

Abstract
Purpose of review
About 40% of the neuroendocrine tumors pheochromocytomas and paragangliomas (PPGLs) are caused by an inherited mutation. Diagnostic genetic screening is recommended for patients and their families. However, the number of susceptibility genes involved is high and continues to grow, making conventional sequencing costly and burdensome. Next-generation sequencing (NGS) enables accurate, thorough, and cost-effective identification of inherited mutations. Here we review recent successes, limitations, and the future of NGS for diagnosis of pheochromocytoma and paraganglioma syndromes.
Recent findings
NGS-based screen of genetic disorders in the clinical setting shows improved diagnostic rates over conventional tests. Both broad, whole-exome sequencing, and targeted NGS approaches have been tested for screening of PPGLs, with accurate mutation detection, higher speed, and reduced costs compared with current assays. Flexibility to expand the targeted gene set is immediate in whole-exome sequencing, and adjustable in targeted NGS, but both methods have limitations.
Summary
The high degree of genetic heterogeneity and heritability of PPGLs make NGS an ideal medium for their diagnostic screening. However, improved detection of large genomic defects and underrepresented gene areas are needed before NGS can fully realize its potential as the premier option for routine genetic testing of these syndromes.
Keywords: diagnostics, genetic screening, next-generation sequencing, paragangliomas, pheochromocytomas
INTRODUCTION
Pheochromocytomas and paragangliomas (PPGLs) are neural crest tumors derived from catecholamine secreting cells of the adrenal medulla or extra-adrenal sympathetic paraganglia, respectively [1,2]. Two striking features of these tumors are their genetic heterogeneity and their high degree of heritability (40% of the cases). Recent Clinical Practice Guide lines set forth by the American Endocrine Society recommends that genetic testing be performed for certain groups at high risk for hereditary PPGL, as detailed below, but that it should be considered for all PPGL patients [3]. The American Society of Clinical Oncology directions reach further by suggesting that all patients with a risk of heritability higher than 10% should undergo testing [4]. Therefore, PPGLs fall well into the category of diseases for which genetic screening is advised. However, as the number of susceptibility genes increases, currently spanning over 200 exons, so does the complexity of genetic testing. Next generation sequencing (NGS) methodology has dramatically changed the field of genetics in the past decade. NGS use has broadened widely since its inception, with improvements in the technology and decrease in costs. NGS methods have now been applied in multiple clinical diagnostic settings, including inherited developmental disorders and cancers, in many cases with greater success rate compared with conventional sequencing techniques [5–11]. Over the past few years, a picture of the state of NGS use in the field of PPGL started to emerge. In this review, we will discuss these studies and address the advantages and challenges of distinct NGS approaches for inherited PPGL diagnosis. Although the use of NGS testing for diagnosis of somatic variants is recognizably relevant from a clinical perspective, these studies will not be extensively discussed here.
THE COMPLEX GENETICS OF PHEOCHROMOCYTOMAS AND PARAGANGLIOMAS
Much progress has been made on our understanding of the genetic basis of PPGLs in the past decades and many familial forms of the disease are now recognized (Table 1). Excellent reviews describing unique clinical features of these various inherited disorders have been published recently [12–14]. PPGLs are arguably the most heritable human tumors. Familial PPGL is usually inherited as an autosomal dominant trait, so the offspring of a mutation carrier will have a 50% chance of having inherited the relevant PPGL gene mutation [1,2]. Genetic testing is recommended for individuals at high risk for susceptibility, which includes positive family history, presence of syndromic features, early onset disease, presence of multiple tumors, malignancy, paraganglioma location, or a combination of some of these characteristics, whereby the pretest probability of mutation detection is high [3]. Many diagnostic stepwise algorithms have been proposed to streamline the increasingly burdensome and costly process of genetic screening of PPGLs [12,15–18]. These algorithms incorporate clinical features to guide the prioritization of the gene for screen and are particularly effective for high-risk groups. However, there is a strong argument for extending genetic testing to all PPGL patients, based on the recognition that at least 10% of ‘low risk’ cases may carry predisposing mutations [13]. In these cases, low penetrance of the mutant allele, the existence of parent-of-origin effects on disease penetrance (in SDHD, SDHAF2 and MAX mutations) or de-novo mutations in the index patient can obscure the diagnosis of inherited PPGL [2]. In nonsyndromic cases, the number of screened genes expands, which makes the process lengthy, if genes are analyzed individually and/or, very costly, if they are tested simultaneously.
Table 1.
Main clinical, biological and genetic features of known pheochromocytoma and paraganglioma susceptibility genes
| Category | Parameters | NF1 | RET | VHL | TMEM127 | MAX | SDHA | SDHB | SDHC | SDHD | SDHAF2 | FH | HIF2/EPAS1 | KIF1B | PHD2/EGLN1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clinicol | Inherited syndrome designation | Neurofibromatosis type 1 | MEN 2A, MEN 2B, Familial MTC, Hirschsprung disease | von Hippel-Lindau disease, Chuvash polycythemia | TMEM127-related pheochromocytoma | MAX-related pheochromocytoma | F-PGL, Mitochondrial complex II deficiency | F-PGL type 4, familial RCC, Carney–Stratakis syndrome, complex II deficiency | FPGL type 3, Carney–Stratakis syndrome, complex II deficiency | F-PGL type 1, Carney–Stratakis syndrome, complex II deficiency | F-PGL type 2, complex II deficiency | Hereditary leiomyoma renal cell carcinoma | Familial erythrocytosis type 4 | Charcot Marie Tooth Disease 2A1 | Familial erythrocytosis type 3 |
| Prototypical presentation | Single pheo, café au lait spots, neurofibromas, family history of neurofibromatosis | Bilateral pheo, Medullary thyroid carcinoma, family history | Young age, bilateral pheo, renal cell carcinoma and CNS heman-gioblastoma, family history | >35 years old, pheo, family history less frequent | Pheo, family history less frequent, paternal transmission | Pheo or PGL | Single PGL, malignant features, occasional family history | Head and neck PGL, family history less frequent | Multiple PGLs, predominantly head and neck, family history, paternal transmission | Multiple PGLs, predominantly head and neck, family history, paternal transmission | Multiple pheo or PGLs, malignant features frequent | PGLs, polycythemia, somato-statinoma | 2 cases only (one familial, bilateral pheo, second single, sporadic pheo) | 1 case only (PGL, polycythemia) | |
| Other manifestations or other conditions associated with mutations ir these genes | Neurofibromas, malignant peripheral nervous sheath tumors, gliomas | MTC, hyperparathyroidism, marfanoid habitus | RCCs, CNS hemangioblastomas | RCC | None reported | GISTs | GISTs, RCCs | GISTs | GISTs | None reported | Uterine leiomyoma | Polycythemia, somatostatinomas | Neuroblastoma, lung cancera | Polycythemia | |
| Biological | Transcription clustera | 2 | 2 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | U |
| Methylation clusterb | 3 | 3 | 2 | 3 | 3 | 1 | 1 | 1 | 1 | U | 1 | U | U | U | |
| Genetic | Mutation type | S>G | G>S | G>S | G | G>S | G | G | G | G | G | G | S + M | G | G |
| Inheritance (autosomal dominant = AD, P = parent of origin effect,U = unknown, N/A = not applicable) <delete?> | AD | AD | AD | AD | AD-P | AD | AD | AD | AD-P | AD-P | AD | U | AD | AD |
HRAS mutations were only detected somatically, not in the germline and have therefore not been included in this table. AD, autosomal dominant; CNS, central nervous system; FH, fumarate hydratase; G, germline; GIST, gastrointestinal stromal tumor; M, mosaic; MTC, medullary thyroid carcinoma; P, parent of origin effect (paternal transmission); PGL, paraganglioma; PHEO, pheochromocytoma; RCC, renal cell carcinoma; S, somatic; U, unknown.
