

Abstract
DNA 5-methylcytosine (5mC)-specific mapping has been hampered by severe DNA degradation and the presence of 5-hydroxymethylcytosine (5hmC) using the conventional bisulfite sequencing approach. Here, we present a 5mC-specific whole-genome amplification method (5mC-WGA), with which we achieved 5mC retention during DNA amplification from limited input down to 10 pg scale with limited interference from 5hmC signals, providing DNA 5mC methylome with high reproducibility and accuracy.
DNA 5mC methylation, as the main postreplication epigenetic mark in mammals, plays crucial roles in various biological pathways including gene expression,1 developmental regulation,2 and tumorigenesis.3 Insights into the functionality of DNA methylation mainly rely on the gold standard bisulfite sequencing (BS-seq), which allows single-base resolution mapping of 5mC with quantification of the methylation level at each site.4,5 However, the bisulfite treatment is harsh and degrades most DNA.6 Bisulfite-free methods have been developed in recent years to avoid severe DNA degradation.7 Alternatively, a method that can faithfully amplify the 5mC pattern during DNA amplification would be highly desirable for subsequent sequencing such as BS-seq or loci-specific detection approaches. In addition, the conventional BS-seq cannot discriminate 5-hydroxymethylcytosine (5hmC), the oxidative product of 5mC, from the sole 5mC methylation, which restrains studies of DNA methylation dynamics.
Oxidative bisulfite sequencing (oxBS-seq) has been developed for specific 5mC methylome mapping,8 but its application has been hampered by severe DNA degradation under the oxidative conditions and cannot be applied for limited input samples. Genome-wide mapping of 5hmC, when combined with bisulfite sequencing for analysis, also makes the 5mC-specific detection possible.9,10 However, 5hmC exists in lower abundance and can be highly dynamic. Simple subtraction of 5hmC signals from the whole-genome bisulfite sequencing may introduce additional variations into the DNA 5mC detection. Because all these methods still rely on bisulfite treatment, which degrades most DNA, 5mC-specific sequencing or detection from limited input is still a challenge.
DNA methylation is maintained by three canonical DNA methyltransferases, namely, DNMT1, DNMT3A, and DNMT3B. While DNMT3A and DNMT3B are responsible for the de novo methylation, DNMT1 installs methyl groups donated by S-adenosylmethionine (SAM) on cytosines that are located on hemimethylated CpG sites during DNA replication.11 Inspired by the conservative methylation regulation during DNA replication, we developed a whole-genome amplification method with specific retention of 5mC starting from a limited input amount, a conceptually “simple” but potentially highly useful approach by preamplifying the limited DNA sample to a higher abundance with retention of 5mC, which we named 5mC-retained whole-genome amplification (5mC-WGA). Amplified products are compatible with most downstream detection methods, with 5mC modification replicated faithfully during the amplification. Followed with the standard bisulfite sequencing, our method can identify 5mC sites with limited interference from 5hmC, as DNMT1 cannot replicate 5hmC during our isothermal amplification.
Our design started with the incorporation of the strand displacement amplification with the DNMT1 methylation (Figure S1a,b). Isothermal DNA amplification allows DNMT1 to work under its optimal temperature.12 We started our tests with human brain genomic DNA with well-defined CpG methylation information already available. With an optimized combination of DNA polymerase phi29 and DNMT1, we successfully amplified input DNA by at least 100-fold (Figure S2), ranging from 10 ng down to 10 pg with 5mC marks retained faithfully (Table S1). We picked both a known 5mC hypermethylated region (a locus in NFATC1) and a nonmethylated region (a locus in MAPK8IP2) for low-throughput validation13 (Figure S3a). The amplified products were subjected to bisulfite treatment, PCR amplification with predesigned primers, and Sanger sequencing. The results showed that 5mC-WGA amplified the hypermethylated sites with high accuracy but did not produce methylation within the nonmethylated region (Figure 1a). The amplified products were further subjected to MeDIP-seq (methylated DNA immunoprecipitation and sequencing) along with bulk control. The results again demonstrated faithful retention of the DNA methylation during amplification (Figures 1b and S3b).
Figure 1.
