterations were defined in 207 (n = 207/372, 55.6%) samples, and 288 alterations were identified excluding variants of uncertain significance and synonymous mutations. Alterations were identified in at least one sample from 184 patients; TP53‐associated genes (n = 71, 38.6%), KRAS (n = 33, 17.9%), APC (n = 14, 7.6%), EGFR (n = 12, 6.5%), BRAF (n = 11, 5.9%), NF1 (n = 10, 5.4%), MYC (n = 9, 4.9%), GNAS (n = 8, 4.3%), MET (n = 6, 3.3%), PIK3CA (n = 5, 2.7%), and ATM (n = 5, 2.7%). Other low‐frequency but clinically relevant genomic alterations were as follows: AR (n = 4, 2.2%), TERT (n = 4, 2.2%), ERBB2 (n = 4, 2.2%), SMAD4 (n = 3, 1.6%), CDK4 (n = 2, 1.1%), NRAS (n = 2, 1.1%), FGFR1 (n = 2, 1.1%), FGFR2 (n = 2, 1.1%), PTEN (n = 2, 1.1%), RB1 (n = 2, 1.1%), and CDK6, CDKN2A, BRCA1, BRCA2, JAK2, IDH2, MAPK, NTRK1, CDH1, ARID1A, and PDGFRA (n = 1, 0.5%).
Conclusion
Evaluation of ctDNA is feasible among patients with AC. The frequency of genomic alterations is similar to that previously reported in tissue NGS. Liquid biopsies are not invasive and can provide personalized options for targeted therapies in patients with AC.
Implications for Practice
The complexity of appendiceal cancer and its unique genomic characteristics suggest that customized combination therapy may be required for many patients. Theoretically, as more oncogenic pathways are discovered and more targeted therapies are approved, customized treatment based on the patient's unique molecular profile will lead to personalized care and improve patient outcomes. Liquid biopsies are noninvasive, cost‐effective, and promising methods that provide patients with access to personalized treatment.
Keywords: Appendiceal Cancer, Next‐generation sequencing, ctDNA
Short abstract
In this report, circulating tumor DNA (ctDNA) through next‐generation sequencing from patients with a diagnosis of appendiceal cancer across various histological subtypes was evaluated. The aim was to confirm the feasibility of next‐generation sequencing using ctDNA in low‐grade mucinous appendiceal cancer and to characterize common alterations in the genomic profile, with the goal of identifying molecular alterations leading to the identification of potential actionable targets and combinations.
Introduction
Appendiceal cancers (ACs) are rare and account for 0.5% of all gastrointestinal neoplasms 1. ACs comprise different histologies, of which neuroendocrine origin (65%) and mucinous and nonmucinous adenocarcinomas (20%) are the most common. Other histologies include goblet and ex‐goblet cell tumors, lymphomas, and mesenchymal sarcomas 1. Treatments for AC depend on grade and stage. Surgical resection or debulking is the standard therapy for low‐grade mucinous tumors. The role of systemic therapy (adjuvant, neoadjuvant, or palliative) is controversial, with several series demonstrating inferior outcome for chemotherapy‐treated patients with low‐grade tumors. For high‐grade tumors, chemotherapy is based on results of trials with colorectal cancer. The lack of prospective trials in ACs has contributed significantly to this controversy 2.
Historically, AC treatment decisions are extrapolated from colorectal carcinoma (CRC) because of similarities in location and pathogenic features. Published literature suggests that some pathways, including point mutations in the KRAS proto‐oncogene, mutations and/or deletions in the TP53 gene (chromosome 17p), truncating mutations or deletions in the adenomatous polyposis coli (APC) gene (chromosome 5q), and mutations in the beta‐catenin gene 3, 4, 5, 6, are common in both CRC and ACs. Given the rarity of the disease, its heterogeneous nature, and the absence of clinical trials and genomic data for ACs, it is important to evaluate common genomic alterations that these cancers carry because clear molecular differences exist between ACs and CRCs 7, 8, 9. A study conducted by Raghav et al. demonstrated that, compared with CRC, mutations in BRAF, EGFR, and c‐KIT are less frequent in AC, PI3K mutations occur with similar frequency, and KRAS mutations occur at a higher rate 9. The molecular profile supports the assumption that, despite their anatomic similarities, AC and CRC are two molecularly distinct tumor types 9. Consequently, therapy for appendiceal tumors extrapolated from CRC regimens is unjustified 8.