Mutant tumors belong to one of two main transcriptional clusters: 1, pseudohypoxia; 2, kinase-related signaling.
Mutant tumors belong to three main methylation clusters: 1: SDH/FH-mutant tumors; 2: predominantly VHL-mutant tumors; 3: NF1/RET/MAX/TMEM127-mutant and some sporadic tumors.
NEXT-GENERATION SEQUENCING PLATFORM OF CHOICE
Similar to other hereditary disorders, in particular those in which allelic heterogeneity is extensive, the use of NGS, also referred to as massively parallel sequencing, has increased exponentially over the past decade and has begun to replace conventional (Sanger) sequencing in many clinical contexts [6,19,20]. Methodological details of the techniques are beyond the scope of this review. Instead, here we discuss the NGS approaches that have been applied to PPGLs and how these findings will shape the future of genetic testing in these tumors. Table 2 summarizes the context, study design, results, and limitations of these published studies. Most of the NGS studies of PPGLs, a few preceding the review period but included because of their relevance, were performed for purposes of gene discovery and employed whole exome sequencing (WES) [21,22,23▪,24▪▪,25▪▪,26▪, 28▪▪,30▪,32–34▪▪,35]. Targeted NGS analyses were also reported [27▪▪,29,31▪▪,36▪▪]. Two studies directly compared NGS with conventional sequencing for diagnosis of germline mutations in known PPGL genes [27▪▪,28▪▪].
Table 2.
Summary of the results of published next generation sequencing studies in pheochromocytomas and paragangliomas
| Study | Reference | n | Phenotype | Mutation in known susceptibility gene ? | Sample type | Method | Enrichment platform/library generation | Read direction/fragment length | Average depth of coverage | Sequencing platform | Genetic analysis approach | Main genetic outcomes/discoveries | Accuracy | Notes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Comino-Mendez et al. [21] | 3 | Transcriptionally clustered pheochromocytomas with no known mutation | No | Blood | WES | Agilent SureSelect Human All Exon | 2X75PE | PPGL genes were covered at a minimum of 10X (94–96%), overall average not available | Illumina Genome Analyzer II | Broad discovery analysis | MAX identified as a novel PPGL susceptibility gene | n/a | |
| 2 | Qi et al. [22] | 6 relatives | MEN2 and FMTC family | No | Blood | WES | Agilent SureSelect Biotinylated RNA Library | 2X90PE | min 5OX (RET) | Illumina HiSeq2000 | Analysis focused on RET | compound RET mutations associated with modified phenotypes | NA | |
| 3 | Toledo et al. [23▪] | 6 | PPGLs of unknown genetic cause | No | Paired blood and tumor [4] and two paired tumors from the same patient | WES | Agilent Sure Select 44Mbp | 2X54PE (2 tumors), 2X90PE (4 pairs) | 52X (tumor-tumor pair); 52–55X(blood-tumor pairs) | Illumina HiSeq2000 | Broad discovery analysis | EPAS1 mutation discovery in one sample and 3 additional EPAS1 mutations in 167 sporadic PPGLs screened by Sanger | NA | |
| 4 | Crona et al. [24▪▪] | 4 | Benign and sporadic PPGLs | No | Tumor | WES | SureSelect Human All Exon 50 Mb kit | 2XPE, size? | Illumina HiSeq2000 | Broad discovery analysis | HRAS somatic mutations identified in PPGLs | NA | ||
| 5 | Letouze et al. [25▪▪] | 1 | SDH-like pheochromocytoma, but without a germline mutation in SDH genes | No | Paired blood and tumor | WES | Agilent SureSelect Human All Exon Kit v4+UTR | 2X75PE | 8OX (sample is included in Castro-Vega et al. | Illumina HiSeq2000 | Broad discovery analysis | FH identified as a novel PPGL susceptibility gene | NA | |
| 6 | Cron a et al. [26▪] | 3 | Sporadic | No | Tumor | WES | Agilent SureSelect | ? | ? | Illumina HiSeq2000 | Analysis focused on SDHA, SDHB, SDHC, SDHD, SDHAF2, VHL, EPAS1, RET, NF1, TMEM127 and MAX | One NF1 variant and one RET variant | NA | |
| 7 | Ratten berry et al. [27▪▪] | 205 | Familial and sporadic PPGLs | 85 | Blood | PCR + NGS sequencing | 48.48 Access Array system (Fluidigm) | 310–460bp amplicon size range | min 3OX | GS Junior NGS sequencer Roche 454 | Analysis focused on MAX, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2J-MEM127, VHL | Multiple novel and known mutations of PPGL genes | 98.5% NGS assay sensitivity | 454 sequencing technology is prone to false positives and sequence context-based errors (e.g., repeat regions) |
| 8 | Mclnerney-Leo et al. [28▪▪] | 11 | Familial (germline mutations in VHL, RET, SDHB, SDHC or SDHD) | Yes | Blood | WES | HluminaTruSeq (7 cases); NimbleGen-SeqCap EZ v3.0 (5 cases) + computa-computational analysis of PPGL genes in WES from reference samples of five capture kits: Agilent SureSelectXT; Nimblegen-SeqCap EZ v3.0; HluminaTruSeqT-MExome Enrichment Kit v2.0; NexteraTM Rapid Capture Expanded Exome; Illumina-NexteraTM Rapid Capture Exome | 2X100PE | ? | Illumina HiSeq2000 | Analysis focused on RET, NF1, VHL, SDHx, SDHAF2,- KIF1B, TMEM127, EGLN1, MAX | At least one exon was not captured in SDHA/D (1/5 platforms), MAX/KIF1B (2/5), NF1/SDHC (3/5) | 6/7 (85.7%) using HluminaTruSeq; 5/5 (100%) mutations detected using Nim-bleGenSeqCap EZ v3.0 | Appropriate choice of capture platform is critical to ensure adequate coverage of all exons of known PPGL genes, especially SDHA/C/D |