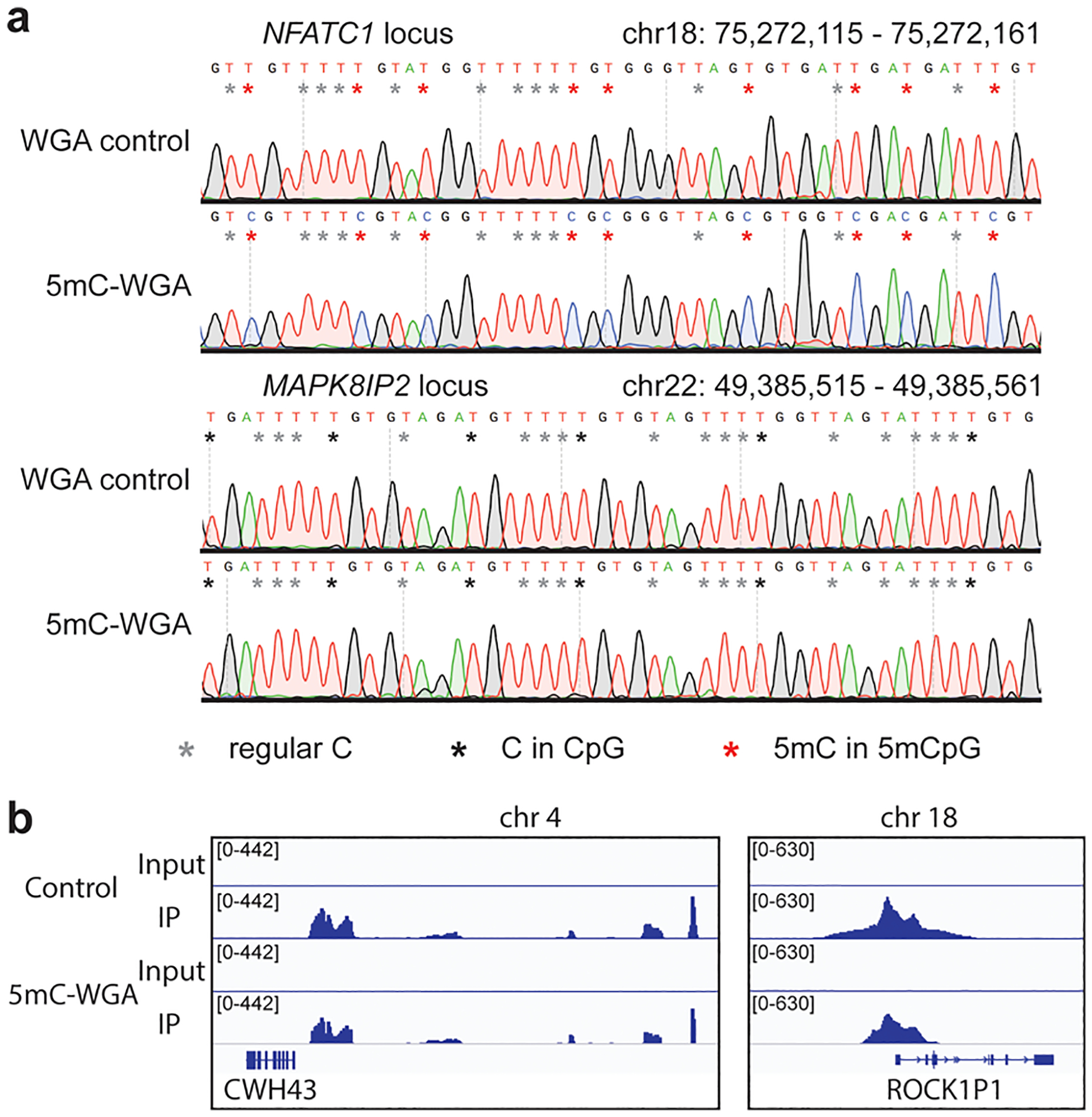
Method preliminary test. (a) Sanger sequencing for loci- specific detection. The upper two panels refer to the known 5mC hypermethylated locus and the lower two refer to the nonmethylated locus. The WGA control that was amplified without DNMT1 showed no 5mC retention, but the 5mC-WGA showed highly faithful retention at the hypermethylated region. 5mC-WGA and WGA control both showed no de novo signals. (b) Examples for MeDIP-seq validation. 5mC-WGA exhibits a methylation retention similar to that of the bulk control.
