

Abstract
Ras proteins are small GTPases which regulate cellular proliferation, differentiation, and apoptosis. Constitutively active mutant Ras are expressed in ~15-20% human cancers, and K-Ras mutations account for ~85% of all Ras mutations. Despite the significance of Ras proteins in refractory cancers, there is no anti-Ras drug available in clinic. Since K-Ras must interact with the plasma membrane (PM) for biological activity, inhibition of the K-Ras/PM interaction is a tractable approach to block oncogenic K-Ras activity. Here, we discovered chalcones 1 and 8 exhibit anti-K-Ras activity, and show that the compounds mislocalize K-Ras from the PM and block oncogenic K-Ras signal output. Also, 1 inhibits the growth of K-Ras-driven human cancer cells. Our data suggest that 1 could be a promising starting point for developing anti-K-Ras cancer drug.
Keywords: K-Ras, Trimethoxy chalcone, Plasma membrane, Mislocalization, K-Ras-driven cancer
Graphical Abstract

Ras proteins are small, membrane-bound GTPases that oscillate between active GTP-bound and inactive GDP-bound states through interaction with guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs), respectively.1 Ras proteins stimulate multiple downstream effectors which regulate cell proliferation, differentiation, survival and apoptosis.2 Four Ras isoforms, K-Ras4A and K-Ras4B (both encoded by KRAS), as well as H-Ras and N-Ras are ubiquitously expressed in cells and must interact primarily with the inner leaflet of the plasma membrane (PM) for conducting signal transduction.2 For this requisite stable PM interaction, Ras undergoes a series of posttranslational modification at the C-terminal CAAX motif (where C= Cys, A = aliphatic amino acids, and X = Met or Ser). First, the Cys is farnesylated by a cytosolic farnesyltransferase, which allows Ras to bind to the cytosolic leaflet of the ER. RCE1 (Ras converting CAAX endopeptidase 1) then cleaves the AAX tripeptide, followed by the methylation of the now C-terminal farnesylated Cys by ICMT (isoprenylcysteine carboxyl methyltransferase).3-5 N-, H-, and K-Ras4A (the alternative splicing variant of K-Ras) are further modified with addition of palmitic acids on one or two other Cys near the farnesylated Cys, allowing Ras to interact with the PM. For K-Ras4B (hereafter referred to as K-Ras), a polybasic domain made of six lysine residues near the farnesylated Cys allows K-Ras to interact with anionic phospholipids in the PM through electrostatic interaction.6, 7
Constitutively active mutant Ras are found in ~15-20% of all human cancers.8 Of those, oncogenic mutant K-Ras is expressed in ~98% of pancreatic, 45% of colorectal, and 30% of all lung cancers, but as of yet there are no anti-K-Ras therapies available in clinic.9, 10 One approach for blocking oncogenic K-Ras activity is to dissociate K-Ras from the PM, since K-Ras must interact primarily with the PM.9-11 Removal of oncogenic K-Ras from the PM inhibits the growth of K-Ras-driven cancer cells in vitro and in vivo.12-14 Therefore, disrupting K-Ras/PM interaction is a tractable target for blocking oncogenic K-Ras activity.