The role of genomic profiling in patients with AC to develop and implement matched targeted therapies 10, 11, 12, 13 has not been studied. The implementation of this approach in the clinical setting relies on the ability to biopsy tumors, perhaps multiple times, prior to the selection of new treatment regimens 14. Tissue biopsies are invasive, expensive, associated with potential complications, and may not be feasible in the setting of peritoneal spread of the disease 14. Spatial and temporal heterogeneity has been established between primary cancers and their metastatic lesions 15, 16, and thus primary tumor biopsies may not be the best source of material for genomic characterization of the disease 15, 17, 18, 19. Cancer molecular profile might change with time after exposure to different cytotoxic or targeted therapies 20, which translates to the need for repeated tumor sampling, which is not feasible 15, 17, 18, 19. All these challenges highlight the need for a method that is easily repeatable and minimally invasive such as next‐generation sequencing (NGS) of circulating tumor DNA (ctDNA) 21. ctDNA is secreted into the circulation by cancer cells, thus representing a source of tumor material representative of all disease sites. Blood testing for NGS presents a real‐time, easily accessible tool for the identification of molecular biomarkers 18, 19, 22. Because of the minimally invasive nature of a blood test, as opposed to a tissue biopsy, the reproducibility of liquid biopsies has been used for several proof‐of‐concept studies predicting response and resistance to treatment 23, 24, as well as prognosis and recurrence 25, 26, 27.
In this report, analysis of ctDNA through blood‐based Guardant360 NGS from patients with a diagnosis of AC across various histological subtypes was evaluated. The aim was to confirm the feasibility of NGS using ctDNA in low‐grade mucinous AC and characterize common alterations in the genomic profile. Furthermore, we aimed to identify whether the molecular alterations lead to the identification of potential actionable targets and combinations.
Subjects, Materials, and Methods
This was a retrospective review evaluating the molecular alterations in ctDNA samples from patients who had a diagnosis of AC and underwent Guardant360 clinical‐grade NGS across multiple institutions. Samples from patients with AC between the years 2014 and 2018 were analyzed. Patient‐specific covariates included sex and age. Ethical approval was not required for the study; patient identity protection was maintained throughout the study in a deidentified database through a data transfer agreement between Guardant Health and Emory University, and existing data were collected in accordance with the Emory University institutional review board guidelines. All the authors contributed to reviewing and approving the final manuscript. Some patients had samples analyzed more than once but at different times. Data regarding histologic subtypes of AC were unavailable.
Next‐Generation Sequencing
NGS of plasma ctDNA (liquid biopsies) was done by Guardant Health (Guardant360, http://www.guardanthealth.com/), a College of American Pathologists‐accredited and Clinical Laboratory Improvement Amendments‐certified laboratory. Next‐generation sequencing data were interpreted by N‐of‐One, Inc. (Lexington, MA; www.n‐of‐one.com). This is a highly analytically and clinically sensitive and specific test, able to detect single molecules of tumor DNA in 10 mL blood samples and ˃85% of single‐nucleotide polymorphisms found in tumors of patients with advanced cancers, with an analytic specificity of ˃99.9999% 28. At the time of this study, the test detected single nucleotide alterations (e.g., mutations, fusions, copy number change) in a panel of 54–73 genes. This panel detects point mutations (single nucleotide variations [SNVs]) in 73 genes, indels in 23 genes, amplifications in 18 genes, and fusions in 6 genes (supplemental online Table 1). Sequencing covered all cell‐free DNA, including germline found in the bloodstream (e.g., as a result of immune lysis), as well as the somatic ctDNA 28. Germline alterations were filtered out and not reported. Guardant Health uses an internal database, COSMIC v77, dbSNP build 147, and ExAC version 0.3.1 to call pathogenic or likely pathogenic somatic mutations. Gene amplifications were reported by absolute copy number detected in plasma, as compared with normal controls from healthy patients included in each run 28.
Results
Patient Demographics
Between the years 2014 and 2018, a total of 303 patients with ACs underwent Guardant360 testing using clinical‐grade NGS of ctDNA across multiple institutions, and 184 (61%) patients had at least one sample with alterations. The median age was 56.8 years (range, 25–83), with a female preponderance (56%). ctDNA NGS testing was performed on 372 plasma samples; 48 patients had testing performed twice, 9 patients had testing performed three times, and 1 patient four times. Genomic alterations were defined in 207 (n = 207/372, 55.6%) samples with a total of 288 alterations identified after excluding variants of uncertain significance and synonymous mutations.