We then proceeded to the high-throughput sequencing for more accurate validation of our method with amplified products starting from 10 pg, 100 pg, and 1 ng genomic DNA, respectively. We still detected expected retention of methylation when we amplified targeted loci. However, we did observe relatively low genome coverage with lower input down to 10 pg, which is also commonly observed with single-cell bisulfite sequencing. A careful analysis on the high-throughput data revealed that the decrease in genome coverage is mainly due to the biased priming with random hexamer during the strand displacement amplification. With primer-derived artifacts and sequence-dependent hybridization kinetics, amplification bias would be introduced when the input amount is limited.14 Therefore, to minimize priming bias, we incorporated a unique DNA primase into our amplification system. While most primases use NTPs as substrates to produce RNA primers, TthPrimPol uses dNTPs to synthesize DNA primers during polymerization. A recent study applied TthPrimPol in DNA amplification together with the highly processive strand displacement polymerase phi29 to initiate a true random priming process.15 Such combination accomplished near-complete whole-genome amplification with high reproducibility and superb genome coverage, which could partially solve the priming bias issue for 5mC-WGA.
We therefore performed preliminary tests on the primer-free system. Early trials showed that TthPrimPol, phi29, and DNMT1 work compatibly in our reaction buffer. With a welltuned amplification system and an optimal methylation condition (Figure 2), we can acquire similar 5mC-retained amplification verified by our low-throughput analysis on specific loci. We then proceeded to test the three-enzyme whole-genome amplification system.
Figure 2.
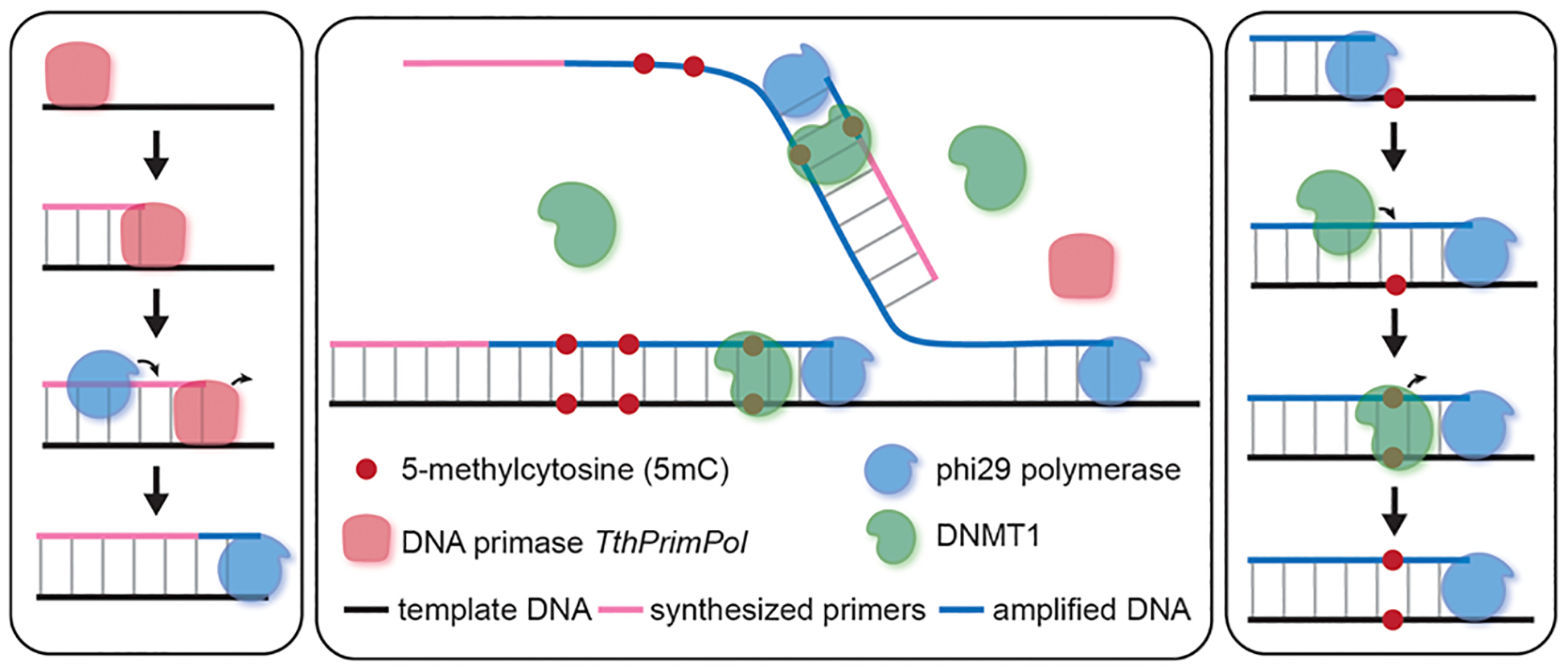
Method design. Scheme demonstrating the design for primer-free amplification system. Left: the DNA primase TthPrimPol synthesizes DNA primers for strand displacement amplification by phi29. Right: DNMT1 adds a methyl group to the unmethylated CpG site on the hemimethylated locus. Middle: the entire three-enzyme primer-free amplification system with methylation retention.