Recently, we performed a cell-based high content screening to identify novel compounds capable of dissociating K-Ras from the PM. Identification and characterization of such compounds will provide deeper insights into K-Ras trafficking to and interaction with the PM, which might offer a starting point for the development of novel anti-K-Ras cancer therapies. Given the historical and ongoing challenges associated with anti-K-Ras drug discovery efforts, it is not surprising that there has been a strong reliance on the examination of large compound libraries. From a practical standpoint, hits obtained from such repositories might include a number of unrelated chemical classes, or complex core structures which may present no obvious starting-off point for library expansion by rational drug design. Moreover, since the target responsible for inducing disruption of K-Ras-PM localization is largely unknown, molecules of so-called privileged classes warrant attention as they afford the advantages of impacting a number of potentially relevant biological pathways along with the expected benefit of high hit rate per library size.15, 16
Thus, in lieu of such large exploratory compound libraries, it was reasoned that if an easily synthesized, structurally simple privileged scaffold were to be employed, upon identification of a putative pharmacophore on either ring, rapid diversification and structure activity relationships (SAR) might be easily developed about that core structure. The chalcone privileged scaffold (1,3-diaryl-2-propen-1-one),17 consisting of two aromatic rings A and B linked by a conjugated carbonyl system, served as an excellent starting point, allowing for a highly-optimizable class of compounds that boasts facile synthesis and a wide range of biological activities. Design considerations involved in the selection of a small exploratory panel for this study centered on the inclusion of functionalities shown to be of importance to the anticancer properties of chalcones, broadly defined.18 Of these, the trimethoxyphenyl motif is probably the most common pharmacophore investigated for anticancer properties, with the 3’,4’,5’-pattern on ring A notably associated with cytotoxic/antiproliferative effects arising from tubulin interaction.19,20 So as to minimize any possible confounds attributable to this mechanism of action in this initial screening set, it was decided to focus upon chalcones featuring the inverse substitution pattern, e.g., 3,4,5-trimethoxy on ring B (Table 1).
Table 1.
The IC50 and Emax values of chalcones synthesized from Scheme 1 for K-Ras dissociation from the PM
 |
|||||||
|---|---|---|---|---|---|---|---|
| Compound | X | R1 | R2 | IC50 (μM) | S.E.M. | Emax | S.E.M. |
| 1 | C-H | 4’-NO2 | 3,4,5-OCH3 | 7.01 | 0.92 | 0.57 | 0.04 |
| 2 | C-H | 4’-NH2 | 3,4,5-OCH3 | 9.17 | 3.12 | 0.54 | 0.03 |
| 3 | N | H | 3,4,5-OCH3 | 52.97 | 2.96 | 0.45 | 0.02 |
| 4 | C-H | 2’,6’-F2 | 3,4,5-OCH3 | 8.20 | 1.80 | 0.45 | 0.03 |
| 5 | C-H | 4’-F | 3,4,5-OCH3 | 15.90 | 8.23 | 0.39 | 0.02 |
| 6 | C-H | 4’-Br | 3,4,5-OCH3 | 24.64 | 2.39 | 0.47 | 0.02 |
| 7 | C-H | 4’-OCH3 | 3,4,5-OCH3 | 8.29 | 2.19 | 0.45 | 0.05 |
| 8 | C-H | 4’-N(CH3)2 | 3,4,5-OCH3 | 7.42 | 0.53 | 0.58 | 0.03 |
| 9 | C-H | 3’,4’,5’-OCH3 | 4-NO2 | 8.20 | 2.87 | 0.46 | 0.02 |
| 10 | C-H | 3’,4’,5’-OCH3 | 2,6-F2 | 7.49 | 2.69 | 0.45 | 0.01 |
IC50: 50% inhibitory concentration for K-RasG12V PM dissociation
Emax: Maximal effects elicited by the compounds
S.E.M.: Standard error of mean
Though generally less studied and regarded as having less cytotoxic potential when positioned on the B-ring,21 we were intrigued by the report that the 4’-nitro-3,4,5-trimethoxyphenyl derivative 122 exhibited anti-inflammatory, antioxidant and anticancer cancer properties.23 The nontoxic nature of this substrate, in addition to its multitarget potential, provided the impetus for the development of this initial flight of chalcones for the mislocalization of K-Ras. Moreover, to eliminate any conjecture that the strongly electron-withdrawing (EW) 4’-nitro group might be bio-reductively converted to the corresponding electron-donating (ED) 4’-amino derivative 2 under the screening conditions, this analogue was also included.24,25 To complete this library based upon the 3,4,5-trimethoxy pattern on ring B, most of the remaining compounds in this study included functionalities running the gamut of EW (-F, -Br) to ED (-OCH3, -N(CH3)2).19-26-28 Notable exceptions entail the 