Molecular Alterations
TP53 associated genes were most commonly altered (n = 71, 38.6%), followed by KRAS (n = 33, 17.9%), APC (n = 14, 7.6%), EGFR (n = 12, 6.5%), BRAF (n = 11, 5.9%), NF1 (n = 10, 5.4%), MYC (n = 9, 4.9%), GNAS (n = 8, 4.3%), MET (n = 6, 3.3%), PIK3CA (n = 5, 2.7%), and ATM (n = 5, 2.7%). Other genomic alterations of low frequency, but clinical relevance, included AR (n = 4, 2.2%), TERT (n = 4, 2.2%), ERBB2 (n = 4, 2.2%), SMAD4 (n = 3, 1.6%), CDK4 (n = 2, 1.1%), NRAS (n = 2, 1.1%), FGFR1 (n = 2, 1.1%), FGFR2 (n = 2, 1.1%), PTEN (n = 2, 1.1%), and RB1 (n = 2, 1.1%). Alterations in CDK6, CDKN2A, BRCA1, BRCA2, JAK2, IDH2, MAPK, NTRK1, CDH1, ARID1A, and PDGFRA were all reported once (n = 1, 0.5%; Fig. 1).
Figure 1.
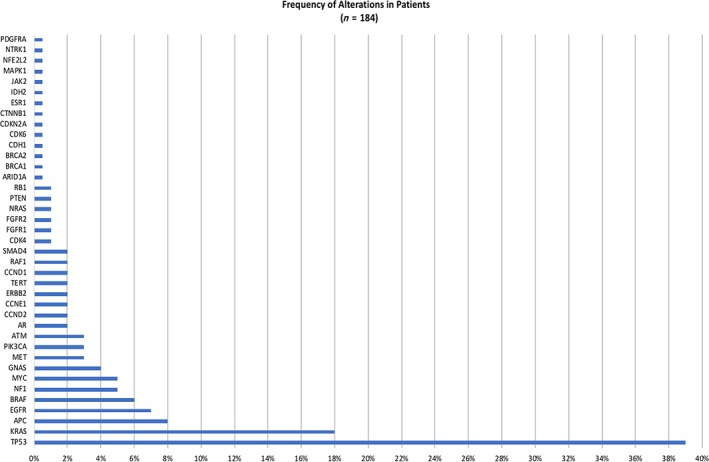
Prevalence of genomic alterations (variants of uncertain significance excluded).
The alterations seen in BRAF with their respective frequencies are as follows: V504V (1), D594G (1), AMP (3), N581S (1), G466V (1), and V600E (1). The alterations seen in KRAS with their respective frequencies are as follow: E63K (1), G13D (1), Q61H (2), G12D (2), D132I (1), G12V (4), AMP (2), and G12C (1).
Regarding the types of alterations identified, no fusions were found. Only one patient had an ERBB2 copy number variation (CNV) or amplification, as the other ERBB2 mutations identified were SNVs. Of the BRAF SNVs identified, all were activating, but only one was V600E. Four patients had MET amplifications (CNVs; Fig. 2). Table 2 summarizes potential actionable mutations in this analysis.
Figure 2.
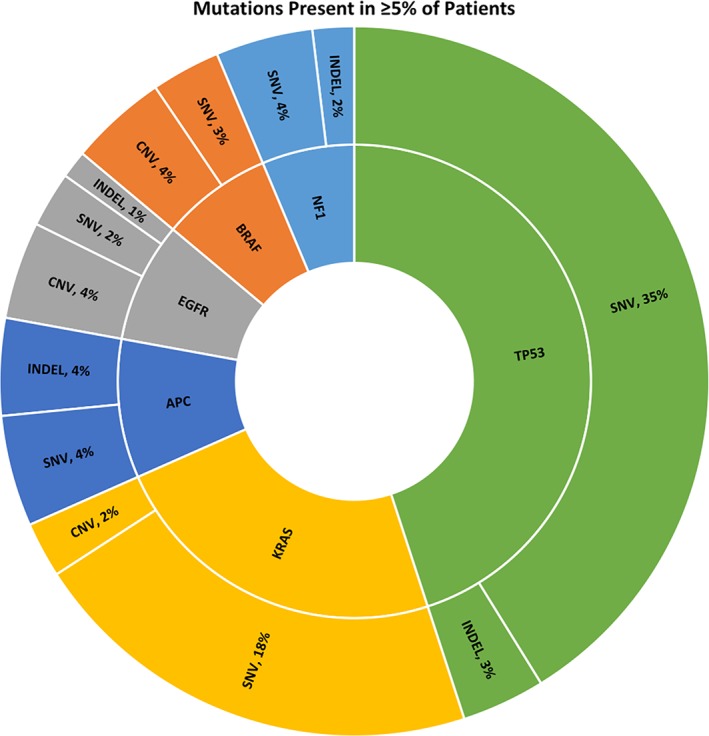
Types of alterations in the most commonly identified mutations (≥5% of patients, excluding variants of uncertain significance).Abbreviations: CNV, copy number variation; SNV, single nucleotide variation.
Table 2.