We amplified 10 pg of genomic DNA purified from mES cells, and the products were subjected to bisulfite treatment followed by library construction, which altogether we named as 5mC-WGA-BS. We analyzed our sequencing results together with two control samples: one as 10 pg of gDNA without any treatment and the other amplified without DNMT1 under the same condition. When aligned to the bulk positive control, our amplified samples all showed methylation retention with a little lower methylation level compared to the direct bisulfite bulk control sample, while all CHG and CHH levels remained low (Figure 3a). Our negative control without DNMT1 yielded a quite low methylation level, which confirmed that DNMT1 is responsible for methylation maintenance during amplification. The GC content bias analysis did not show any significant bias toward certain GC contents, suggesting a universal amplification of 5mC-containing regions. Compared with the standard commercial library construction kits, our method did not incorporate bias during amplification (Figure S4). Hierarchical clustering based on CpG methylation levels revealed high reproducibility among our replicate libraries generated from low inputs (Figure 3b). The high reproducibility was also verified by the high correlation among three different samples compared with one another (Figure 3c). The well-overlaid methylation metagene plots showed a typical methylation pattern where TSS regions show low methylation while gene body regions enrich DNA methylation (Figure 3d). In contrast to the random-hexamer-assisted amplification we used at the beginning, the primer-free system achieved higher specificity and reliability in 5mC retention (Figure S5).
Figure 3.
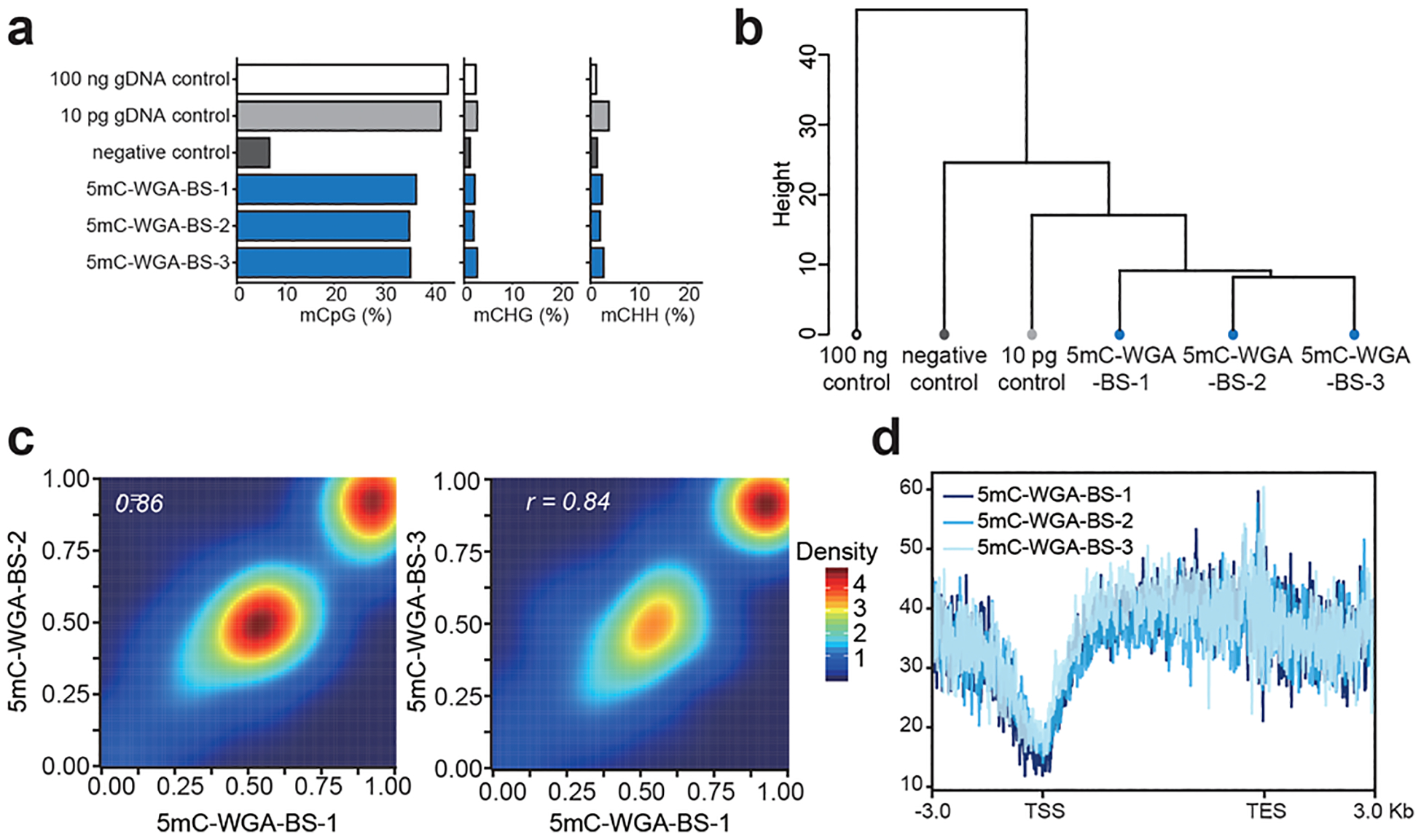
Method evaluation. (a) Methylation levels at CpG, CHG, and CHH sites of the bulk control, the limited input control, the negative control that was amplified without DNMT1, and three replicates of 5mC-WGA-BS, listing from the top to the bottom. (b) Hierarchical clustering based on CpG methylation levels showing high reproducibility of 5mC-WGA-BS. (c) Correlation analysis showing high correlation among replicates. Single-base resolution data were analyzed. (d) Metagene plot of 5mC-WGA-BS showing low methylation levels at TSS regions and high methylation levels at gene body regions for all three replicates.