| 9 | Casey et al. [29] | 31 | Mostly sporadic PPGLs (28/31] | No | Blood | PCR + NGS sequencing | information unavailable | ? | ? | Illumina sequencer ? | VHL, SDHA, SDHB, SDHC, SDHD, TMEM127, MAX, and RET | WES NGS allows PPGL screen to include less frequently studied PPGL genes (e.g., SDHA and KIF1B), improving detection of rarer mutations | NA | |
| 10 | Clark et al. [30▪] | 1 | Child pheochromocytoma of unknown genetic cause | No | Blood | WES | Agilent SureSelect All Exon 50Mb Target Enrichment System | 2X76PE | ? | Illumina GA Analyser-IIx | Broad discovery analysis | FH mutation discovery in proband and one additional mutation in 71 samples screened by Sanger | NA | |
| 11 | Welander et al.[31▪▪] | 86 | 18 familial/syndromic; 56 apparently sporadic | 18/86 | Tumor | PCR + NGS sequencing | IlluminaTruSeq custom amplicon | 2X150PE; 272 amplcons | i-915X | IlluminaMiSeq | Analysis focused on EGLNl, EPASl, KIF1B, MAX, MENl, NFl, RET, SDHA, SDHB,SDHC, SDHD, SDHAF2, TMEM127, VHL | 265/272 amplicons (97%) yieldec sequence reads, with a mean depth of 915 per amplicon and sample. | 100% sensitivity and specificity in known cases | Target sequencing of PPGLs genes carries high sensitivity and specificity although some exons could not be multiplexed |
| 12 | Flynn et al. [32▪▪] | 40 | PPGLs of both known and unknown genetic cause | Yes, in 14 cases | Paired blood and tumor (40 for WES and 39 tumors for RNAseq) | WES and RNAseq | Nimblegen V2 (Nim-blegen, Roche, WI, USA) or the Agilent SureSelect V5 exome capture | 2X100PE | 120X (WES); 60–80M reads (RNAseq) | Illumina HiSeq2000 | Broad discovery analysis | Novel somatic mutations in multiple genes, major structural defects, gene fusions | all 14 previously known mutations were confirmed by WES and/or RNAseq | |
| 13 | Fishbein et al. [33▪▪] | 21 (+ 103] | PPGLs of unknown genetic cause | No | Paired blood and tumor | WES + targeted NGS for validation | Agilent SureSelect All Exon v3 | 2X100PE | 84X(tumor); 85X (germline); unknown depth of validation cohort | Illumina HiSeq2000; IlluminaMiSeq for targeted sequencing | Broad discovery analysis | somatic ATRX mutations associated with malignant PPGLs | NA | |
| 14 | Castro-Vega et al. [34▪▪] | 31 | PPGLs of both known and unknown genetic cause | Yes, in 17 cases | 30 paired blood and tumor, 1 trio (blood, primary tumor and metastasis) | WES | SureSelect Human All Exon Kit v4 + UTR, Human All Exon v4 + UTR-70Mb | 2X75PE | 80X | Illumina HiSeq2000 | Broad discovery analysis | Novel mutations in multiple genes and structural defects, few recurrent mutations | NA |
FH, fumarate hydratase; FMTC, familial medullary thyroid carcinoma; MEN2A, multiple endocrine neoplasia type 2A; NGS, next-generation sequencing; PE, paired-end; PGL, paraganglioma; PHEO, pheochromocytoma; PPGL, pheochromocytoma and paraganglioma; RNAseq, RNA sequencing; WES, whole-exome sequencing; ?, information not provided. Next-generation sequencin
Whole-exome sequencing
In this method, fragmented DNA samples are hybridized to oligonucleotide probes representing coding regions of the genome, the exome, and high throughput sequenced [6]. Approximately, 85% of disease-causing mutations are expected to occur within the exome, which represents 1–2% of the whole-genome region. As a result, WES has become the NGS method of choice in multiple studies of cancer and other hereditary conditions [20]. The advantages of WES, especially in comparison with whole-genome sequencing (WGS) are multiple: costs are lower; the smaller target sequence greatly simplifies sample processing and analysis, the requirements of sample quantity are not too stringent. Furthermore, the existence of genome-wide coverage facilitates the analysis of novel candidate genes as they are uncovered, without the need to reprocess the sample.
McInerney-Leo et al. [28▪▪] tested the efficiency of two different commercial WES platforms for diagnosis of a small cohort of hereditary PPGLs: one mutation was missed by one of the platforms, but detected in the other. Also, by specifically comparing the coverage of 12 PPGL genes across reference data from five exome enrichment kits, it was noticed that only one of them showed complete coverage of all coding sequences of interest, with SDH genes showing the highest degree of variation in the depth of reads. The poor representation of some PPGL-related exons was in part due to low depth of sequence of the reference dataset used, and may be resolved by increasing the depth in actual samples. However, more problematic is the issue of incomplete coverage of the length of some exons, which should be a consideration when selecting the enrichment platform for WES-based screen. Individual PPGL exon coverage and depth is not available from other WES studies in PPGLs but this information could help in developing future guidelines and standards for WES-based screening, as discussed below.