Frequency of actionable mutations
| Row labels | Count of gene |
|---|---|
| EGFR | 15 |
| NF1 | 13 |
| PIK3CA | 7 |
| ATM | 6 |
| MET | 6 |
| AR | 4 |
| ERBB2 | 4 |
| CDK4 | 2 |
| FGFR1 | 2 |
| FGFR2 | 2 |
| BRCA1 | 1 |
| BRCA2 | 1 |
| CDK6 | 1 |
| CDKN2A | 1 |
| IDH2 | 1 |
| JAK2 | 1 |
| NTRK1 | 1 |
| PDGFRA | 1 |
Alts prevalence (no variants of uncertain significance).
Plasma‐Derived ctDNA for Longitudinal Disease Monitoring
Among the 303 patients studied, 48 had testing performed twice, 9 had testing performed three times, and 1 patient had testing performed four times. By analyzing these longitudinal blood samples, we found that appendiceal tumors can gain new mutations over time that could potentially be targeted. With serial testing, we identified two patients that gained mutations in BRCA2, two patients that gained mutations in MTOR, two patients that gained mutations in ERBB2, one patient that gained a mutation in IDH2, and one patient that gained a mutation in FGFR3, which could all be targeted. Loss of mutations was identified in 11 of the 19 patients. These include CCND1, CCND2, PDGFRa (3), ERBB2, MAP2K2, APC, RET, AR, CDK4, IDH2, FGFR2, TP53 (2), and DDR2 (Table 1).
Table 1.
Longitudinal study of genomic alterations in repeated blood samples
| Patient number (n = 19) | Year | Gene | Alteration | Percentagea |
|---|---|---|---|---|
| 1 | June 2015 | KRAS | G12V | 1.65 |
| TP53 | V272L | 0.95 | ||
| April 2017 | BRCA2 | E1110K | 0.23266482 | |
| KRAS | G12V | 0.33588519 | ||
| NF1 | p.Val2259fs | 0.11896277 | ||
| V1453D | 0.55910126 | |||
| TP53 | V272L | 0.26401373 | ||
| 2 | May 2016 | NF1 | G629R | 0.31 |
| August 2016 | BRCA2 | L474L | 0.15 | |
| CCND2 | AMP | 0 | ||
| NF1 | G629R | 0.13 | ||
| PDGFRA | T223T | 0.19 | ||
| December 2016 | BRCA2 | L474L | 0.38428861 | |
| CCNE1 | NA | 0.329072 | ||
| MTOR | N161S | 0.23338677 | ||
| NF1 | G629R | 0.3031467 | ||
| 3 | March 2017 | ATM | D2708N | 3.1544311 |
| ERBB2 | I949T | 0.49880917 | ||
| April 2017 | ATM | D2708N | 1.6990995 | |
| 4 | September 2015 | ATM | R3008C | 0.23 |
| MAP2K2 | K68K | 0.23 | ||
| TP53 | R248W | 0.55 | ||
| November 2015 | PDGFRA | S851S | 0.23 | |
| TP53 | R248W | 0.44 | ||
| August 2017 | ATM | p.Gly3019fs | 0.176305 | |
| TP53 | R248W | 2.24065 | ||
| 5 | March 2016 | ARAF | P200L | 0.22 |
| NF1 | R1204W | 0.47 | ||
| PDGFRA | L261L | 0.36 | ||
| June 2017 | ARAF | P200L | 0.372306 | |
| NF1 | R1204W | 0.359565 | ||
| 6 | June 2016 | CCND1 | AMP | 0 |
| July 2016 | ERBB2 | T278T | 0.12 | |
| 7 | July 2016 | APC | H2532Y | 0.14 |
| ARID1A | P225L | 1.38 | ||
| TP53 | G245V | 0.47 | ||
| July 2016 | ARID1A | P225L | 1.14 | |
| CDH1 | L71L | 0.1 | ||
| TP53 | G245V | 0.96 | ||
| 8 | August 2016 | PDGFRA | R981H | 0.38 |
| August 2016 | PDGFRA | R981H | 0.22 | |
| 9 | September 2016 | TP53 | R273H | 0.24 |
| March 2017 | TP53 | G325 | 0.11297166 | |
| R273H | 0.32542678 | |||
| 10 | September 2016 | RET | G592E | 0.12 |
| October 2016 | ERBB2 | P525S | 0.19 | |
| NF1 | Q1341 | 0.15 | ||
| RB1 | S114L | 0.23 | ||
| 11 | October 2016 | AR | Y514C | 0.31 |
| CDK4 | A220A | 0.15 | ||
| IDH2 | R140Q | 0.19 | ||
| February 2017 | MTOR | L573L | 0.35728586 | |
| 12 | November 2016 | FGFR2 | P582H | 0.15 |
| November 2016 | RB1 | R661W | 0.10031596 | |
| 13 | November 2016 | APC | p.Thr1556fs | 0.37954538 |
| KRAS | G12A | 0.19769047 | ||