2-azachalcone derivative 3,29 which was included due to the electron deficient nature of the pyridine ring as well as to identify any effects which might be attributable to metal binding by chelation to the proximate ring nitrogen and carbonyl group of the 2-pyridyl ketone. Additionally, as many of the biological effects of chalcones have been ascribed to the Michael-acceptor properties of the α,β-unsaturated carbonyl system linking the two aromatic rings, the 2,6-difluoro moieties of compounds 4 and 10 were designed for the putative enhanced acceptor abilities due to orthogonality induced by such bis-ortho substituents.30 Finally, although not strictly axiomatic, it has often been noticed that so-called “reverse chalcones” wherein the carbonyl and ethylene groups are interchanged, display similar biological activities as the original. Thus, the 3’,4’,5’-ring A analog of compound 1 (compound 9)19 was included, especially as this substrate was apparently devoid of tubulin binding effects.31
Chalcones 1-10 were prepared by base-catalyzed Claisen-Schmidt condensation utilizing commercially available benzaldehydes and aryl methyl ketones with ethanol as the solvent and aqueous NaOH (10%) as the base (Scheme 1). Characterization of compounds were accomplished by GC/MS and 1H and 13C NMR analysis with acceptable purities >96%. The conjugated carbonyl system of chalcones was verified to be the trans-isomers in all cases by 1H NMR wherein two doublets with coupling constants from 15.5-16.1 Hz were observed. Melting points and NMR spectra of known compounds were compared to those previously published in literature; however, due to questionable NMR data existing in some previous publications, all characterization data and full spectra are provided in the Supplementary Data. To the best of our knowledge, this is the first time chalcones 4 and 10 have been reported in literature, and their identities were verified by high-resolution mass spectrometry.
Scheme 1. Reagents: ethanol, aqueous NaOH (10%).
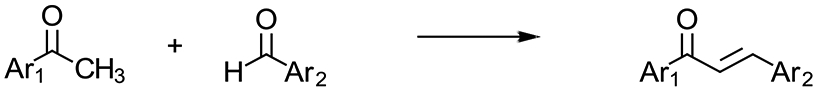
To examine the effect of chalcone compounds on K-Ras interaction with the plasma membrane (PM), we performed quantitative confocal microscopy. Madin-Darby kidney (MDCK) cells stably co-expressing mGFP-tagged oncogenic mutant K-Ras (K-RasG12) and mCherry-CAAX, a generic endomembrane marker32,33 were treated with different concentrations of chalcone compounds for 48h, and cells were fixed and imaged by a confocal microscope. To quantitate the extent of K-RasG12V dissociation from the PM, we used Manders coefficient, which calculates the fraction of mCherry-CAAX colocalizing with mGFP-K-RasG12V. The greater the value of the Manders coefficient, the more extensive the displacement of K-RasG12V from the PM.33,34 The potency (IC50) and efficacy (Emax) of each compound were further derived from the Manders coefficient values. Our data show that compounds 1 and 8 are most potent and effective at dissociating K-Ras from the PM, indicated by the lowest IC50 with the highest Emax values, respectively (Table 1 and Figure 1A and B). These data suggest that 3,4,5-trimethoxy moiety on the B ring of chalcones might represent a possible pharmacophore for K-Ras PM mislocalization. Though further work remains to establish a reliable SAR, it is interesting to note that the trends observed roughly parallel those reported in the publication that inspired the choice of the prototype compound 1.23 For instance, while the most active analogue in both instances possess an EW 4’-nitro substituent, the next most active entity had a strongly ED substituent at that site (e.g., dimethylamino derivative 8 here but ─OCH3 in the Bandgar publication).23 The lower activity of the 4’-amino analogue 2 relative to 1 suggests that the biological activity of the 4’-nitro derivative is not an artifact due to adventitious reduction under the conditions of the assay. In the case of halogens at the C4’-position, a slight size effect is noticeable in both cases in that the smaller fluoro substituent is more active than the corresponding bromo atom in this study, whereas in the earlier publication a chloro group was found superior to a bromo atom.23 These comparisons are not meant to imply any similarity in mechanism of action or cancer type, but merely to note possible trends in the anticancer properties of 3,4,5-trimethoxyphenyl chalcones.
Figure 1. 1 and 8 are most potent and effective at dissociating K-Ras from the PM.