Overall, WES-based screen is the favored platform for comprehensive, yet analytically manage able genetic screen amenable to entering the mainstream of PPGLs diagnostic testing (Table 3). However, further improvements in the efficiency of exome capture methods are needed to ensure that all target exons are represented through their entire length and at adequate depth of coverage. Enhanced alignment and base calling algorithms are also needed to ensure accuracy of the sequencing. Other technical shortcomings are discussed in a separate section, below.
Table 3.
Summary of distinguishing features of whole xome sequencing, targeted next generation sequencing, and conventional Sanger sequencing in pheochromocytomas and paragangliomas
| Feature | WES | Targeted NGS panel | Conventional testing (Sanger or MLPA) |
|---|---|---|---|
| Detection of known PPGL genes | Yesa | Yes | Yes |
| Need to process some PPGL exons separately (by conventional sequencing) | High | High | NA |
| Detection of novel genes | Yes | Nob | Nob |
| Detection of large genomic or copy number defects | Low | Yesb | Yesc |
| Fast turnaround time | Yes | Yes | Nod |
| Low costs | Yes | Yes | Noe |
| Complexity of bioinformatic analysis | High | Low | NA |
| Sequencing error rates | High | High | Low |
| Incidental findings | Yes | No | NA |
| VUS | High | High | Low |
| Performed in a stepwise manner | No | No | Yes |
| Individual lab autonomy for sequencing | No | Yes | Yes |
| Scalability | Lowf | High | NA |
MLPA, multiplex ligation-dependent probe amplification, method used to detect copy number changes in PPGL genes; NA, not applicable; PPGL, pheochromocytomas and paragangliomas; VUS, variants of unknown significance; WES, whole-exome sequencing.
Detection of some PPGL gene exons may be incomplete in current platforms.
New assay design required or use of a broad targeted panel.
By MLPA assay.
Exception when first clinically driven test identifies mutated gene.
High costs if multistep gene analysis is required.
Increase in WES scale can only occur at the expense of reduced sequencing depth per sample – not recommended.
Targeted next generation sequencing
In this approach, the sequencing analysis is limited to known genes and exons, and next generation sequencing is performed in samples amplified by PCR. Custom primers are designed to target whole or specific areas (often exons) of genes of interest. Barcodes are attached to individual samples during library generation allowing for a high degree of multiplexing, which improves the throughput of sequence processing and reduces costs. Targeted NGS has many valuable features: primers can be individually designed and adjusted to achieve similar efficiency across the gene(s), samples are sequenced at much deeper coverage (200–1000), the instrumentation is simplified and affordable by individual labs, and the analysis pipeline is straight forward and customizable. Furthermore, targeted NGS may be the only viable approach for samples with limited amounts of DNA of suboptimal quality. Rattenberry et al. [27▪▪] performed a feasibility study of nine PPGL genes in a large sample cohort and found high degree of diagnostic concordance with conventional sequencing (Table 2). However, several problems were highlighted, including sequence errors in repeat areas (instrument-biased) and the inability to multiplex exons with high GC content, which had to be analyzed separately through Sanger sequencing. Limitations in multiplexing were also noted by Welander et al. [31▪▪] using a different platform and instrumental setting, although all mutations in 18 known hereditary PPGLs were identified by their approach. Despite the relative simplicity of targeted NGS, in practice achieving optimal and uniform multiplexing of all desired exons in every sample is not straightforward. Furthermore, the number and speed with which novel genes are identified and then incorporated in the targeted screen design can pose technical and economic challenges for implementation. In targeted NGS, unlike WES, addition of new genes to existing panels requires the generation of new libraries and new sample sequencing (Table 3).
Other NGS platforms
WGS is the most comprehensive genome-wide option, as it includes noncoding regions in addition to exons. These areas are increasingly recognized as relevant for diagnosis of genetic disorders and cancers [19,37]. Additional advantages are the ability to identify gene translocations and copy number gains or losses. Major limitations of WGS are the costs, and importantly, the complexity of the bioinformatic analysis. The NIH-sponsored TCGA (The Cancer Genomic Atlas) effort in PPGLs (https://tcgadata.nci.nih.gov/tcga/tcgaCancerDetails.jsp?diseaseType=PCPG&diseaseName=Pheochromocytoma%20and%20Paraganglioma), which is at its final stages of completion, includes WGS analysis of a large sample collection. Although not meant as a clinical diagnostic tool, WGS data from TCGA is certain to provide insights into genomic alterations that could not have been detected by other NGS methodologies and will contribute to gauging the added value of WGS for diagnostic purposes in PPGLs.
RNA sequencing (RNAseq) differs from other modalities by utilizing RNA (preferentially from tumor tissue), instead of DNA, for analysis, which limits considerably its use in large scale for diagnostic purposes. However, this approach has important attributes: it provides a combination of sequence data and quantification of gene expression in a single methodology; enables an immediate view of the transcription consequences of mutations that occur at splice sites or those that involve gene fusions resulting from translocations or rearrangements, and identifies preferential allelic expression of coding variants. However, analytical pipelines are more complex than WES and targeted NGS. In PPGL, gene fusions and intrachromosomal breakpoints that may be biologically consequential were recently identified by RNAseq [32▪▪]. Further investigation of the frequency of these events will provide new insights into the biology of PPGLs and whether this approach would be of use in the diagnostic arena.
Currently, effective analysis of coding regions of target genes is likely to encompass the great majority of causative mutations in hereditary PPGLs. However, it is difficult to estimate how much has been missed by confining the analysis to exons and exon-intron boundaries. Data outside of these constraints are essentially nonexistent. As research advances, the real contribution of other defects, including large genomic gains or losses, translocations, fusions, and noncoding mutations, to PPGL pathogenesis will become better known. This information will be relevant to determine the method of choice for comprehensive testing of these disorders.