| December 2016 | TP53 | S241F | 0.17788795 | |
| January 2017 | IDH2 | R149W | 0.15263173 | |
| 14 | November 2016 | EGFR | L619L | 0.10810312 |
| p.Cys582fs | 0.09437097 | |||
| PIK3CA | G865S | 1.4246402 | ||
| TP53 | A159P | 1.3637159 | ||
| C242S | 0.21164862 | |||
| E258D | 0.44184776 | |||
| P152Q | 0.17850897 | |||
| V203M | 0.39819554 | |||
| Y205C | 0.12467888 | |||
| December 2016 | AR | R630Q | 0.30100751 | |
| EGFR | p.Cys582fs | 0.06360528 | ||
| FGFR3 | P300P | 0.09369497 | ||
| PIK3CA | G865S | 0.79638684 | ||
| TP53 | A159P | 1.2249438 | ||
| A159V | 0.10088058 | |||
| C242S | 0.21596307 | |||
| E258D | 0.19042433 | |||
| V203M | 0.25117232 | |||
| 15 | January 2017 | PTEN | N49S | 0.17563186 |
| June 2017 | EGFR | F795V | 0.15857818 | |
| PTEN | N49S | 0.2780047 | ||
| 16 | February 2017 | EGFR | A750E | 0.57628694 |
| March 2017 | EGFR | A750E | 0.42459659 | |
| 17 | June 2017 | APC | p.Glu1157fs | 0.36905797 |
| July 2017 | APC | p.Glu1157fs | 0.660704 | |
| 18 | August 2017 | KRAS | G12V | 0.988142 |
| TP53 | G245S | 0.657264 | ||
| November 2017 | KRAS | G12V | 0.215241 | |
| 19 | December 2017 | DDR2 | I798F | 0.168744 |
| EGFR | E967 | 0.113921 | ||
| March 2018 | EGFR | E967 | 0.526277 |
Percentage refers to the number of genomic alterations and/or variants found in a particular gene out of the total number of genomic alterations detected in 184 patients.
Correlation Between Age and Sex with Respect to NGS Results
Age and sex did not seem to correlate with mutation findings in this study. KRAS mutations occurred equally among men and women with a mean age of 53.5 years. Prevalence of BRAF mutations and ATM mutations were also similar between men and women with a mean age of 58.9 years and 51.7 years, respectively. In this study, BRCA1 and BRCA2 mutations were only seen in women with a mean age of 51 years and 60 years, respectively (supplemental online Table 2). These results need to be validated by a larger sample size in future studies to reach a statistically significant correlation.
Mutation Frequency by Histology
For this analysis, 63 of 303 patients had known histology per medical records included with the Guardant360 sample. These 63 patients had a total of 109 samples. The majority of patients had mucinous histology (52.4%), followed by adenocarcinoma (22.2%) and atypical goblet cell carcinoma (22.2%; Table 3). The number of mutation observations by gene was counted across all patients and stratified by histology (Fig. 3). Synonymous mutations, variants of uncertain significance, and genes with only one mutation across the cohort were excluded, and only alterations with potential functional and/or clinical significance were included. For patients with multiple samples, a given mutation was counted one time over the course of their sampling to include all samples in the analysis without inflating the mutation frequencies due to mutations that persist over time.
Table 3.
Mutation frequency by histology
| Histology | Samples, n (%) |
|---|---|
| Mucinous | 33 (52.4) |
| Adenocarcinoma | 14 (22.2) |
| Atypical GCC | 14 (22.2) |
| GCC | 1 (1.6) |
| NET typical | 1 (1.6) |
Abbreviations: GCC, goblet cell carcinoma; NET, neuroendocrine tumors.
Figure 3.
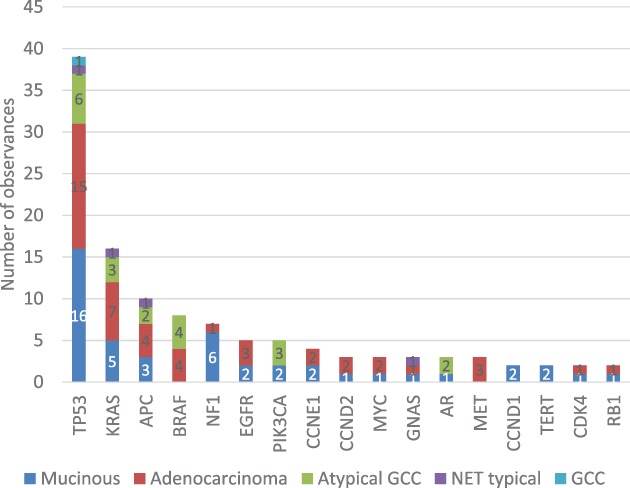
Mutation frequencies in genes with at least two observations across the cohort. Abbreviations: GCC, goblet cell carcinoma; NET, neuroendocrine tumors.