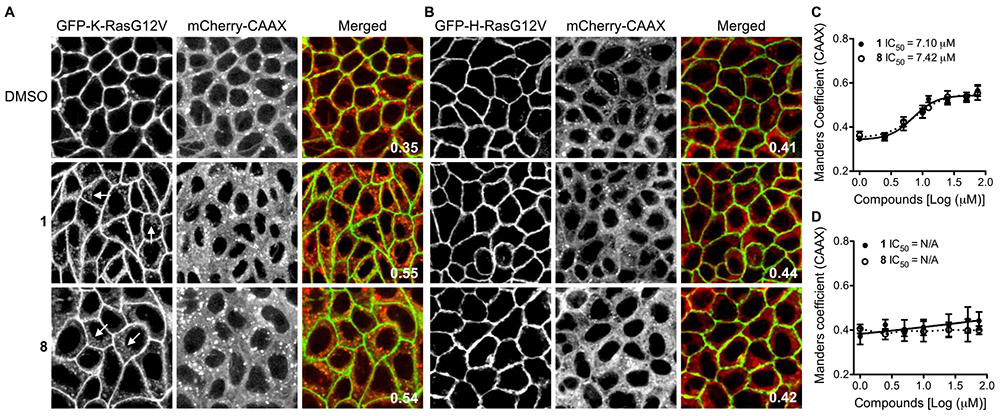
MDCK cells stably co-expressing GFP-K-RasG12V or -H-RasG12V with mCherry-CAAX were treated with chalcone compounds for 48h. Cells were fixed with 4% paraformaldehyde (PFA) and imaged by confocal microscopy. (A and B) Representative images are shown after treatment with 50 μM of 1 or 8. GFP-RasG12V dissociated from the PM is indicated by arrowheads. Values in merged panels represent the fraction of mCherry-CAAX co-localized with GFP-RasG12V calculated by Manders coefficient. (C and D) IC50 of 1 and 8 for K-RasG12V and H-RasG12V were estimated from the dose-response plots from three independent experiments.
To examine the effect of substrates 1 and 8 on the PM interaction of other Ras isoforms, MDCK cells stably co-expressing mGFP-tagged oncogenic H-Ras (H-RasG12V) and mCherry-CAAX were treated with 1 or 8 for 48h, and cells were fixed and imaged using a confocal microscope. Our data show that 1 and 8 did not mislocalize H-RasG12V from the PM, suggesting the effects of these compounds are K-Ras-specific (Fig. 1C and D).
Since dissociation of Ras from the PM blocks Ras signal transduction,33-36 we further examined the effect of 1 on oncogenic Ras signal output. MDCK cells stably expressing mGFP-K-RasG12V or -H-RasG12V were treated with 1 for 48h, and cell lysates were immunoblotted with anti-phospho-ERK and Akt (Ser473) antibodies. The levels of phosphorylated ERK and Akt are measured to study Ras activity since active Ras induces phosphorylation of ERK and Akt.2 Our data show that 1 significantly reduced the phosphorylation of ERK and Akt in K-RasG12V-expressing cells, but not in H-RasG12V-expressing cells (Fig. 2A - D). Together with confocal microscopy data, our data suggest that 1 selectively dissociates the PM interaction and signal transduction of oncogenic mutant K-Ras.
Figure 2. Compound 1 inhibits oncogenic K-Ras signaling and growth of K-Ras-driven cancers.