To examine class heterogeneity, we analyzed all unmethylated, methylated, and hydroxymethylated C sites derived from 5mC-WGA-BS, oxBS-seq, and TAB-seq (a direct 5hmC readout method). The oxBS-seq data were generated from our same input, while TAB-seq data were acquired from a published data set.16 All methods were analyzed along with bisulfite sequencing from bulk input to simultaneously estimate methylation levels and hydroxymethylation levels by extracting information from both BS-seq and 5mC-WGA-BS, or oxBS-seq, or TAB-seq based on maximum likelihood methylation levels (MLML).17 While our method showed a similar pattern to that of oxBS-seq, TAB-seq demonstrated a different trend as expected (Figure 4a). Our method demonstrated a higher correlation with oxBS-seq in 5mC detection than TAB-seq as expected. In addition, subtraction of whole-genome bisulfite sequencing with 5mC-WGA-BS also detects most 5hmC sites (Figure S6). 5mC-WGA-BS showed a high correlation with oxBS-seq in 5mC spotting (Figure 4b). 5hmC tends to exist at lower abundance than 5mC and marks more dynamic 5mC sites. All the replicates showed a high correlation to detected 5hmC sites (Figure S7a), confirming 5hmC as a derivative of 5mC. In addition, because of the known enrichment of 5hmC on gene bodies, we did notice a decrease in the 5mC methylation level detected using 5mC-WGA-BS compared to that of the conventional bisulfite sequencing (Figure 4c). The 5hmC sites revealed from 5mC-WGA-BS (subtraction from conventional BS-seq, see Supporting Information for details) showed high correlation with results from 5hmC-Seal that captures 5hmC-containing DNA fragments (Figure S7b). Examples are plotted to show results from different approaches, confirming that 5mC-WGA-BS faithfully uncovers 5mC sites and can help extract 5hmC information (Figures 4d and S8).
Figure 4.