Discussion
The challenge for molecularly targeted personalized therapy in oncology, include the need for effective novel molecular tests 29 and potent targeted therapies. Extensive research demonstrates that detecting biomarker mutations regardless of cancer primary site allows for successful changes in treatments 2. Liquid biopsies offer several advantages for molecular testing, including feasibility of repeat testing, representation of all disease sites, low expense, and minimal invasiveness. The results of ctDNA analysis have led to further research in real‐time prognostics and drug‐cycling applications, such as the use of KRAS mutations for prognosis in advanced pancreatic cancer 30, serial ctDNA biopsies to track treatment‐conditional clonal evolution to cycle EGFR inhibitors in CRC 31, and cell‐free plasma exome sequencing to identify new pathways of acquired resistance to targeted therapeutics in cancers 23.
The present analysis is the first report exploring the genetic mutations in patients with AC using ctDNA derived from liquid biopsy. The genes commonly found to have characterized alterations in patients with AC included genes known to be commonly altered in ACs and CRC such as TP53, KRAS, APC, PIK3CA, and BRAF. The frequencies of genomic alterations in ctDNA were similar to those previously reported in tissue NGS 9 and those previously reported in a study by Riviere et al. evaluating 42 AC liquid biopsies 14. These findings confirm the feasibility and reliability of using ctDNA in profiling ACs.
In addition, although KRAS, APC, PIK3CA, and BRAF genes are commonly altered in CRC, the frequency of these alterations differ in ACs. Specific alterations, such as BRAF mutations present in 15% of CRCs, and are associated with poor prognosis 32. In this study, BRAF mutation was reported in 6% of the samples of ACs. In a study of 183 patients with AC analyzed by NGS, high mutation rates were observed in KRAS (55%), TP53 (40%), GNAS (31%), SMAD4 (16%), and APC (10%) 33. As opposed to our findings, the later study concluded that ACs exhibited higher mutation rates in KRAS and GNAS and lower mutation rates in TP53, APC, and PIK3CA (6%) than CRC 33. When compared with CRC 9, our findings suggest that KRAS mutation is much less prevalent in ACs, at a rate of 18% in this study. In addition, several unique genetic alterations were observed in our series: APC (7.6%), EGFR (6.5%), NF1 (5.4%), MYC (4.9%), GNAS (4.3%), PIK3CA (2.7%), MET (3.3%), and ATM (2.7%). Other genomic alterations found at low frequencies include AR (2.2%), TERT (2.2%), ERBB2 (2.2%), SMAD4 (1.6%), CDK4 (1.1%), FGFR1 (1.1%), FGFR2 (1.1%), PTEN (1.1%), and RB1 (1.1%). Alterations in CDK6, CDKN2A, BRCA1, BRCA2, JAK2, IDH2, NTRK1, CDH1, and PDGFRA were all reported once (0.5%). Some of these alterations are in clinical development as potential targets. These findings highlight the unique molecular profile of AC and necessity to research AC independent of CRC. In our population of patients with AC (n = 303), 21.2% (n = 61/288) of alterations identified could potentially be targeted by drugs approved for other cancers. This frequency is relatively high when compared with commonly profiled tumors, such as non‐small cell lung cancer or CRC. Examples of these mutations with therapeutic implications include EGFR, NF1, PIK3CA, MET, ATM, AR, ERBB2, CDK4, FGFR1, FGFR2, CDK6, CDKN2A, BRCA1, BRCA2, JAK2, IDH2, NTRK1 and PDGFRA (Table 2). The identification of these abnormalities would have justified using pan‐kinase, cyclin‐D, PARP, JAK25, and IDH inhibitors, which are not commonly used in ACs. The absence of standard evidence‐based therapies in ACs highlights the importance of genomic profiles in identifying therapeutic options for patients.
Repeat sampling is a unique advantage of liquid biopsies over tissue based assays. In this series, 48 patients had serial profiling of ctDNA. Analysis showed gain and loss of mutations with time. This could be related to type of therapy received. A limitation to this analysis is the unavailability of the exact treatments patients received. Some of the gained mutations are targetable via agents not commonly used in ACs. The gained mutations included BRCA2, mTOR, ERBB2, IDH2, and FGFR3. Age and sex do not seem to be correlated with specific mutations. Liquid biopsies are easy accessible and cost‐effective. Tissue biopsies are invasive and costly, with needle biopsies documented to cost over $10,000, based on a population‐based national Medicare sample 34. Furthermore, biopsy site could be a challenge to obtain a representative sample for tissue testing specifically with histologies like mucinous cancers, which are common in ACs. In lung cancer, liquid biopsies have become a standard diagnostic procedure for targetable treatments 35. In addition, it is currently established that there is molecular heterogeneity within the primary tumor and its metastatic tumors in the same patient 15 and even within the same tumor (depending on sampling area). Therefore, ctDNA results may reflect genomic aberrations in DNA shed from the primary site and metastatic sites 10.