MDCK cells stably expressing GFP-K-RasG12V or -H-RasG12V were treated with 1 for 48h, and cell lysates were immunoblotted with anti-ppERK or -pAkt (S473) antibodies. (A and B) Representative blots from three independent experiments are shown. Total ERK and Akt were used as loading controls. (C and D) The graphs show the means ± S.E.M. from three independent experiments. Significant differences between control (DMSO-treated) cells and 1-treated cells were assessed using a one-way ANOVA test (* p<0.05; N.S. – not significant). A panel of pancreatic ductal adenocarcinoma (PDAC) (E) and non-small cell lung cancer cells (NSCLC) (F) were plated on a 96-well plate and treated with various concentrations of 1 for 4 days. Complete growth medium with the drug was replaced every 24h. Cell proliferation was analyzed using CyQuant proliferation assay kit. The graph shows the mean cell proliferation ± S.E.M. from three independent experiments relative to that for the control cells (DMSO-treated). Cell lines expressing oncogenic mutant K-Ras or wild-type K-Ras are shown in red and black, respectively. (G and H) The graphs represent the mean cell proliferation ± S.E.M. relative to that for the control cells (DMSO-treated) after treatment with 50 μM for 4 days. Open and closed bars represent cancer cells expressing wild-type K-Ras and oncogenic K-Ras, respectively. Significant differences between 1-treated and control cells were assessed using Mann-Whitney U test (* p<0.05, ** p<0.001, N.S. – not significant).
To further characterize the effect of 1 on oncogenic K-Ras signaling, we performed cell proliferation assay on human pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung cancer (NSCLC) cells harboring oncogenic mutant K-Ras. For control cells, we used human cancer cells expressing wild-type (WT) K-Ras (BxPC3 for PDAC, and H1299, H1975 and H522 for NSCLC). Our data show that 1 significantly inhibited the growth of PDAC cells expressing oncogenic K-Ras (Fig. 2F and H). For NSCLC cells, 1 had a greater inhibitory activity on H441 and H358 cells over cells expressing WT K-Ras (Fig. 2E and G). A549 however, showed no growth inhibition, albeit it expressed oncogenic mutant K-Ras. The growth of cancer cells expressing WT K-Ras was not inhibited by 1 (Fig. 2E - H). Previous studies reported that growth of H441 and H358, but not A549 cells, and PDAC cells harboring oncogenic mutant K-Ras tested in our experiment is highly dependent on oncogenic K-Ras activity, a phenomenon called K-Ras addiction.37, 38 Taken together, our data suggest that 1 inhibits growth of K-Ras-addicted human cancer cells.
Protein kinase C (PKC) can phosphorylate K-Ras at Ser181, and the phosphorylated K-Ras is redistributed from the PM to other cellular membranes.39 To examine whether 1 dissociates K-Ras from the PM through activating PKC, we measured the level of phosphorylated myristoylated alanine-rich C-kinase substrate (MARCKS), a PKC substrate. A NSCLC cell line, H358, of which growth was highly sensitive to 1 (Fig. 2F and H) or MDCK cells stably expressing GFP-K-RasG12V were treated with various concentrations of 1 and phosphorylated MARCKS, an indicator of PKC activity was measured. Our data show that 1 increases MARCKS phosphorylation in a dose-dependent manner (Fig. 3). To further validate our data, we mutated K-Ras Ser181 residue to Ala (K-RasG12V S181A), which is insensitive to PKC-mediated phosphorylation, and examined the effect of 1 in the PM localization of K-RasG12V S181A. Our data show that K-RasG12V S181A is not dissociated from the PM after 1 treatment, suggesting 1 dissociates K-Ras from the PM through phosphorylating Ser181 residue. Taken together, our data suggest that 1 dissociates oncogenic K-Ras from the PM by PKC-mediated phosphorylation at Ser181. This in turn, results in inhibition of oncogenic K-Ras activity and the growth of K-Ras-addicted human cancer cells.
Figure 3. 1 dissociates K-Ras from the PM through PKC-mediated K-Ras phosphorylation.

H358, a NSCLC cell line (A) or MDCK cells stably expressing GFP-K-RasG12V (B) were treated with various concentrations of 1 for 48h, and cell lysates were immunoblotted for phosphorylated MARCKS (p-MARCKS) and quantified (means ± S.E.M.). Representative blots from three independent experiments are shown. An GAPDH blots were used as loading controls. (C) MDCK cells co-expressing GFP-K-RasG12V S181A and mCherry-CAAX were treated with 50μM 1 for 48h and images by confocal microscopy. Representative images of GFP-K-RasG12V S181A from three independent experiments are shown. Inserted values represent the fraction of mCherry-CAAX co-localized with GFP-K-RasG12V S181A calculated by Manders coefficient.