ANALYTICAL AND TECHNICAL SHORTCOMINGS
Analysis of the sequence data produced by these different NGS modalities is beyond the scope of this review, but this is clearly one of the bottlenecks for rapid implementation of NGS methods in clinical practice. Understandably, the analytical complexity of WES, RNAseq and WGS is higher than that of targeted sequencing [6]. The importance of rigorous bioinformatic analysis and interpretation standards cannot be overstated.
NGS technologies have recognizably higher raw base error rates than Sanger sequencing [5,6]. However, since its inception, technological advances in instrumentation, sample processing, and algorithms, coupled with higher depth of sequencing coverage in most study designs, have led to improved accuracy in base calling. For WES and WGS an average sequencing depth of 50–100 of bidirectional (or paired-end) reads is usually considered sufficient to detect most germline single-nucleotide variants accurately [6,38–40]. However, these conservative numbers can, and should be, increased under specific circumstances. Other technical limitations to NGS methods recognized in the setting of PPGLs, discussed above, and off-target sequencing and misalignment to homologous regions, such as paralogs or pseudogenes, can also lead to reduced sensitivity and specificity of variant detection. The rate of alignment errors have substantially decreased with longer read lengths (~50 vs. 100 bp or more in recent pipelines), higher depth of sequencing, and improved base call algorithms.
NGS exhibits much greater sensitivity and specificity for detection of substitutions than it does for other sequence changes [5,41,42]. For detection of insertions, deletions, larger copy number, and structural changes, specific analytical algorithms are required for accurate calling. Leveraging high depth of coverage of the test samples and targeted NGS designs involving whole genes instead of exon-only enrichments can significantly improve detection of these large genomic defects. Several targeted NGS screening platforms for clinical diagnosis of hereditary breast, ovarian, and colon cancer have been developed [41,43–45], in which such designs were implemented coupled with high depth of coverage and robust analytical pipelines led to successful detection of a wide range of deletion or duplication lengths. The ability to identify larger structural defects should be a goal of NGS-based screens in PPGLs because as much as 10% of the defects involving SDH and VHL can result from whole or partial gene deletions.
Beyond coding sequence mutations and large structural defects of target genes, gene inactivation mediated by epigenetic, but not genetic events (epimutations), have been recently reported in SDHC of Carney triad syndrome patients [46,47], in whom paragangliomas are associated with pulmonary chondroma and gastrointestinal stromal tumors (GIST). In these patients, the SDHC gene is hypermethylated and hypoexpressed in tumor tissues, but also in nontumoral tissues, suggesting possible mosaicism. These findings, which were considered to be primary drivers of the tumorigenesis in these patients, have not been examined more generally in other PPGL cases, but if confirmed, may indicate that all-encompassing screening of these tumors may require an expansion of the current techniques to identify hypermethylated areas on target genes.
Hence, no single platform currently fulfills the requirements of an ideal PPGL screening test (Table 3). One reasonable expectation is that an improved version of WES, with more uniform and completecoverage of all targetexons relevant to PPGL should be the more immediate goal for implementing a comprehensive primary platform for genetic testing. A separate test, WGS-based or utilizing specially designedtargeted NGS panelsor high-resolution copy number analysis, may be required for detection of larger structural defects or analysis of noncoding variants in patients for which a mutation is not identified in the first test. Furthermore, tests to detect mosaic epimutations may need to be developed if these events are found more generally in PPGLs.
DATA REPORTING
In the setting of genetic disorders, the availability of a clinical summary on the test order form is often a prerequisite to interpreting the results of NGS testing [10,11]. This is not an absolute requirement in PPGLs as the clinical diagnosis is often straightforward. However, information on family history, tumor location, recurrence, malignancy status, and existence of other conditions known to be related to PPGL-related syndromes can be invaluable to improve accuracy of diagnostic reports.
Interpreting the results of NGS, especially WES, can be more complex than conventional testing due to the massive amounts of data generated. However, on a diagnostic setting, the analysis can be restricted to the known disease genes by computational selection. Typically, appropriate filters are applied using similar criteria to those already employed in conventional clinical diagnosis to exclude common variants (for example, those occurring in less than 5 or even 1% of the general population), variants that lead to a synonymous change (exceptions are those that generate or abolish a splice site), intronic variants beyond the canonical 2-bases surrounding exons and other, context-specific filtering. The variants that remain after these filters are then classified as follows: benign, deleterious (previously reported in hereditary PPGLs, as referenced in the literature and/or online mutation databases available for various PPGL genes), potentially pathogenic variants (conserved amino acids, nonsense or frameshift), or variants of unknown clinical significance (VUS, further discussed below). The final report of a diagnostic genetic test should be the result of careful analysis and discussion with geneticists or other PPGL experts and extensive literature searches to determine the classification of variants [10,11,44]. It is important to keep in mind that despite technical improvements in design and analytical algorithms, some variants may still require confirmation by conventional sequencing due to poor coverage or to an alignment-challenging sequence context. In fact, although NGS-based tests are still under development, we believe that positive tests should be validated by Sanger sequencing before results are reported to the patient.
In the current scenario of autosomal dominantly inherited PPGLs, pathogenic mutations are expected to be represented as heterozygous variants, and thus the estimated variant frequency threshold (VFT) for heterozygous mutations should be close to 50%. However, in practice, there are instances of allelic strand sequence preference, when VFTs deviate from this pattern, as reported by Rattenberry et al. [27▪▪], and also seen in our own experience. VFT values can be variable in mosaic mutations. This is the case in EPAS1, wherein a postzygotic de-novo mutation can lead to increased risk of PPGLs, polycythemia and occasionally somatostatinomas of the duodenum [36▪▪]. Determining the risks of germline transmission of mosaic diseases can be challenging and impractical [48], therefore genetic counseling should play a dominant role, more than the genetic test itself, in the discussion of transmission risks of patients with EPAS1 mutations at tumor level.