The current study is the largest population‐based study that incorporates patients with AC who have undergone liquid biopsies. There are several limitations to our study inherent to all retrospective analyses. First, genomics data were obtained from a deidentified database and, hence, only limited clinical information was available. There were no data available regarding whether samples were obtained prior to or after medical treatment and/or surgery. There are no data to compare tissue genomics with liquid testing in this analysis. In addition, no survival data were available, and the data were limited by the coding of physicians at the different institutions. Another limitation is the different subtypes and histologies of AC, which are pathologically challenging to classify and entail different treatments. There is no standardized classification for mucinous ACs, and the challenge lies in pathologic classification of goblet and ex‐goblet cancers. For the samples analyzed in this study, histology coding was only available for 63 of 303 patients, and blood‐based test for microsatellite instability at the time of the analysis was not yet validated. Other mutations that are not accounted for in Guardant360 might be present. These mutations are under study regarding the optimization of detection through ctDNA. Despite these limitations, our findings have important implications. The complexity of AC and its unique genomic characteristics suggest that customized combination therapy may be required for many patients 14. Theoretically, as more oncogenic pathways are discovered and more targeted therapies are approved, customized treatment based on the patient's unique molecular profile will lead to personalized care and improve patient outcomes 2.
Conclusion
Evaluation of ctDNA was feasible and reliable among individuals with AC. The frequency of genomic alterations detected by ctDNA testing is similar to that previously reported in tissue NGS. Liquid biopsies are noninvasive, cost‐effective, and promising methods that provide patients with access to personalized treatment. Liquid biopsies merit investigation in prospective clinical trials, specifically in rare tumors such as AC in which traditional drug development paradigms are not feasible.
Author Contributions
Conception/design: Walid L. Shaib, Katerina Zakka
Provision of study material or patients: Ali Roberts
Collection and/or assembly of data: Walid L. Shaib, Katerina Zakka, Ali Roberts
Data analysis and interpretation: Walid L. Shaib, Katerina Zakka, Ali Roberts
Manuscript writing: Walid L. Shaib, Katerina Zakka, Ali Roberts
Final approval of manuscript: Walid L. Shaib, Charles Staley III, Katerina Zakka, Ali Roberts, Mehmet Akce, Christina Wu, Olatunji B. Alese, Bassel F. El‐Rayes
Disclosures
Ali Roberts: Guardant Health (E, OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Tables
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Shaib WL, Assi R, Shamseddine A et al. Appendiceal mucinous neoplasms: Diagnosis and management. The Oncologist 2017;22:1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Borazanci E, Millis SZ, Kimbrough J et al. Potential actionable targets in appendiceal cancer detected by immunohistochemistry, fluorescent in situ hybridization, and mutational analysis. J Gastrointestinal Oncol 2017;8:164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marra G, Boland CR. Hereditary nonpolyposis colorectal cancer: The syndrome, the genes, and historical perspectives. J Natl Cancer Inst 1995;87:1114–1125. [DOI] [PubMed] [Google Scholar]
- 4. Perucho M. Microsatellite instability: The mutator that mutates the other mutator. Nat Med 1996;2:630–631. [DOI] [PubMed] [Google Scholar]
- 5. Chung DC. The genetic basis of colorectal cancer: Insights into critical pathways of tumorigenesis. Gastroenterology 2000;119:854–865. [DOI] [PubMed] [Google Scholar]
- 6. Kabbani W, Houlihan PS, Luthra R et al. Mucinous and nonmucinous appendiceal adenocarcinomas: Different clinicopathological features but similar genetic alterations. Mod Pathol 2002;15:599–605. [DOI] [PubMed] [Google Scholar]
- 7. Jesinghaus M, Konukiewitz B, Foersch S et al. Appendiceal goblet cell carcinoids and adenocarcinomas ex‐goblet cell carcinoid are genetically distinct from primary colorectal‐type adenocarcinoma of the appendix. Mod Pathol 2018;31:829–839. [DOI] [PubMed] [Google Scholar]