KRAS is one of the top ten genes mutated in human cancers harboring loss-of-function mutations for seven of the PKC isozymes,40 suggesting that PKC may suppress oncogenic K-Ras signaling such that loss of PKC would be required for K-Ras to exert its full oncogenic potential.40 PKC isozymes comprise three classes: conventional (α, β, γ), novel (δ, ε, η, θ), and atypical (ζ, ι). Several studies have reported that loss of PKCδ is found in cancers harboring oncogenic K-Ras. PKCδ protein levels are lower in endometrial cancer cells harboring oncogenic K-Ras than that of wild-type K-Ras.41 Also, total PKC activity is significantly lower in human colorectal cancers compared to normal mucosa because of decreased PKCβ and PKCδ.42 Moreover, approximately 40% of PKCδ loss-of-function mutations found in a large panel of human cancers are in pancreatic cancers,40 and patients with lung adenocarcinomas harboring oncogenic mutant K-Ras show an increased overall survival rate when they also have higher PKCδ mRNA levels.43 These studies suggest that PKCδ may have an anti-cancer activity in cancers expressing oncogenic mutant K-Ras. Taking these studies together with our data, we propose that 1 has anti-K-Ras activity through stimulating PKCδ. The exact molecular mechanism of 1-mediated PKCδ stimulation needs to be further elucidated.
A previous study demonstrated the effect of chalcones on cancer cell lines harboring oncogenic K-Ras, where a class of indolyl-tetralone chalcones induced apoptosis of A549 by cell cycle blockage.44 Although the two sets of compounds are not strictly comparable, it is interesting to note that a chalcone with the 3,4,5-trimethoxy motif on ring B lacked any inhibition in that study. Here, we show that compound 1 inhibits the growth of K-Ras-addicted human cancers, but not A549, which does require oncogenic mutant K-Ras activity for its growth.37 Taken together, we propose that unlike the indolyl-tetralone chalcones, our compounds are specifically targeting oncogenic K-Ras, and that the mechanism of cell death induced by indolyl-tetralone chalcones is likely independent of K-Ras inhibition. Therefore, we can conclude that chalcones with the 3,4,5-trimethoxy moiety are a novel class of compounds for oncogenic mutant K-Ras.
In summary, we identified that chalcone-based compounds 1 and 8 selectively dissociate oncogenic K-Ras from the PM through PKC-mediated phosphorylation. Upon further investigation, we determined 1 capable of inhibiting oncogenic K-Ras signal output, as well as selectively inhibiting the growth of K-Ras-addicted cancers. Our data suggest that 1 could be a great starting point to develop anti-K-Ras therapies. Future work will include thoroughly exploring the efficacy of the trimethoxy pharmacophore towards the association of oncogenic K-Ras and the PM in greater detail.
Supplementary Material
Acknowledgments
K-J.C wishes to acknowledge the National Cancer Institute [R00-CA188593].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294(5545): 1299–1304. [DOI] [PubMed] [Google Scholar]
- 2.Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4(5): 373–384. [DOI] [PubMed] [Google Scholar]
- 3.Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57(7): 1167–1177. [DOI] [PubMed] [Google Scholar]
- 4.Gutierrez L, Magee AI, Marshall CJ, Hancock JF. Post-translational processing of p21ras is two-step and involves carboxyl-methylation and carboxy-terminal proteolysis. EMBO J. 1989;8(4): 1093–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke S, Vogel JP, Deschenes RJ, Stock J. Posttranslational modification of the Ha-ras oncogene protein: evidence for a third class of protein carboxyl methyltransferases. Proc Natl Acad Sci U S A. 1988;85(13): 4643–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y, Prakash P, Liang H, Cho KJ, Gorfe AA, Hancock JF. Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell. 2017;168(1–2): 239–251 e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. Membrane phosphatidylserine regulates surface charge and protein localization. Science. 2008;319(5860): 210–213. [DOI] [PubMed] [Google Scholar]