VUSs are detected in unprecedented numbers by NGS-based screening and represent a common challenge for test reporting. However, limitations in the ability to distinguish pathogenic from nonpathogenic mutations are likely to gradually decrease, as reference databases become more complete and our knowledge of PPGLs improves, with more mutations being recurrently detected and their functional effects tested. Although there has been some debate as to whether VUSs should be reported due to the uncertainty of their value, the predominant view is that the benefits of reporting outweigh risks of, for example, not revealing variants that may be eventually proven to be pathogenic [10,20]. The approach in PPGL should follow the lead of clinical genetics, in which extensive consultation between laboratory personnel with the attending physician and medical geneticists for cases in which the diagnostic classification is uncertain takes place before the results are disclosed to patients [10,49,50]. Laboratories and attending physicians should regularly review the status of VUS cases as more research data become available. In this realm, new government-sponsored initiatives are being developed to make clinically relevant information publicly searchable [51]. Patients should be clearly informed of the significance and potential change in VUS status and are encouraged to seek regular updates from their attending physicians.
Other challenges of NGS based testing involve interpreting alternative modes of inheritance or co-occurrence of multiple variants with potential pathogenic effects in single samples. Appropriate interpretation of these findings will require further research and functional validation of novel variants. Guidelines for application of NGS to genetic diagnostics are still under development [20,49,50,52]; we believe that a framework of PPGL screening standards should be devised (Fig. 1). These recommendations should attempt to integrate these emerging guidelines with established PPGL testing routines and regulatory requirements unique to different centers and countries. The challenges imposed by the complexity of interpreting results demand the inclusion of a medical geneticist or certified genetic counselor in the testing process [10,41]. Leading organizations in the PPGL field, including the investigator-driven Pheochromocytoma Support Organization, the Endocrine Society, the European Network for the Study of Adrenal Tumors, and other worldwide associations, are encouraged to come together to develop standardized practices and policies for NGS-based tests as they begin to enter the mainstream of clinical practice in many countries. Input from various patient advocacy groups (Pheo Para Troopers, Pheo-Para Alliance, VHL Alliance, and others) should also be sought. Another relevant aspect of NGS-based testing, specifically genome wide approaches, involves ‘incidental findings’, the identification of variants unrelated to the phenotype of interest but which may have clinical significance. Reporting of these findings is an area of broad debate in NGS-related clinical applications [10,20,49,50, 53,54] and should be extended to the PPGL field. The framework devised by the clinical genetics field is an excellent starting point to initiate this discussion.
FIGURE 1.
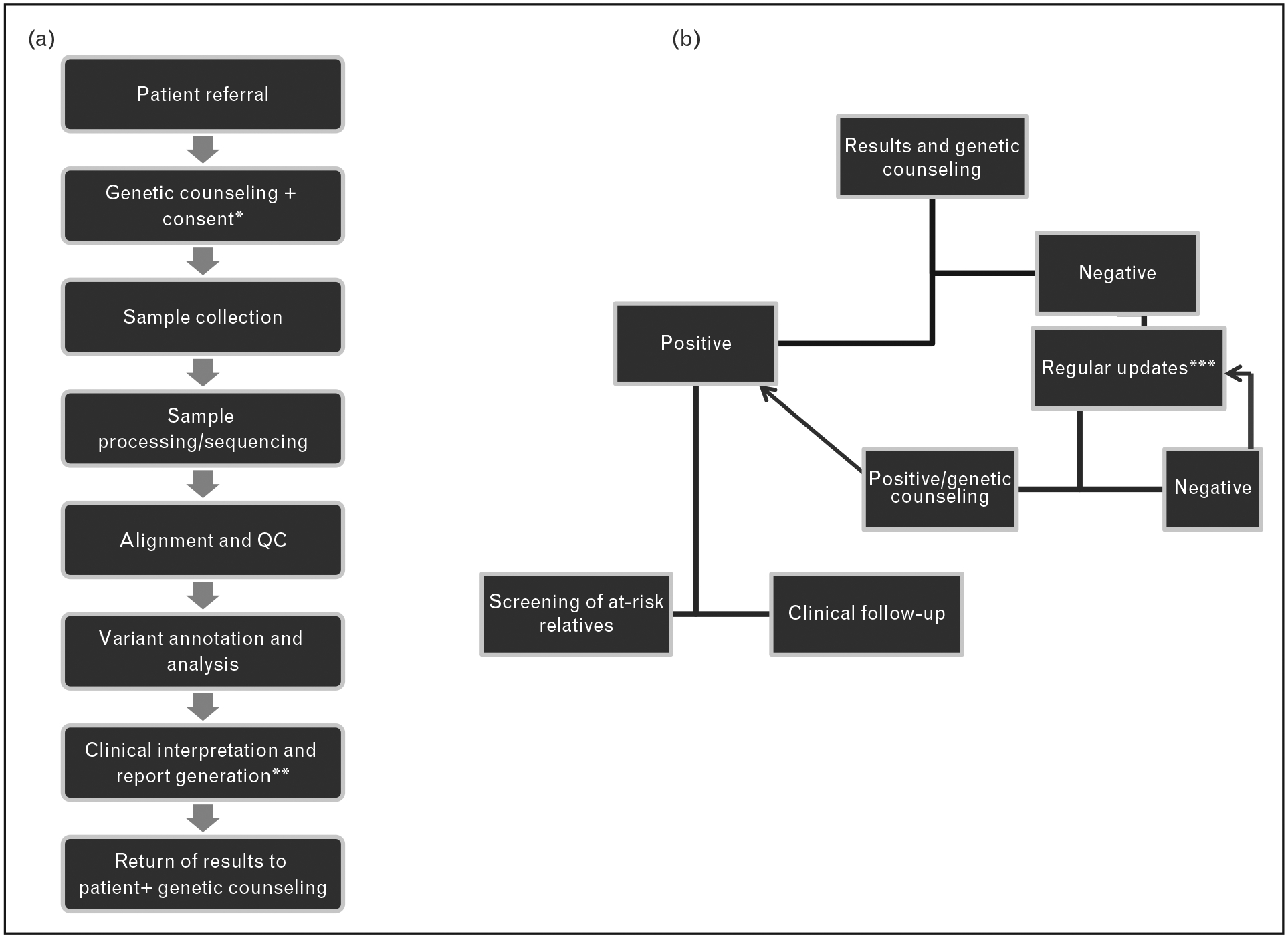
Proposed workflow of a genome-wide next-generation sequencing-based screen of patients with pheochromocytoma or paraganglioma. (a) Initial steps of the screening process involve genetic counseling and informed consent (apatient must opt in or out of ‘incidental finding’ reporting and decisions of future evaluation of the collected sequence data for future updates – see text for additional details). bIt is suggested that the final report be the consensus interpretation of physicians, researchers and clinical geneticists. (b) Results are returned to the patient at a genetic counseling session. Unquestionably, positive results follow the current route of clinical follow-up for index patient and screening of at-risk relatives. Negative results may include lack of a clearly pathogenic mutation in a known susceptibility gene or detection of variants of unknown significance (VUS). Regular updates on the status of VUS or evaluation of novel susceptibility genes from collected data are performed. If a new pathogenic variant is detected or pathogenic status of VUS is established, based on new research data, the patient will be offered genetic counseling and follow procedures for a ‘positive’ mutation carrier. If there are no changes in the genetic screening status, the process of regular updates may continue.