- 8. Levine EA, Blazer DG 3rd, Kim MK et al. Gene expression profiling of peritoneal metastases from appendiceal and colon cancer demonstrates unique biologic signatures and predicts patient outcomes. J Am Coll Surg 2012;214:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raghav KP, Shetty AV, Kazmi SM et al. Impact of molecular alterations and targeted therapy in appendiceal adenocarcinomas. The Oncologist 2013;18:1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schwaederle M, Husain H, Fanta PT et al. Detection rate of actionable mutations in diverse cancers using a biopsy‐free (blood) circulating tumor cell DNA assay. Oncotarget 2016;7:9707–9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wheler JJ, Janku F, Naing A et al. Cancer therapy directed by comprehensive genomic profiling: A single center study. Cancer Res 2016;76:3690–3701. [DOI] [PubMed] [Google Scholar]
- 12. Tsimberidou AM, Iskander NG, Hong DS et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin Cancer Res 2012;18:6373–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Von Hoff DD, Stephenson JJ Jr, Rosen P et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 2010;28:4877–4883. [DOI] [PubMed] [Google Scholar]
- 14. Riviere P, Fanta PT, Ikeda S et al. The mutational landscape of gastrointestinal malignancies as reflected by circulating tumor DNA. Mol Cancer Ther 2018;17:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gerlinger M, Rowan AJ, Horswell S et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. New Engl J Med 2012;366:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yap TA, Gerlinger M, Futreal PA et al. Intratumor heterogeneity: Seeing the wood for the trees. Sci Transl Med 2012;4:127ps110. [DOI] [PubMed] [Google Scholar]
- 17. Martelotto LG, Ng CK, Piscuoglio S et al. Breast cancer intra‐tumor heterogeneity. Breast Cancer Res 2014;16:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bidard FC, Weigelt B Reis‐Filho JS. Going with the flow: From circulating tumor cells to DNA. Sci Transl Med 2013;5:207ps214. [DOI] [PubMed] [Google Scholar]
- 19. De Mattos‐Arruda L, Cortes J, Santarpia L et al. Circulating tumour cells and cell‐free DNA as tools for managing breast cancer. Nat Rev Clin Oncol 2013;10:377–389. [DOI] [PubMed] [Google Scholar]
- 20. Lee DH. Practical issues of biomarker‐assisted targeted therapy in precision medicine and immuno‐oncology era. ESMO Open 2018;3(suppl 1):e000370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Mattos‐Arruda, L , Weigelt B, Cortes J et al. Capturing intra‐tumor genetic heterogeneity by de novo mutation profiling of circulating cell‐free tumor DNA: A proof‐of‐principle. Ann Oncol 2014;25:1729–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parsons HA, Beaver JA, Park BH. Circulating Plasma Tumor DNA. Adv Exp Med Biol 2016;882:259–276. [DOI] [PubMed] [Google Scholar]
- 23. Murtaza M, Dawson SJ, Tsui DW et al. Non‐invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013;497:108–112. [DOI] [PubMed] [Google Scholar]
- 24. Xia S, Kohli M, Du M et al. Plasma genetic and genomic abnormalities predict treatment response and clinical outcome in advanced prostate cancer. Oncotarget 2015;6:16411–16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dawson SJ, Tsui DW, Murtaza M et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. New Engl J Med 2013;368:1199–1209. [DOI] [PubMed] [Google Scholar]
- 26. Shaw JA, Page K, Blighe K et al. Genomic analysis of circulating cell‐free DNA infers breast cancer dormancy. Genome Res 2012;22:220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goldstein DA, Elvin JA, Wang K et al. Comprehensive genomic profiling of cancer of the appendix to reveal new routes to targeted therapies. J Clin Oncol 2015;33:608a. [Google Scholar]
- 28. Lanman RB, Mortimer SA, Zill OA et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell‐free circulating tumor DNA. PloS One 2015. 10:e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lyman GH, Moses, HL Biomarker tests for molecularly targeted therapies—The key to unlocking precision medicine. New Engl J Med 2016;375:4–6. [DOI] [PubMed] [Google Scholar]
- 30. Tjensvoll K, Lapin M, Buhl T et al. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol 2016;10:635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Siravegna G, Mussolin B, Buscarino M et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med 2015. ;21 :827. [DOI] [PubMed] [Google Scholar]
- 32. Chen KH, Lin YL, Liau JY et al. BRAF mutation may have different prognostic implications in early‐ and late‐stage colorectal cancer. Med Oncol 2016;33:39. [DOI] [PubMed] [Google Scholar]
- 33. Tokunaga R, Xiu J, Johnston C et al. Molecular profiling of appendiceal adenocarcinoma and comparison with right‐sided and left‐sided colorectal cancer. Clin Cancer Res 2019;25:3096–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lokhandwala T, Bittoni MA, Dann RA et al. Costs of diagnostic assessment for lung cancer: A Medicare claims analysis. Clin Lung Cancer 2017;18:e27–e34. [DOI] [PubMed] [Google Scholar]
- 35. Liquid biopsy holds its own in NSCLC. Cancer Discov 2019;9:570–571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Tables