- 8.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10): 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papke B, Der CJ. Drugging RAS: Know the enemy. Science. 2017;355(6330): 1158–1163. [DOI] [PubMed] [Google Scholar]
- 10.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nature reviews. 2014; 13(11): 828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorfe AA, Cho KJ. Approaches to inhibiting oncogenic K-Ras. Small GTPases. 2019: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van der Hoeven D, Cho KJ, Zhou Y, et al. Sphingomyelin Metabolism Is a Regulator of K-Ras Function. Mol Cell Biol. 2018;38(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimmermann G, Papke B, Ismail S, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497(7451): 638–642. [DOI] [PubMed] [Google Scholar]
- 14.Leung ELH, Luo LX, Liu ZQ, et al. Inhibition of KRAS-dependent lung cancer cell growth by deltarasin: blockage of autophagy increases its cytotoxicity. Cell Death Dis. 2018;9(2): 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J, Kim H, Park SB. Privileged Structures: Efficient Chemical "Navigators" toward Unexplored Biologically Relevant Chemical Spaces. Journal of the American Chemical Society. 2014;136(42): 14629–14638. [DOI] [PubMed] [Google Scholar]
- 16.Evans BE, Rittle KE, Bock MG, et al. METHODS FOR DRUG DISCOVERY - DEVELOPMENT OF POTENT, SELECTIVE, ORALLY EFFECTIVE CHOLECYSTOKININ ANTAGONISTS. Journal of Medicinal Chemistry. 1988;31(12): 2235–2246. [DOI] [PubMed] [Google Scholar]
- 17.Zhuang CL, Zhang W, Sheng CQ, Zhang WN, Xing CG, Miao ZY. Chalcone: A Privileged Structure in Medicinal Chemistry. Chemical Reviews. 2017;117(12): 7762–7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahapatra DM, Bharti SK, Asati V. Anti-cancer chalcones: Structural and molecular target perspectives. European Journal of Medicinal Chemistry. 2015;98: 69–114. [DOI] [PubMed] [Google Scholar]
- 19.Edwards ML, Stemerick DM, Sunkara PS. CHALCONES - A NEW CLASS OF ANTIMITOTIC AGENTS. Journal of Medicinal Chemistry. 1990;33(7): 1948–1954. [DOI] [PubMed] [Google Scholar]
- 20.Ducki S The development of chalcones as promising anticancer agents. Idrugs. 2007;10(1): 42–46. [PubMed] [Google Scholar]
- 21.Bhat BA, Dhar KL, Puri SC, Saxena AK, Shanmugavel M, Qazi GN. Synthesis and biological evaluation of chalcones and their derived pyrazoles as potential cytotoxic agents. Bioorganic & Medicinal Chemistry Letters. 2005;15(12): 3177–3180. [DOI] [PubMed] [Google Scholar]
- 22.Gupta RA, Kaskhedikar SG. Synthesis, antitubercular activity, and QSAR analysis of substituted nitroaryl analogs: chalcone, pyrazole, isoxazole, and pyrimidines. Medicinal Chemistry Research. 2013;22(8): 3863–3880. [Google Scholar]
- 23.Bandgar BP, Gawande SS, Bodade RG, Totre JV, Khobragade CN. Synthesis and biological evaluation of simple methoxylated chalcones as anticancer, anti-inflammatory and antioxidant agents. Bioorganic & Medicinal Chemistry. 2010;18(3): 1364–1370. [DOI] [PubMed] [Google Scholar]
- 24.Pati HN, Holt HL, LeBlanc R, et al. Synthesis and cytotoxic properties of nitro- and aminochalcones. Medicinal Chemistry Research. 2005;14(1): 19–25. [Google Scholar]
- 25.Ahmed FF, Abd El-Hafeez AA, Abbas SH, Abdelhamid D, Abdel-Aziz M. New 1,2,4-triazole-Chalcone hybrids induce Caspase-3 dependent apoptosis in A549 human lung adenocarcinoma cells. European Journal of Medicinal Chemistry. 2018;151: 705–722. [DOI] [PubMed] [Google Scholar]
- 26.Rojas J, Paya M, Dominguez JN, Ferrandiz ML. The synthesis and effect of fluorinated chalcone derivatives on nitric oxide production. Bioorganic & Medicinal Chemistry Letters. 2002;12(15): 1951–1954. [DOI] [PubMed] [Google Scholar]