CONCLUSION
The emerging body of evidence in the field of NGS based genetic screen on PPGLs suggests that multiple sequencing approaches (targeted, WES, and WGS) are likely to find applications in the routine diagnostic setting.
Diagnostic panels for subsets of PPGL genes can already be found commercially in the USA. In academic centers worldwide, the transition from conventional methods to NGS is advancing at a rapid pace. Current limitations of targeted NGS and WES require that these methods are complemented by independent analysis of poorly covered gene areas and copy number analysis for a comprehensive, all-encompassing screen. Further progress in the methodology with longer sequence reads, higher-depth of sequencing, careful target primer design, barcoding and multiplexing, and the possibility of using whole-genome methods to address deletions will likely aid in overcoming the current limitations and further increasing sensitivity. Furthermore, the ability to incorporate other susceptibility targets as they are discovered and added to the list of PPGL genes offers enormous advantage to NGS-based screens, especially the genome-wide methods. As different design and platform options continue to be perfected, a consensus set of guidelines should be developed, at least in the academic setting, to fulfill basic diagnostic and quality control standards for both technical processing and interpretation of the results. These platforms would also be amenable to use in other clinically relevant applications beyond germline diagnosis, including tumor screening for detection of potentially therapeutically targetable somatic mutations.
KEY POINTS.
PPGLs are genetically heterogeneous and often inherited (40% carry a germline susceptibility mutation).
NGS technology, now broadly available and cost effective, has been successfully implemented in clinical diagnosis of multiple inherited disorders.
Pilot studies have shown feasibility of both WES and targeted NGS for diagnosis of inherited pheochromocytomas and paragangliomas.
Technical fine-tuning, including improved and uniform coverage of all target exons and detection of large copy number changes will be required to improve sensitivity and specificity of NGS for its use in pheochromocytoma and paraganglioma diagnosis.
A consensus set of guidelines and standards for NGS-based testing in PPGL should be developed in the near future.
Acknowledgements
We thank our funding agencies for support of our work.
We apologize to colleagues whose work was not cited due to space limitations or oversight.
Financial support and sponsorship
P.L.M.D. is funded by the Department of Defense (D.O.D.-C.D.M.R.P.), Cancer Prevention and Research Institute of Texas (C.P.R.I.T.) and the Voelcker Fund.
Footnotes
Conflicts of interest
P.L.M.D. is a nonpaid member of the executive committee and a founding member of the nonprofit P.R.E.S.S.O.R. (Pheochromocytoma Research Organization).
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪of special interest
▪▪of outstanding interest
- 1.Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer 2014; 14:108–119. [DOI] [PubMed] [Google Scholar]
- 2.Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol 2015; 11:101–111. [DOI] [PubMed] [Google Scholar]
- 3.Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014; 99:1915–1942. [DOI] [PubMed] [Google Scholar]
- 4.Robson ME, Storm CD, Weitzel J, et al. American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol 2010; 28:893–901. [DOI] [PubMed] [Google Scholar]
- 5.Ku CS, Cooper DN, Polychronakos C, et al. Exome sequencing: dual role as a discovery and diagnostic tool. Ann Neurol 2012; 71:5–14. [DOI] [PubMed] [Google Scholar]
- 6.Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 2011; 12:745–755. [DOI] [PubMed] [Google Scholar]
- 7.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science 2008; 322:881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sikkema-Raddatz B, Johansson LF, de Boer EN, et al. Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum Mutat 2013; 34:1035–1042. [DOI] [PubMed] [Google Scholar]
- 9.Jacob HJ, Abrams K, Bick DP, et al. Genomics in clinical practice: lessons from the front lines. Sci Transl Med 2013; 5:194cm5. [DOI] [PubMed] [Google Scholar]
- 10.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013; 369:1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. J Am Med Assoc 2014; 312:1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jafri M, Maher ER. The genetics of phaeochromocytoma: using clinical features to guide genetic testing. Eur J Endocrinol 2012; 166:151–158. [DOI] [PubMed] [Google Scholar]
- 13.Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res 2012; 44:328–333. [DOI] [PubMed] [Google Scholar]
- 14.Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer 2011; 18:R253–R276. [DOI] [PubMed] [Google Scholar]
- 15.Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 2005; 23:8812–8818. [DOI] [PubMed] [Google Scholar]
- 16.Erlic Z, Rybicki L, Peczkowska M, et al. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res 2009; 15:6378–6385. [DOI] [PubMed] [Google Scholar]
- 17.Cascon A, Lopez-Jimenez E, Landa I, et al. Rationalization of genetic testing in patients with apparently sporadic pheochromocytoma/paraganglioma. Horm Metab Res 2009; 41:672–675. [DOI] [PubMed] [Google Scholar]
- 18.Mannelli M, Castellano M, Schiavi F, et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab 2009; 94:1541–1547. [DOI] [PubMed] [Google Scholar]
- 19.Gonzaga-Jauregui C, Lupski JR, Gibbs RA. Human genome sequencing in health and disease. Annu Rev Med 2012; 63:35–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacArthur DG, Manolio TA, Dimmock DP, et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014; 508:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 2011; 43:663–667. [DOI] [PubMed] [Google Scholar]
- 22.Qi XP, Ma JM, Du ZF, et al. RET germline mutations identified by exome sequencing in a Chinese multiple endocrine neoplasia type 2A/familial medullary thyroid carcinoma family. PLoS One 2011; 6:e20353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.▪.Toledo RA, Qin Y, Srikantan S, et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr RelatCancer 2013; 20:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of somatic EPAS1 mutations in PPGLs.