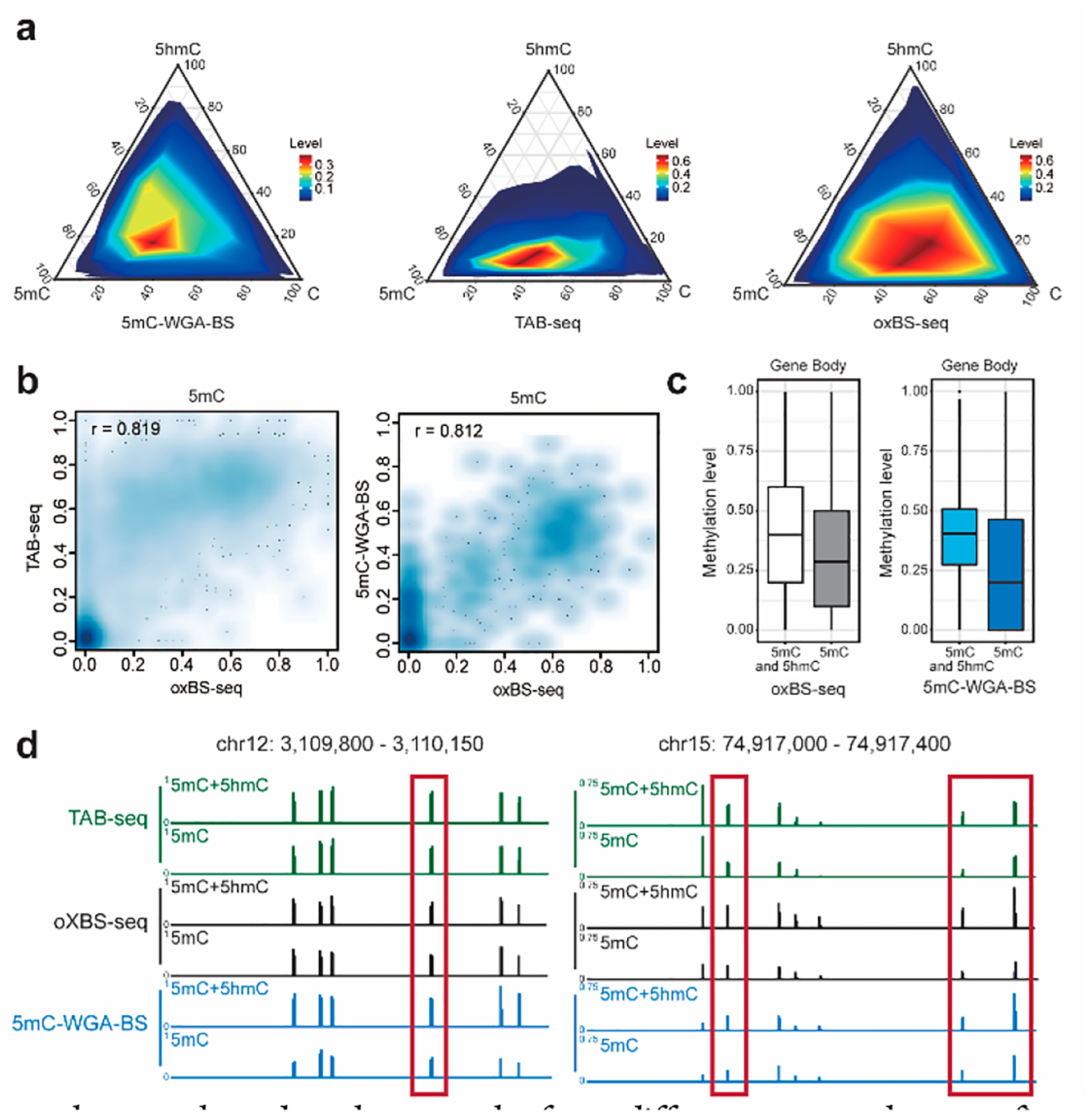
Method comparison. (a) Ternary plots of C, 5mC, and 5hmC levels at single-base resolution sites for 5mC-WGA-BS, TAB- seq, and oxBS-seq. 5hmC sites of 5mC-WGA-BS and oxBS-seq were extracted based on direct readout of 5mC sites and bulk bisulfite sequencing. 5mC sites of TAB-seq were obtained from a subtraction of 5hmC sites from the bulk bisulfite sequencing. 5mC-WGA-BS signals showed a similar pattern to oxBS-seq signals, while TAB-seq mainly detected 5hmC sites at lower modification levels as expected. (b) Correlation analysis for both TAB-seq and 5mC-WGA-BS compared with oxBS-seq. (c) Methylation levels across Ensembl gene bodies by oxBS-seq or 5mC-WGA-BS showing reduced methylation level compared to traditional bisulfite sequencing. (d) Examples (highlighted with red frames) showing 5mC-sepecific methylation detected by 5mC-WGA-BS.
This 5mC-WGA-BS method works well with 10 pg of isolated genomic DNA. Preliminary tests using five cells also showed the retention of 5mC as expected, with CHG and CHH levels remaining low (Figure S9a). The metagene plots demonstrated a similar pattern when the input was amplified in the presence of DNMT1 but no methylation pattern when amplified without DNMT1 (Figure S9b). The ternary plot and the correlation analysis with oxBS-seq both corresponded to results from the bulk genomic DNA libraries (Figure S9c,d). Our trials at the single-cell level suggested a requirement of a further optimized lysis condition to eliminate potential genomic DNA degradation but still ensure full denaturation of the chromatin to be ready for amplification. Perhaps a microfluidics device or other procedures could help in the future. Because phi29 effectively amplifies only long DNAs, the method currently could be employed only to genomic DNA rather than synthetic probes or short DNA fragments.