- 27.Vaidya SS, Vinaya H, Mahajan SS. Microwave-assisted synthesis, pharmacological evaluation, and QSAR studies of 1,3-diaryl-2-propen-1-ones. Medicinal Chemistry Research. 2012;21(12): 4311–4323. [Google Scholar]
- 28.Custodio JMF, Faria ECM, Sallum LO, et al. The Influence of Methoxy and Ethoxy Groups on Supramolecular Arrangement of Two Methoxy-chalcones. Journal of the Brazilian Chemical Society. 2017;28(11): 2180–2191. [Google Scholar]
- 29.Tran TD, Nguyen TTN, Do TH, Huynh TNP, Tran CD, Thai KM. Synthesis and Antibacterial Activity of Some Heterocyclic Chalcone Analogues Alone and in Combination with Antibiotics. Molecules. 2012;17(6): 6684–6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Batt DG, Goodman R, Jones DG, et al. 2'-SUBSTITUTED CHALCONE DERIVATIVES AS INHIBITORS OF INTERLEUKIN-1 BIOSYNTHESIS. Journal of Medicinal Chemistry. 1993;36(10): 1434–1442. [DOI] [PubMed] [Google Scholar]
- 31.Salum LB, Altei WF, Chiaradia LD, et al. Cytotoxic 3,4,5-trimethoxychalcones as mitotic arresters and cell migration inhibitors. European Journal of Medicinal Chemistry. 2013;63: 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choy E, Chiu VK, Silletti J, et al. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 1999;98(1): 69–80. [DOI] [PubMed] [Google Scholar]
- 33.Cho KJ, Park JH, Piggott AM, et al. Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. J Biol Chem. 2012;287(52): 43573–43584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho KJ, Casteel DE, Prakash P, et al. AMPK and Endothelial Nitric Oxide Synthase Signaling Regulates K-Ras Plasma Membrane Interactions via Cyclic GMP-Dependent Protein Kinase 2. Mol Cell Biol. 2016;36(24): 3086–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho KJ, van der Hoeven D, Zhou Y, et al. Inhibition of Acid Sphingomyelinase Depletes Cellular Phosphatidylserine and Mislocalizes K-Ras from the Plasma Membrane. Mol Cell Biol. 2016;36(2): 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willumsen BM, Christensen A, Hubbert NL, Papageorge AG, Lowy DR. The p21 ras C-terminus is required for transformation and membrane association. Nature. 1984;310(5978): 583–586. [DOI] [PubMed] [Google Scholar]
- 37.Singh A, Greninger P, Rhodes D, et al. A gene expression signature associated with "K-Ras addiction" reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15(6): 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayes TK, Neel NF, Hu C, et al. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell. 2016;29(1): 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bivona TG, Quatela SE, Bodemann BO, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21(4): 481–493. [DOI] [PubMed] [Google Scholar]
- 40.Antal CE, Hudson AM, Kang E, et al. Cancer-associated protein kinase C mutations reveal kinase's role as tumor suppressor. Cell. 2015;160(3): 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reno EM, Haughian JM, Dimitrova IK, Jackson TA, Shroyer KR, Bradford AP. Analysis of protein kinase C delta (PKC delta) expression in endometrial tumors. Hum Pathol. 2008;39(1): 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Craven PA, DeRubertis FR. Loss of protein kinase C delta isozyme immunoreactivity in human adenocarcinomas. Dig Dis Sci. 1994;39(3): 481–489. [DOI] [PubMed] [Google Scholar]
- 43.Ohm AM, Tan AC, Heasley LE, Reyland ME. Co-dependency of PKCdelta and K-Ras: inverse association with cytotoxic drug sensitivity in KRAS mutant lung cancer. Oncogene. 2017;36(30): 4370–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang YQ, Hedblom A, Koerner SK, et al. Novel synthetic chalcones induce apoptosis in the A549 non-small cell lung cancer cells harboring a KRAS mutation. Bioorganic & Medicinal Chemistry Letters. 2016;26(23): 5703–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.