- 24.▪▪.Crona J, Delgado Verdugo A, Maharjan R, et al. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J Clin Endocrinol Metab 2013; 98:E1266–E1271. [DOI] [PubMed] [Google Scholar]; Identification of somatic HRAS mutations in PPGLs.
- 25.▪▪.Letouze E, Martinelli C, Loriot C, et al. SDH mutations establish a hyper-methylator phenotype in paraganglioma. Cancer Cell 2013; 23:739–752. [DOI] [PubMed] [Google Scholar]; Identification of FH mutation as a susceptibility gene in PPGL.
- 26.▪.Crona J, Verdugo AD, Granberg D, et al. Next-generation sequencing in the clinical genetic screening of patients with pheochromocytoma and paraganglioma. Endocr Connect 2013; 2:104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of known PPGL gene mutations in tumor samples from unclassified PPGLs.
- 27.▪▪.Rattenberry E, Vialard L, Yeung A, et al. A comprehensive next generation sequencing-based genetic testing strategy to improve diagnosis of inherited pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2013; 98:E1248–E1256. [DOI] [PubMed] [Google Scholar]; Direct testing of targeted NGS for clinical diagnosis of seven PPGL genes.
- 28.▪▪.McInerney-Leo AM, Marshall MS, Gardiner B, et al. Whole exome sequencing is an efficient and sensitive method for detection of germline mutations in patients with phaeochromcytomas and paragangliomas. Clin Endocrinol (Oxf) 2014; 80:25–33. [DOI] [PubMed] [Google Scholar]; This study provides comparison of WES and conventional sequencing in PPGL samples and of PPGL gene performance across exome capture platforms.
- 29.Casey R, Garrahy A, Tuthill A, et al. Universal genetic screening uncovers a novel presentation of an SDHAF2 mutation. J Clin Endocrinol Metab 2014; 99:E1392–E1396. [DOI] [PubMed] [Google Scholar]
- 30.▪.Clark GR, Sciacovelli M, Gaude E, et al. Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab 2014; 99:E2046–E2050. [DOI] [PubMed] [Google Scholar]; Identification of germline FH mutations in PPGL.
- 31.▪▪.Welander J, Andreasson A, Juhlin CC, et al. Rare germline mutations identified by targeted next-generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2014; 99:E1352–E1360. [DOI] [PMC free article] [PubMed] [Google Scholar]; Targeted NGS of 12 PPGL genes for screening of PPGL tumor samples.
- 32.▪▪.Flynn A, Benn D, Clifton-Bligh R, et al. The genomic landscape of phaeo-chromocytoma. J Pathol 2014. [Google Scholar]; Identification of multiple somatic mutations and rearrangements in PPGLs using WES and RNAseq
- 33.▪▪.Fishbein L, Khare S, Wubbenhorst B, et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun 2015; 6:6140. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of somatic ATRX mutations in metastatic PPGls with or without known germline mutations.
- 34.▪▪.Castro-Vega LJ, Letouze E, Burnichon N, et al. Multiomics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. NatCommun 2015; 6:6044. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of somatic mutations and copy number changes of multiple novel cancer genes in PPGLs.
- 35.Cao M, Sun F, Huang X, et al. Analysis of the inheritance pattern of a Chinese family with phaeochromocytomas through whole exome sequencing. Gene 2013; 526:164–169. [DOI] [PubMed] [Google Scholar]
- 36.▪▪.Buffet A, Smati S, Mansuy L, et al. Mosaicism in HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab 2014; 99:E369–E373. [DOI] [PubMed] [Google Scholar]; Identification of mosaic EPAS1 mutations in polycythemia-paraganglioma syndrome.
- 37.Garraway LA, Lander ES. Lessons from the cancer genome. Cell 2013; 153:17–37. [DOI] [PubMed] [Google Scholar]
- 38.Ku CS, Cooper DN, Ziogas DE, et al. Research and clinical applications of cancer genome sequencing. Curr Opin Obstet Gynecol 2013; 25:3–10. [DOI] [PubMed] [Google Scholar]
- 39.Ku CS, Cooper DN, Wu M, et al. Gene discovery in familial cancer syndromes by exome sequencing: prospects for the elucidation of familial colorectal cancer type X. Mod Pathol 2012; 25:1055–1068. [DOI] [PubMed] [Google Scholar]
- 40.ten Bosch JR, Grody WW. Keeping up with the next generation: massively parallel sequencing in clinical diagnostics. J Mol Diagn 2008; 10:484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walsh T, Lee MK, Casadei S, et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci U S A 2010; 107:12629–12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013; 31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pritchard CC, Smith C, Salipante SJ, et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J Mol Diagn 2012; 14:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walsh T, Casadei S, Lee MK, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A 2011; 108:18032–18037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brownstein Z, Friedman LM, Shahin H, et al. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in Middle Eastern families. Genome Biol 2011; 12:R89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Killian JK, Miettinen M, Walker RL, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med 2014; 6:268ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haller F, Moskalev EA, Faucz FR, et al. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocr Relat Cancer 2014; 21:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet 2013; 14:307–320. [DOI] [PubMed] [Google Scholar]
- 49.Rehm HL, Bale SJ, Bayrak-Toydemir P, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med 2013; 15:733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gargis AS, Kalman L, Berry MW, et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol 2012; 30:1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lander ES. Cutting the Gordian Helix: Regulating Genomic Testing in the Era of Precision Medicine. N Engl J Med 2015; 372:1185–1186. [DOI] [PubMed] [Google Scholar]
- 52.Worthey EA. Analysis and annotation of whole-genome or whole-exome sequencing-derived variants for clinical diagnosis. Curr Protoc Hum Genet 2013;79:Unit 9.24. [DOI] [PubMed] [Google Scholar]
- 53.Kim SC, Jung Y, Park J, et al. A high-dimensional, deep-sequencing study of lung adenocarcinoma in female never-smokers. PLoS One 2013; 8:e55596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bowdin S, Ray PN, Cohn RD, Meyn MS. The genome clinic: a multidisciplinary approach to assessing the opportunities and challenges of integrating genomic analysis into clinical care. Hum Mutat 2014; 35:513–519. [DOI] [PubMed] [Google Scholar]