With our high-throughput results, we also investigated the methylation activity of DNMT1 in the presence of opposite CpG or 5hmCpG. DNMT1 was reported to exhibit an enzymatic methylation activity opposite to CpGs, which is about 1/10 of that opposite to 5mCpGs and around 1/3 opposite to 5hmCpGs compared with that opposite to 5mCpGs.18 These results are consistent with the percentages of the potential reactive sites for methylation based on our analyses. However, the interference on quantitative 5mCpGs detection is quite limited, as we only observed low methylation levels that stemmed from these activities (Figure S10a,b). While potential de novo sites account for about 10% of the detected methylated sites, they tend to be low in their methylation levels. Thus, the interference from de novo methylation is limited to lower than 5% (Figure S10a). Our method showed little activity with CpH methylation, especially when compared with CpG methylation (Figure S10c). All these analyses suggested that our approach is a reliable 5mCpG-specific detection method for limited input materials. Future studies may further elucidate potential activities of DNMT1 at unmethylated CpGs, 5hmCpGs, and CpHs.
In conclusion, we present a 5mC-specific whole-genome amplification system for simultaneous DNA amplification and methylation in a one-pot, primer-free reaction. The amplified products could be subjected to bisulfite sequencing or other detection platforms for faithful methylome mapping or detection using limited input materials. This methylation-retained amplification approach could enable facile detection of 5mC in clinical samples such as DNA from biopsy or cell-free DNA in plasma in the future.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. G. Brett Robb and Dr. Mala Samaranayake at New England Biolabs for their generosity in providing the concentrated DNMT1 used in the study. We thank Dr. Pieter W. Faber and the staff at the Genomics Facility and Comprehensive Cancer Center sequencing facility at the University of Chicago for performing the Sanger and NGS sequencing measurements. This work was supported by NIH HG008935 (C.H.). C.H. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b12707.
Experimental details and preliminary test design and other supportive details of method validation and evaluation, including Figures S1–S10 and Tables S1 and S2 (PDF)
The authors declare the following competing financial interest(s): C.H. is a scientific founder and scientific advisory board member of Accent Therapeutics, Inc. and a shareholder of Epican Genetech.
Contributor Information
Chang Liu, Department of Chemistry, Department of Biochemistry and Molecular Biology, and Institute for Biophysical Dynamics and Howard Hughes Medical Institute, The University of Chicago, Chicago, Illinois 60637, United States.
Xiaolong Cui, Department of Chemistry, Department of Biochemistry and Molecular Biology, and Institute for Biophysical Dynamics and Howard Hughes Medical Institute, The University of Chicago, Chicago, Illinois 60637, United States.
Boxuan Simen Zhao, Department of Genetics, Stanford University, Stanford, California 94305, United States.
Pradnya Narkhede, Department of Chemistry, University of Cambridge, Cambridge CB2 0SP, U.K..
Yawei Gao, Clinical and Translational Research Center of Shanghai First Maternity and Infant Hospital, Shanghai Key Laboratory of Signaling and Disease Research, School of Life Sciences and Technology, Tongji University, Shanghai 200092, China.
Jun Liu, Department of Chemistry, Department of Biochemistry and Molecular Biology, and Institute for Biophysical Dynamics and Howard Hughes Medical Institute, The University of Chicago, Chicago, Illinois 60637, United States.
Xiaoyang Dou, Department of Chemistry, Department of Biochemistry and Molecular Biology, and Institute for Biophysical Dynamics and Howard Hughes Medical Institute, The University of Chicago, Chicago, Illinois 60637, United States.
Qing Dai, Department of Chemistry, Department of Biochemistry and Molecular Biology, and Institute for Biophysical Dynamics and Howard Hughes Medical Institute, The University of Chicago, Chicago, Illinois 60637, United States.
Li-Sheng Zhang, Department of Chemistry, Department of Biochemistry and Molecular Biology, and Institute for Biophysical Dynamics and Howard Hughes Medical Institute, The University of Chicago, Chicago, Illinois 60637, United States.
Chuan He, Department of Chemistry, Department of Biochemistry and Molecular Biology, and Institute for Biophysical Dynamics and Howard Hughes Medical Institute, The University of Chicago, Chicago, Illinois 60637, United States;.
REFERENCES
- (1).Luo C; Hajkova P; Ecker JR Dynamic DNA methylation: In the right place at the right time. Science 2018, 361 (6409), 1336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bourc’his D; Greenberg MVC The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Bio 2019, 20, 590–607. [DOI] [PubMed] [Google Scholar]
- (3).Robertson KD DNA methylation and human disease. Nat. Rev. Genet 2005, 6, 597–610. [DOI] [PubMed] [Google Scholar]
- (4).Guo H; Zhu P; Yan L; Li R; Hu B; Lian Y; Yan J; Ren X; Lin S; Li J; Jin X; Shi X; Liu P; Wang X; Wang W; Wei Y; Li X; Guo F; Wu X; Fan X; Yong J; Wen L; Xie SX; Tang F; Qiao J The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–10. [DOI] [PubMed] [Google Scholar]
- (5).Smallwood SA; Lee HJ; Angermueller C; Krueger F; Saadeh H; Peat J; Andrews SR; Stegle O; Reik W; Kelsey G Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Olova N; Krueger F; Andrews S; Oxley D; Berrens RV; Branco MR; Reik W Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome Biol 2018, 19, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Liu Y; Siejka-Zielinska P; Velikova G; Bi Y; Yuan F;Tomkova M; Bai C; Chen L; Schuster-Böckler B; Song CX Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol 2019, 37, 424–9. [DOI] [PubMed] [Google Scholar]
- (8).Booth MJ; Branco MR; Ficz G; Oxley D; Krueger F; Reik W; Balasubramanian S Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012, 336 (6083), 934–7. [DOI] [PubMed] [Google Scholar]
- (9).Yu M; Hon GC; Szulwach KE; Song CX; Zhang L; Kim A; Li X; Dai Q; Shen Y; Park B; Min JH; Jin P; Ren B; He C Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012, 149 (6), 1368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Nazor KL; Boland MJ; Bibikova M; Klotzle B; Yu M; Glenn-Pratola VL; Schell JP; Coleman RL; Cabral-da-Silva MC; Schmidt U; Peterson SE; He C; Loring JF; Fan JB Application of a low cost array-based technique — TAB-Array — for quantifying and mapping both 5mC and 5hmC at single base resolution in human pluripotent stem cells. Genomics 2014, 104 (5), 358–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lyko F The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet 2018, 19 (2), 81–92. [DOI] [PubMed] [Google Scholar]
- (12).Yan L; Zhou J; Zheng Y; Gamson AS; Roembke BT; Nakayama S; Sintim HO Isothermal amplified detection of DNA and RNA. Mol. BioSyst 2014, 10 (5), 970–1003. [DOI] [PubMed] [Google Scholar]
- (13).Maunakea AK; Nagarajan RP; Bilenky M; Ballinger TJ; D’Souza C; Fouse SD; Johnson BE; Hong C; Nielsen C; Zhao Y; Turecki G; Delaney A; Varhol R; Thiessen N; Shchors K; Heine VM; Rowitch DH; Xing X; Fiore C; Schillebeeckx M; Jones SJ; Haussler D; Marra MA; Hirst M; Wang T; Costello JF Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466 (7303), 253–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hansen KD; Brenner SE; Dudoit S Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res 2010, 38 (12), No. e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Picher ÁJ; Budeus B; Wafzig O; Krüger C; García-Gomez S; Martínez-Jimenez MI; Díaz-Talavera A; Weber D; Blanco L; Schneider A TruePrime is a novel method for whole-genome amplification from single cells based on TthPrimPol. Nat. Commun 2016, 7, 13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hon GC; Song CX; Du T; Jin F; Selvaraj S; Lee AY; Yen CA; Ye Z; Mao SQ; Wang BA; Kuan S; Edsall LE; Zhao BS; Xu GL; He C; Ren B 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol. Cell 2014, 56 (2), 286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Qu J; Zhou M; Song Q; Hong EE; Smith AD MLML: consistent simultaneous estimates of DNA methylation and hydroxymethylation. Bioinformatics 2013, 29 (20), 2645–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Seiler CL; Fernandez J; Koerperich Z; Andersen MP; Kotandeniya D; Nguyen ME; Sham YY; Tretyakova NY Maintenance DNA methyltransferase activity in the presence of oxidized forms of 5-methylcytosine: structural basis for ten eleven translocation-mediated DNA demethylation. Biochemistry 2018, 57 (42), 6061–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.